

Abstract
Peripheral injection of bacterial endotoxin lipopolysaccharide (LPS) induces brain mRNA expression of the proinflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor-α and the cytokine-responsive immediate-early gene IκBα. Peripheral LPS also increases levels of plasma glucocorticoids. Whether the induction of IκBα mRNA in the brain after peripheral LPS injection is caused by the feedback action of glucocorticoids has not been determined. In this study, we examined the mRNA expression of IκBα and IL-1β in the rat brain by in situ hybridization histochemistry. Injection of the glucocorticoid agonist dexamethasone induced IκBα mRNA expression in the brain in a pattern identical to that of LPS injection. LPS but not dexamethasone also induced IL-1β mRNA expression. Pretreatment with dexamethasone 30 min before LPS injection enhanced the expression of IκBα mRNA in the brain in a dose-dependent manner. Immobilization of rats for 2 hr (which raises glucocorticoid levels) also induced IκBα mRNA expression without inducing the expression of IL-1β. Brain IκBα expression induced by peripheral LPS injection was attenuated by pretreatment of rats with the glucocorticoid antagonist RU-486. Finally, increased expression of IL-1β mRNA in the brain was observed at 4 hr after peripheral LPS injection in adrenalectomized rats compared with sham-operated rats. These results reveal that in the brain glucocorticoids selectively induce IκBα mRNA expression, which serves as a negative feedback mechanism for peripheral LPS-induced synthesis of proinflammatory cytokines. Such an inhibitory control mechanism may be important for preventing prolonged expression of proinflammatory cytokines in the brain after peripheral immune challenge.
Keywords: glucocorticoid, IκBα, lipopolysaccharide, brain, inflammatory cytokines, interleukin-1
We have shown previously that peripheral lipopolysaccharide (LPS) injection induces the expression of IκBα mRNA in the brain (Quan et al., 1997). IκBα is an immediate-early gene that can be induced by LPS and proinflammatory cytokines (Miyamoto and Verma, 1995). The induction of IκBα mRNA first occurred at structures of the blood–brain barrier (BBB; choroid plexus, blood vessels, meninges, and circumventricular organs) and later propagated to cells within the brain parenchyma. This pattern of IκBα mRNA expression is consistent with the idea that proinflammatory cytokines are produced by cells of the BBB after peripheral immune challenge and then act on cells inside the BBB (Quan, 1998). Actions of proinflammatory cytokines on cells of the CNS are known to relay signals of peripheral immune challenge to the CNS (Rothwell et al., 1996).
Another potent stimulator of IκBα mRNA expression is the adrenal glucocorticoids. It has been shown that glucocorticoids can induce IκBα expression in vitro, which serves as an immunosuppressive mechanism by binding to nuclear factor-κB (NF-κB) and consequently blocking NF-κB-mediated synthesis of many immunoregulatory genes including proinflammatory cytokines (Auphan et al., 1995; Scheinman et al., 1995). It is well known that peripheral LPS injection activates the hypothalamus—pituitary–adrenal axis and can result in a dramatic increase in the plasma levels of glucocorticoids (Smith et al., 1994; Whiteside et al., 1999). Therefore, the induction of IκBα mRNA expression in the brain after peripheral LPS injection may be caused, at least in part, by glucocorticoids.
The objective of this study is to determine whether IκBα mRNA can be induced in the brain by glucocorticoids and, if so, whether the induction of IκBα by glucocorticoids is a mechanism for inhibiting proinflammatory cytokine expression in the brain after peripheral immune challenge.
MATERIALS AND METHODS
Animals. Male Sprague Dawley rats (175–200 gm; Harlan Sprague Dawley, Indianapolis, IN) were group housed (three per cage) with food and water available ad libitum in a light- (6:00 A.M. to 6:00 P.M.) and temperature-controlled environment (20–22°C). All procedures were approved by the Ohio State University Animal Care and Use Committee.
Experimental procedures. The animals were divided into 17 experimental groups (n = 4 in each group). For fresh-frozen tissue collection, all animals were killed by decapitation, and their brains were removed for analysis. Experiment 1 comprised two groups. Animals were injected intraperitoneally with PBS (group 1) or 10 mg/kg dexamethasone (DEX; Sigma, St. Louis, MO; group 2). They were killed at 2 hr after the injection. In experiment 2, groups 3–6 were used. Group 3 served as home-cage controls. They received an intraperitoneal injection of 0.5 ml of dimethylsulfoxide (DMSO; the vehicle for RU-486) and were killed 2.5 hr later. Group 4 was injected with DMSO 30 min before they were subjected to immobilization stress for 2 hr and killed immediately after the stress. Animals were immobilized by placing the head through plastic restrainers and taping each limb to a plastic platform. Group 5 was injected with RU-486 (50 mg/kg, i.p.; Sigma; dissolved in DMSO) 30 min before animals were stressed for 2 hr by immobilization. Group 6 received a RU-486 injection and was killed at 2.5 hr after the injection to control for the effects of RU-486. In experiment 3, groups 7–9 were used. Group 7 was injected intraperitoneally with 100 μg/kg LPS (serotype 055:B5; Sigma). Group 8 was injected with 100 μg/kg LPS and 10 mg/kg DEX. Group 9 was injected with 100 μg/kg LPS and 50 mg/kg DEX. In experiment 4, groups 10 and 11 were used. Group 10 was injected with 1 mg/kg LPS and received an injection of 0.5 ml of DMSO 30 min before the LPS injection. Group 11 was injected with 1 mg/kg LPS and received 50 mg/kg RU-486 30 min before the LPS injection. In experiment 5, groups 12 and 13 were used. These animals were adrenalectomized surgically under anesthesia (sodium pentobarbital, 50 mg/kg). The adrenalectomy was performed bilaterally after an incision was made. The approximate duration of surgery was 25 min. Group 12 was given a PBS injection, group 13 was injected with 1 mg/kg LPS immediately after the surgery, and both groups were killed at 2 hr after the injections. In experiment 6, groups 14–17 were used. Groups 14 and 15 were sham-operated, and groups 16 and 17 were adrenalectomized. Immediately after surgery, they were injected with 1 mg/kg LPS. Groups 14 and 16 were killed at 2 hr after the injection, and groups 15 and 17 were killed at 4 hr after the LPS injection. Animals that received any surgical procedure were anesthetized throughout the entire procedure and remain anesthetized until they were killed.
Brain section collection. Brains were removed immediately after decapitation, frozen by immersion in 2-methylbutane at −30°C, and stored at −70°C before sectioning. They were then cryostat-cut to 15-μm-thick coronal sections and thaw-mounted onto pretreated adhesive slides (Superfrost Slides; Fisher Scientific, Houston, TX), dried, and stored at −80°C until further processing. Levels collected were the organum vasculosum of the lamina terminalis (−0.02 mm relative to bregma), the subfornical organ (SFO; −0.92 mm), the central nucleus of the amygdala containing also the arcuate nucleus and median eminence (−3.3 mm), and the area postrema containing the nucleus of the solitary tract (−13.7 mm) (Paxinos and Watson, 1986).
In situ hybridization. The in situhybridization protocols were performed as described previously for ribonucleotide (cRNA) probes (Whitfield et al., 1990; Quan et al., 1997). First, tissue sections were processed by fixation with 4% formaldehyde solution, acetylation with 0.25% acetic anhydride in 0.1m triethanolamine-HCl, pH 8.0, solution, dehydration with ethanol, and delipidation with chloroform.
Second, the antisense probes directed against the full-length (1.05 kb) rat IκBα cDNA inserted into the pBluescript plasmid (generously provided by Dr. Rebecca Taub, University of Pennsylvania) were transcribed by the use of the Riboprobe System (Promega, Madison, WI) with T7 RNA polymerase and α-35S-UTP (specific activity > 1000 Ci/mmol; New England Nuclear, Boston, MA) after linearization with BamHI restriction enzyme (Promega). To control for the specificity of the probe, sense probes of rat IκBα were also generated by transcribing the same plasmid with T3 RNA polymerase after linearization with HindIII restriction enzyme (Promega). To generate antisense probes for interleukin-1β (IL-1β), a DNA fragment containing 500 bp of the IL-1β mRNA sequence was generated by PCR amplification of the rat IL-1β cDNA that had been inserted into the pET plasmid (generously provided by Dr. Ronald Hart, State University of New Jersey). The two PCR primers flanked the IL-1β sequence between bases 443 and 952, and the reverse primer was attached with a T7 promoter sequence. A 500 nucleotide antisense ribonucleotide (cRNA) probe was then transcribed from this DNA fragment with T7 RNA polymerase and35S-UTP. Radiolabeled probes were diluted in the riboprobe hybridization buffer and applied to brain sections (500,000 cpm/section). After overnight incubation at 55°C in a humidified chamber, slides containing brain sections were washed first in 20 μg/ml RNase solution and then in 2× SSC and 0.2× SSC (55 and 60°C, respectively) solutions to reduce nonspecific binding of the probe. The slides were then dehydrated with ethanol and air-dried for autoradiography.
Autoradiography. Slides and 14C plastic standards containing known amounts of radioactivity (American Radiolabeled Chemicals, St. Louis, MO) were placed in x-ray cassettes, apposed to film (BioMax MR; Eastman Kodak, Rochester, NY) for 4–8 d (slides from the same experiment were exposed for the same amount of time), and developed in an automatic film developer (X-OMAT; Eastman Kodak).
Data analysis. Autoradiographic film images of brain sections and standards were digitized on a Macintosh computer-based image analysis system with IMAGE software (Wayne Rasband, Research Services Branch, National Institute of Mental Health, Bethesda, MD). Light transmittance through the film was measured by outlining the structure on the television monitor. A density-slice function was applied to each structure to select densities greater than film background and thus measured transmittance that was confined to the cellular sources of the radioactivity. The density so obtained was used to represent the relative amount of mRNA expression. The intensity of mRNA labeling is expressed in the unit of counts per minute per milligram of plastic. The experiments were conducted sequentially, and the results between different experiments were not compared. Statistical analysis was done by ANOVA followed by post hocStudent's t test.
Combined immunohistochemistry and hybridization histochemistry. Two cell type-specific antibodies were used to determine the phenotypes of IκBα mRNA-producing cells: anti-GFAP (ICN) and anti-Iba1 (generously donated by Dr. Y. Imai, Department of Neurochemistry, National Institute of Neuroscience, Tokyo, Japan). These antibodies specifically mark astrocytes and microglia (Ohsawa et al., 1997), respectively. For double-label experiments, rats were adrenalectomized and injected with 1 mg/kg LPS. Two hours after the injection, animals were perfusion-fixed with 300 ml of 4% paraformaldehyde in 0.1 m phosphate buffer. After the brains were removed, they were post-fixed in 4% paraformaldehyde in 0.2 m phosphate buffer overnight at 4°C and then treated in 30% sucrose solution overnight. Brains were then snap-frozen in cold 2-methylbutane for 10 sec. Brain sections were cut on a cryostat from these samples. To achieve precise cellular localization of IκBα mRNA labeling, in situ hybridization of IκBα mRNA was performed with digoxygenin-labeled antisense riboprobes and was visualized by the conventional alkaline phosphatase-catalyzed color reaction. The immunocytochemical procedures were performed on these sections after in situ hybridization using anti-GFAP (1:200) and anti-Iba1 (1:500) and were visualized by the conventional avidin–biotin immunoperoxidase protocol. The in situhybridization signal appears in blue color, and the immunocytochemical labeling appears in brown color.
RESULTS
Representative film autoradiographs from experiment 1 are shown in Figure 1. Sections from the levels of the SFO and paraventricular nuclei of the hypothalamus (PVN) are shown because they represent the features of the IκBα mRNA expression pattern throughout the entire brain. IκBα mRNA expression in PBS-injected control animals was scarcely detectable throughout the brain except in the hippocampus and piriform cortex where variable levels of constitutive expression of IκBα mRNA were observed in neurons (data not shown). Injection of DEX (10 mg/kg, i.p) induced widespread expression of IκBα mRNA throughout the brain at 2 hr. The highest expression levels were found in the circumventricular organs, choroid plexus, meninges, and PVN. That the induced expression of IκBα did not occur in neuronal cells of the brain was concluded because the mRNA labeling was found only in small cells (<8 μm in diameter; data not shown). Quantitative analysis of the density of IκBα mRNA reveals significant elevation at both the SFO (more than a fivefold increase over control value) and PVN (threefold increase) in DEX-injected animals (see Fig. 5A).
Fig. 1.

Representative film autoradiographs at the levels of the SFO (A, C) andPVN (B, D) show patterns of IκBα mRNA hybridization in animals killed at 2 hr after either saline (PBS; A, B) or dexamethasone (DEX; 10 mg/kg; C, D) injection. Scale bar, 0.5 cm.
Fig. 5.

Means and SEMs of mRNA levels for IκBα in the SFO and PVN from experiments 1–4 (see Figs. 1-4) are shown [n = 4; *,p < 0.05 by t test; comparisons were made between experimental groups and control groups (open bars in each graph)]. Veh, Vehicle.
Representative film autoradiographs from experiment 2 are compiled in Figure 2. Compared with home-cage control animals, animals receiving immobilization stress for 2 hr had elevated expression levels of IκBα mRNA in the brain. The increased expression of IκBα mRNA was completely blocked by treating the animals with the glucocorticoid antagonist RU-486 30 min before the immobilization stress. Quantitative analysis of these results is shown (see Fig. 5B). Injection of RU-486 alone did not induce any IκBα mRNA expression (data not shown).
Fig. 2.
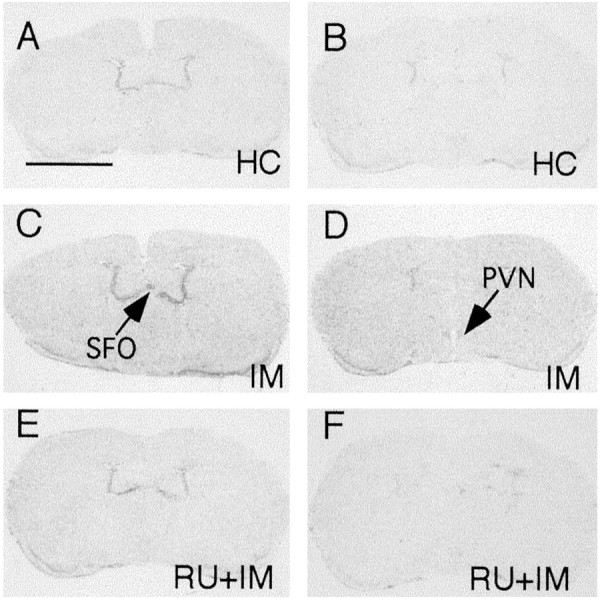
Representative film autoradiographs at the levels of the SFO (A, C, E) andPVN (B, D, F) show patterns of IκBα mRNA hybridization in home-cage (HC) control animals (A, B), in animals killed at the end of 2 hr of immobilization (IM) stress (C, D), and in IM animals that received 50 mg/kg RU-486 30 min before stress (RU+IM; E, F). Scale bar, 0.5 cm.
In experiment 3, injection of 100 μg/kg LPS induced the same patterns of IκBα mRNA expression that were induced by DEX injection (Fig.3). Injection of 100 μg/kg LPS together with 10 mg/kg DEX (Fig. 3, LPS+D10) significantly increased the expression levels of IκBα mRNA over those induced by LPS alone. Injection of 100 μg/kg LPS together with 50 mg/kg DEX (Fig. 3,LPS+D50) induced the highest levels of IκBα mRNA expression observed in experiment 3. Quantitative analysis of IκBα mRNA expression in this experiment is shown (see Fig.5C).
Fig. 3.
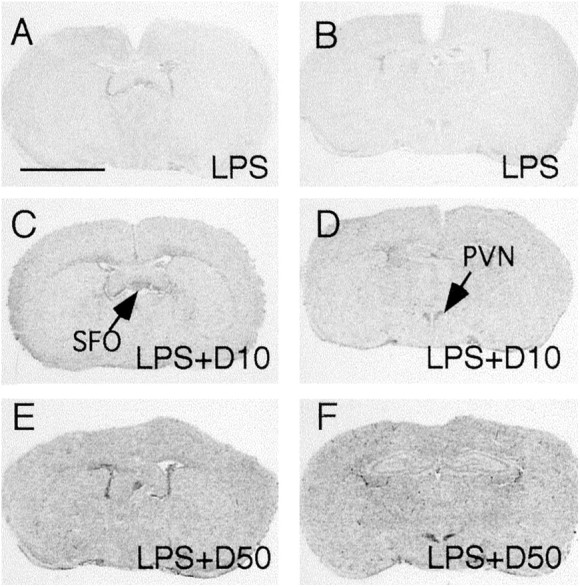
Representative film autoradiographs at the levels of the SFO (A, C, E) andPVN (B, D, F) show patterns of IκBα mRNA hybridization in animals killed at 2 hr after they received an intraperitoneal injection of 100 μg/kg LPS (LPS; A, B), in animals killed at 2 hr after they were injected with 100 μg/kg LPS + 10 mg/kg DEX (LPS+D10; C, D), and in animals killed at 2 hr after they were injected with 100 μg/kg LPS + 50 mg/kg DEX (LPS+D50; E, F). Scale bar, 0.5 cm.
In experiment 4, high-dose LPS (1 mg/kg) injection induced strong IκBα mRNA expression throughout the brain, and pretreatment with 50 mg/kg RU-486 significantly attenuated the levels of IκBα expression in the brain (Figs. 4,5D). This dose of LPS injection also induced IκBα mRNA expression in the brain parenchyma of anesthetized adrenalectomized animals in experiment 5 (Fig.6A), whereas IκBα mRNA was not induced in anesthetized adrenalectomized animals that received a PBS injection (data not shown). Representative high-magnification microphotographs show that IκBα mRNA expression in the brain colocalizes with microglial cells (Fig.6B) and endothelial cells (Fig. 6C). No convincing double labeling of IκBα mRNA and GFAP was found (Fig.6D).
Fig. 4.

Representative film autoradiographs at the levels of the SFO (A, C) andPVN (B, D) show patterns of IκBα mRNA hybridization in animals killed at 2 hr after they received an intraperitoneal injection of DMSO + 1 mg/kg LPS [LPS (hi); A, B] and in animals killed at 2 hr after they were injected with 50 mg/kg RU-486 + 1 mg/kg LPS [RU+LPS (hi); C, D]. Scale bar, 0.5 cm.
Fig. 6.

Representative bright-field microphotographs show labeling of IκBα mRNA in individual cells in the brain parenchyma of adrenalectomized animals killed at 2 hr after they received an intraperitoneal injection of 1 mg/kg LPS. A, Low-magnification microphotograph shows single-labeled IκBα mRNA-expressing cells (arrowheads) in the brain.B, Colocalization of IκBα mRNA expression and microglial immunostaining (arrow) is shown.C, Labeled IκBα mRNA-expressing cells (blue color) in endothelial cells (arrows; microglia were labeled by brown color) are shown.D, Labeled IκBα mRNA-expressing (arrow) cells do not colocalize with GFAP-stained (brown color) astrocytes. Scale bars: A, 50 μm; B–D, 10 μm.
Neither DEX nor immobilization stress induced any detectable expression of IL-1β mRNA in the brain (data not shown). Injection of LPS at 100 μg/kg or 1 mg/kg induced IL-1β mRNA at the meninges, blood vessels, and circumventricular organs (CVOs) at 2 hr after the injection as we reported previously (Quan et al., 1998). Neither RU-486 nor adrenalectomy affected the LPS-induced expression of IL-1β at this time point (data not shown). At 4 hr after LPS injection, the IL-1β mRNA expression was significantly increased in the CVOs and in areas within and adjacent to the PVN in anesthetized adrenalectomized animals compared with the anesthetized sham-operated animals (experiment 6; Fig. 7). Densitometric measurements of the IL-1β hybridization signal reveal a significant increase, 69 ± 6% at the SFO (p < 0.05,t test) and 84 ± 7% (p < 0.05, t test) at the PVN, in anesthetized adrenalectomized animals compared with anesthetized sham-operated animals.
Fig. 7.
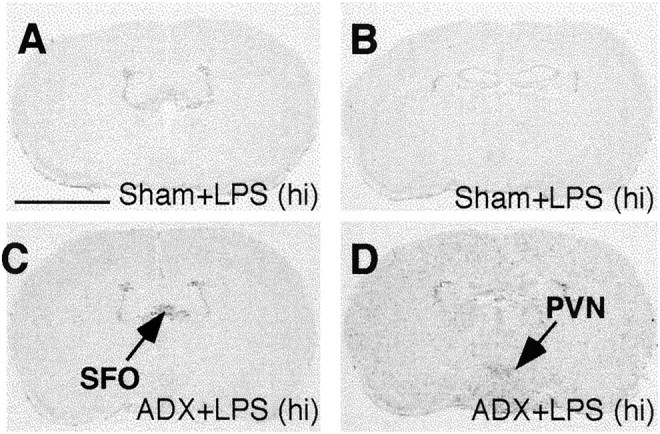
Representative film autoradiographs at the levels of the SFO and PVN show patterns of IL-1β mRNA hybridization in animals killed at 4 hr after they received an intraperitoneal injection of 1 mg/kg LPS. Sections from sham-operated (A, B) and adrenalectomized (ADX; C, D) animals are shown. Scale bar, 0.5 cm.
DISCUSSION
The results of the present study show that injection of the exogenous glucocorticoid agonist DEX induces the expression of IκBα mRNA in non-neuronal cells of the brain. The expression of IκBα mRNA in the brain was also induced by immobilization stress, which elevates endogenously produced glucocorticoids (Nemeth et al., 1977). Administration of DEX enhanced the expression of IκBα mRNA induced by peripheral LPS injection, whereas administration of the glucocorticoid antagonist RU-486 attenuated LPS-induced IκBα mRNA expression. DEX and immobilization stress did not induce IL-1β expression in the brain. Peripheral LPS injection also induced IκBα mRNA expression in adrenalectomized animals. Expression of IL-1β in the brain induced by peripheral LPS was increased in adrenalectomized animals at 4 hr, but not at 2 hr, after LPS injection. Taken together, these data show that glucocorticoids can induce IκBα expression in the brain and suggest that induction of IκBα expression may be a negative feedback control mechanism for the induction of proinflammatory cytokine synthesis by peripheral immune challenge.
The fact that we were unable to detect IL-1β mRNA expression byin situ hybridization after 2 hr of immobilization stress (data not shown) should not be interpreted as indicating that IL-1β expression in the brain is not inducible by stress. Inconsistent results have been reported regarding whether stress can induce IL-1β production in the brain. Using in situ hybridization,Yabuuchi et al. (1996) showed that stress produced by subcutaneous injection of formalin induces IL-1β mRNA expression in the hypothalamus. Plata-Salaman et al. (2000), on the other hand, using reverse transcription-PCR did not find increased expression of IL-1β mRNA in the brain of predator-stressed rats. A report by Minami et al. (1991) showed that immobilization stress induces a transient increase in IL-1β mRNA expression in the hypothalamus. Analyzing protein expression, Nguyen et al. (2000) found that IL-1β is induced in the brain of rats after they received inescapable shock. Therefore, it seems that the detection of IL-1 expression in the brain after stress depends on the severity and nature of the stress, the timing of brain sample collection relative to the end of stress, and the sensitivity of the assay method. In the present study, because neither DEX nor immobilization stress induced IL-1β mRNA expression in the brain, the results do indicate that increased glucocorticoids are not an inducer of brain IL-1β expression.
The results of the present study are at variance with a report (Unlap and Jope, 1997) showing that DEX administration attenuated NF-κB DNA-binding activity without altering IκBα levels in the brain. The IκBα levels in that study were estimated by measuring the amount of cytosolic IκBα protein in the cortex and hippocampus. It is known, however, that free IκBα protein is intrinsically unstable and rapidly degraded unless it associates with NF-κB (Rice and Ernst, 1993). In addition, we found variable constitutive expression of IκBα mRNA in neurons of the piriform cortex and hippocampus (data not shown). Therefore, measurement of IκBα protein in the cortex and hippocampus may not be sensitive to functional changes in IκBα in the brain. The results of the present study, on the other hand, are consistent with in vitrostudies of Scheinman et al. (1995) and Auphan et al. (1995) that showed that glucocorticoids activate the transcription of IκBα mRNA. These studies also found that transcriptional activation of IκBα was primarily manifested at the mRNA level, not at the protein level.
The induction of IκBα mRNA expression in the brain after immobilization was completely blocked by the administration of the glucocorticoid antagonist RU-486 (Fig. 5B), suggesting that IκBα induction in this experiment was mediated by glucocorticoids. In contrast, LPS-induced IκBα in the brain was only attenuated by RU-486 (Fig. 5D). This result is not caused by an insufficient dose of the RU-486 treatment because LPS injection induced IκBα mRNA expression even in adrenalectomized animals. In addition, this dose of RU-486 has been used to block fully many glucocorticoid-mediated immune suppressions during viral infection (Sheridan et al., 1998). Therefore, mediators other than glucocorticoids are partly responsible for IκBα induction in the brain by LPS. Further analysis of LPS-injected adrenalectomized animals found induced IκBα mRNA expression in both endothelial and microglial cells. This result is important because it shows that in the absence of increased production of glucocorticoids, which can cross the BBB, peripheral LPS is able to induce signal molecules inside the BBB to act on the IκBα-expressing microglia. We have suggested previously that inflammatory cytokines produced by cells of the BBB are secreted inside the BBB as a mechanism that relays peripheral immune signal to the brain parenchyma (Quan et al., 1997). In agreement with this idea, Nadeau et al. (2000) showed recently that tumor necrosis factor-α acting in the brain parenchyma induces IκBα expression in microglia.
High doses (10–50 mg/kg) of the glucocorticoid agonist DEX were administered in this study to ensure that DEX can act on cells inside the BBB. Cole et al. (2000) have shown that DEX injected at doses <10 μg/kg only affects glucocorticoid receptors outside of the BBB. That the induction of IκBα mRNA in the brain by the high-dose DEX injection truly mimicked the action of endogenous glucocorticoid was supported by our result that IκBα mRNA was similarly induced by immobilization stress. It has been reported that there is a >10-fold increase in plasma glucocorticoid levels in rats after similar LPS injection (Whiteside et al., 1999) and a 3–4-fold increase after immobilization stress (Al-Mohaisen et al., 2000). Therefore, it is possible that the induction of IκBα in the brain by glucocorticoids only occurs when the levels of plasma glucocorticoids are highly elevated.
Induction of IL-1β mRNA expression was enhanced at 4 hr, but not at 2 hr, after peripheral injection of 1 mg/kg LPS in anesthetized adrenalectomized animals. We have shown previously that injection of the same dose of LPS induces a peak IL-1β mRNA expression in the CVOs at 2 hr after the injection and that the induced mRNA levels rapidly decline thereafter (Quan et al., 1998). The present results suggest that glucocorticoids do not affect the initial induction of IL-1β mRNA expression by LPS but rather inhibit further induction of IL-1β mRNA after its peak expression. It has been shown that, different from peripheral tissue, the production of inflammatory cytokines by cells in the brain is not countered by sufficient production of anti-inflammatory cytokines (Wong et al., 1997). Chronic infusion of IL-1 into the parenchyma of the CNS results in chronic expression of IL-1β in the brain (Plata-Salaman and ffrench-Mullen, 1992). Therefore, one can envision that after IL-1 expression is triggered by peripheral immune challenge, chronic IL-1 expression in the brain may be precipitated if no inhibitory mechanisms come into play. The present results suggest that the induction of glucocorticoids and the consequent induction of IκBα may contribute significantly to prevent prolonged expression of proinflammatory cytokines after acute peripheral immune challenge.
The present results do not exclude other modalities by which glucocorticoids might inhibit the synthesis of proinflammatory cytokines in the brain. Interestingly, although glucocorticoids are known to inhibit the gene expression of numerous proinflammatory cytokines, the binding of glucocorticoid receptors to the glucocorticoid-responsive element (GRE) is often not the mechanism for the inhibitory effects of the glucocorticoids (Barnes, 1998). Besides IκB induction, it has been shown that glucocorticoids can stimulate glucocorticoid receptors to bind directly to NF-κB in cells of the brain (Unlap and Jope, 1997), thereby inhibiting the transcription of proinflammatory cytokines. These two mechanisms allow glucocorticoids to inhibit the transcription of NF-κB-controlled genes without the presence of GRE in their promoter. In addition, Nguyen et al. (1998)have shown that the IL-1β protein level increases in the brain in adrenalectomized and acutely stressed animals. We did not observe increased IL-1β mRNA expression in adrenalectomized animals that received immobilization stress (data not shown). Therefore, it is likely that IL-1β protein expression in the brain after stress may also be modulated by glucocorticoids post-transcriptionally. If these results are taken together, glucocorticoids appear to inhibit the synthesis of proinflammatory cytokines in the brain by multiple mechanisms. The results of the present study demonstrate that induction of IκBα expression by glucocorticoids serves as a negative feedback mechanism for brain proinflammatory cytokine expression induced by peripheral immune challenge.
Footnotes
This study is supported by Ohio State University and the Intramural Program of the National Institute of Mental Health.
Correspondence should be addressed to Dr. Ning Quan, 2214 Postle Hall, 305 West 12th Avenue, Department of Oral Biology, Ohio State University, Columbus, OH 43210. E-mail: quan.14@osu.edu.
REFERENCES
- 1.Al-Mohaisen M, Cardounel A, Kalimi M. Repeated immobilization stress increases total cytosolic glucocorticoid receptor in rat liver. Steroids. 2000;65:8–15. doi: 10.1016/s0039-128x(99)00076-8. [DOI] [PubMed] [Google Scholar]
- 2.Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosuppression by glucocorticoids: inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- 3.Barnes PJ. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin Sci (Colch) 1998;94:557–572. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 4.Cole MA, Kim PJ, Kalman BA, Spencer RL. Dexamethasone suppression of corticosteroid secretion: evaluation of the site of action by receptor measures and functional studies. Psychoneuroendocrinology. 2000;25:151–167. doi: 10.1016/s0306-4530(99)00045-1. [DOI] [PubMed] [Google Scholar]
- 5.Minami M, Kuraishi Y, Yamaguchi T, Nakai S, Hirai Y, Satoh M. Immobilization stress induces interleukin-1β mRNA in the rat hypothalamus. Neurosci Lett. 1991;123:254–256. doi: 10.1016/0304-3940(91)90944-o. [DOI] [PubMed] [Google Scholar]
- 6.Miyamoto S, Verma IM. Rel/NF-κB/I κB story. Adv Cancer Res. 1995;66:255–292. [PubMed] [Google Scholar]
- 7.Nadeau S, Rivest S. Role of microglial-derived tumor necrosis factor in mediating CD14 transcription and nuclear factor κ B activity in the brain during endotoxemia. J Neurosci. 2000;20:3456–3468. doi: 10.1523/JNEUROSCI.20-09-03456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemeth S, Vigas M, Kvetnansky R, Orlicky, Mikulaj J. The effect of repeated immobilization on the level of plasma corticosterone and on the activity of several liver enzymes in rats. Endokrinologie. 1977;69:87–93. [PubMed] [Google Scholar]
- 9.Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF. Exposure to acute stress induces brain interleukin-1β protein in the rat. J Neurosci. 1998;18:2239–2246. doi: 10.1523/JNEUROSCI.18-06-02239.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen KT, Deak T, Will MJ, Hansen MK, Hunsaker BN, Fleshner M, Watkins LR, Maier SF. Timecourse and corticosterone sensitivity of the brain, pituitary, and serum interleukin-1beta protein response to acute stress. Brain Res. 2000;859:193–201. doi: 10.1016/s0006-8993(99)02443-9. [DOI] [PubMed] [Google Scholar]
- 11.Ohsawa K, Imai Y, Nakajima K, Kohsaka S. Generation and characterization of a microglial cell line, MG5, derived from a p53-deficient mouse. Glia. 1997;21:285–298. [PubMed] [Google Scholar]
- 12.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; Orlando, FL: 1986. [DOI] [PubMed] [Google Scholar]
- 13.Plata-Salaman CR, ffrench-Mullen JM. Intracerebroventricular administration of a specific IL-1 receptor antagonist blocks food and water intake suppression induced by interleukin-1β. Physiol Behav. 1992;51:1277–1279. doi: 10.1016/0031-9384(92)90321-r. [DOI] [PubMed] [Google Scholar]
- 14.Plata-Salaman CR, Ilyin SE, Turrin NP, Gayle D, Flynn MC, Bedard T, Merali Z, Anisman H. Neither acute nor chronic exposure to a naturalistic (predator) stressor influences the interleukin-1β system, tumor necrosis factor-α, transforming growth factor-β1, and neuropeptide mRNAs in specific brain regions. Brain Res Bull. 2000;51:187–193. doi: 10.1016/s0361-9230(99)00204-x. [DOI] [PubMed] [Google Scholar]
- 15.Quan N. Brain cytokine expression in response to peripheral infection. In: Oomura Y, Hori T, editors. Taniguchi symposia on brain science, Vol 21. Karger; New York: 1998. pp. 125–141. [Google Scholar]
- 16.Quan N, Whiteside M, Kim L, Herkenham M. Induction of inhibitory factor κBα mRNA in the central nervous system after peripheral lipopolysaccharide administration: an in situ hybridization histochemistry study in the rat. Proc Natl Acad Sci USA. 1997;94:10985–10990. doi: 10.1073/pnas.94.20.10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quan N, Whiteside M, Herkenham M. Time course and localization patterns of interleukin-1β messenger RNA expression in brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience. 1998;83:281–293. doi: 10.1016/s0306-4522(97)00350-3. [DOI] [PubMed] [Google Scholar]
- 18.Rice NR, Ernst MK. In vivo control of NF-kappa B activation by I kappa B alpha. EMBO J. 1993;12:4685–4695. doi: 10.1002/j.1460-2075.1993.tb06157.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rothwell NJ, Luheshi G, Toulmond S. Cytokines and their receptors in the central nervous system: physiology, pharmacology, and pathology. Pharmacol Ther. 1996;69:85–95. doi: 10.1016/0163-7258(95)02033-0. [DOI] [PubMed] [Google Scholar]
- 20.Scheinman RI, Cogswell PC, Lofquist AK, Baldwin AS., Jr Role of transcriptional activation of I kappa B alpha in mediation of immunosuppression by glucocorticoids. Science. 1995;270:283–286. doi: 10.1126/science.270.5234.283. [DOI] [PubMed] [Google Scholar]
- 21.Sheridan JF, Dobbs C, Jung J, Chu X, Konstantinos A, Padgett D, Glaser R. Stress-induced neuroendocrine modulation of viral pathogenesis and immunity. Ann NY Acad Sci. 1998;840:803–808. doi: 10.1111/j.1749-6632.1998.tb09618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith T, Hewson AK, Quarrie L, Leonard JP, Cuzner ML. Hypothalamic PGE2 and cAMP production and adrenocortical activation following intraperitoneal endotoxin injection: in vivo microdialysis studies in Lewis and Fischer rats. Neuroendocrinology. 1994;59:396–405. doi: 10.1159/000126683. [DOI] [PubMed] [Google Scholar]
- 23.Unlap MT, Jope RS. Dexamethasone attenuates NF-kappa B DNA binding activity without inducing I kappa B levels in rat brain in vivo. Brain Res Mol Brain Res. 1997;45:83–89. doi: 10.1016/s0169-328x(96)00240-9. [DOI] [PubMed] [Google Scholar]
- 24.Whiteside MB, Quan N, Herkenharn M. Induction of pituitary cytokine transcripts by peripheral lipopolysaccharide. J Neuroendocrinol. 1999;11:115–120. doi: 10.1046/j.1365-2826.1999.00297.x. [DOI] [PubMed] [Google Scholar]
- 25.Whitfield HJ, Jr, Brady LS, Smith MA, Mamalaki E, Fox RJ, Herkenham M. Optimization of cRNA probe in situ hybridization methodology for localization of glucocorticoid receptor mRNA in rat brain: a detailed protocol. Cell Mol Neurobiol. 1990;10:145–157. doi: 10.1007/BF00733641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wong M-L, Bongiorno PB, Rettori V, McCann SM, Licinio J. Interleukin (IL) 1β, IL-1 receptor antagonist, IL-10, and IL-13 gene expression in the central nervous system and anterior pituitary during systemic inflammation: pathophysiological implications. Proc Natl Acad Sci USA. 1997;94:227–232. doi: 10.1073/pnas.94.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yabuuchi K, Maruta E, Minami M, Satoh M. Induction of interleukin-1 beta mRNA in the hypothalamus following subcutaneous injections of formalin into the rat hind paws. Neurosci Lett. 1996;207:109–112. doi: 10.1016/0304-3940(96)12505-2. [DOI] [PubMed] [Google Scholar]