

Abstract
Activation of group III metabotropic glutamate receptors (mGluR4, mGluR6, mGluR7, and mGluR8) has been established to be neuroprotectivein vitro and in vivo. To disclose the identity of the receptor subtype(s) that exert(s) the protective effect, we have used group III agonists in combination with mGluR4 subtype-deficient mice (−/−). In cortical cultures prepared from wild-type (+/+) mice and exposed to a toxic pulse of NMDA, the selective group III agonist (+)-4-phosphonophenylglycine [(+)-PPG] reversed excitotoxicity with an EC50 value of 4.9 μm, whereas its enantiomer (−)-PPG was inactive. This correlated closely with the potency of (+)-PPG in activating recombinant mGluR4a. In cortical neurons from −/− mice, (+)-PPG showed no protection against the NMDA insult up to 300 μm, whereas group I/II mGluR ligands still retained their protective activity. Classical group III agonists (l-2-amino-4-phosphonobutyrate andl-serine-O-phosphate) were also substantially neuroprotective against NMDA toxicity in +/+ and heterozygous (+/−) cultures but were inactive in −/− cultures. Interestingly, −/− cultures were more vulnerable to low concentrations of NMDA and showed higher extracellular glutamate levels compared with +/+ cultures.
We have also examined neurodegeneration induced by intrastriatal infusion of NMDA in wild-type or mGluR4-deficient mice. Low doses of (R,S)-PPG (10 nmol/0.5 μl) substantially reduced NMDA toxicity in +/+ mice but were ineffective in −/− mice. Higher doses of (R,S)-PPG were neuroprotective in both strains of animals. Finally, microdialysis studies showed that intrastriatal infusion of NMDA increased extracellular glutamate levels to a greater extent in −/− than in +/+ mice, supporting the hypothesis that the mGluR4 subtype is necessary for the maintenance of the homeostasis of extracellular glutamate levels.
Keywords: neurodegeneration, knock-out mice, cortical cultures, metabotropic glutamate receptors, gene targeting, excitotoxicity
Metabotropic glutamate receptors form a family of currently eight subtypes (mGluR1–GluR8), subdivided into three groups (I–III) on the basis of their amino acid sequence identities, pharmacological profiles, and signal transduction pathways (Nakanishi, 1994; Conn and Pin, 1997). Group III includes mGluR4, mGluR6, mGluR7, and mGluR8 subtypes, which are negatively coupled to adenylate cyclase in heterologous expression systems. In the mammalian brain, mGluR4, mGluR7, and mGluR8 are presynaptically localized and are thought to mediate presynaptic depression of glutamatergic synaptic potentials, most likely via inhibition of voltage-gated calcium entry and regulation of glutamate release (Trombley and Westbrook, 1992; Conn and Pin, 1997; Shigemoto et al., 1997). In contrast, mGluR6 appears to be exclusively expressed in retinal ON bipolar cells in which it couples to a cGMP-phosphodiesterase and amplifies visual transmission (Nakanishi, 1994). l-AP-4,l-serine-O-phosphate (l-SOP), (R,S)- 4-phosphonophenylglycine [(R,S)-PPG], and close analogs are the only selective agonists known for group III mGluRs, with low micromolar potency (EC50 values of 0.1–7 μm) at mGluR4, mGluR6, and mGluR8; mGluR7 can only be activated at concentrations higher than 100 μm (Okamoto et al., 1994; Johansen et al., 1995; Conn and Pin, 1997; Flor et al., 1997; Wu et al., 1998; Gasparini et al., 1999a). Selective activation of group III mGluRs results in neuroprotection in vitro: agonists such asl-AP-4, l-SOP, and (R,S)-PPG promote survival of rat cerebellar granule cells and protect cultured cortical and cerebellar neurons against toxic insults, such as prolonged β-amyloid peptide exposure, transient ionotropic glutamate receptor activation, or mechanical damage (Graham and Burgoyne, 1994; Copani et al., 1995; Bruno et al., 1996; Faden et al., 1997; Gasparini et al., 1999a). In addition to those in vitro studies, the group III agonist (R,S)-PPG was recently found neuroprotective and anticonvulsive in vivo (Gasparini et al., 1999a). However, l-AP-4,l-SOP, and (R,S)-PPG are not receptor subtype-specific drugs, and they share properties that cannot always be reconciled with the activation of group III mGluRs, such as the noncompetitive inhibition of excitatory amino acid-induced polyphosphoinositide hydrolysis in brain tissue (Nicoletti et al., 1986; Schoepp et al., 1990) and the binding to the Ca2+/Cl−-dependent glutamate transporter (Fagg et al., 1982; Gasparini et al., 1999a). Because of that, some concern remains on the role of group III mGluRs in neuroprotection.
Here we attempt to disclose the contribution of individual receptor subtypes to neuroprotection induced by group III agonists. Therefore, we used various group III agonists, including the two purified enantiomers of (R,S)-PPG [(+)-PPG and (−)-PPG], and we compared agonist activity at cloned receptors with neuroprotective effects in cortical cultures, which were prepared from wild-type (+/+), heterozygous (+/−), and mGluR4 subtype-deficient (−/−) mice. We have also performed in vivo studies by injecting NMDA into the caudate nucleus of +/+ or −/− mice.
MATERIALS AND METHODS
Chemical synthesis of (R,S)-PPG, (+)-PPG and (−)-PPG.(R,S)-PPG was synthesized in four steps starting from 4-hydroxybenzaldehyde as described previously (Gasparini et al., 1999a). (+)- and (−)-PPG were purified from (R,S)-PPG as starting material; enantiomeric resolution was realized by chromatographic separation of the fully protected enantiomers. Details will be described elsewhere (F. Gasparini, manuscript in preparation).
Breeding of wild-type (CD-1), heterozygous, and homozygous mGluR4-deficient mutant mice. Two breeding pairs of homozygous mGluR4-deficient mice (Pekhletski et al., 1996) were shipped from the University of Toronto (Toronto, Canada) to Novartis Pharma AG (Basel, Switzerland), and mating procedures of homozygotes and CD-1 wild types were performed according to standard procedures. The genotype at the mGluR4 locus was confirmed by Southern blotting and multiple primer PCR performed on tail biopsy samples of parent mice (for a detailed description, see Pekhletski et al., 1996).
Preparation of cultured cortical cells from wild-type, heterozygous, and homozygous mGluR4-deficient mice. Mixed cultures of cortical cells containing both neurons and astrocytes were prepared from fetal mice (14–16 d of gestation), as described previously (Bruno et al., 1996, 1997), and were used 13–14 d after plating.
Assessment of NMDA toxicity in culture. Mixed cortical cultures were exposed to 100 μm NMDA for 10 min at room temperature in a HEPES-buffered salt solution. After extensive washing, cultures were incubated for 22–24 hr at 37°C in MEM–Eagle's buffer (Life Technologies, Basel, Switzerland) supplemented with 25 mmNaHCO3 and 21 mm glucose. Neuronal toxicity was examined by phase-contrast microscopy and quantitated after staining with trypan blue. Stained neurons were counted from three random fields per well. Lactate dehydrogenase (LDH) release into the medium was also measured either as described previously (Bruno et al., 1996) or by using a commercially available kit (Promega, Madison, WI).
d-[3H]Aspartate uptake in cortical cultures. Mixed cortical cultures derived from wild-type or mGluR4-deficient mice were washed twice in Locke's solution and exposed for 15 min to 60 nCi/well ofd-[2,3-3H]aspartate (specific activity, 20 Ci/mmol; Amersham Pharmacia Biotech, Milano, Italy) in the absence or presence of group III mGluR agonists. At the end of the exposure, cultures were washed three times in Locke's solution and lysed in 0.5 m NaOH. The lysate was counted by scintillation spectrometry.
Determination of extracellularl-glutamate levels. Mixed cultures of cortical neurons were prepared, and excitotoxicity was performed as described above. Analysis of glutamate was performed by precolumn derivatization with o-phthalaldehyde and mercaptoethanol, followed by HPLC with fluorescence detection. Culture medium was collected at the end of the NMDA exposure (100 μm in +/+ culture and 60 μm in −/− culture) in the presence or absence of l-AP-4 (100 μm), (R,S)-PPG (100 μm), and (R,S)-α-methyl-4-phosphonophenylglycine (MPPG) (100 μm). One hundred microliter sample aliquots were diluted with 0.1 N HCl and mixed with equal volumes of fluorescent reagent. The mixture was kept at room temperature for 1 min to derivatize the sample before being injected into the column by a 200 μl loop. The system used an autosampler 507 (Beckman Instruments, Fullerton, CA), a programmable solvent module 126 (Beckman Instruments), an analytical reverse phase C-18 column at 30°C (Ultrasphere ODS 3 μm Spherical, 80 Å pore, 2 × 250 mm; Beckman Instruments), an RF-551 spectrofluorimetric detector (Shimadzu, Tokyo, Japan), and a computer running a Gold Nouveau software (Beckman Instruments). The excitation and emission wavelengths were set at 360 and 450 nm, respectively. The mobile phase consisted of (A) 50 mm sodium phosphate, pH 7.2, containing 10% methanol and (B) 50 mm sodium phosphate, pH 7.2, containing 70% methanol, at a flow rate of 0.3 ml/min. Both buffers were filtered through a 0.45 μm filter and degassed under vacuum for 5 min. Gradient elution consisted of 98% A and 2% B initially for 16 min, was then increased to 98% B over 1 min, maintained for 12 min to elute other substances, and then returned to the initial conditions before running the next sample. From peak areas, culture medium concentrations of glutamate were calculated by the use of external standards.
Stable mammalian cell lines for cloned mGluR subtypes and pharmacological assay for cloned group III mGluRs. Generation, culture, and pharmacological characterization of stable cell lines for human (h) mGluR4a, mGluR6, mGluR7b, and mGluR8a have been described previously (Flor et al., 1995b, 1996, 1997; Laurie et al., 1997; Gasparini et al., 1999). Measurements of cAMP accumulation was performed essentially as described previously (Flor et al., 1995a,b, 1997).
Assessment of in vivo neuronal injury. CD-1 wild types or homozygous mGluR4-deficient mice weighing 24–28 gm were used for all the experiments. Mice were injected, under ketamine (100 mg/kg, i.p.) plus xylazine (10 mg/kg, i.p.) anesthesia, with NMDA (50 nmol/0.5 μl) or NMDA (50 nmol/0.5 μl) plus (R,S)-PPG (10 or 100 nmol/0.5 μl) in a stereotaxic frame. The site of injection was the left anterior striatum with the following coordinates: 0.6 mm anterior to the bregma, 1.7 lateral from the midline, and 3.5 mm ventral from the surface of skull, according to the atlas of Franklin and Paxinos (1997). After surgery, mice were housed in separated cages in a temperature-controlled environment on a 12 hr light/dark cycle, with access to water and food ad libitum. The animals were allowed 7 d to develop an excitotoxic striatal neuronal death induced by NMDA. In animals injected with NMDA or NMDA plus (R,S)-PPG, neuronal injury was assessed by performing histological analysis. Serial frontal sections including the whole caudate nucleus were Nissl-stained and examined by phase-contrast microscopy.
Microdialysis in freely moving animals. CD-1 wild types or homozygous mGluR4-deficient mice weighing 24–28 gm were implanted with microdialysis intracerebral guides (CMA/7 Guide Cannula; CMA Microdialysis, Stockholm, Sweden), under ketamine (100 mg/kg, i.p.) plus xylazine (10 mg/kg, i.p.) anesthesia, in a Kopf stereotaxic frame (David Kopf Instruments, Tujunga, CA). The site of implantation was the left striatum [coordinates: 0.6 mm anterior to the bregma, 1.7 mm lateral to the midline, and 2.5–5.5 mm ventral from the surface of skull, according to the atlas of Franklin and Paxinos (1997)]. After surgery, mice were housed in separate cages in a temperature-controlled environment on a 12 hr light/dark cycle, with access to water and foodad libitum, and were allowed to recover for 4 d before the experiment. On the evening before the experiment, a probe was inserted into the intracerebral guide, after removing a dummy, and mice were transferred to a plastic bowl cage with a moving arm (CMA/120 System for Freely Moving Animals; CMA Microdialysis) with access to water and food ad libitum. Concentric vertical microdialysis probes 2 mm long and 0.24 mm in outer diameter having a cuprophane membrane with a molecular cutoff of 6000 Da (CMA/7 Microdialysis Probe; CMA Microdialysis) were used. The probes were perfused continuously with artificial CSF (ACSF), at a flow rate of 1.5 μl/min, using a microinjection pump (Bioanalytical Systems Inc., Congleton, UK) The ACSF contained (in mm): 150 NaCl, 3 KCl, 1.7 CaCl2, and 0.9 MgCl2. This solution was not buffered, and the pH was typically 6.5. On the following morning, 30 μl (20 min) consecutive perfusate sample fractions were continuously collected by a fraction collector (CMA/142 Microfraction Collector; CMA Microdialysis). After four sample fractions, used to determine the basal levels of glutamate, NMDA (37.5 mm) was perfused for 7 min. When needed, (R,S)-PPG (5 mm) was perfused for the 20 min preceding the NMDA infusion. Sample fractions of perfusate were collected for the next 2 hr. Analysis of glutamate was performed by HPLC with fluorescence detection.
Statistics. Significant differences were estimated using the two-tailed Dunnett's t test by comparing multiple test–drug groups with one control group (Dunnett, 1964). Values of 2p < 0.05 were considered statistically significant.
Materials. Tissue culture reagents were from Life Technologies and Sigma (Buchs, Switzerland). Forskolin, 3-isobutyl-1-methylxanthine, and l-SOP were obtained from Sigma. l-AP-4, NMDA, DNQX, and (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801) were purchased from Tocris Cookson (Anawa Trading SA, Zürich, Switzerland). 2-Methyl-6-(phenylethynyl)-pyridine (MPEP) and 7-hydroxyiminocyclopropan[b]chromen-1a-carboxylic acid ethyl ester (CPCCOEt) were synthesized at Novartis Pharma AG (Gasparini et al., 1999b; Litschig et al., 1999). All other chemicals were of reagent grade and were obtained from Fluka (Buchs, Switzerland), Merck (Darmstadt, Germany), Serva (Heidelberg, Germany), or Sigma.
RESULTS
In vitro studies
Neuroprotective activity of the active isomer of (R,S)-PPG in cultured cortical cells
(R,S)-PPG activates mGluR4, mGluR6, mGluR7, and mGluR8 and is neuroprotective against excitotoxic death (Gasparini et al., 1999). To provide a stronger pharmacological evidence that activation of group III mGluRs is neuroprotective, we prepared the pure (+) and (−) stereoisomers of PPG and compared their activities at heterologously expressed mGluR4 with their effects on NMDA toxicity. Toxicity was induced by exposing mixed cultures of cortical cells to a 10 min pulse with NMDA. As shown in Figure1A, only the (+) isomer of PPG was neuroprotective, whereas the (−) isomer was inactive up to 100 μm. The calculated EC50 value for (+)-PPG-evoked neuroprotection was essentially identical to the EC50 value of the compound to inhibit cAMP formation in Chinese hamster ovary (CHO) cells expressing recombinant human mGluR4a (Fig.1B,C), but it was substantially different from the reported potency of (R,S)-PPG at hmGluR7b- or mGluR8a-expressing cells (Gasparini et al., 1999). (−)-PPG had no activity at recombinantly expressed mGluR4 up to 2000 μm (Fig. 1C). These observations confirmed the protective role for group III mGluRs and suggested that mGluR4 rather than mGluR7 or mGluR8 mediates the protective activity of group III mGluR agonists in cultured cortical cells (Bruno et al., 1995; Gasparini et al., 1999).
Fig. 1.

Neuroprotective activity of the two purified PPG enantiomers and correlation with agonist activity at mGluR4.A, Neuronal degeneration in mixed cortical cultures (CD-1 wild-type mice) was induced by 100 μm NMDA (resulting in ∼80% of maximal NMDA toxicity; set to 100%) and assessed by trypan blue staining. Statistically significant neuroprotection by (+)-PPG is indicated by asterisks(2p < 0.01; Dunnett's t test;n = 6; 2 independent experiments).B, Concentration–protection relationship of (+)-PPG against NMDA (100 μm)-induced degeneration in mixed cortical cultures assessed by trypan blue staining. The value measured with 100 μm NMDA alone was taken as 100%. Mean ± SEM values from three independent experiments (n ≥ 7) are shown. C, Concentration–response curves for inhibition of forskolin (10 μm)-stimulated cAMP accumulation by (+)- and (−)-PPG at CHO cells stably expressing human mGluR4a (Flor et al., 1995). Each data point represents the mean ± SEM values from two independent experiments; n≥ 4. To determine the EC50 values, sigmoidal curves were fit using the GraphPad Prism program (GraphPad Software, Inc., San Diego, CA).
Lack of neuroprotective activity of group III mGluR agonists in cultures from mGluR4-deficient mice
To test the above hypothesis, we have prepared mixed cortical cultures from mGluR4-deficient (−/−) embryonic mice (Pekhletski et al., 1996). The lack of functional mGluR4 was confirmed by Southern blotting and multiple primer PCR performed on tail biopsy samples of parent mice (for a detailed description, see Pekhletski et al., 1996; data not shown). When microscopically examined at 13 din vitro (i.e., when toxicity experiments are usually performed), cultures from −/− mice showed a normal morphology of neuronal cell bodies, although the neuritic connections between clusters of neurons were less pronounced than in wild-type (+/+) cultures. Neuronal viability under basal conditions, as well as toxicity in response to high concentrations of NMDA (100 μm), were not significantly different between cultures prepared from +/+, heterozygous (+/−), or −/− mutant mice (Fig. 2A). However, the potency of NMDA in inducing neuronal toxicity was slightly higher in cultures from −/− than in cultures from +/+ mice (EC50 values of 42 ± 2 and 73 ± 4 μm, respectively;means ± SEM) (Fig. 2B), suggesting that cortical neurons from mGluR4-deficient mice are more vulnerable to excitotoxic damage. That endogenous activation of mGluR4 regulates neuronal vulnerability is strengthened by the observation that low/intermediate concentrations of NMDA (30 or 60 μm) produced a greater extent of neuronal death in +/+ cultures when coincubated with MPPG (Table1), a drug that preferentially antagonizes group III mGluRs (Jane et al., 1996).
Fig. 2.
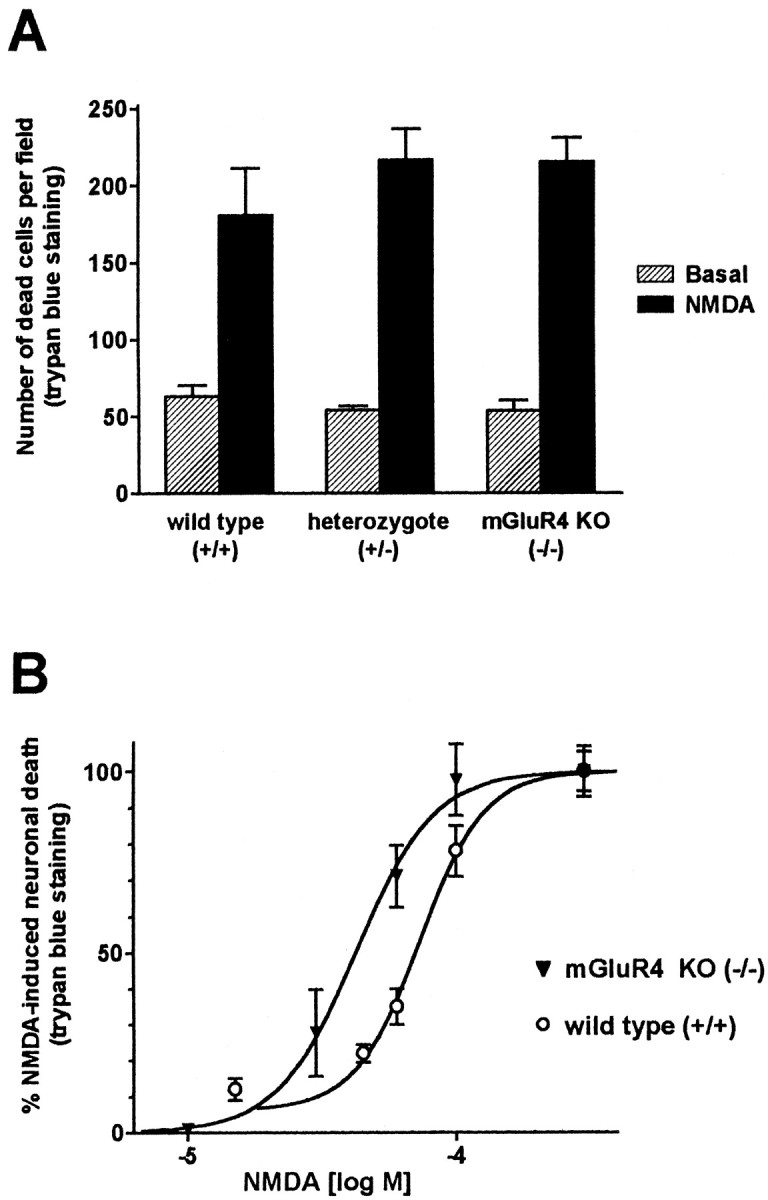
NMDA-induced neuronal toxicity in mixed cortical cultures from wild-type (+/+), heterozygous (+/−), and mGluR4 knock-out (KO; −/−) mice. A, The number of dead cells per microscopic field (250× magnification) was determined after trypan blue staining. Means ± SEM from at least five independent determinations per group are shown; NMDA was applied at 100 μm. Experimental procedures for obtaining the basal values differed from NMDA values only by omitting the toxin.B, Concentration–response relationships for NMDA-induced neurotoxicity in mixed cortical cultures from wild-type and mGluR4 knock-out mice. The maximal extent of toxicity, obtained with 300 μm NMDA, was set to 100%; each experiment was performed three times; n ≥ 8. Sigmoidal curves were fit using the GraphPad Prism program (GraphPad Software, Inc.).
Table 1.
NMDA toxicity in wild-type mouse cortical cultures in the absence (control) or presence of MPPG (100 μm)
| Treatment | Number of dead cells (trypan blue staining) | |
|---|---|---|
| Control | MPPG | |
| Basal | 24 ± 8 | 31 ± 3 |
| NMDA (30 μm) | 77 ± 7 | 144 ± 13* |
| NMDA (60 μm) | 139 ± 5 | 177 ± 5* |
| NMDA (100 μm) | 185 ± 3 | 217 ± 9 |
Values are means ± SEM of four to six independent determinations.
indicates 2p < 0.01 versus the respective control values.
We examined the effect of various mGluR-selective compounds on NMDA toxicity in cortical cultures from the three different mouse genotypes. As shown in Figure 3, the group III agonists l-AP-4, l-SOP, and (R,S)-PPG (all at 100 μm) were substantially neuroprotective against NMDA toxicity in +/+ or +/− cultures (55–80% protection in all cases), but they were all inactive in −/− cultures. The pure enantiomer (+)-PPG tested against NMDA toxicity in −/− cultures showed no significant neuroprotection up to 300 μm (2p > 0.05, Dunnett's t test, n = 6 for each concentration tested). In contrast, (S)-4-carboxy-3-hydroxyphenylglycine (4C3HPG) (a mixed group I antagonist/group II mGluR agonist), showed significant neuroprotection in +/+, +/−, and −/− genotypes (Fig. 3). Similarly, the selective group I mGluR antagonists CPCCOEt (Litschig et al., 1999) and MPEP (Gasparini et al., 1999b), as well as the NMDA receptor antagonist MK-801, were all effective as neuroprotective agents in cultures from mGluR4-deficient mice (Table2).
Fig. 3.

Protection against NMDA-induced neuronal toxicity in mixed cortical cultures from wild type (+/+), heterozygous (+/−), and mGluR4 knock-out (KO; −/−) mice. Neuronal degeneration in wild-type and heterozygous cultures was induced by 100 μm NMDA. In knock-out cultures, 60 μm NMDA was used (resulting in ∼80% of maximal NMDA toxicity for each genotype). This level of neuronal death was set to 100%. Statistically significant neuroprotection by the different mGluR ligands is indicated by asterisks (2p < 0.01; Dunnett'st test; n ≥ 9; 3 independent experiments).
Table 2.
NMDA-induced neuronal death in the presence of different mGluR ligands and NMDA receptor antagonists applied to mixed cortical cultures prepared from mGluR4 knock-out (KO, −/−) or wild-type (+/+) mice
| Compound | (Read-out) | % of NMDA-induced neuronal death | |
|---|---|---|---|
| mGluR4 KO (−/−) | Wild-type (+/+) | ||
| l-AP-4 | (trypan blue staining) | 103 ± 5 | 38 ± 3 |
| (LDH release) | 94 ± 8 | 41 ± 5 | |
| (R,S)-PPG | (trypan blue staining) | 98 ± 6 | 45 ± 5 |
| (LDH release) | 82 ± 12 | 55 ± 5 | |
| 4C3HPG | (trypan blue staining) | 63 ± 4 | 45 ± 5 |
| (LDH release) | 56 ± 9 | 36 ± 3 | |
| CPCCOEt | (LDH release) | 64 ± 16 | 79 ± 5 |
| MPEP | (LDH release) | 5 ± 5 | 32 ± 6 |
| MK-801 | (trypan blue staining) | 27 ± 4 | 12 ± 7 |
| (LDH release) | 18 ± 8 | 15 ± 3 | |
l-AP-4, (R,S)-PPG, and 4C3HPG were applied at a final concentration of 100 μm and CPCCOEt, MPEP, and MK-801 at 30, 3, and 10 μm, respectively. All compounds were coapplied with the NMDA pulse and were maintained in the medium for the following 24 hr. Values are means ± SEM of six to nine independent determinations.
Determination of extracellular l-glutamate levels in cultured cortical cells
Searching for a mechanism responsible for the increased vulnerability of −/− cultures to NMDA toxicity, we measured extracellular glutamate levels because NMDA-stimulated glutamate release contributes to excitotoxic damage (Monyer et al., 1992) and presynaptic group III mGluRs are known to inhibit glutamate release. Interestingly, the basal levels of glutamate (i.e., levels measured in cultures incubated for 10 min in serum- and glutamine-free medium stock but without NMDA) were more than fourfold higher in −/− than in +/+ cultures. The toxic pulse with NMDA increased extracellular glutamate levels to the same extent (approximately twofold over the respective basal levels) in both cultures (Table 3).l-AP-4 or (R,S)-PPG (both at 100 μm) coapplied with NMDA reduced extracellular glutamate levels in both +/+ and −/− cultures but did not normalize the difference in basal glutamate levels between the two cultures. Application of l-AP-4 or (R,S)-PPG with NMDA in −/− cultures never reduced extracellular glutamate below the levels found in +/+ cultures treated with NMDA alone (Table 3).
Table 3.
Extracellular glutamate levels in mGluR4 knock-out (KO, −/−) and wild-type (+/+) mice cultures treated with NMDA and/or group III mGluR agonists or antagonists
| Treatment | l-Glutamate (nm) | |
|---|---|---|
| mGluR4 KO (−/−) | Wild-type (+/+) | |
| Basal | 267 ± 41 | 57 ± 5 |
| l-AP-4 (100 μm) | 167 ± 183-150 | 68 ± 3 |
| (R,S)-PPG (100 μm) | n.d. | 63 ± 9 |
| MPPG (100 μm) | n.d. | 93 ± 73-150 |
| NMDA (60 or 100 μm)3-160 | 470 ± 343-150 | 152 ± 213-150 |
| NMDA plus l-AP 4 | 210 ± 253-159 | 59 ± 133-159 |
| NMDA plus (R,S)-PPG | 150 ± 213-159 | 73 ± 83-159 |
Values are means ± SEM of 6-13 independent determinations.
F3-150: indicates 2p < 0.05 versus the respective basal values.
F3-159: indicates 2p < 0.05 versus the respective NMDA values;
F3-160: 60 μm was used for −/− and 100 μm for +/+ cultures. n.d., Not determined.
Measurement of d-[3H]aspartate uptake in cultures
To exclude that the difference in extracellular glutamate between cultures from wild-type and knock-out mice was attributable to changes in the activity of glutamate transporters, we have measured the uptake of d-[3H]aspartate (a common substrate for all known glutamate transporters) in culture. Time-dependent studies showed thatd-[3H]aspartate was at plateau after 20 min, when 25–30% of radioactivity was incorporated into the cells. When assessed by HPLC, all intracellular radioactivity coeluted with authentic d,l-aspartate, confirming thatd-[3H]aspartate was entirely taken up as such and was not metabolized intracellularly. Membrane binding of d-[3H]aspartate to NMDA receptors did not contribute to the overall cellular radioactivity, because the intracellular amount ofd-[3H]aspartate did not differ when incubations were performed in the presence of 1 mm NMDA or 1 mm of the competitive NMDA receptor antagonist 2-amino-5-phosphonopentanoic acid. In contrast, d-[3H]aspartate uptake was reduced by ∼75% when cultures were incubated in the presence of the two inhibitors of the Na+-dependent glutamate transporter,l-trans-pyrrolidine-2,4-dicarboxylic acid and l-threo-3-hydroxyaspartic acid (both at 0.5 mm) (data not shown) As shown in Table4, there was no substantial difference ind-[3H]aspartate uptake between cultures prepared from wild-type and mGluR4-deficient mice. Table 4 also shows that group III agonists did not affectd-[3H]aspartate uptake in both types of cultures.
Table 4.
Group III mGluR agonists do not differentially affectd-[3H]aspartate uptake in mGluR4 knock-out (KO, −/−) and wild-type (+/+) mice cultures
| Treatment | Counts per minute/dish | |
|---|---|---|
| mGluR4 KO (−/−) | Wild-type (+/+) | |
| Basal | 26970 ± 436 | 28734 ± 537 |
| l-AP-4 (100 μm) | 27507 ± 294 | 27829 ± 542 |
| (R,S)-PPG (10 μm) | 26447 ± 665 | 27981 ± 875 |
| (R,S)-PPG (100 μm) | 27613 ± 604 | 27172 ± 465 |
Values are means ± SEM of four independent determinations.
In vivo studies
Assessment of NMDA toxicity in the striatum of wild-type and mGluR4-deficient mice
To examine the role for mGluR4 in regulating excitotoxic deathin vivo, we assessed NMDA toxicity in the caudate nucleus, which receives extensive glutamatergic innervation from the cerebral cortex. A single unilateral infusion of low doses of NMDA (50 nmol/0.5 μl) in the caudate nucleus produced the same extent of neuronal degeneration in wild-type and mGluR4-deficient mice. However, the two groups of animals substantially differed when NMDA was combined with 10 nmol of (R,S)-PPG. In knock-out mice, this dose of (R,S)-PPG did not produce any detectable neuroprotection, and the extension of the lesion was similar to that observed after infusion of NMDA alone (789 ± 159 vs 835 ± 122 μm along the anteroposterior axis; means ± SEM; n = 4). In contrast, the extension of the lesion was markedly reduced in wild-type mice treated with NMDA plus 10 nmol of (R,S)-PPG (149 ± 82 μm) compared with mice treated with NMDA alone (802 ± 79 μm; means ± SEM; n = 4) (Fig.4). At higher doses (100 nmol), (R,S)-PPG continued to be protective in +/+ mice (extension of the lesion, 286 ± 78 μm) but was also partially protective in −/− mice (extension, 470 ± 35 μm; means ± SEM; n = 4).
Fig. 4.
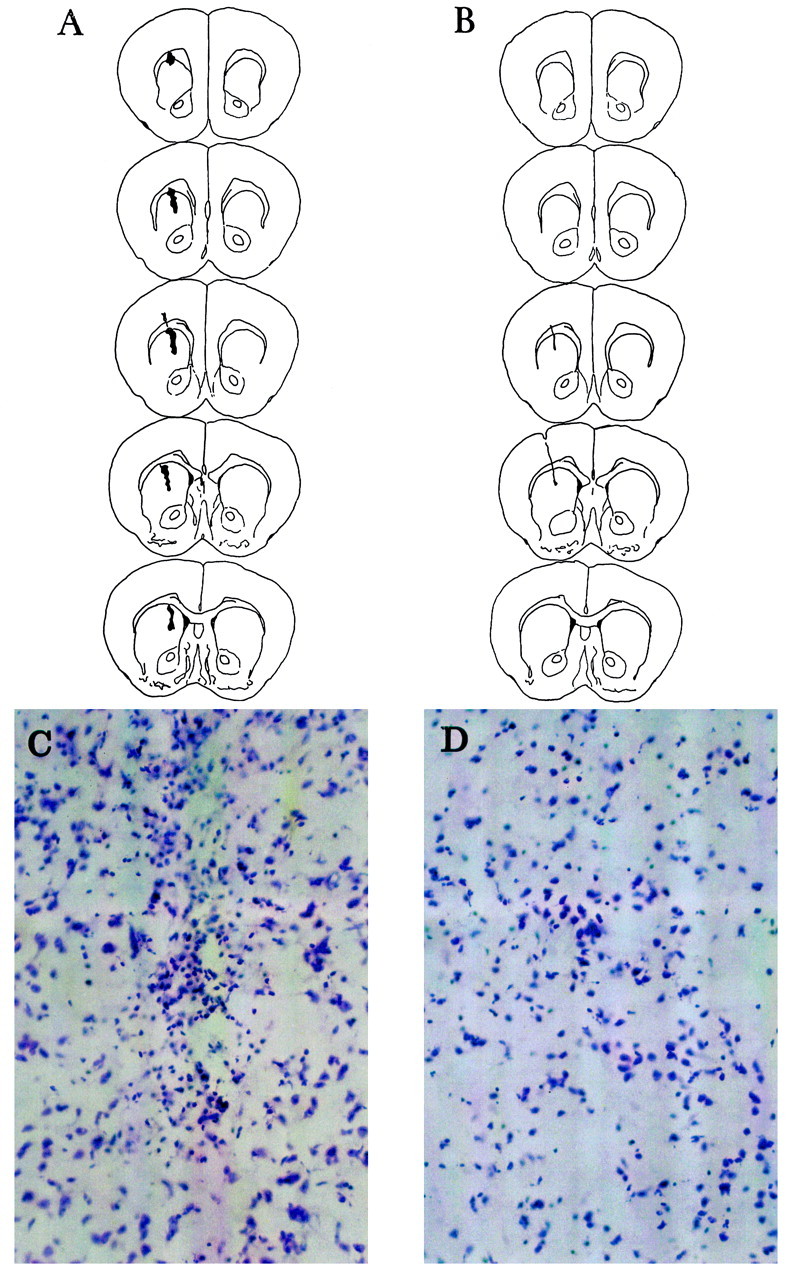
Serial sections across the extension of the caudate nucleus from a representative mGluR4 knock-out (A) or wild-type (B) mouse locally injected with NMDA (50 nmol) plus PPG (10 nmol). Necrotic areas are in black. Microphotographs at the injection site of the −/− and +/+ mouse are shown in C andD, respectively. Note the absence of reactive gliosis inD. NMDA alone in +/+ or −/− mice produced a lesion virtually identical to that shown in A (i.e., in −/− mice treated with NMDA plus 10 nmol PPG).
Determination of extracellular glutamate levels in freely moving wild-type or mGluR4-deficient mice
We have performed microdialysis studies in wild-type and mGluR4 knock-out mice to extend the analysis of glutamate release to anin vivo model. The extracellular fluid was collected from the left caudate nucleus both under basal conditions and after infusion of NMDA through the microdialysis probe. In contrast to what was observed in cultured cortical cells, the basal extracellular levels of glutamate did not differ between wild-type and mGluR4 knock-out mice. However, NMDA infusion led to a much greater increase in glutamate levels in −/− than in +/+ mice (more than 10-fold vs twofold to threefold, respectively) (Fig. 5). We have also tested the effect of (R,S)-PPG infused at concentrations of 5 mm in the microdialysis probe. At this concentration, (R,S)-PPG reduced NMDA-stimulated glutamate release in both +/+ and −/− mice (Fig. 5). We did not extend the study to further concentrations of (R,S)-PPG.
Fig. 5.

Extracellular levels of glutamate in the striatum of wild-type (+/+) and mGluR4-deficient (−/−) freely moving mice. Animals were perfused with NMDA (37.5 mm) in the presence or absence of (R,S)-PPG (5 mm). Values are mean ± SEM of three animals.
DISCUSSION
Metabotropic glutamate receptor subtypes mediate distinct, facilitatory (group I subtypes) or inhibitory (group II and group III subtypes) actions on acute or chronic neurodegenerative processes. Drugs interacting with mGluR subtypes are expected to influence both the induction and progression of neuronal degeneration without blocking the efficiency of fast excitatory synaptic transmission. For these reasons, mGluR subtypes may be activated or inhibited without producing severe side effects typical for drugs interacting with ionotropic glutamate receptors (Nicoletti et al., 1996; Wood, 1998). The role of postsynaptic group I mGluRs in neurodegeneration is still controversial, although mixed mGluR1/5 receptor antagonists were reported to be protective in in vitro and in vivomodels of excitotoxic or hypoxic–ischemic neuronal death (Strasser et al., 1998; Bruno et al., 1999; Pellegrini-Giampietro et al., 1999). In contrast, agonists of group II and group III mGluRs were found to be neuroprotective but through different mechanisms. Protection by group II mGluR agonists in vitro requires the presence of astrocytes, involves a novel form of glial–neuronal interaction, and may be mediated by the release of growth factors such as TGF-β (Bruno et al., 1997, 1998). In vivo, LY354740 (Monn et al., 1997), a systemically active group II mGluR agonist, showed potential antiparkinsonian properties (Konieczny et al., 1998).
Group III agonists are equally protective in mixed cortical cultures (containing astrocytes and neurons) and in cultures of pure neurons. Therefore, this neuroprotection is not expected to be mediated by astrocyte factors, but rather involves one or more receptor subtypes expressed on neuronal structures. Here, we have searched for the identity of the neuroprotective group III mGluR subtype using mixed cortical cultures. This model was particularly suitable for our study because cortical neurons of mixed cultures express all currently known group III mGluR subtypes (Faden et al., 1997). A possible role in neuroprotection for one or more group III mGluR subtype(s), most likely with low micromolar affinity for the known agonists, was initially suggested by the use of the novel phenylglycine derivative (R,S)-PPG (Gasparini et al., 1999a). We have separated the stereoisomers of (R,S)-PPG and found that all protective activity is harbored in the (+) isomer. (+)-PPG was neuroprotective against NMDA toxicity with an EC50 value of ∼5 μm. This value coincides with that found for the activation of recombinant mGluR4 but differs by at least 25-fold from the potency of (R,S)-PPG at mGluR7 and mGluR8 (Gasparini et al., 1999a). In the present study, we provide strong evidence for a critical role of mGluR4 in mediating neuroprotection via the use of mixed cortical cultures prepared from mGluR4 subtype-deficient mice (−/−) in which all group III agonists (i.e.,l-AP-4, (R,S)-PPG, andl-SOP) failed to protect against NMDA toxicity. This was in contrast to the protective effects of group III agonists in wild-type and heterozygous neurons and did not reflect a general refractoriness of −/− knock-out neurons to mechanisms of protection, because the group II agonist 4C3HPG and the group I antagonists CPCCOEt and MPEP (Gasparini et al., 1999b; Litschig et al., 1999) retained their protective activity in knock-out cultures.
We have extended the study to an in vivo model of excitotoxic degeneration by unilaterally injecting NMDA plus (R,S)-PPG into the caudate nucleus of wild-type or mGluR4-deficient mice. This brain region has been selected because it receives an extensive glutamatergic innervation from the cerebral cortex. Low doses of (R,S)-PPG (10 nmol), which may preferentially activate mGluR4 over mGluR7 receptors, were neuroprotective in +/+ mice but were totally inactive in −/− mice. In contrast, −/− mice were partially sensitive to higher doses of (R,S)-PPG (100 nmol), which are expected to recruit mGluR7. The prominent role for mGluR4 in mediating agonist-induced neuroprotection is consistent with the evidence that this particular receptor subtype contributes substantially to the high-affinity binding of [3H]l-AP-4 in many regions of mouse brain (Thomsen and Hampson, 1999).
Interestingly, cultures from mGluR4 −/− mice could be effectively damaged by low concentrations of NMDA, which produced small effects in cultures from +/+ mice. Searching for a reason responsible for the increased vulnerability of −/− cultures, we have focused on extracellular levels of glutamate because of the following reasons: (1) mGluR4 is known to be, at least primarily, presynaptically located and its activation may inhibit glutamate release (Conn and Pin, 1997;Shigemoto et al., 1997); (2) cultured cortical cells respond to a toxic NMDA pulse with an enhanced release of glutamate, which in turn contributes to the development of excitotoxic neuronal death (Monyer et al., 1992; Bruno et al., 1995); and finally (3) inhibition of glutamate release is a validated mechanism for drugs of potential use in neurodegenerative disorders, such as riluzole (Doble, 1999). We have found a striking difference in the regulation of extracellular steady-state glutamate concentrations between +/+ and mGluR4 −/− cultures. Basal glutamate levels in −/− cultures were at least four times as high as levels in +/+ cultures and, in some experiments, they were even higher than in wild-type cultures exposed to NMDA. The higher glutamate levels in −/− cultures may be a direct consequence of the lack of mGluR4 because (1) no changes in glutamate transport (measured as d-[3H]aspartate uptake) were found between +/+ and −/− cultures, and (2) pharmacological inhibition of group III mGluRs by MPPG increased glutamate levels in +/+ cultures. In −/− cultures, (R,S)-PPG andl-AP-4 reduced both basal and NMDA-stimulated glutamate release, but only partially, leaving abnormally high levels of extracellular glutamate. This partial reduction may reflect the recruitment of mGluR7 or mGluR8 by group III agonists. mGluR8 is a better candidate because it accounts for most of the residual [3H]l-AP-4 binding in the brain of mGluR4 knock-out mice (Thomsen and Hampson, 1999). We speculate that one of the functions of mGluR4 is to maintain extracellular glutamate levels below a “toxic” threshold during synaptic activity. Elevated extracellular glutamate levels may contribute to NMDA toxicity by activating ancillary receptors, such as AMPA receptors or group I mGluRs (Table 2, neuroprotection by CPCCOEt and MPEP). In +/+ cultures, group III mGluR agonists might exert part of their protective activity by reducing NMDA-stimulated glutamate release, thus eliminating an amplifying component of NMDA toxicity. In −/− cultures, the higher basal levels of glutamate may well account for the increased vulnerability to NMDA, and the inability of agonists to reduce glutamate levels below a critical threshold (which in this model may be set at 150–160 nm) may contribute to explain the lack of neuroprotection. The assessment of extracellular glutamate levels in freely moving animals by microdialysis confirmed a critical role for mGluR4 in regulating extracellular glutamate levels. Accordingly, in mGluR4 −/− mice, intrastriatal NMDA infusion induced a greater increase in extracellular glutamate than in +/+ mice. In this particular model, basal glutamate levels did not significantly differ between the two strains of mice, perhaps because of compensatory mechanisms that might have occurred during the development of −/− mice. The reduction of glutamate release observed with high doses of (R,S)-PPG in microdialysis studies may again reflect the activation of mGluR7 or mGluR8. We decided not to titrate (R,S)-PPG to disclose a possible difference between the two strains of mice because the amount of (R,S)-PPG diffusing from the probe to the tissue cannot be accurately estimated. Comparative studies in mGluR4-, mGluR7-, and mGluR8-deficient mice (or studies in double knock-out mice) should be performed to determine the relative contribution of these receptor subtypes in neurodegeneration–neuroprotection and in the regulation of glutamate release. Although we favor the hypothesis that activation of mGlu4 receptors is neuroprotective by maintaining ambient glutamate concentrations below a critical threshold, other mechanisms that are only tangentially related to glutamate release could also be relevant. Similar to other group III mGluRs (Saugstad et al., 1996, 1997; Corti et al., 1998), mGluR4 receptors positively couple to inwardly rectifying K+ channels in amphibian oocytes (Sharon et al., 1997). l-AP-4 can also activate K+ channels in CNS neurons (Holmes et al., 1996; Dutar et al., 1999). Through this mechanism, activation of mGluR4 might affect neuronal sensitivity to excitotoxic damage independently of changes in glutamate release.
In conclusion, our results support the neuroprotective activity of group III mGluRs and indicate that, both in in vitro andin vivo models, neuroprotection is primarily mediated by mGluR4. This encourages studies aiming at synthesizing mGluR4-selective agonists, which may have a high chance of success in experimental models of acute and chronic neurodegenerative disorders.
Footnotes
Correspondence should be addressed to Dr. Peter J. Flor, Nervous System Research, Novartis Pharma AG, K-125.6.08, CH-4002 Basel, Switzerland. E-mail: peter_josef.flor@pharma.novartis.com.
REFERENCES
- 1.Bruno V, Battaglia G, Copani A, Giffard RG, Raciti G, Raffaele R, Shinozaki H, Nicoletti F. Activation of class II or class III metabotropic glutamate receptors protects cultured cortical neurons against excitotoxic degeneration. Eur J Neurosci. 1995;7:1906–1913. doi: 10.1111/j.1460-9568.1995.tb00712.x. [DOI] [PubMed] [Google Scholar]
- 2.Bruno V, Copani A, Bonanno L, Knöpfel T, Kuhn R, Roberts PJ, Nicoletti F. Activation of group III metabotropic glutamate receptors is neuroprotective in cortical cultures. Eur J Pharmacol. 1996;310:61–66. doi: 10.1016/0014-2999(96)00358-5. [DOI] [PubMed] [Google Scholar]
- 3.Bruno V, Sureda FX, Storto M, Casabona G, Caruso A, Knöpfel T, Kuhn R, Nicoletti F. The neuroprotective activity of group-II metabotropic glutamate receptors requires new protein synthesis and involves a glial-neuronal signaling. J Neurosci. 1997;17:1891–1897. doi: 10.1523/JNEUROSCI.17-06-01891.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruno V, Battaglia G, Casabona G, Copani A, Caciagli F, Nicoletti F. Neuroprotection by glial metabotropic glutamate receptors is mediated by transforming growth factor-β. J Neurosci. 1998;18:9594–9600. doi: 10.1523/JNEUROSCI.18-23-09594.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruno V, Battaglia G, Kingston A, O'Neill MJ, Catania MV, Di Grezia R, Nicoletti F. Neuroprotective activity of the potent and selective mGlu1a metabotropic glutamate receptor antagonist, (+)-2-methyl-4-carboxyphenylglycine (LY367385): comparison with LY357366, a broader spectrum antagonist with equal affinity for mGlu1a and mGlu5 receptors. Neuropharmacology. 1999;38:199–207. doi: 10.1016/s0028-3908(98)00159-2. [DOI] [PubMed] [Google Scholar]
- 6.Conn PJ, Pin J-P. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 7.Copani A, Bruno V, Battaglia G, Leanza G, Pellitteri R, Russo A, Stanzani S, Nicoletti F. Activation of metabotropic glutamate receptors protects cultured neurons against apoptosis induced by beta-amyloid peptide. Mol Pharmacol. 1995;47:890–897. [PubMed] [Google Scholar]
- 8.Corti C, Restituito S, Rimland JM, Brabet I, Corsi M, Pin JP, Ferraguti F. Cloning and characterization of alternative mRNA forms for the rat metabotropic glutamate receptors mGluR7 and mGluR8. Eur J Neurosci. 1998;12:3629–3641. doi: 10.1046/j.1460-9568.1998.00371.x. [DOI] [PubMed] [Google Scholar]
- 9.Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999;81:163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 10.Dunnett CW. New tables for multiple comparisons with a control. Biometrics. 1964;20:482–491. [Google Scholar]
- 11.Dutar P, Vu HM, Perkel DJ. Pharmacological characterization of an unusual mGluR-evoked neuronal hyperpolarization mediated by activation of GIRK channels. Neuropharmacology. 1999;38:467–475. doi: 10.1016/s0028-3908(98)00206-8. [DOI] [PubMed] [Google Scholar]
- 12.Faden AI, Ivanova SA, Yakovlev AG, Mukhin AG. Neuroprotective effects of group III mGluR in traumatic neuronal injury. J Neurotrauma. 1997;14:885–895. doi: 10.1089/neu.1997.14.885. [DOI] [PubMed] [Google Scholar]
- 13.Fagg GE, Foster AC, Mena EE, Cotman CW. Chloride and calcium ions reveal a pharmacologically distinct population of l-glutamate binding sites in synaptic membranes: correspondence between biochemical and electrophysiological data. J Neurosci. 1982;2:958–965. doi: 10.1523/JNEUROSCI.02-07-00958.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Flor PJ, Lindauer K, Püttner I, Rüegg D, Lukic S, Knöpfel T, Kuhn R. Molecular cloning, functional expression and pharmacological characterization of the human metabotropic glutamate receptor type 2. Eur J Neurosci. 1995a;7:622–629. doi: 10.1111/j.1460-9568.1995.tb00666.x. [DOI] [PubMed] [Google Scholar]
- 15.Flor PJ, Lukic S, Rüegg D, Leonhardt T, Knöpfel T, Kuhn R. Molecular cloning, functional expression and pharmacological characterization of the human metabotropic glutamate receptor type 4. Neuropharmacology. 1995b;34:149–155. doi: 10.1016/0028-3908(94)00149-m. [DOI] [PubMed] [Google Scholar]
- 16.Flor PJ, Gomeza J, Tones MA, Kuhn R, Pin JP, Knöpfel T. The C-terminal domain of the mGluR1 metabotropic glutamate receptor affects sensitivity to agonists. J Neurochem. 1996;67:58–63. doi: 10.1046/j.1471-4159.1996.67010058.x. [DOI] [PubMed] [Google Scholar]
- 17.Flor PJ, Van Der Putten H, Rüegg D, Lukic S, Leonhardt T, Bence M, Sansig G, Knöpfel T, Kuhn R. A novel splice variant of a metabotropic glutamate receptor, human mGluR7b. Neuropharmacology. 1997;36:153–159. doi: 10.1016/s0028-3908(96)00176-1. [DOI] [PubMed] [Google Scholar]
- 18.Gasparini F, Bruno V, Battaglia G, Lukic S, Leonhardt T, Inderbitzin W, Laurie D, Sommer B, Varney MA, Hess SD, Johnson EC, Kuhn R, Urwyler S, Sauer D, Portet C, Schmutz M, Nicoletti F, Flor PJ. (R,S)-4-phosphonophenylglycine, a potent and selective group III metabotropic glutamate receptor agonist is anticonvulsive and neuroprotective in vivo. J Pharmacol Exp Ther. 1999a;290:1678–1687. [PubMed] [Google Scholar]
- 19.Gasparini F, Lingenhöhl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Veliçelebi G, Kuhn R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999b;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 20.Graham ME, Burgoyne RD. Activation of metabotropic glutamate receptors by l-AP4 stimulates survival of rat cerebellar granule cells in culture. Eur J Pharmacol. 1994;288:115–123. doi: 10.1016/0922-4106(94)90016-7. [DOI] [PubMed] [Google Scholar]
- 21.Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- 22.Holmes KH, Keele NB, Arvanov VL, Shinnick-Gallagher P. Metabotropic glutamate receptor agonist-induced hyperpolarization in rat basolateral amygdala neurons: receptor characterization and ion channels. J Neurophysiol. 1996;76:3059–3069. doi: 10.1152/jn.1996.76.5.3059. [DOI] [PubMed] [Google Scholar]
- 23.Jane DE, Thomas NK, Tse HW, Watkins JC. Potent antagonists at the l-AP4- and (1S,3S)-ACPD-sensitive presynaptic metabotropic glutamate receptors in the neonatal rat spinal cord. Neuropharmacology. 1996;35:1029–1035. doi: 10.1016/s0028-3908(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 24.Johansen PA, Chase LA, Sinor AD, Koerner JF, Johnson RL, Robinson MB. Type 4a metabotropic glutamate receptor: identification of new potent agonists and differentiation from the l-(+)-2-amino-4-phosphonobutanoic acid-sensitive receptor in the lateral perforant pathway in rats. Mol Pharmacol. 1995;48:140–149. [PubMed] [Google Scholar]
- 25.Konieczny J, Ossowska K, Wolfarth, Pilc A. LY354740, a group II metabotropic glutamate receptor agonist with potential antiparkinsonian properties in rats. Naunyn Schmiedebergs Arch Pharmacol. 1998;358:500–502. doi: 10.1007/pl00005284. [DOI] [PubMed] [Google Scholar]
- 26.Laurie DJ, Schoeffter P, Wiederhold KH, Sommer B. Cloning, distribution and functional expression of the human mGlu6 metabotropic glutamate receptor. Neuropharmacology. 1997;36:145–152. doi: 10.1016/s0028-3908(96)00172-4. [DOI] [PubMed] [Google Scholar]
- 27.Litschig S, Gasparini F, Rüegg D, Munier N, Flor PJ, Vranesic I-T, Prezeau L, Pin J-P, Thomsen C, Kuhn R. CPCCOEt, a noncompetitive metabotropic glutamate receptor 1 antagonist, inhibits receptor signaling without affecting glutamate binding. Mol Pharmacol. 1999;55:453–461. [PubMed] [Google Scholar]
- 28.Monn JA, Valli MJ, Massey SM, Wright RA, Salhoff CR, Jonhson BG, Howe T, Alt CA, Rhodes GA, Robey RL, Griffey KR, Tizzano JP, Kallmann MJ, Helton DR, Schoepp DD. Design, synthesis, and pharmacological characterization of (+)-2-aminobicyclo[3.1.0]hexane-2,6-dicarboxylic acid (LY354740): a potent, selective, and orally active group 2 metabotropic glutamate receptor agonist possessing anticonvulsant and anxiolytic properties. J Med Chem. 1997;40:528–537. doi: 10.1021/jm9606756. [DOI] [PubMed] [Google Scholar]
- 29.Monyer H, Giffard RG, Hartley DM, Dugan LL, Goldberg MP, Choi DW. Oxygen or glucose deprivation-induced neuronal injury in cortical cell cultures is reduced by tetanus toxin. Neuron. 1992;8:967–973. doi: 10.1016/0896-6273(92)90211-u. [DOI] [PubMed] [Google Scholar]
- 30.Nakanishi S. Metabotropic glutamate receptors. Neuron. 1994;13:1031–1037. doi: 10.1016/0896-6273(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 31.Nicoletti F, Wroblewski JT, Iadarola MJ, Costa E. Serine-O-phosphate, an endogenous metabolite, inhibits the stimulation of inositol phospholipid hydrolysis elicited by ibotenic acid in rat hippocampal slices. Neuropharmacology. 1986;25:335–338. doi: 10.1016/0028-3908(86)90262-5. [DOI] [PubMed] [Google Scholar]
- 32.Nicoletti F, Bruno V, Copani A, Casabona G, Knöpfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996;1:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- 33.Okamoto N, Hori S, Akazawa C, Hayashi Y, Shigemoto R, Mizuno N, Nakanishi S. Molecular characterization of a new metabotropic glutamate receptor mGluR7 coupled to inhibitory cyclic AMP signal transduction. J Biol Chem. 1994;269:1231–1236. [PubMed] [Google Scholar]
- 34.Pekhletski R, Gerlai R, Oversteet LS, Huang X-P, Agopyan N, Slater NT, Abramow-Newerly W, Roder JC, Hampson DR. Impaired cerebellar synaptic plasticity and motor performance in mice lacking the mGluR4 subtype of metabotropic glutamate receptor. J Neurosci. 1996;16:6364–6373. doi: 10.1523/JNEUROSCI.16-20-06364.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pellegrini-Giampietro DE, Cozzi A, Peruginelli F, Leonardi P, Meli E, Pellicciari R, Moroni F. 1-Aminoindan-1,5-dicarboxylic acid and (S)-(+)-2-(3′-carboxybicyclo[1.1.1] pentyl)-glycine, two mGlu1 receptor-preferring antagonists, reduce neuronal death in in vitro and in vivo models of cerebral ischaemia. Eur J Neurosci. 1999;11:3637–3647. doi: 10.1046/j.1460-9568.1999.00786.x. [DOI] [PubMed] [Google Scholar]
- 36.Saugstad JA, Segerson TP, Westbrook GL. Metabotropic glutamate receptors activate G-protein-coupled inwardly rectifying potassium channels in Xenopus oocytes. J Neurosci. 1996;16:5979–5985. doi: 10.1523/JNEUROSCI.16-19-05979.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saugstad JA, Kinzie JM, Shinohara MM, Segerson TP, Westbrook GL. Cloning and expression of rat metabotropic glutamate receptor 8 reveals a distinct pharmacological profile. Mol Pharmacol. 1997;51:119–125. doi: 10.1124/mol.51.1.119. [DOI] [PubMed] [Google Scholar]
- 38.Schoepp DD, Johnson BG, Smith EC, McQuaid LA. Stereoselectivity and mode of inhibition of phosphoinositide-coupled excitatory amino acid receptors by 2-amino-3-phosphonopropionic acid. Mol Pharmacol. 1990;38:222–228. [PubMed] [Google Scholar]
- 39.Sharon D, Vorobiov D, Dascal N. Positive and negative coupling of the metabotropic glutamate receptors to a G protein-activated K+ channel, GIRK, in Xenopus oocytes. J Gen Physiol. 1997;109:477–490. doi: 10.1085/jgp.109.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strasser U, Lobner D, Berhens MM, Canzoniero LM, Choi DW. Antagonists of group-I mGluRs attenuate exicitotoxic neuronal death in cortical cultures. Eur J Neurosci. 1998;10:2848–2855. doi: 10.1111/j.1460-9568.1998.00291.x. [DOI] [PubMed] [Google Scholar]
- 42.Thomsen C, Hampson DR. Contribution of metabotropic glutamate receptor mGluR4 to l-2-[H-3]amino-4-phosphonobutyrate binding in mouse brain. J Neurochem. 1999;71:835–840. doi: 10.1046/j.1471-4159.1999.0720835.x. [DOI] [PubMed] [Google Scholar]
- 43.Trombley PQ, Westbrook GL. l-AP4 inhibits calcium currents and synaptic transmission via a G-protein-coupled glutamate receptor. J Neurosci. 1992;12:2043–2050. doi: 10.1523/JNEUROSCI.12-06-02043.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wood PL. NMDA antagonists for stroke and head trauma: current status. Exp Opin Invest Drugs. 1998;7:1505–1508. doi: 10.1517/13543784.7.9.1505. [DOI] [PubMed] [Google Scholar]
- 45.Wu S, Wright RA, Rockey PK, Burgett SG, Arnold JS, Rosteck PR, Jr, Johnson BG, Schoepp DD, Belagaje RM. Group III human metabotropic glutamate receptors 4, 7 and 8: molecular cloning, functional expression, and comparison of pharmacological properties in RGT cells. Mol Brain Res. 1998;53:88–97. doi: 10.1016/s0169-328x(97)00277-5. [DOI] [PubMed] [Google Scholar]