

Abstract
It is widely accepted that peripheral injury increases spinal inducible cyclooxygenase (COX-2) expression and prostaglandin E2 (PGE2) formation as key mediators of nociceptive sensitization. Here, we used inducible nitric oxide synthase (iNOS) gene-deficient (iNOS−/−) mice to determine the contribution of iNOS-derived nitric oxide (NO) to this process. iNOS−/− mice exhibited reduced thermal hyperalgesia after zymosan injection. Spinal NO and PGE2 formation both remained at baseline levels, in contrast to wild-type (wt) mice. In wt mice reduced hyperalgesia similar to that seen in iNOS−/− mice was induced by local spinal, but not by systemic treatment with the iNOS inhibitorl-NIL, suggesting that the reduced heat sensitization in iNOS−/− mice was attributable to the lack of spinal rather than peripheral iNOS. Two additional observations indicate that the antinociceptive effects of iNOS inhibition are dependent on a loss of stimulation of PG synthesis. First, intrathecal injection of the COX inhibitor indomethacin, which exerted pronounced antinociceptive effects in wt mice, was completely ineffective in iNOS−/− mice. Second, treatment with the NO donor RE-2047 not only completely restored spinal PG production and thermal sensitization in iNOS−/− mice but also its sensitivity to indomethacin. In both types of mice induction of thermal hyperalgesia was accompanied by similar increases in COX-1 and COX-2 mRNA expression. The stimulation of PG production by NO therefore involves an increase in enzymatic activity, rather than an alteration of COX gene expression. These results indicate that NO derived from spinal iNOS acts as a fast inductor of spinal thermal hyperalgesia.
Keywords: nitric oxide, inducible nitric oxide synthase, zymosan, thermal hyperalgesia, paw edema, spinal microdialysis, l-NIL, RE-2047, prostaglandins, cyclooxygenase
Acute tissue damage is often accompanied by the fast development of hyperalgesia and allodynia (Andrew and Greenspan, 1999). Both peripheral mechanisms at the site of injury and central processes particularly in the spinal cord contribute to this phenomenon. Prostaglandins (PGs) (Yaksh and Malmberg, 1993;Brune, 1994) as well as nitric oxide (NO) (Lawand et al., 1997) are produced in response to tissue damage peripherally and centrally. Whereas PGs are generally accepted to play a dominant role in nociceptive sensitization (Bley et al., 1998), the role of NO is less clear. Also some authors claim of an anti-nociceptive action of NO (Goettl and Larson, 1996; Hamalainen and Lovick, 1997), most favor a pro-nociceptive activity (Malmberg and Yaksh, 1993; Kawabata et al., 1994; Chen and Levine, 1999). Part of this controversy may arise from the existence of three different isoenzymes of NO synthase (NOS) (Gonzalez-Hernandez and Rustioni, 1999), which may have distinct effects on nociception, and from the lack of specific inhibitors for these different isoforms. In the CNS including the spinal cord, NO is thought to be primarily produced by the neuronal isoform of NOS (nNOS) (Downen et al., 1999). However, endothelial NOS is also found in neurons (Wei et al., 1999) and under certain conditions, e.g., after tissue damage (Sinz et al., 1999), inducible NOS (iNOS) can be expressed in the CNS (Meller et al., 1994; Lee and Brosnan, 1996;Barker et al., 1998). Therefore, all three NO-generating isoenzymes appear to be possible sources of NO in the CNS.
To define the role of NO in spinal processing of nociceptive information more clearly, we investigated nociceptive sensitization in genetically modified mice deficient in the iNOS isoenzyme (iNOS−/− mice). For this purpose, we adapted the Hargreaves model of thermal hyperalgesia (Hargreaves et al., 1988) to mice and developed a technique for spinal microdialysis in mice. We showed that iNOS−/− mice exhibit a delay in thermal sensitization and lack the rise in spinal PG production, which is normally observed in response to peripheral nociceptive stimulation.
MATERIALS AND METHODS
Assessment of thermal hyperalgesia. A modified Hargreaves plantar test (Hargreaves et al., 1988) was used to assess thermal hyperalgesia in mice. A metal grid bottom instead of a glass floor in the observation cage and 10.5 × 13.0 × 4.5 cm boxes to restrict animal movement were used. Zymosan A (Sigma, Deisenhofen, Germany) was injected subcutaneously into the plantar side of right hindpaws, and paw withdrawal latencies (PWL) were determined on exposure of the paws to a defined thermal stimulus were measured using a commercially available apparatus (Hargreaves Test Ugo Basile Biological Research Apparatus, Comerio, Italy).
Mice were kept in the test cages for 1 d to allow accommodation. On day 2, each mouse was tested several times to gain baseline PWL. On day 3 thermal hyperalgesia was assessed for 8 hr starting 15 min after subcutaneous zymosan injection (3.0 mg/ml in 20 μl of PBS, containing NaCl 8 gm/l, Na2HPO4 2.9 gm/l, KCl 0.2 gm/l, KH2 PO4 0.24 gm/l). Experiments were performed in air conditioned rooms (22°C) between 12 A.M. and 8 P.M. In some experiments the assessment of thermal hyperalgesia was continued for 7 d (one measurement per day). Right (injected) and left (noninjected) paws were measured alternately in intervals of 5–10 min. At 1 hr intervals, PWL were averaged.
Under control conditions, PWL were identical in wt (10.40 ± 0.35 sec; n = 40) and iNOS−/−mice (10.25 ± 0.20 sec;n = 18). In an initial set of experiments, zymosan (20 μl) was tested in concentrations of 12.0, 6.0, or 3.0 mg/ml. Zymosan injection caused a dose-dependent increase in areas [PWL × observation interval [seconds × hours]; calculated using the linear trapezoidal rule for each mouse] between right and left hindpaw PWL from 0.17 ± 1.75 (PBS) to 10.10 ± 1.81 (3.0 mg/ml) to 17.47 ± 2.55 (6.0 mg/ml) and to 23.30 ± 2.22 (12.0 mg/ml). Injection of vehicle did not affect nociceptive behavior in any of the experiments. For all subsequent tests an intermediate zymosan concentration of 3.0 mg/ml was used for the detection of pro-nociceptive and anti-nociceptive effects.
Animals. Male iNOS−/− mice weighing 26.7 (22.9–36.8) gm [mean (range)] with the genetic background of C57/Bl6 mice and male C57/Bl6 mice (wt) weighing 21.2 (19.6–26.1) gm were used for all experiments. Breeding pairs of iNOS −/− mice (Laubach et al., 1995) were obtained from The Jackson Laboratory (Bar Harbor, ME). INOS −/− mice show no major abnormalities (Laubach et al., 1995; MacMicking et al., 1995; Wei et al., 1995). Mice were housed under a 12 hr light/dark cycle and cared for according to the guidelines of the Institutional Animal Care and Use Committee. Water and food were given ad libitum.
Application of drugs. All drugs were dissolved in isotonic, physiological solvents. Indomethacin was dissolved as described elsewhere (Shen and Winter, 1977). Briefly, for a 10 mmsolution, 17.9 mg of indomethacin, 15.3 mg of Na2CO3 × 10 H2O, and 5 ml of artificial CSF (ACSF) consisting of (in mm): 151.1 Na+, 2.6 K+, 0.9 Mg2+, 1.3 Ca2+, 122.7 Cl−, 21.0 mmHCO3−, 2.5 mmHPO4−, and 3.5 dextrose, pH 7.20, was used. Intraperitoneal drug or vehicle (PBS) injections (50 μl) were given into the lower left abdominal quadrant. Intrathecal injections were performed according to Hylden and Wilcox (1980). In brief, mice were anesthetized with isoflurane, and 5 μl of drug containing solutions or vehicle (ACSF) were injected into the spinal subarachnoid space between L5 and L6 30 min before the administration of zymosan using a 26 gauge needle mated to a 10 μl Hamilton syringe. Mice showing neurological abnormalities were excluded. We added 1% black ink (Pelikan, Hannover, Germany) to all solutions used for intrathecal injections. Proper intrathecal injections were verified by inspection of slices of the spinal cord after lumbar laminectomy.
Tissue samples. After completion of the Hargreaves test, mice were killed under CO2 anesthesia by intracardial puncture and decapitation. Hindpaws and the thoracolumbar segment of the spinal cord were removed for morphological and biochemical analyses. After intra-articular disconnection at the ankle joint, right and left hindpaws were weighed. Differences in paw weight (Δ PW) were used to measure edema formation.
Spinal microdialysis. The dialysis tube was constructed from a Cuprophan hollow fiber (outer diameter, 216 μm) with a 36 kDa molecular weight cutoff (Hospal, Nuernberg, Germany). This fiber was connected at one side to a polyethylene (PE) tube (inner diameter, 0.4 mm; outer diameter, 0.8 mm) using cyanacrylate glue (number 448 Stabiloplast; Renfert, Chemietechnik, GmbH). A small metal spike was inserted into the other side and fixed with a quick-setting cyanacrylate glue (UHU, Buehl, Germany).
Mice were deeply anesthetized with isoflurane (1.5–2.0% vol; Abbott GmbH, Wiesbaden, Germany) and placed on an electronically controlled heating pad (37°C; CMA/Microdialysis, Stockholm, Sweden). After cutaneous incision of the thoracolumbar region, superficial and deep dorsal lumbar fascia were slit, and muscle tissue was removed from the vertebrae T12–L1. The dialysis tube was introduced through the intervertebral joints between the thoracic and lumbar segments. All accessible parts of the dialysis tube were covered with cyanacrylate glue. After cutting the spike, the free end of the hollow fiber was connected to another PE tube using the same cyanacrylate adhesive. Afterward mice were surgically sewed and permanently anesthetized with urethane (∼750 mg/kg, i.p.).
The PE tube was connected to a microdialysis pump (CMA 100; CMA/Microdialysis), and ACSF was perfused at a flow rate of 3 μl/min. ACSF was bubbled with carbogen (5% CO2 and 95% O2) and kept at 37°C during the experiments. Samples were collected at 30 min intervals in Eppendorf cups kept on ice and finally stored at −70°C for subsequent analysis of PGE2, as well as NO2− and NO3−(NOx) as the breakdown products of NO. After a washout period of 30 min, baseline samples were collected for 1.5 hr every 30 min. Thereafter, 20 μl of zymosan (3.0 mg/ml) was injected subcutaneously into the right hindpaw, and samples were collected for another 4 hr. After mice were killed, the proper placement of microdialysis tubes was verified by perfusion with black ink (Pelikan), and subsequent microscopic examination.
NOx and PGE2measurements. NO production was assessed indirectly by determining NO degradation products after reduction of NO3− to NO2− with nitrate reductase (Cytochrome; Sigma) by the Griess reaction-dependent method described elsewhere (Green et al., 1981). We incubated 50 μl of perfusion samples with 50 μl of modified Griess reagent (Sigma), and the absorption was recorded at 540 nm (Flow Titertec, Multiscan Plus MK11; ICN Biochemicals, Frankfurt, Germany).
Tissue samples obtained from the spinal cord and from the hindpaws were weighed, transferred into 99.5% methanol (1 mg of tissue to 10 μl of methanol), and shaken for 2 hr at room temperature. We transferred 100 μl of the supernatants into Eppendorf cups, and methanol was evaporated. The remaining pellet was dissolved in 100 μl of enzyme immunoassay (EIA) buffer. We incubated 20 μl of the microdialysis perfusion samples with 80 μl of enzyme immunoassay (EIA) buffer for PGE2 measurements.
All further steps were performed as described in the Cayman Chemical Company PGE2 EIA Kit - Monoclonal, calibration range: 1000–7.8 pg/ml (Cayman Chemicals, Ann Arbor, MI). Measurement was completed by using an ELISA reader (Flow Titertec, Multiscan Plus MK11; ICN Biochemicals) with an absorbency maximum at 405 nm.
RT-PCR. Immediately after preparation, tissue samples of the right hindpaw and spinal cord segment L4 were snap frozen with 800 μl of lysis buffer (Qiagen, Hilden, Germany) in liquid nitrogen, stored at −70°C, and homogenized with a microshredder. RNA was isolated using the RNeasy-kit (Qiagen). Real-time RT-PCR was used to determine expression of mouse-actin, COX-1, and COX-2 mRNA. TaqMan probes were labeled at the 5′ end with the reporter dye molecule FAM (6-carboxy-fluorescein; emission λ, 518 nm), and at the 3′ end with the quencher dye molecule TAMRA (6-carboxy-tetramethyl-rhodamine, emission λ, 582 nm). In addition, the 3′ end was phosphorylated to prevent extension of the probe during PCR.
We used 25 μl of reaction mixture, which contained 2 μl of template, 5 μl of 10 × PCR buffer (100 mm Tris, pH 8.3, 500 mm KCl), 3 μl of Mn(OAc)2, 0.3 μl of dATP, dCTP, dGTP, and dUTP, 0.1 μl of Diagonal DNA polymerase, 0.05 μl of each primer, 0.05 μl of the TaqMan probe, and 14.55 μl of sterile water for the PCR. For RT-PCR and detection of fluorescence signals the ABI Prism 7700 SDS analytical thermal cycler (Perkin-Elmer, Foster City, CA) was used. Thermal cycle conditions were: 2 min at 50°C, 30 min 60°C, 5 min 95°C, and then 45 cycles (15 sec at 94°C, 1 min at 60°C).
The emission of the reporter dye was compared with that of the quenching dye during PCR amplification and the increase of fluorescence signals, ΔRn, was calculated as: ΔRn = (Rn+) − (Rn−) [Rn+= ratio of reporter and quencher dye at any given time during a reaction; Rn− = ratio of reporter and quencher baseline emission]. Referring to a standard RNA a threshold was defined indicating the exponential phase of the fluorescent signal increase. The CT value, which correlates to the number of RNA copies present at the start of PCR according to the references of PE Applied Biosystems (User Bulletin 2; ABI PRISM 7700 Sequence Detection System, 1997) was determined as the amplification cycle number when the ΔRn of a sample intersected this threshold value. The quantity of mRNA was given as ΔCT, which was calculated as ΔCT = CTgene of interest − CTβ-actin mRNA.
iNOS mRNA transcripts were analyzed in spinal cord tissue (thoracolumbar segment) homogenates according to a protocol previously described in detail (Deckert-Schluter et al., 1995). Primer sequences were described previously (Deckert-Schluter et al., 1998; compare Table1) and were synthesized by TIB MOLBIOL (Berlin, Germany). Briefly, total mRNA was extracted as described above. After reverse transcription of mRNA using the Superscript RT kit (Life Technologies, Frederick, MD) PCR reactions were conducted in a final volume of 25 μl of using the AmpliTaq Gold kit from PE Biosystems (Weiterstadt, Germany). Conditions are: 1 × 2 min 95°C; 45 × 1 min 95°C, 1 min 58°C, 1 min 72°C; 1 × 7 min 72°C. The PCR product was visualized by electrophoresis on 1.5% agarose gels.
Table 1.
Primer sequences
| β-actin forward | -TCACCCACACTGTGCCCATCTACGA |
| β-actin reverse | -GGATGCCACAGGATTCCATACCCA |
| β-actin probe | -(6FAM)TATGCTC(TAMRA)TCCCTCACGCCATCCTGCGT |
| COX-2 forward | -TTTGTTGAGTCATTCACCAGACAGAT |
| COX-2 reverse | -CAGTATTGAGGAGAACAGATGGGATT |
| COX-2 probe | -(6FAM)CTACCATGGTC(TAMRA)TCCCCAAAGATAGCATCA |
| iNOS forward | -TCACGCTTGGGTCTTGTTCACT |
| iNOS reverse | -TTGTCTCTGGGTCCTCTGGTCA |
Forward and reverse primer as well as probe sequences for real time RT-PCR of β-actin and COX-2. Forward and reverse primer sequences for RT-PCR of iNOS.
Statistical analysis. Results are expressed as the mean ± SEM. The statistical analyses of the behavioral experiments were performed using one-way ANOVA followed by post hocBonferroni test (the α level was set to 0.05) or by a Student'st test (p < 0.05 was considered statistically significant).
RESULTS
iNOS-derived NO facilitates development of spinal hyperalgesia
A modification of the Hargreaves test was used to assess thermal hyperalgesia in wt and iNOS−/− mice. Both types of mice exhibited a time-dependent sensitization to noxious heat in response to plantar zymosan injection. At the time point of maximum thermal hyperalgesia, PWL were reduced from ∼10 sec under control conditions to ∼3 sec after zymosan injection. In wt mice, however, maximum sensitization occurred after 3 hr, whereas that of iNOS−/− mice was not reached until 8 hr (Fig. 1). Wild type-like thermal sensitization could be restored in iNOS−/− mice by treatment with the NO-donor RE-2047 (3-methyl-N-nitroso-sydnone-5-imine; a generous gift from Prof. Rehse, Department of Pharmaceutical Chemistry, Berlin, Germany) (Rehse and Ciborski, 1995) in a dose-dependent manner. At the highest dose of RE-2047 hyperalgesia in iNOS−/− mice was indistinguishable from that of wt mice, confirming that the reduced thermal sensitization in iNOS−/− mice was caused by the absence of NO generated by iNOS (Fig. 2).
Fig. 1.

Time course of zymosan-induced thermal hyperalgesia in iNOS−/− mice. Development with time of PWL in iNOS−/− (n = 12; open circles) and wt mice (n = 12; black circles) after intraperitoneal administration of PBS and after injection of 3.0 mg/ml zymosan subcutaneously into the right hindpaw. Left PWLs (black lines) were similar for both lines of mice. Different PWL (*p < 0.05) indicate reduced heat sensitization after zymosan injection in iNOS−/− mice. Data are expressed as means ± SEM.
Fig. 2.

NO donor restores thermal hyperalgesia in iNOS−/− mice. Intraperitoneal injection of RE-2047, a metabolic NO-donor, in doses increasing from 5 (black diamonds) to 15 (gray diamonds) and 45 mg/kg (open diamonds) antagonized the restored heat sensitization of iNOS−/− mice. The difference of heat sensitization between wt (black circles) and iNOS−/− mice (open circles) after intraperitoneal administration of PBS is drawn as dotted lines.
To determine whether the lack of iNOS in the peripheral tissue or in the CNS was responsible for reduced early thermal hyperalgesia, we used the selective iNOS inhibitor l-NIL (l-N6-(I-iminoethyl)-lysine hydrochloride; Alexis Biochemicals, Gruenberg, Germany; Moore et al., 1994) and took advantage of its inability to penetrate the blood–brain barrier in significant amounts (U. Werner, B. Layh, K. Brune, H. and Guehring, unpublished observations). In this series of experiments we compared the effects of l-NIL after systemic peripheral (intraperitoneal) and local spinal (intrathecal) injection in wt mice. After intraperitoneal injection at doses ranging from 15.0 to 135.0 mg/kg l-NIL effectively reduced edema and PGE2 production in the zymosan-injected paw, but had no effect on thermal hyperalgesia (Fig.3A, Table2). Increases in the weight of the injected paws were 35.2 ± 3.0% and 48.7 ± 2.1% inl-NIL and vehicle-treated mice, respectively. A similar difference was found for the PGE2 content in the zymosan-injected paws, which was 25.6 ± 5.0 pg/mg inl-NIL treated and 54.6 ± 2.7 pg/mg in control mice. As expected, l-NIL was completely ineffective in iNOS−/− mice. Both paw edema formation and PGE2 production inl-NIL-treated mice were not significantly different from those found in iNOS−/− mice (edema, 30.25 ± 4.14%; PGE2, 23.3 ± 1.9 pg/mg).
Fig. 3.
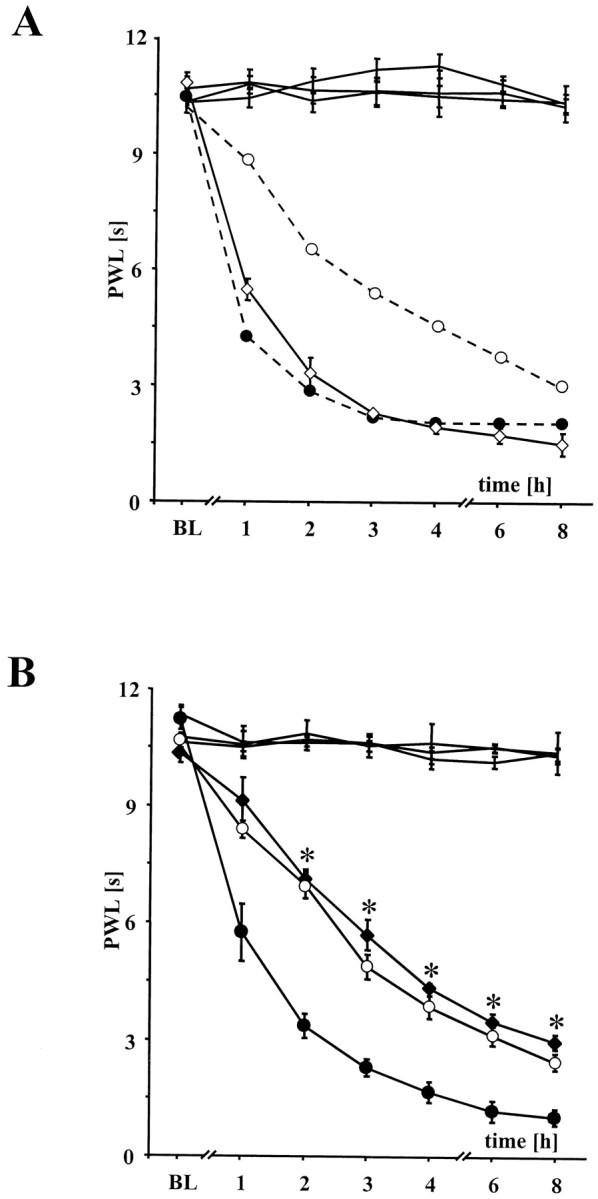
Effects of peripheral versus spinal injection ofl-NIL on thermal hyperalgesia in wt mice. A,In wt mice (open diamonds) the selective iNOS inhibitorl-NIL administered intraperitoneally was without any effect even in doses ranging from 5 to 135 mg/kg. The difference of heat sensitization between wt (black circles) and iNOS−/− mice (open circles) after intraperitoneal administration of PBS is drawn as dotted lines. Data are expressed as mean ± SEM. B, In contrast to Adrugs were administered intrathecally. Intrathecal administration ofl-NIL (0.1 μm) to wt mice (black diamonds) reduced heat sensitization significantly to the level of iNOS−/− mice. Differences in heat sensitization between wt (black circles) and iNOS−/− mice (open circles) were also observed after acute intrathecal injection of ACSF (5 μl). Data are expressed as means ± SEM.
Table 2.
Thermal hyperalgesia after intraperitoneal administration of drugs
| Mouse | Drug mg/kg (i.p.) | Hindpaw stimulus | Area (0–8 hr) | Area (8–168 hr) |
|---|---|---|---|---|
| wt | PBS | Zymosan | 63.0 ± 2.03 | 593.7 ± 56.20 |
| iNOS −/− | PBS | Zymosan | 40.8 ± 2.89 | 586.1 ± 132.21 |
| wt | l-NIL 15 | Zymosan | 60.5 ± 2.53 | |
| wt | l-NIL 45 | Zymosan | 67.6 ± 4.10 | |
| wt | l-NIL 135 | Zymosan | 65.5 ± 3.01 | |
| wt | Diclofenac 5 | Zymosan | 34.8 ± 1.32 | |
| iNOS −/− | RE 5 | Zymosan | 47.3 ± 1.66 | |
| iNOS −/− | RE 15 | Zymosan | 54.7 ± 0.84 | |
| iNOS −/− | RE 45 | Zymosan | 60.7 ± 0.83 | |
| wt | PBS | PBS | 5.0 ± 1.12 | |
| iNOS −/− | PBS | PBS | 3.0 ± 2.50 |
Areas [sec*hr] between right and left hindpaws calculated from PWL in two different time intervals from 0 to 8 hr and from 8 to 168 hr in wt (wt) and iNOS-gene deficient (iNOS−/−) mice. Peripheral inflammation was induced with zymosan, and PBS was used as a control. Data are expressed as means ± SEM.
By contrast, when l-NIL (0.1 μm; 5 μl) was injected intrathecally the development of zymosan-induced sensitization was significantly retarded compared to vehicle (ACSF; 5 μl)-treated animals and indistinguishable from that of iNOS−/− mice (Fig.3B). The latter finding indicates that the lack of spinal rather than of peripheral iNOS was responsible for the observed delay in thermal hyperalgesia. Interestingly, neitherl-NIL nor the nonspecific NOS inhibitorl-NAME (NG-nitro-l-arginine methyl ester; Alexis Biochemicals, Gruenberg, Germany; Amir and English, 1991) affected heat sensitization in iNOS−/− mice (Table3), indicating that nNOS and eNOS did not significantly contribute to zymosan-induced thermal hyperalgesia.
Table 3.
Thermal hyperalgesia after intrathecal administration of drugs
| Mouse | Drug (i.t.) | Area (0–8 hr) |
|---|---|---|
| wt | ACSF | 62.0 ± 1.52 |
| iNOS −/− | ACSF | 45.1 ± 1.26 |
| wt | l-NIL 0.1 μm | 42.1 ± 0.57 |
| iNOS −/− | l-NIL 0.1 μm | 43.0 ± 2.96 |
| iNOS −/− | l-NAME 0.1 μm | 39.5 ± 1.27 |
| wt | Indo 0.1 mm | 60.4 ± 2.22 |
| wt | Indo 1.0 mm | 56.4 ± 2.96 |
| wt | Indo 10.0 mm | 46.3 ± 3.64 |
| iNOS −/− | Indo 0.1 mm | 42.4 ± 0.88 |
| iNOS −/− | Indo 1.0 mm | 42.1 ± 1.58 |
| iNOS −/− | Indo 10.0 mm | 42.8 ± 1.17 |
| iNOS −/− | RE 45 mg/kg, i.p. Indo 10.0 mm, i.t. | 41.5 ± 2.44 |
Areas [sec*hr] between right and left hindpaws calculated from PWL in the time interval from 0 to 8 hr in wt (wt) and iNOS-gene deficient (iNOS−/−) mice. In every mouse peripheral inflammation was induced with zymosan. Indo, Indomethacin. Data are expressed as means ± SEM.
To investigate whether the facilitating action of iNOS on thermal hyperalgesia was related to the COX system in the spinal cord, we compared the effects of spinal COX inhibition in iNOS−/− and wt mice. In wt mice intrathecal injection of the COX inhibitor indomethacin (Sigma) reduced thermal hyperalgesia in a dose-dependent manner (Fig.4A). This antinociceptive effect was completely absent in iNOS−/− mice (Table3, Fig. 4B) but was restored after substitution of NO with RE-2047, suggesting that the increase in spinal PGE2 production after zymosan injection required NO, which in wt mice is produced by spinal iNOS.
Fig. 4.
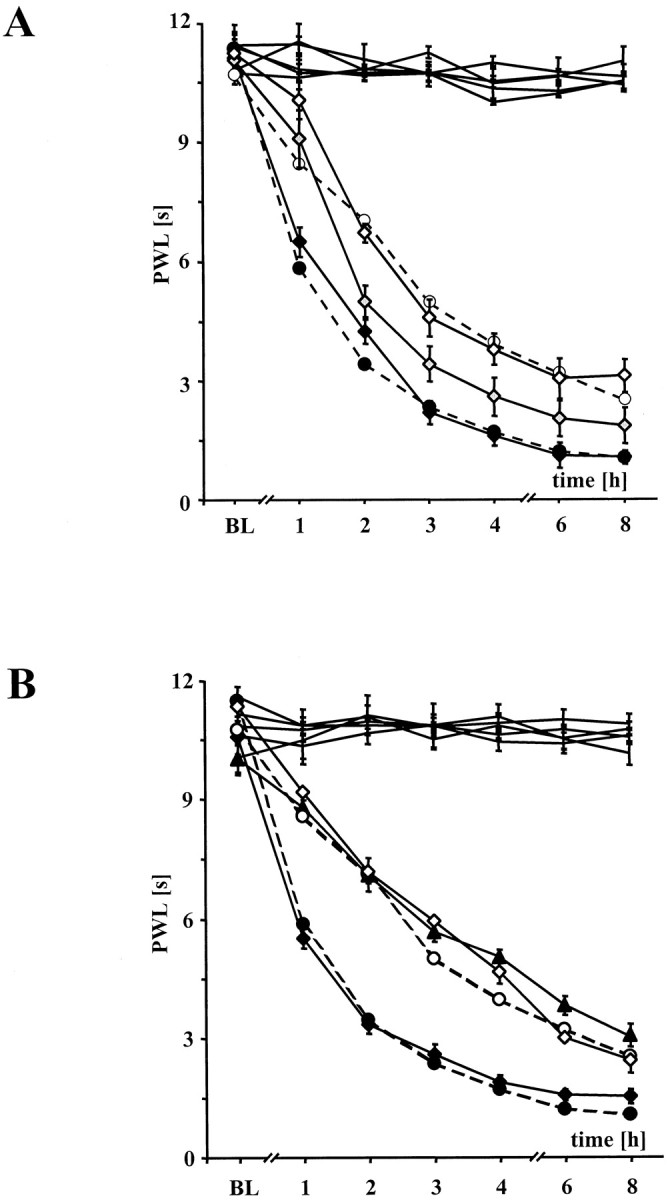
Effects of indomethacin in wt and iNOS−/− mice.A, Intrathecal injection of indomethacin, a nonselective COX inhibitor, in ascending doses of 0.1 (black diamonds) to 1.0 (gray diamonds) and 10.0 mm (open diamonds) decelerated the development of thermal hyperalgesia of wt mice. Intrathecal injection was performed to exclude the possibility of a decreased production of prostaglandins in the inflamed hindpaws during systemic application of COX inhibitors. Wt (black circles) and iNOS−/− mice (open circles) after acute intrathecal injection of ACSF (5 μl). Data are expressed as mean ± SEM. B,Intrathecal injection of indomethacin 10.0 mm (black triangle) antagonized the development of thermal hyperalgesia after intraperitoneal pretreatment with the NO donor RE-2047 (45 mg/kg). iNOS−/− mice after intraperitoneal pretreatment with 45 mg/kg RE-2047 and after intrathecal injection of ACSF (black diamonds) served as controls. Thermal hyperalgesia in iNOS−/− mice was not affected after intraperitoneal pretreatment with PBS and intrathecal pretreatment with indomethacin 10.0 mm(black triangle). Wt (black circles) and iNOS−/− mice (open circles) after acute intrathecal injection of 5 μl of ACSF (dotted lines). Data are expressed as means ± SEM.
Spinal NOx and PGE2 formation
The difference in heat sensitization between wt and iNOS−/− mice was already present 1 hr after zymosan injection. Because iNOS is generally considered not to be constitutively expressed in the CNS, the fast rise in NO production after zymosan injection would require an at least equally fast induction of iNOS expression. We have therefore analyzed the expression of iNOS mRNA during the early phase of hyperalgesia development by RT-PCR (Fig.5). Significant amounts of iNOS mRNA were detected 30 and 60 min after zymosan injection and 120 min after zymosan injection in separate experiments (data not shown). As expected, iNOS mRNA was absent under control conditions and in iNOS−/− mice after zymosan injection.
Fig. 5.

Early iNOS mRNA expression in wt mice. iNOS mRNA expression in spinal cord of wt and iNOS−/− mice after zymosan injection into the right hindpaw. In wt but not in iNOS−/− mice, iNOS mRNA was increased in thoracolumbar segment of spinal cord even 0.5 (3 of 6 mice) and 1 hr (6 of 6 mice) after peripheral inflammation. β-actin RT-PCR product level served as control. This figure represents one of two independent experiments.
To further elucidate the role of iNOS in the spinal mechanisms of heat sensitization and its relation to the COX pathway we performed spinal microdialysis to measure NO and PGE2 production in the spinal cord in response to zymosan injection. In a first series of experiments we tested whether insertion of the microdialysis probe was sufficient to induce spinal PG formation. In these experiments, PBS instead of zymosan was injected subcutaneously into the right hindpaw. Under these conditions only a modest increase in PGE2 was induced in wt mice (from 18.4 ± 1.9 to 26.3 ± 2.5 pg/ml).
When zymosan was injected, both PGE2 and NO degradation products (NOx,, i.e., NO2− and NO3−) increased in wt mice. NOx concentrations reached their maximum already after 1 hr and thus closely paralleled the development of iNOS-dependent hyperalgesia (Fig.6A). By contrast, PGE2 showed a more prolonged increase, exhibiting a continuous rise over the whole observation period of 4 hr (Fig.6B).
Fig. 6.
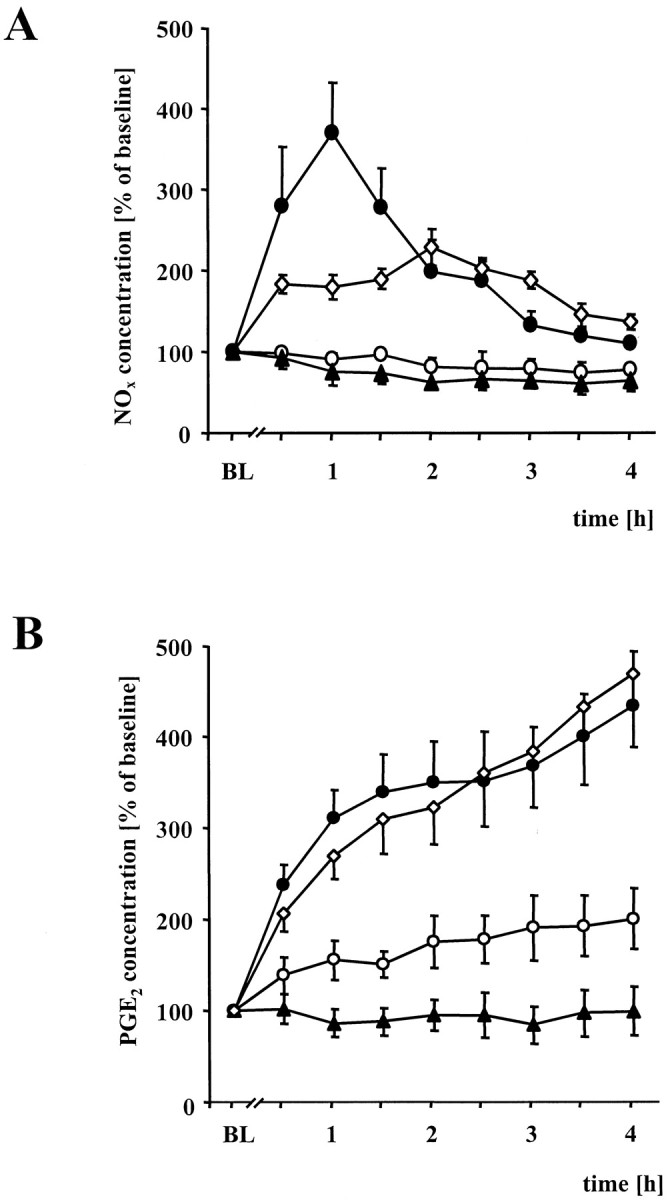
Spinal microdialysis in wt and iNOS−/− mice.A, Time course of percentile NOxlevel changes in the spinal cord of wt mice (black circles) and iNOS−/− mice (open circles) after subcutaneous injection of 3.0 mg/ml zymosan into the right hindpaw. Samples were collected via microdialysis of the thoracolumbar segment. In wt mice zymosan injection led to a rapid increase of breakdown products of NO, whereas only a slight increase of NOx was observed in iNOS−/− mice. After treatment with RE-2047, an NO donor, NOx levels in the spinal cord of iNOS−/− mice increased (open diamonds). Black trianglesrepresent NOx levels in wt mice after injection of PBS instead of zymosan into the right hindpaw. Data are expressed as means ± SEM. B, Time course of the percentile PGE2changes in the spinal cord of wt (black circles) and iNOS−/− mice (open circles). The additional curve (black diamonds) shows the changes of PGE2levels after injection of RE-2047 intraperitoneally into iNOS−/− mice. Black triangles represent wt mice after PBS administration into the right hindpaw instead of zymosan. Data are expressed as means ± SEM.
Next we investigated whether the effects of reduced NO formation on nociceptive sensitization might be secondary to a suppression of PGE2 formation. iNOS−/− mice lacked not only the zymosan-induced increase in NO, but also showed reduced spinal PGE2 formation. The rise in PGE2 production was much smaller compared to that in wt mice. As described above the NO donor RE-2047 reconstituted wt-like hyperalgesia in iNOS−/− mice. As shown in Figure6A, RE-2047 not only increased spinal NOx concentrations, but also restored spinal PGE2 formation (Fig. 6B).
We analyzed whether the expression of COX-1 or COX-2 differs in iNOS−/− and wt mice. Time course of COX-1 and COX-2 mRNA expression and of the PGE2 concentration in spinal cord tissue was followed for 7 d. As shown in Figure7A, increases in PGE2 concentrations reached their maximum 8 hr after zymosan injection, declined, and came back to baseline levels after 7 d. In iNOS −/− mice the rise in PGE2 was again largely suppressed, and only at 8 hr a modest increase in PGE2 was seen.
Fig. 7.
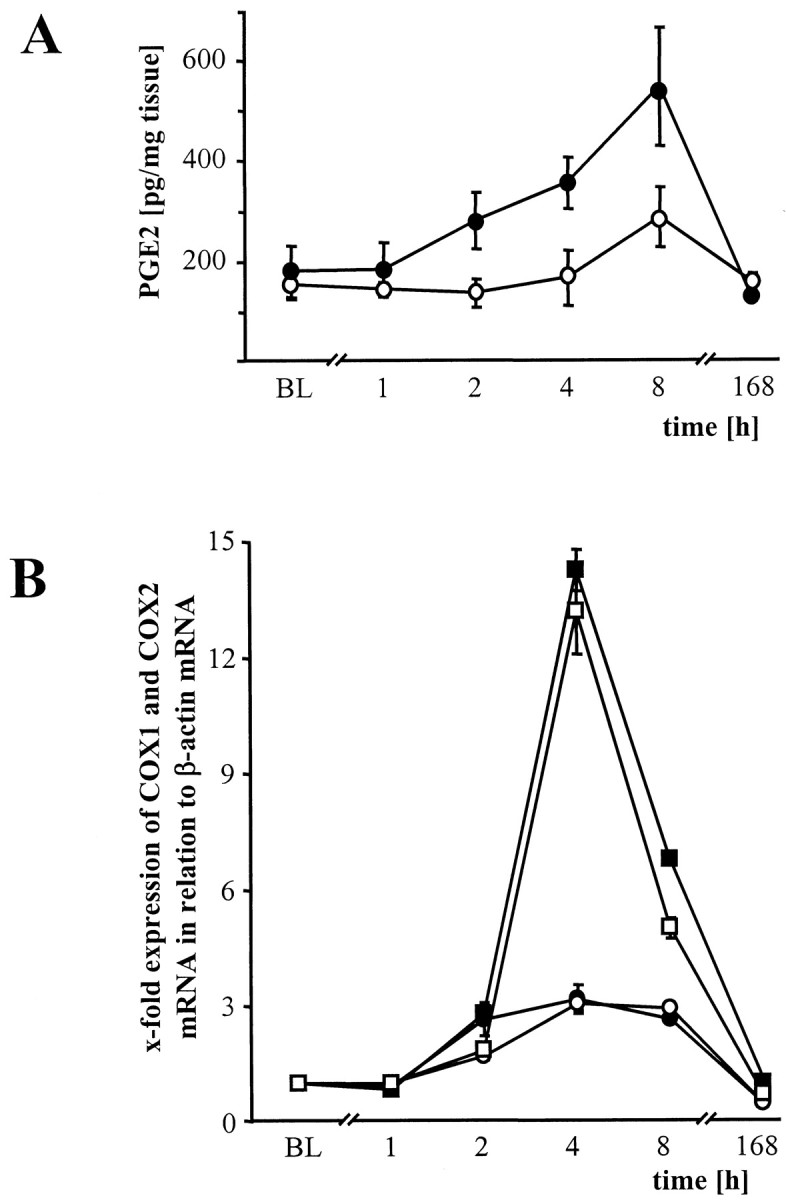
PGE2 concentration and COX-1 and COX-2 mRNA expression in spinal cord tissue. A, Time course of absolute PGE2 levels in the spinal cord tissue samples of wt (black circles) and in iNOS−/− (open circles) mice after zymosan injection into the right hindpaw. PGE2 measurement was performed from tissue samples after perfusion with 100 ml of ice-cold PBS. Data are expressed as means ± SEM. B, Time course of real time RT-PCR measurement of spinal COX-1 (circles) and COX-2 (squares) mRNA expression in wt (black) and iNOS−/− (open) mice after peripheral administered zymosan. There are no differences between COX mRNA expression in both lines of mice. Data are expressed as means ± SEM.
In contrast to the striking difference in PGE2production seen between iNOS−/− and wt mice, COX-1 and COX-2 mRNA expression were very similar in both types of mice. This suggests that the NO-induced rise in PGE2 levels was attributable to an increase in COX-1 and/or COX-2 enzymatic activity, rather than to altered gene expression.
DISCUSSION
Our data indicate that NO production from iNOS in the spinal cord immediately follows the early induction of peripheral tissue damage and inflammation. This process occurs in parallel to the development of thermal hyperalgesia and is required for the increase in spinal PGE2 production. Our data therefore attribute a decisive role to iNOS in the early phase of development of thermal hyperalgesia.
It appears that NO production immediately after peripheral tissue damage is predominantly iNOS-derived. This is corroborated by our results, which show an induction of iNOS mRNA in the spinal cord as early as 30 min after zymosan injection. Furthermore, administration ofl-NAME intrathecally into iNOS−/− mice was without any effect with regard to thermal hyperalgesia. This strongly argues against an extensive contribution of nNOS or eNOS. Observations ofClark et al. (1996), MacNaughton et al. (1998), Haddad et al. (1995), and Salvemini et al. (1995) of an early iNOS expression also support our results. Reconstitution of the fast development of hyperalgesia by the NO donor RE-2047 (Rehse and Ciborski, 1995) demonstrates the importance of early NO production. To relate this to the lack of iNOS genes, we injected the selective iNOS inhibitor l-NIL into wt mice; it reduced development of thermal hyperalgesia. The dose ofl-NIL chosen (0.1 μm; 5 μl) does not affect the other NOS isoforms (Moore et al., 1994). Furthermore, our results support previous studies showing that selective pharmacological inhibition of iNOS attenuates thermal hyperalgesia in rats (Osborne and Coderre, 1999). Only when given intrathecally did l-NIL inhibit NO production in the spinal cord and result in antinociceptive effects. These data indicate that only spinal iNOS-derived NO production correlates with antinociceptive activity. Interestingly, a substantial part of thermal hyperalgesia was insensitive to inhibition of iNOS and is also considered insensitive to COX inhibition. This insensitivity becomes dominant during the late phase of thermal sensitization ∼8 hr after zymosan injection. It is likely that both peripheral mechanisms such as sensitization of capsaicin receptors (Caterina et al., 1997; Kress and Zeilhofer, 1999) and central mechanisms independent from PG and NO contribute to this late phase.
In wt mice, COX inhibition by indomethacin and disruption of the iNOS gene resulted in virtually indistinguishable antinociceptive effects. In iNOS−/− mice, indomethacin failed to display an antinociceptive effect. Antinociceptive activity could be restored in these mice by adding back NO with the NO donor RE-2047. These results demonstrate that the production of spinal PGE2 requires spinal NO and that spinal iNOS-derived NO appears to mediate thermal hyperalgesia largely if not solely via an increase in spinal PGE2 formation. In this respect, our data support previous evidence that PGs are key mediators of thermal hyperalgesia (Minami et al., 1994; Ferreira and Lorenzetti, 1996; Yamamoto and Nozaki-Taguchi, 1997). We also confirmed findings that hindpaw inflammation produces enhanced PG levels in the spinal cord (Yang et al., 1996; Hay and de Belleroche, 1997; Ichitani et al., 1997; Yamamoto and Nozaki-Taguchi, 1997; Dirig and Yaksh, 1999; Ebersberger et al., 1999).
In line with several studies, we could show that PG production in the spinal cord is modulated positively by NO release and is diminished by lack of NO production (Salvemini et al., 1994, 1995; Salvemini, 1997; but see also Hamilton and Warner, 1998). Substitution of NO by RE-2047 completely reconstituted PGE2 production in iNOS−/− mice as well as nociceptive responses in the behavioral tests, indicating once again that iNOS-derived NO amplifies PG production at spinal level. Moreover, the constitutive baseline mRNA expression (Beiche et al., 1996, 1998) and zymosan-induced expression of COX-2 were similar in both types of mice in the dorsal horn of the spinal cord. Consequently, it appears plausible to attribute to iNOS-derived NO a pivotal role as a trigger of spinal enzymatic PG production.
PGE2 and NO production did not run in parallel. NO exhibited a fast peak within ∼1 hr, whereas increases in PGE2 occurred at a markedly prolonged time scale. Our results suggest that PGE2 production is not attributable to increased COX-1 or COX-2 expression. It may be speculated that the enhanced early PG production is caused by a free radical driven modulation of phospholipase A2activity and therefore of the concentration of arachidonic acid, which is the rate-limiting substrate of PG production (but see alsoZingarelli et al., 1997; Sahnoun et al., 1998). However, a direct or indirect interaction of NO or one of its metabolites with the COX-1 or COX-2 proteins by enhancing their enzymatic activity is also possible (Salvemini, 1997). In either case one would assume that the NO-induced change in COX enzyme activity outlasts the rise in spinal NO, e.g., via sustained changes in enzyme activity of COX-1, COX-2, or phospholipase A2.
An intriguing question raised by our study is whether inhibition of iNOS provides a novel target for analgesic therapy. Our experiments have shown that inhibition of iNOS via gene disruption or by selective drugs almost completely abolishes the PG-mediated part of thermal hyperalgesia. With the possible exception of inhibition of resistance to certain microbial agents, iNOS−/− mice are not only viable but also show no major abnormalities indicating that pharmacological inhibition of iNOS may be considered as largely safe. The use of iNOS inhibitors as analgesics would be largely limited by the fact that they would have to be administered very early, before spinal PG production has been triggered.
Our data also indicate that NO donors reaching the CNS may be enhancers of pain development and in this respect be problematic. This observation is in line with findings of Urban et al. (1999), Inoue et al. (1997), and Aley et al. (1998) (but see also Lauretti et al., 1999) and should be subjected to further research.
Our data offer no clues indicating whether the decrease of NO production seen in our animals before hyperalgesia reaches its peak is of importance for the later normalization of hyperalgesia that goes along with healing processes in the damaged tissue (Reichner et al., 1999).
In summary, we have shown that NO generated by iNOS not only plays an important role as a mediator of tissue inflammation acting in peripheral tissue, but that it also serves an important role in the spinal cord, where it triggers PGE2 formation, enhances COX-2 activity, and facilitates the development of thermal and possibly other forms of hyperalgesia.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (SFB 353, A1). We thank Dr. Ivan Otterness, visiting professor of the department, for the helpful suggestions and corrections. We acknowledge the professional help of Tanja Mittmann, Alexandra Schmauss, and Isabella Kolberg.
H.G. and M.G. contributed equally to this work.
Correspondence should be addressed to Dr. Hans Guehring, Department of Experimental and Clinical Pharmacology and Toxicology, Fahrstrasse 17, D-91054 Erlangen, Germany. E-mail:guehring@pharmakologie.uni-erlangen.de.
REFERENCES
- 1.Aley KO, McCarter G, Levine JD. Nitric oxide signaling in pain and nociceptor sensitization in the rat. J Neurosci. 1998;18:7008–7014. doi: 10.1523/JNEUROSCI.18-17-07008.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amir S, English AM. An inhibitor of nitric oxide production, NG-nitro-l-arginine-methyl ester, improves survival in anaphylactic shock. Eur J Pharmacol. 1991;203:125–127. doi: 10.1016/0014-2999(91)90800-6. [DOI] [PubMed] [Google Scholar]
- 3.Andrew D, Greenspan JD. Mechanical and heat sensitization of cutaneous nociceptors after peripheral inflammation in the rat. J Neurophysiol. 1999;82:2649–2656. doi: 10.1152/jn.1999.82.5.2649. [DOI] [PubMed] [Google Scholar]
- 4.Barker JE, Strangward HM, Brand MP, Hurst RD, Land JM, Clark JB, Heales SJ. Increased inducible nitric oxide synthase protein but limited nitric oxide formation occurs in astrocytes of the hph-1 (tetrahydrobiopterin deficient) mouse. Brain Res. 1998;804:1–6. doi: 10.1016/s0006-8993(98)00603-9. [DOI] [PubMed] [Google Scholar]
- 5.Beiche F, Scheuerer S, Brune K, Geisslinger G, Goppelt-Struebe M. Up-regulation of cyclooxygenase-2 mRNA in the rat spinal cord following peripheral inflammation. FEBS Lett. 1996;390:165–169. doi: 10.1016/0014-5793(96)00604-7. [DOI] [PubMed] [Google Scholar]
- 6.Beiche F, Brune K, Geisslinger G, Goppelt-Struebe M. Expression of cyclooxygenase isoforms in the rat spinal cord and their regulation during adjuvant-induced arthritis. Inflamm Res. 1998;47:482–487. doi: 10.1007/s000110050362. [DOI] [PubMed] [Google Scholar]
- 7.Bley KR, Hunter JC, Eglen RM, Smith JA. The role of IP prostanoid receptors in inflammatory pain. Trends Pharmacol Sci. 1998;19:141–147. doi: 10.1016/s0165-6147(98)01185-7. [DOI] [PubMed] [Google Scholar]
- 8.Brune K. Spinal cord effects of antipyretic analgesics. Drugs. 1994;5:21–27. doi: 10.2165/00003495-199400475-00005. [DOI] [PubMed] [Google Scholar]
- 9.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway [see comments]. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 10.Chen X, Levine JD. NOS inhibitor antagonism of PGE2-induced mechanical sensitization of cutaneous C-fiber nociceptors in the Rat. J Neurophysiol. 1999;81:963–966. doi: 10.1152/jn.1999.81.3.963. [DOI] [PubMed] [Google Scholar]
- 11.Clark RS, Kochanek PM, Schwarz MA, Schiding JK, Turner DS, Chen M, Carlos TM, Watkins SC. Inducible nitric oxide synthase expression in cerebrovascular smooth muscle and neutrophils after traumatic brain injury in immature rats. Pediatr Res. 1996;39:784–790. doi: 10.1203/00006450-199605000-00007. [DOI] [PubMed] [Google Scholar]
- 12.Deckert-Schluter M, Albrecht S, Hof H, Wiestler OD, Schluter D. Dynamics of the intracerebral and splenic cytokine mRNA production in Toxoplasma gondii-resistant and -susceptible congenic strains of mice. Immunology. 1995;85:408–418. [PMC free article] [PubMed] [Google Scholar]
- 13.Deckert-Schluter M, Bluethmann H, Rang A, Hof H, Schluter D. Crucial role of TNF receptor type 1 (p55), but not of TNF receptor type 2 (p75), in murine toxoplasmosis. J Immunol. 1998;160:3427–3436. [PubMed] [Google Scholar]
- 14.Dirig DM, Yaksh TL. In vitro prostanoid release from spinal cord following peripheral inflammation: effects of substance P, NMDA and capsaicin. Br J Pharmacol. 1999;126:1333–1340. doi: 10.1038/sj.bjp.0702427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Downen M, Zhao ML, Lee P, Weidenheim KM, Dickson DW, Lee SC. Neuronal nitric oxide synthase expression in developing and adult human CNS. J Neuropathol Exp Neurol. 1999;58:12–21. doi: 10.1097/00005072-199901000-00002. [DOI] [PubMed] [Google Scholar]
- 16.Ebersberger A, Grubb BD, Willingale HL, Gardiner NJ, Nebe J, Schaible HG. The intraspinal release of prostaglandin E2 in a model of acute arthritis is accompanied by an up-regulation of cyclo-oxygenase-2 in the spinal cord. Neuroscience. 1999;93:775–781. doi: 10.1016/s0306-4522(99)00164-5. [DOI] [PubMed] [Google Scholar]
- 17.Ferreira SH, Lorenzetti BB. Intrathecal administration of prostaglandin E2 causes sensitization of the primary afferent neuron via the spinal release of glutamate. Inflamm Res. 1996;45:499–502. doi: 10.1007/BF02311085. [DOI] [PubMed] [Google Scholar]
- 18.Goettl VM, Larson AA. Nitric oxide mediates long-term hyperalgesic and antinociceptive effects of the N-terminus of substance P in the formalin assay in mice. Pain. 1996;67:435–441. doi: 10.1016/0304-3959(96)03155-7. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez-Hernandez T, Rustioni A. Expression of three forms of nitric oxide synthase in peripheral nerve regeneration. J Neurosci Res. 1999;55:198–207. doi: 10.1002/(SICI)1097-4547(19990115)55:2<198::AID-JNR7>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 20.Green LC, Tannenbaum SR, Goldman P. Nitrate synthesis in the germfree and conventional rat. Science. 1981;212:56–58. doi: 10.1126/science.6451927. [DOI] [PubMed] [Google Scholar]
- 21.Haddad EB, Liu SF, Salmon M, Robichaud A, Barnes PJ, Chung KF. Expression of inducible nitric oxide synthase mRNA in Brown Norway rats exposed to ozone: effect of dexamethasone. Eur J Pharmacol. 1995;293:287–290. doi: 10.1016/0926-6917(95)00032-1. [DOI] [PubMed] [Google Scholar]
- 22.Hamalainen MM, Lovick TA. Involvement of nitric oxide and serotonin in modulation of antinociception and pressor responses evoked by stimulation in the dorsolateral region of the periaqueductal gray matter in the rat. Neuroscience. 1997;80:821–827. doi: 10.1016/s0306-4522(97)00124-3. [DOI] [PubMed] [Google Scholar]
- 23.Hamilton LC, Warner TD. Interactions between inducible isoforms of nitric oxide synthase and cyclo-oxygenase in vivo: investigations using the selective inhibitors, 1400W and celecoxib. Br J Pharmacol. 1998;125:335–340. doi: 10.1038/sj.bjp.0702077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 25.Hay C, de Belleroche J. Carrageenan-induced hyperalgesia is associated with increased cyclo- oxygenase-2 expression in spinal cord. NeuroReport. 1997;8:1249–1251. doi: 10.1097/00001756-199703240-00038. [DOI] [PubMed] [Google Scholar]
- 26.Hylden JL, Wilcox GL. Intrathecal morphine in mice: a new technique. Eur J Pharmacol. 1980;67:313–316. doi: 10.1016/0014-2999(80)90515-4. [DOI] [PubMed] [Google Scholar]
- 27.Ichitani Y, Shi T, Haeggstrom JZ, Samuelsson B, Hokfelt T. Increased levels of cyclooxygenase-2 mRNA in the rat spinal cord after peripheral inflammation: an in situ hybridization study. NeuroReport. 1997;8:2949–2952. doi: 10.1097/00001756-199709080-00028. [DOI] [PubMed] [Google Scholar]
- 28.Inoue T, Mashimo T, Shibuta S, Yoshiya I. Intrathecal administration of a new nitric oxide donor, NOC-18, produces acute thermal hyperalgesia in the rat. J Neurol Sci. 1997;153:1–7. doi: 10.1016/s0022-510x(97)00188-3. [DOI] [PubMed] [Google Scholar]
- 29.Kawabata A, Manabe S, Manabe Y, Takagi H. Effect of topical administration of L-arginine on formalin-induced nociception in the mouse: a dual role of peripherally formed NO in pain modulation. Br J Pharmacol. 1994;112:547–550. doi: 10.1111/j.1476-5381.1994.tb13108.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kress M, Zeilhofer HU. Capsaicin, protons and heat: new excitement about nociceptors. Trends Pharmacol Sci. 1999;20:112–118. doi: 10.1016/s0165-6147(99)01294-8. [DOI] [PubMed] [Google Scholar]
- 31.Laubach VE, Shesely EG, Smithies O, Sherman PA. Mice lacking inducible nitric oxide synthase are not resistant to lipopolysaccharide-induced death. Proc Natl Acad Sci USA. 1995;92:10688–10692. doi: 10.1073/pnas.92.23.10688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lauretti GR, Lima IC, Reis MP, Prado WA, Pereira NL. Oral ketamine and transdermal nitroglycerin as analgesic adjuvants to oral morphine therapy for cancer pain management. Anesthesiology. 1999;90:1528–1533. doi: 10.1097/00000542-199906000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Lawand NB, Willis WD, Westlund KN. Blockade of joint inflammation and secondary hyperalgesia by l-NAME, a nitric oxide synthase inhibitor. NeuroReport. 1997;8:895–899. doi: 10.1097/00001756-199703030-00016. [DOI] [PubMed] [Google Scholar]
- 34.Lee SC, Brosnan CF. Cytokine regulation of iNOS expression in human glial cells. Methods. 1996;10:31–37. doi: 10.1006/meth.1996.0075. [DOI] [PubMed] [Google Scholar]
- 35.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, Chen H, Mudgett JS. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 36.MacNaughton WK, Aurora AR, Bhamra J, Sharkey KA, Miller MJ. Expression, activity and cellular localization of inducible nitric oxide synthase in rat ileum and colon post-irradiation. Int J Radiat Biol. 1998;74:255–264. doi: 10.1080/095530098141645. [DOI] [PubMed] [Google Scholar]
- 37.Malmberg AB, Yaksh TL. Spinal nitric oxide synthesis inhibition blocks NMDA-induced thermal hyperalgesia and produces antinociception in the formalin test in rats. Pain. 1993;54:291–300. doi: 10.1016/0304-3959(93)90028-N. [DOI] [PubMed] [Google Scholar]
- 38.Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 39.Minami T, Uda R, Horiguchi S, Ito S, Hyodo M, Hayaishi O. Allodynia evoked by intrathecal administration of prostaglandin E2 to conscious mice. Pain. 1994;57:217–223. doi: 10.1016/0304-3959(94)90226-7. [DOI] [PubMed] [Google Scholar]
- 40.Moore WM, Webber RK, Jerome GM, Tjoeng FS, Misko TP, Currie MG. L-N6-(1-iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J Med Chem. 1994;37:3886–3888. doi: 10.1021/jm00049a007. [DOI] [PubMed] [Google Scholar]
- 41.Osborne MG, Coderre TJ. Effects of intrathecal administration of nitric oxide synthase inhibitors on carrageenan-induced thermal hyperalgesia. Br J Pharmacol. 1999;126:1840–1846. doi: 10.1038/sj.bjp.0702508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rehse K, Ciborski T. New no-donors with antithrombotic and vasodilating activities, X: antiplatelet and antithrombotic effects of 3-methylsydnone-5- nitrosimine (RE 2047) in combination with ASA, pentoxifylline, and ticlopidine. Arch Pharmacol (Weinheim) 1995;328:71–78. doi: 10.1002/ardp.19953280113. [DOI] [PubMed] [Google Scholar]
- 43.Reichner JS, Meszaros AJ, Louis CA, Henry WL, Jr, Mastrofrancesco B, Martin BA, Albina JE. Molecular and metabolic evidence for the restricted expression of inducible nitric oxide synthase in healing wounds. Am J Pathol. 1999;154:1097–1104. doi: 10.1016/S0002-9440(10)65362-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahnoun Z, Jamoussi K, Zeghal KM. Free radicals and antioxidants: physiology, human pathology and therapeutic aspects (part II). Therapie. 1998;53:315–339. [PubMed] [Google Scholar]
- 45.Salvemini D. Regulation of cyclooxygenase enzymes by nitric oxide. Cell Mol Life Sci. 1997;53:576–582. doi: 10.1007/s000180050074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salvemini D, Seibert K, Masferrer JL, Misko TP, Currie MG, Needleman Endogenous nitric oxide enhances prostaglandin production in a model of renal inflammation. J Clin Invest. 1994;93:1940–1947. doi: 10.1172/JCI117185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salvemini D, Manning PT, Zweifel BS, Seibert K, Connor J, Currie MG, Needleman P, Masferrer JL. Dual inhibition of nitric oxide and prostaglandin production contributes to the anti-inflammatory properties of nitric oxide synthase inhibitors. J Clin Invest. 1995;96:301–308. doi: 10.1172/JCI118035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen TY, Winter CA. Chemical and biological studies on indomethacin, sulindac and their analogs. Adv Drug Res. 1977;12:90–245. [PubMed] [Google Scholar]
- 49.Sinz EH, Kochanek PM, Dixon CE, Clark RS, Carcillo JA, Schiding JK, Chen M, Wisniewski SR, Carlos TM, Williams D, DeKosky ST, Watkins SC, Marion DW, Billiar TR. Inducible nitric oxide synthase is an endogenous neuroprotectant after traumatic brain injury in rats and mice. J Clin Invest. 1999;104:647–656. doi: 10.1172/JCI6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Urban MO, Coutinho SV, Gebhart GF. Involvement of excitatory amino acid receptors and nitric oxide in the rostral ventromedial medulla in modulating secondary hyperalgesia produced by mustard oil. Pain. 1999;81:45–55. doi: 10.1016/s0304-3959(98)00265-6. [DOI] [PubMed] [Google Scholar]
- 51.Wei G, Dawson VL, Zweier JL. Role of neuronal and endothelial nitric oxide synthase in nitric oxide generation in the brain following cerebral ischemia. Biochim Biophys Acta. 1999;1455:23–34. doi: 10.1016/s0925-4439(99)00051-4. [DOI] [PubMed] [Google Scholar]
- 52.Wei XQ, Charles IG, Smith A, Ure J, Feng GJ, Huang FP, Xu D, Muller W, Moncada S, Liew FY. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature. 1995;375:408–411. doi: 10.1038/375408a0. [DOI] [PubMed] [Google Scholar]
- 53.Yaksh TL, Malmberg AB. Spinal actions of NSAIDS in blocking spinally mediated hyperalgesia: the role of cyclooxygenase products. Agents Actions Suppl. 1993;41:89–100. [PubMed] [Google Scholar]
- 54.Yamamoto T, Nozaki-Taguchi N. Role of spinal cyclooxygenase (COX)-2 on thermal hyperalgesia evoked by carageenan injection in the rat. NeuroReport. 1997;8:2179–2182. doi: 10.1097/00001756-199707070-00018. [DOI] [PubMed] [Google Scholar]
- 55.Yang LC, Marsala M, Yaksh TL. Characterization of time course of spinal amino acids, citrulline and PGE2 release after carrageenan/kaolin-induced knee joint inflammation: a chronic microdialysis study. Pain. 1996;67:345–354. doi: 10.1016/0304-3959(96)03106-5. [DOI] [PubMed] [Google Scholar]
- 56.Zingarelli B, Southan GJ, Gilad E, O'Connor M, Salzman AL, Szabo C. The inhibitory effects of mercaptoalkylguanidines on cyclo-oxygenase activity. Br J Pharmacol. 1997;120:357–366. doi: 10.1038/sj.bjp.0700892. [DOI] [PMC free article] [PubMed] [Google Scholar]