

Abstract
The inhibitory synapse of the crayfish neuromuscular junction was used to examine mechanisms underlying the F2 component of synaptic facilitation. Because previous studies have shown accelerated transmitter release during facilitation, we examined whether an activity-dependent plasticity in ICa could underlie this acceleration. We established that fluorescent transients generated by Magnesium Green can resolve small differences in presynaptic Ca2+ influx that correlate with changes in IPSC waveform. However, there was no change in Ca2+ transients associated with the accelerated release. Analyzing the initial rise of IPSC and the duration of the presynaptic spike yielded a depolarization–release coupling plot that captures the impact of spike waveform on the initial rate of release. We conclude that accelerated release during F2 facilitation cannot be attributed to plasticity of ICa or modulation of spike waveform. Kinetic analysis showed a reduction in synaptic delay during facilitation only when broad action potentials were used. In unfacilitated release, synaptic delay increased as spike duration lengthened. We propose that small single Ca2+ channel currents during the plateau phase of broad action potentials raise local Ca2+concentration only enough to fill a high-affinity site. Occupation of this site in itself, or events downstream, would convert a vesicle from control to facilitated state. If the conversion were a slow process, it could explain the changes in synaptic delay reported here. This hypothesis can also account for a number of observations related to Ca2+ cooperativity and synaptic facilitation.
Keywords: synaptic delay, synapse, crayfish inhibitor, neuromuscular junction, facilitation, calcium indicator
Synaptic facilitation is a common phenomenon observed in both central and peripheral synapses. It is a short-term synaptic enhancement that persists for up to several hundred milliseconds and may contribute to the integration of synaptic signals. Facilitation is subdivided into two distinct kinetic components, F1 and F2, with decay time constant of tens and hundreds of milliseconds, respectively (Magleby, 1987). Although it has long been established that Ca2+ influx elicited by conditioning stimuli is essential for the activation of facilitation (Katz and Miledi, 1968; Zucker, 1989), specific underlying mechanisms have not yet been fully elucidated. Considerable insight, however, has been obtained by mathematical modeling in which one of the Ca2+ binding sites of the release process is assumed to have a high affinity and underlie the facilitation process (Yamada and Zucker, 1992; Winslow et al., 1994; Bertram et al., 1996). These models also predict a small change in release kinetics during facilitation, although the change was considered too small to be detectable. These predictions are consistent with experimental work that has uncovered no change in release kinetics during facilitation (Datyner and Gage, 1980; Parnas et al., 1989).
The crayfish neuromuscular junction is a well established preparation for the study of synaptic facilitation, having exceptionally large magnitudes for all components of short-term synaptic enhancement (Atwood and Wojtowicz, 1986; Bittner, 1989). A previous report using this preparation showed an acceleration in release kinetics during F2 facilitation when transmitter release was elicited by prolonged presynaptic depolarization (Vyshedskiy and Lin, 1997c). This observation appears to be at odds with earlier studies, prompting further examination of plasticity in presynapticICa. Plasticity inICa has been shown to play a role in facilitation at the calyx of Held (Borst and Sakmann, 1998; Cuttle et al., 1998) but does not appear to be the general mechanism underlying facilitation (Charlton et al., 1982). Here, we use Ca2+-sensitive dyes to monitor the activity of presynaptic ICa during facilitation.
If accelerated release is not mediated by plasticity inICa, it might then be attributable to changes in the release process itself. Until recently, modulation of release kinetics had been a rare observation. However, it now has been demonstrated that blocking or deletingN-ethylmaleimide-sensitive factor retards release kinetics (Schweizer et al., 1998; Kawasaki et al., 1998) and that synaptic depression at the calyx of Held is associated with decelerated release (Wu and Borst, 1999). Moreover, serotonin can modulate release kinetics in Aplysia (Klein, 1994) and crayfish (Vyshedskiy et al., 1998). Augmentation is associated with an increase in hypertonic shock-induced release (Stevens and Wesseling, 1999). Finally, an increase in action potential duration can lengthen synaptic delay (Datyner and Gage, 1980; Sabatini and Regehr, 1996). Although the mechanisms underlying such changes in release kinetics may differ, these observations suggest that the secretion process could be an important site for modulating synaptic strength. Here, we use synaptic delay to probe changes in release kinetics during facilitation.
MATERIALS AND METHODS
Preparation and electrophysiology. Crayfish,Procambarus clarkii, were obtained from Carolina Biological (Burlington, NC). Animals were maintained at room temperature, 23°C, until use. All experiments using action potential-based protocols were performed at 23°C. Experiments using the presynaptic voltage control method were performed at 15°C. The typical size of the animals was ∼6 cm, head to tail. The opener muscle of the first walking leg was used for all experiments. A presynaptic electrode penetrated the major branch point of the inhibitory axon (inhibitor) to record action potential and inject dye. The major branch point was ∼100–300 μm from the terminals on a central muscle at which fluorescence transients were measured. A suction electrode was used to stimulate the inhibitor. Two postsynaptic electrodes, 5 mΩ with 3 mKCl, penetrated a muscle fiber. A two-electrode voltage-clamp amplifier (GeneClamp 500; Axon Instruments, Foster City, CA) was used to record IPSC and filtered at 2 kHz. We monitored chloride equilibrium potential (ECl) during experiments to control for possible long-term changes in IPSC attributable to driftingECl (Vyshedskiy and Lin, 1997a).
When the presynaptic voltage control was performed, the current electrode was inserted at the primary branch point of the inhibitor, and the voltage electrode was inserted at a point at which a tertiary branch emerged from a secondary branch. This configuration allowed for optimal control of presynaptic potential in varicosities near the voltage electrode (Vyshedskiy and Lin, 1997a). Limited space under the water immersion lens allowed us to use only three microelectrodes simultaneously. A single electrode was used to record IPSP from a central muscle fiber.
Control saline contained (in mm): 195 NaCl, 5.4 KCl, 13.5 CaCl2, 2.6 MgCl2, and 10 HEPES, titrated to pH 7.4 by NaOH. When tetraethylammonium (TEA) chloride was introduced into the control saline, an equal amount of NaCl was removed. All chemicals were purchased from Sigma (St. Louis, MO). Morphological examination of the terminal branches that innervated the recorded muscle fiber was performed after each experiment, by sketching or photography (Vyshedskiy and Lin, 1997a).
Photometric measurement of calcium transients. The inhibitory axon was penetrated at the major branch point by an electrode containing 1.25–5 mmmembrane-impermeable Magnesium Green, K+salt, dissolved in 400 mm KCl and 20 mm K-HEPES, pH 7.4, with a final resistance of 20–50 MΩ [see Vyshedskiy and Lin (2000) for reasons for selecting Magnesium Green over other Ca2+ indicators]. The dye was injected by a hyperpolarizing current of 2–8 nA, until varicosities close to the injection site were clearly visible. Experiments commenced after the fluorescence level had stabilized, ∼30 min after dye injection was completed. It is not possible to accurately estimate the concentration of injected dye. However, a previous study has established that the injection protocol did not interfere with intrinsic Ca2+ buffering or transmitter release (Vyshedskiy and Lin, 2000).
Photometric measurement of Ca2+ transients in this preparation has been described previously (Vyshedskiy and Lin, 2000). Briefly, a photomultiplier tube (HC124–06; Hamamatsu, Bridgewater, NJ) was used to record fluorescence transients on an upright microscope (Axioskop; Zeiss, Oberkochen, Germany) with 40 or 60× water immersion lenses. A 100 W tungsten lamp was powered by a stabilized power supply (ATE 15–15DM; Kepco, Flushing, NY). Illumination was gated by a shutter (Uniblitz; Vinsent Associates) with a typical duration of 600 msec and repeated at 0.2 Hz. The specifications of the filter set were as follows: excitation, 485DF15; dichroic, 505DRLP; emission, 530DF30 (Omega Optical, Brattleboro, VT). The area of illumination was restricted by an iris diaphragm custom-milled to allow an opening of 20∼50 μm in diameter, which typically encompassed one to five varicosities on the upper surface of a central muscle fiber. The output of the photomultiplier tube was filtered with an eight-pole Bessel filter (902LPP; Frequency Devices, Haverhill, MA) at fc of 1 kHz and digitized at 10 kHz. Fluorescence transients are presented as ΔF/F = (F(t) −Frest)/Frest* 100%, where Frest represents the fluorescence intensity of stained varicosities in the absence of activity. Typically, background fluorescence levels in an unstained region were <10% of Frest and were therefore not corrected for.
RESULTS
Calcium transients during F2 facilitation investigated by presynaptic voltage control protocols
Changes in the kinetics of transmitter release during F2 facilitation are best illustrated with the presynaptic voltage-control technique. F2 facilitation was activated by a series of eight identical conditioning pulses, 5 msec in duration at 20 Hz (Fig.1A, inset). The amplitude of conditioning pulses was set to just below the threshold level that activated a detectable IPSP. Facilitation was monitored by a 20 msec step, depolarized to 0 mV, delivered 150 msec after the last of the conditioning pulses. This protocol activated a near-maximal level of F2 facilitation but completely avoided possible complications associated with transmitter depletion (Vyshedskiy and Lin, 1997b). Facilitated IPSP (Fig. 1A, solid line) shifts to the left of control IPSP (Fig.1A, dotted line) and suggests accelerated release kinetics (Vyshedskiy and Lin, 1997c). The acceleration is not likely to be attributable to changes in the characteristics of presynaptic voltage control or plasticity of presynapticICa because both presynaptic test steps (Fig. 1C) and Ca2+transients (Fig. 1B) remain unchanged before (dotted line) and during (solid line) facilitation. Results similar to those shown in Figure 1 were obtained in four additional preparations. However, it remains possible that our photometric measurement is not sensitive enough to detect small changes in ICa, which could modulate release kinetics.
Fig. 1.
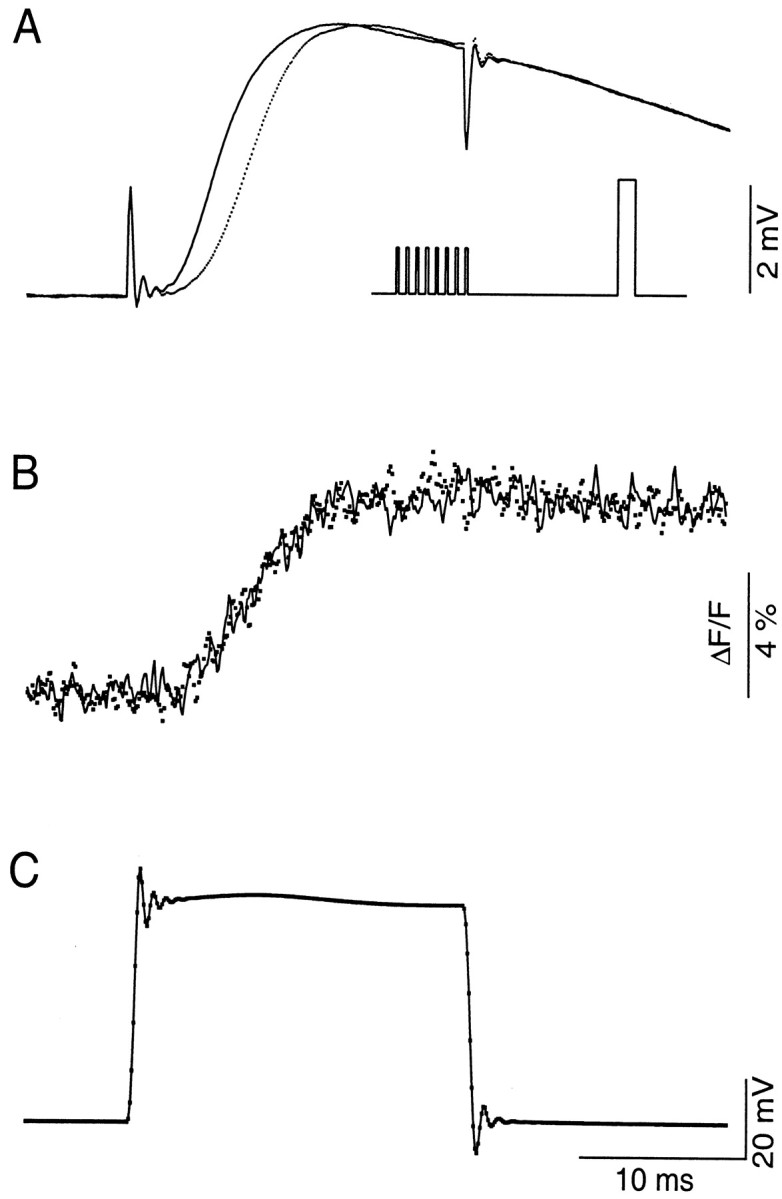
The presynaptic voltage control method shows that accelerated release during facilitation is not associated with a change in the presynaptic Ca2+ transient. Simultaneously recorded IPSPs (A), Ca2+transients (B), and presynaptic voltage steps (C) are displayed on the same time scale. Control (dotted line) and facilitated (solid line) recordings are superimposed. The inset inA illustrates the protocol used to activate F2 facilitation (see Results for details). The time courses of presynaptic voltage steps with (solid line) or without (dotted line) preceding conditioning stimuli are identical, whereas that of facilitated IPSP (A,solid line) is accelerated. Meanwhile, Ca2+ transients activated by control and test steps (B) are also superimposable. This experiment was performed in the presence of 1 mm 4-AP, 40 mmTEA, and 100 nm TTX. Each represents the average of 30 trials. This experiment was performed at 15°C.
Acceleration in release kinetics during F2 facilitation investigated by action potential-based protocols
To generalize the observations shown above and to take into account the sensitivity of our photometric measurement, action potential-based protocols were used to investigate the kinetics of release during facilitation. Because the accelerated release kinetics are best uncovered by prolonged presynaptic depolarization, a paired-pulse protocol was performed in the presence of 1 mm4-AP and 20 mm TEA. Figure 2illustrates presynaptic spikes (A3), Ca2+ transients (A2), and IPSCs (A1) activated by a paired stimulation with an interpulse interval of 100 msec. Superimposition of the first and second spikes shows that the duration of the second action potential (Fig. 2B3, solid line) is significantly shorter than that of the first one (Fig.2B3, dotted line). A corresponding difference in Ca2+transients (Fig. 2B2,solid and dotted lines) is also observed. Interestingly, facilitated IPSC (Fig.2B1, solid line) exhibits not only a greater amplitude but also an earlier rising phase than the control IPSC (Fig.2B1, dotted line). The reduced duration of the second action potential, however, prevents us from directly comparing the kinetics of the release process. (Shorter duration of the second spike was a consistent finding in all preparations, >100. The underlying mechanism of this finding remains unexplored.)
Fig. 2.

Facilitation activated by paired action potentials with prolonged duration. A1,A2, and A3illustrate IPSCs, presynaptic Ca2+ transients, and presynaptic action potentials, respectively. B, The first (dotted line) and second (solid line) IPSCs (B1), Ca2+ transients (B2), and action potentials (B3) are superimposed and displayed at a higher time resolution. Although the second IPSC is facilitated, the duration of the second action potential is shorter than that of the first one. The Ca2+ transient activated by the second action potential also has a lower amplitude. This experiment was performed in the presence of 1 mm 4-AP and 20 mm TEA. Each trace represents the average of 100 trials.
The sensitivity of our photometric measurements was evaluated empirically. We analyzed the time course of Ca2+ transients activated by action potentials with different durations. As the shape of the action potentials varies, the time course of Ca2+influx should change as well. Therefore, if the photometric measurement is sensitive enough, the shapes of Ca2+transients and action potentials should correlate in a predictable manner. In addition, the shape of IPSC, which is dictated by the time course of Ca2+ influx, can provide an independent verification for detected changes in Ca2+ transients. Figure3 illustrates how this objective was accomplished. A reference action potential, recorded in 10 mm TEA (Fig. 3C, broken line), exhibits a more rapid initial repolarization than both the first (dotted line) and second (solid line) of the paired action potentials recorded in 20 mm TEA. The corresponding Ca2+ transients show that the reference action potential elicits a faster rising Ca2+ influx (Fig. 3B,broken line) than those activated by the paired stimuli (Fig. 3B, dotted and solid lines), presumably because of a larger Ca2+driving force associated with the rapid repolarization. The difference in the rising phase of the Ca2+ transients is also reflected in the rising phases of their corresponding IPSCs (Fig. 3A, broken and dotted lines). Specifically, the reference IPSC takes off faster than the control IPSC, although the latter “crosses” the former later and exhibits a higher amplitude. Thus, our photometric measurement can detect small changes in Ca2+ influx that result in the crossing between the reference and control IPSCs. The difference in rising phase between facilitated and control IPSCs is much larger than that between reference and control IPSCs. Consequently, if the accelerated time course of facilitated IPSC is attributable to a faster activation of presynaptic Ca2+ influx, the Ca2+ transients should be capable of reflecting this change. Results similar to those shown in Figure 3 were observed in four of five preparations analyzed. Therefore, it is reasonable to conclude that accelerated transmitter release during F2 facilitation is unlikely to be attributable to an accelerated Ca2+ influx.
Fig. 3.

Photometric measurement of Ca2+transients can detect small changes in presynaptic Ca2+ influx. A, The first (dotted line) and second (solid line) IPSCs activated by a paired-pulse protocol are aligned to show that facilitation is primarily associated with a leftward shift in the IPSC rising phase. Also superimposed is a reference IPSC recorded in 10 mm TEA. The reference IPSC (broken line) exhibits a faster rising phase than the control IPSC (dotted line) but has a lower peak amplitude. B, The Ca2+ transient corresponding to the reference IPSC (broken line) shows a faster rising phase than the transients elicited by the first and second action potentials of the paired-pulse protocol. (The trace styles are matched to those in A.) Thus, the resolution of the photometric measurement is such that it can detect changes in Ca2+ influx that result in crossing between the reference and control IPSCs shown in A.C, Action potentials recorded simultaneously withtraces shown in A and B, with matched trace patterns. The arrow indicates a point 1.5 msec after the peak of the action potentials. Eachtrace represents the average of 100 trials.
An alternative and more quantitative approach, which circumvents the complications arising from changing spike duration, is to analyze the IPSC time course before the first and second action potentials significantly diverge. To implement this analysis, one needs to establish a quantitative description of the relationship between the IPSC time course and the shape of action potentials in control, unfacilitated responses. Figure4A3illustrates a family of action potentials recorded as K+ channel block progressed with increasing TEA concentration. The corresponding IPSCs are shown in Figure 4A1. The broadest spike (dotted line) activates an IPSC with the slowest rising phase, although its peak amplitude is larger than that of all other IPSCs (Fig. 4A1, dotted line). Ca2+ transients follow a similar trend (Fig. 4A2). The slow initial rise of the IPSC associated with the broadest spike can be attributed to a small initial Ca2+ influx because of a small driving force. We constructed a “depolarization–release” (D–R) coupling relationship by plotting IPSC integral (∫IPSC) against membrane potential at the falling phase of presynaptic spike. Specifically, presynaptic membrane potential was measured 1.5 msec after the peak of an action potential (Fig. 4A3,arrow). IPSC was integrated between 1.5 and 2.5 msec after the peak of the presynaptic spike (Fig.4A1, double arrows). [The reason for using IPSC integral, rather than amplitude at a single time point, is to better incorporate differences in their initial trajectories. The choice of the integration time window is based on knowledge of synaptic delay at the crayfish neuromuscular junctions (Augustine et al., 1985; Vyshedskiy and Lin, 1997a).] Data measured from traces in Figure4A, after ∫IPSCs are normalized, are plotted in Figure 4B (open circles) (see figure legends for details of data normalization). The data points are distributed along a bell-shaped curve, similar to the depolarization–release coupling plots obtained in other preparations (Katz and Miledi, 1967; Llinás et al., 1981; Lin and Llinás, 1993). Results measured from three additional preparations are also shown by different symbols. The bell-shaped distribution defines the range of ∫IPSC, which can be modulated by varying action potential time course. When the same measurements are made for the second spike and facilitated IPSC applied to the broadest action potentials, the data points cluster around an area well above the bell-shaped curve (Fig. 4B,filled symbols) (note that the y-axis is on a logarithmic scale). Because the difference in amplitude between the first and second spikes is <1 mV, 1.5 msec after the peak of the action potential (Fig. 3C, arrow), the bell-shaped curve dictates that this small difference would not be able to generate a change in Ca2+ influx that is large enough to account for the increase in facilitated IPSC. Therefore, changes in action potential waveform observed with the paired-pulse protocol is not likely to contribute to the accelerated release during facilitation. Measurements obtained from two additional preparations in which only facilitation associated with the broadest spike were analyzed are also shown (filled diamonds,plus signs).
Fig. 4.

Systematic changes in the IPSC time course and synaptic delay as the duration of action potential increases.A1, A2, andA3 illustrate a series of IPSCs, Ca2+ transients, and action potentials recorded simultaneously as the block of K+ channels becomes more pronounced. The dotted traces represent responses activated by the broadest action potential. The Ca2+transient and IPSC elicited by the broadest action potential exhibit the slowest initial rate of rise. The double arrows inA1 define the time window within which the integral of IPSC was calculated (∫IPSC). The arrow inA3 identifies the point at which presynaptic membrane potential was measured, 1.5 msec after the peak (Vpre). Each tracerepresents the average of 100 trials. B, A depolarization–release coupling plot obtained by plotting ∫IPSC against Vpre. Results measured from the preparation shown in A are in open circles. Three additional preparations are also shown using different symbols. ∫IPSCs calculated in each preparation were normalized by scaling their averaged values to 1. Facilitated ∫IPSCs, shown as filled symbols, were then scaled according to their corresponding control ≥IPSCs. Two of the preparations (filled diamonds, plus signs) only include data obtained with the broadest action potentials. In these cases, their control ∫IPSCs were scaled to fall on the smooth curve, which is drawn by hand.C, A systematic increase in synaptic delay as the duration of presynaptic action potential increases. Action potential duration was measured at 0 mV (see Results for the measurement of synaptic delay). Results obtained from different preparations are shown using different symbols. Action potential duration ranging from 2 to 3.5 msec is not available because action potentials broaden in a large step as Ca2+ spikes appear.
In addition, traces in Figure4A1 suggest a gradual increase in synaptic delay as the duration of action potential increases. Because this change will be interpreted below in the context of accelerated release during F2 facilitation, a quantitative analysis is provided here. An ideal definition of synaptic delay would require a latency histogram of quantal events (Katz and Miledi, 1965; Barrett and Stevens, 1972; Parnas et al., 1989). However, it is not possible to observe unitary IPSCs because of the small chloride driving force. Instead, we choose to define synaptic delay as the time interval between the peak of presynaptic spike and the point at which an IPSC crosses a threshold set to 5 SDs, of the background noise level, beyond baseline. When synaptic delay is plotted against the duration of the spike at 0 mV, the two parameters are closely correlated (correlation coefficient, 0.85; p < 0.001) (Fig.4C).
A decrease in synaptic delay during F2 facilitation
Assuming facilitated transmitter release is mediated by processes downstream of Ca2+ influx, what are the characteristics of the facilitated release? To address this question, we examined the synaptic delay of facilitated IPSCs. Figure5A shows control (top dotted line) and facilitated (top solid line) IPSCs with their corresponding presynaptic spikes (bottom traces) recorded in 20 (A), 4 (B) and 1 mm TEA. [To compare the changes in synaptic delay measured in different levels of K+channel block, it was necessary to ensure that the magnitude of facilitation remained constant. This goal was accomplished by adjusting the number of conditioning action potentials such that the peaks of the fluorescence transients, and therefore the Ca2+ concentration, activated by conditioning stimuli remained relatively constant (see figure legend) (Vyshedskiy and Lin, 1997b, 2000).]
Fig. 5.

A reduction in synaptic delay during F2 facilitation. Traces in A–C were recorded in 20, 4, and 1 mm TEA, respectively (1 mm 4-AP was present during all recordings). Control (dotted lines) and test (solid lines) action potentials are shown in the bottom panels, and control and facilitated IPSC with matching trace stylesare shown in the top panels. Insetsenlarge the area in which IPSCs start to deviate from baseline. Thehorizontal lines in the insets represent the 5 SD level at which the synaptic delay is measured. Broken traces in the inset represent control IPSCs after they have been scaled (see Results and Fig. 6 for details of scaling). There is a significant increase in the difference between the synaptic delays of control and facilitated IPSCs as the duration of action potential is increased. This trend is consistent regardless of whether the control IPSC is scaled or not. To examine changes in synaptic delay under comparable levels of synaptic facilitation as the duration of action potentials varied, 1 (A), 7 (B), and 13 (C) conditioning action potentials were elicited to activate a relatively constant peak amplitude of fluorescence transients, 15, 22, and 18% respectively. The broad second peak of the control action potential inB was attributable to the average of jittering second spikes. Traces in all panels represent the average of 120 trials.
The synaptic delay of facilitated IPSC, measured at the 5 SD level, is shorter than that of control IPSC by 0.34 msec (Fig. 5A,inset). (Horizontal lines in Fig. 5,insets, represent the 5 SD levels as defined in Fig.4C.) Such a reduction in synaptic delay, however, is only apparent with broad action potentials. The change is significantly smaller in lower levels of K+ channel block (Fig. 5B,C,inset). A systematic change in synaptic delay is apparent when the difference in the synaptic delays of control and facilitated IPSCs (Δdelay) is plotted against the duration of control spikes measured at 0 mV (Fig. 6). (Data measured from traces in Fig. 5 are shown as open crosses.) Results measured from 11 additional preparations are also shown as different open symbols. The correlation between Δdelay and the duration of action potential is statistically significant (correlation coefficient, 0.83; p < 0.001). This plot, however, is not ideal in that the 5 SD criterion overestimates the difference in synaptic delay for narrow spikes. For example, in some preparations, IPSCs recorded in control saline were so small that they peaked at approximately the 5 SD level.
Fig. 6.

Summary of the difference in synaptic delay between control and facilitated IPSCs. Δdelay represents the difference in synaptic delay between control and facilitated IPSCs. Action potential duration was measured at 0 mV from control action potentials. Δdelays measured without scaling control IPSCs are shown in open symbols. Δdelays measured after scaling control IPSC are shown in filled symbols (see Results and insets in Fig. 5 for details on measuring synaptic delay). Each symbol represents a different preparation. The straight lines are drawn by hand.
An alternative, and more traditional, method for comparing synaptic delay between control and facilitated IPSCs is to scale control IPSCs to the same height as facilitated ones. This criterion, however, cannot be applied to broad action potentials in which the test spike is significantly narrower than the control one. Therefore, we divide our data into two groups. In the first group, which contains those involving narrow spikes, we scaled the peak amplitude of control IPSC to the same height as facilitated IPSC and then compared changes in minimal delay (Fig. 5C, broken line). This approach is justified because action potentials included in this group typically had not diverged when IPSCs peaked (Fig. 5C,arrow). Similar to previous studies (Datyner and Gage, 1980;Parnas et al., 1989), the synaptic delay of facilitated IPSC did not change (Fig. 5C, inset, solid line). In the second group, which contained broad action potentials, we search for a point in the presynaptic spike at which the difference between the control and test action potentials was 2 mV (±0.1 mV) (Fig.5A,B, arrows). IPSCs measured 1 msec after this point (Fig.5A,B, dotted vertical lines) were scaled to the same height for latency comparison (Fig.5A,B, insets,broken lines). The difference in synaptic delay, for both groups, was then measured at the 5 SD level of the unscaled IPSC. Data measured this way are shown as filled symbols in Figure 6 in which Δdelay drops down to nearly zero for narrow action potentials. The trend of increasing Δdelay as action potential duration lengthens remains statistically significant (correlation coefficient, 0.68;p < 0.01). (Variations in the criteria for scaling did not change the statistical significance of the correlation. For example, scaling IPSCs at the time point at which the difference between control and test spikes was 2 mV, instead of introducing a 1 msec delay, yielded a similar result.) Therefore, expression of the F2 component of synaptic facilitation shifts from an increase in amplitude to a reduction in synaptic delay as the duration of presynaptic spike increases.
DISCUSSION
Historically, the F2 component of synaptic facilitation has been studied by action potential-based protocols performed in control or low Ca2+ saline (Magleby, 1987; Bittner, 1989). The main characteristic of this component is its decay time constant, ∼300–500 msec. Because the facilitation shown here exhibits a similar decay time constant (Vyshedskiy and Lin, 1997c), we assume it is F2 facilitation. The use of presynaptic voltage steps or broad spikes to investigate facilitation appears to be a significant departure from action potential-based protocols used traditionally. However, these approaches create two unique conditions that provide insights not possible with regular spikes. Specifically, we can analyze facilitation over a prolonged period of Ca2+ influx with small Ca2+ channel currents compared with the large single-channel currents in the form of brief impulses typically associated with narrow spikes.
Here, we have demonstrated that the accelerated release during F2 facilitation cannot be attributed to plasticity inICa or to changes in Ca2+ influx dictated by spike waveforms. Measurements of synaptic delay showed that this parameter decreased during facilitation but increased as spike duration lengthened. A hypothesis is proposed to explain these changes.
The role of presynaptic Ca2+ influx during facilitation
We have used fluorescent transients to examine possible roles of presynaptic ICa in F2 facilitation. The underlying assumption of this approach is that the time course of Ca2+ transients closely reflects that of Ca2+ influx (Sinha et al., 1997; Sabatini and Regehr, 1998). Using the presynaptic voltage-control protocol, we showed that facilitated IPSP exhibits a significantly earlier rising phase, whereas the Ca2+ transient recorded simultaneously remains unchanged. In addition, we empirically calibrated the sensitivity of fluorescence measurements in individual preparations by correlating them with IPSCs. We found that the initial rate of rise of IPSC activated by broad spikes was slower than that of IPSC activated by narrow reference spikes. This difference in IPSC rising phase was matched by their corresponding Ca2+ transients. This match empirically defines the sensitivity of photometric measurements and provides the basis for our conclusion that accelerated IPSC cannot be attributed to facilitated ICa.
With our empirically defined sensitivity, it remained possible that small changes in the waveform of Ca2+influx might have escaped detection. However, analysis of the D–R coupling relationship also suggested that the dramatic acceleration in facilitated release could not be attributed to changes inICa. Specifically, the bell-shaped D–R coupling curve defines the range of ∫IPSCs that can be modulated by changes in Ca2+ influx associated with varying spike duration. Because facilitated ∫IPSCs lie approximately one order of magnitude above the control D–R coupling curve, variations in spike duration would not be able to create large enough increases in Ca2+ influx to account for the facilitated ∫IPSCs.
Changes in the kinetics of transmitter release during F2 facilitation
Having concluded that F2 facilitation cannot be attributed to plasticity in ICa or variations in spike waveform, a logical next step was to determine which aspect of the release process downstream of Ca2+influx was involved. Because of the different widths of control and test spikes in 20 mm TEA, it was impossible to investigate release kinetics over the entire duration of IPSC. Therefore, we focused on the earliest events of the release process, synaptic delay. We observed an increase in synaptic delay as the action potential duration lengthened. Such an increase has been shown in other preparations (Datyner and Gage, 1980; Sabatini and Regehr, 1996), although the underlying cause was not addressed. The time window relevant to synaptic delay should be the brief period after the peak of an action potential. Within this period, prolonging spike duration should increase the number of open channels and channel open time but decrease single-channel current. Assuming the first two factors are not the cause of the prolonged delay, the role of single-channel current in synaptic delay must be considered. Modeling studies suggest that Ca2+ concentration around channel mouths, <50 nm, reaches a plateau within a few hundred microseconds after channel opening (Yamada and Zucker, 1992; Roberts, 1994; Bertram et al., 1996; Wu et al., 1996; Klingauf and Neher, 1997). Changes in single-channel current dictate local Ca2+concentration but have little effect on the time required to reach the plateau (Roberts, 1994; Bertram et al., 1996). Therefore, it is unlikely that the prolonged synaptic delay associated with broad spikes reflects an increase in the time required for local Ca2+ to plateau. We propose that the low Ca2+ concentration associated with a small single-channel current is insufficient to trigger immediate release of a vesicle in its control state but can do so if the vesicle is in a facilitated state (Vyshedskiy et al., 1998; Stevens and Wesseling, 1999). Specifically, the low local Ca2+concentration would fill only a high-affinity site, which in turn would convert a vesicle from control to facilitated state. Facilitated vesicles would subsequently release in the presence of a low concentration of Ca2+. The prolonged synaptic delay of unfacilitated IPSC observed with broad spikes would be attributable to the conversion process, which is assumed to be slow. A decrease in the synaptic delay of facilitated IPSCs would then be expected if the conversion had already occurred. The capacity of a low Ca2+ level to mediate facilitated release is consistent with a previous observation that small presynaptic pulses that failed to activate detectable release under control conditions could trigger facilitated release (Vyshedskiy and Lin, 1997b). Therefore, we further propose that binding of the high-affinity site could increase the affinity of the remaining Ca2+ binding sites in a way similar to the interaction among oxygen binding sites of hemoglobin molecules. Finally, the slow conversion process provides a possible explanation for the peculiar observation that the peak of F2 facilitation exhibits a detectable delay, rather than occurring immediately after conditioning stimuli (Mallart and Martin, 1967; Bittner, 1989; Regehr et al., 1994) (A. Vyshedskiy and J.-W. Lin, unpublished observations).
If this hypothesis is correct, how could one explain the facilitation observed with narrow spikes in which there is an increase in amplitude but not synaptic delay of postsynaptic responses (Datyner and Gage, 1980; Parnas et al., 1989) (Fig. 5)? Potentially, a large single-channel current, associated with the rapid repolarization of a narrow spike, could increase local Ca2+concentration sufficiently to trigger release from vesicles in the control state with minimal delay. The extra release during facilitation would come from brief single-channel openings, which would raise local Ca2+ concentration high enough to trigger release only from facilitated and not from control vesicles. Under these conditions, one would not expect a detectable change in synaptic delay.
Currently, the most parsimonious interpretation of the conversion process is to assume that occupation of the high-affinity site increases the affinity of the remaining Ca2+ binding sites involved in vesicular secretion. Furthermore, the high-affinity site would contribute to the Ca2+ cooperativity of transmitter release defined experimentally. This interpretation is consistent with findings obtained from the squid giant synapse (Llinás et al., 1981;Augustine et al., 1985; Augustine and Charlton, 1986) in which it is possible to correlate ICa and transmitter release at a high time resolution. It was shown that EPSC onsets were delayed when large presynaptic steps were applied or when external Ca2+ concentration was decreased [see also results from the calyx of Held (Wu and Borst, 1999)]. However, the Ca2+ cooperativity of transmitter release remained remarkably constant under those manipulations of Ca2+ influx. These observations are consistent with each other if we accept the notions that (1) delayed EPSC is attributable to the slow conversion process and (2) the high-affinity site contributes to the Ca2+ cooperativity measured experimentally. A corollary of this reasoning is that facilitated release should exhibit reduced calcium cooperativity when the high-affinity site is occupied. This was indeed observed in previous studies (Carlson and Jacklet, 1986; Stanley, 1986; Wright et al., 1996;Vyshedskiy and Lin, 1997b).
In conclusion, we propose that small single Ca2+ channel currents raise local Ca2+ concentration high enough to fill the high-affinity site, but not the low-affinity sites, involved in the secretion process. Binding of the high-affinity site would then increase the affinity of low-affinity sites. The prolonged synaptic delay observed with broad spikes could be attributable to a slow on rate of the high-affinity site or a slow transition in the affinity of low-affinity sites. Synaptic delay would be shortened during facilitation because the slow step would already have occurred. Furthermore, enhanced transmitter release during facilitation would be attributable to an overall increase in the Ca2+ binding affinity of the release machinery. Mathematical simulation should enable estimation of the slow on rate of the high-affinity site from the prolonged synaptic delay. The off rate calculated from this estimate should theoretically correspond to the decay time constant of facilitation. However, it may be necessary to introduce a separate slow step, such as the conversion of Ca2+ binding affinity, to fully account for the kinetics of F2 facilitation.
Footnotes
This work was supported by National Institutes of Health Grant NS31707 (to J.W.L.). We thank Wade Regehr for advice on photometric recordings and Nicky Schweitzer for correcting our English.
Correspondence should be addressed to Dr. Jen-Wei Lin, Department of Biology, Boston University, 5 Cummington Street, Boston, MA 02215. E-mail: jenwelin@bio.bu.edu.
REFERENCES
- 1.Atwood HL, Wojtowicz JM. Short-term and long-term plasticity and physiological differentiation of crustacean motor synapses. Int Rev Neurobiol. 1986;28:275–362. doi: 10.1016/s0074-7742(08)60111-7. [DOI] [PubMed] [Google Scholar]
- 2.Augustine GJ, Charlton MP. Calcium dependence of presynaptic calcium current and postsynaptic response at the squid giant synapse. J Physiol (Lond) 1986;381:619–640. doi: 10.1113/jphysiol.1986.sp016347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Augustine GJ, Charlton MP, Smith SJ. Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. J Physiol (Lond) 1985;367:163–181. doi: 10.1113/jphysiol.1985.sp015819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrett EF, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. J Physiol (Lond) 1972;227:691–708. doi: 10.1113/jphysiol.1972.sp010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertram R, Sherman A, Stanley EF. Single-domain/bound calcium hypothesis of transmitter release and facilitation. J Neurophysiol. 1996;75:1919–1931. doi: 10.1152/jn.1996.75.5.1919. [DOI] [PubMed] [Google Scholar]
- 6.Bittner GD. Synaptic plasticity at the crayfish opener neuromuscular preparation. J Neurobiol. 1989;20:386–406. doi: 10.1002/neu.480200510. [DOI] [PubMed] [Google Scholar]
- 7.Borst JGG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brain stem. J Physiol (Lond) 1998;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlson CG, Jacklet JW. The exponent of the calcium power function is reduced during steady-state facilitation in neuron R15 of Aplysia. Brain Res. 1986;376:204–207. doi: 10.1016/0006-8993(86)90919-4. [DOI] [PubMed] [Google Scholar]
- 9.Charlton M, Smith SJ, Zucker RS. Role of presynaptic calcium ions and channels in synaptic facilitation and depression at the squid giant synapse. J Physiol (Lond) 1982;323:173–193. doi: 10.1113/jphysiol.1982.sp014067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol (Lond) 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Datyner NB, Gage PW. Phasic secretion of acetylcholine at a mammalian neuromuscular junction. J Physiol (Lond) 1980;303:299–314. doi: 10.1113/jphysiol.1980.sp013286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Katz B, Miledi R. The measurements of synaptic delay, and the time course of acetylcholine release as the neuromuscular junction. Proc R Soc Lond B Biol Sci. 1965;161:483–495. doi: 10.1098/rspb.1965.0016. [DOI] [PubMed] [Google Scholar]
- 13.Katz B, Miledi R. A study of synaptic transmission in the absence of nerve impulses. J Physiol (Lond) 1967;192:407–436. doi: 10.1113/jphysiol.1967.sp008307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Katz B, Miledi R. The role of calcium in neuromuscular facilitation. J Physiol (Lond) 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kawasaki F, Mattiuz A, Ordway RW. Synaptic physiology and ultrastructure in comatose mutants define an in vivo role for NSF neurotransmitter release. J Neurosci. 1998;18:10241–10249. doi: 10.1523/JNEUROSCI.18-24-10241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein M. Synaptic augmentation by 5-HT at rested Aplysia sensorimotor synapses: independence of action potential prolongation. Neuron. 1994;13:159–166. doi: 10.1016/0896-6273(94)90466-9. [DOI] [PubMed] [Google Scholar]
- 17.Klingauf J, Neher E. Modeling buffered Ca2+ diffusion near the membrane: implications for secretion in neuroendocrine cells. Biophys J. 1997;72:674–690. doi: 10.1016/s0006-3495(97)78704-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin J, Llinás R. Depolarization-activated potentiation of the T-fiber synapse in the blue crab. J Gen Physiol. 1993;101:45–65. doi: 10.1085/jgp.101.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Llinás R, Steinberg IZ, Walton K. Relationship between presynaptic calcium current and postsynaptic potential in squid giant synapse. Biophys J. 1981;33:323–352. doi: 10.1016/S0006-3495(81)84899-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magleby KL. Short-term changes in synaptic efficacy. In: Edelman GM, Gall WE, Cowan WM, editors. Synaptic function. Wiley; New York: 1987. pp. 21–56. [Google Scholar]
- 21.Mallart A, Martin AR. An analysis of facilitation of transmitter release at the neuromuscular junction of the frog. J Physiol (Lond) 1967;193:679–694. doi: 10.1113/jphysiol.1967.sp008388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parnas H, Hovav G, Parnas I. Effect of Ca2+ diffusion on the time course of neurotransmitter release. Biophys J. 1989;55:859–874. doi: 10.1016/S0006-3495(89)82885-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Regehr WG, Delaney KR, Tank DW. The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapse. J Neurosci. 1994;14:523–537. doi: 10.1523/JNEUROSCI.14-02-00523.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts WM. Localization of calcium signals by a mobile calcium buffer in frog saccular hair cells. J Neurosci. 1994;14:3246–3262. doi: 10.1523/JNEUROSCI.14-05-03246.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- 26.Sabatini BL, Regehr WG. Optical measurement of presynaptic calcium currents. Biophys J. 1998;74:1549–1563. doi: 10.1016/S0006-3495(98)77867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schweizer FE, Dresbach T, Debello WM, O'Connor V, Augustine GJ, Betz H. Regulation of neurotransmitter release kinetics by NSF. Science. 1998;279:1203–1206. doi: 10.1126/science.279.5354.1203. [DOI] [PubMed] [Google Scholar]
- 28.Sinha SR, Wu L-G, Saggau P. Presynaptic calcium dynamics and transmitter release evoked by single action potentials at mammalian central synapses. Biophys J. 1997;72:637–651. doi: 10.1016/s0006-3495(97)78702-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stanley EF. Decline in calcium cooperativity as the basis of facilitation at the squid giant synapse. J Neurosci. 1986;6:782–789. doi: 10.1523/JNEUROSCI.06-03-00782.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevens CF, Wesseling JF. Augmentation is a potentiation of the exocytotic process. Neuron. 1999;22:139–146. doi: 10.1016/s0896-6273(00)80685-6. [DOI] [PubMed] [Google Scholar]
- 31.Vyshedskiy A, Lin J-W. A study of the inhibitor of the crayfish neuromuscular junction by presynaptic voltage control. J Neurophysiol. 1997a;77:103–115. doi: 10.1152/jn.1997.77.1.103. [DOI] [PubMed] [Google Scholar]
- 32.Vyshedskiy A, Lin J-W. Activation and detection of facilitation as studied by presynaptic voltage control at the inhibitor of the crayfish opener muscle. J Neurophysiol. 1997b;77:2300–2315. doi: 10.1152/jn.1997.77.5.2300. [DOI] [PubMed] [Google Scholar]
- 33.Vyshedskiy A, Lin J-W. Change of transmitter release kinetics during facilitation revealed by prolonged test pulses at the inhibitor of the crayfish opener muscle. J Neurophysiol. 1997c;78:1791–1799. doi: 10.1152/jn.1997.78.4.1791. [DOI] [PubMed] [Google Scholar]
- 34.Vyshedskiy A, Lin J-W. Presynaptic calcium influx at the inhibitor of the crayfish neuromuscular junction: a photometric study at a high time resolution. J Neurophysiol. 2000;83:552–562. doi: 10.1152/jn.2000.83.1.552. [DOI] [PubMed] [Google Scholar]
- 35.Vyshedskiy A, Delaney K, Lin J-W. Neuromodulators enhance transmitter release by two separate mechanisms at the inhibitor of crayfish opener muscle. J Neurosci. 1998;18:5160–5169. doi: 10.1523/JNEUROSCI.18-14-05160.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winslow JL, Duffy SN, Charlton MP. Homosynaptic facilitation of transmitter release in crayfish is not affected by mobile calcium chelators: implications for the residual ionized calcium hypothesis from electrophysiological and computational analysis. J Neurophysiol. 1994;72:1769–1793. doi: 10.1152/jn.1994.72.4.1769. [DOI] [PubMed] [Google Scholar]
- 37.Wright SN, Brodwick MS, Bittner GD. Calcium currents, transmitter release and facilitation of release at voltage-clamped crayfish nerve terminals. J Physiol (Lond) 1996;496:363–378. doi: 10.1113/jphysiol.1996.sp021691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu L-G, Borst JGG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- 39.Wu Y-C, Tucker T, Fettiplace R. A theoretical study of calcium microdomains in turtle hair cells. Biophys J. 1996;71:2265–2275. doi: 10.1016/S0006-3495(96)79429-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamada MW, Zucker RS. Time course of transmitter release calculated from stimulations of a calcium diffusion model. Biophys J. 1992;61:671–682. doi: 10.1016/S0006-3495(92)81872-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]