

Abstract
This study investigated the fluctuations in the membrane potential of cortical neurons and glial cells during the slow sleep oscillation and spike-wave (SW) seizures. We performed dual neuron–glia intracellular recordings together with multisite field potential recordings from cortical suprasylvian association areas 5 and 7 of cats under ketamine–xylazine anesthesia. Electrical stimuli applied to the cortex elicited responses consisting of a biphasic depolarization in glial cells, which was associated with an EPSP–IPSP sequence in neurons. During the slow (<1 Hz) oscillation, extracellular measurements of the potassium concentration revealed periodic increases with an amplitude of 1–2 mm, similar in shape to glial activities. We suggest that, through their uptake mechanisms, glia cells modulate the neuronal excitability and contribute to the pacing of the slow oscillation. The slow oscillation often evolved into SW paroxysms, mimicking sleep-triggered seizures. This transition was associated with increased coupling between the depolarizing events in neurons and glial cells. During seizures, the glial membrane potential displayed phasic negative events related to the onset of the paroxysmal depolarizing shifts in neurons. These events were not voltage dependent and increased their incidence and amplitude with the development of the seizure. It is suggested that the intraglial transient negativities represent field reflections of synchronized neuronal potentials. We propose that the mechanisms underlying the neuron–glia communication include, besides the traditional neurotransmitter- and ion-mediated pathways, field effects crossing their membranes as a function of the state of the cortical network.
Keywords: slow oscillations, spike-wave seizures, intracellular, EEG, in vivo, intraglial negativity
The growing complexity of glial properties and neuron–glia interactions opens new questions regarding the participation of glial cells in the genesis of the electroencephalogram (EEG) as well as their involvement in the activity of neuronal aggregates. Several studies have contributed to the understanding of the mechanisms underlying the genesis of the EEG. The contribution of neurons to the shape of extracellular field potentials has been known for a long time (Hubbard et al., 1969) in view of both synaptic and intrinsic currents (Pedley and Traub, 1990). In contrast, the glial involvement in the genesis of field potentials was rather neglected, although pioneering studies devoted to the origin of sustained potentials have emphasized the role of glia (Kuffler et al., 1966; Orkand et al., 1966; Somjen, 1973) (for review, see Speckmann and Elger, 1999). Furthermore, the widespread glial processes, as well as the direct interglial connections, were proposed to play an amplifying role in the genesis of extracellular field potentials (Somjen and Trachtenberg, 1979; Speckmann and Caspers, 1979). More recent studies have shown that the membranes of glial cells are endowed with a series of intrinsic properties (Duffy et al., 1995;Sontheimer and Ritchie, 1995; Walz, 1995), which may account for the role played by glial cells in the genesis of EEG potentials. Data are also available concerning the mechanisms underlying the neuron-glial exchange of information (Nedergaard, 1994; Parpura et al., 1994;Janigro et al., 1997; Pasti et al., 1997).
Glial behavior during epileptic discharges has received more attention, and it was reported that seizures were accompanied by a long-lasting depolarization in glia of the neocortex (Grossman and Hampton, 1968;Sypert and Ward, 1971) and hippocampus (Dichter et al., 1972). Direct measures of extracellular K+concentration ([K+]o) (Lux and Neher, 1973) during epileptiform seizures (Fertziger and Ranck, 1970; Futamachi et al., 1974; Moody et al., 1974) confirmed the initial demonstration in simple preparations that glia are reliable potassium detectors (Nicholls and Kuffler, 1964; Kuffler et al., 1966). Accumulation of K+ in the extracellular space was considered to be a favorable condition for the onset of seizures (Zuckermann and Glaser, 1968). Janigro et al. (1997)demonstrated that impaired glial uptake of K+ may equally cause epileptiform activity in the hippocampus.
As yet, however, there is no mention of the implication of glial cells in the genesis of short-lasting EEG waves during physiological behavior. At variance with previous studies, which have considered the origin of the EEG from unilateral neuron–EEG or glia–EEG interactions, here we investigated the complex relationship between neurons, glia, and field potentials on the basis of their simultaneous recordings during normal and pathological oscillations in vivo. The normal oscillations were made of a slow (<1 Hz, mainly 0.6–1 Hz) sleep rhythm, which was initially described intracellularly in cat cortical neurons under different anesthetics (Steriade et al., 1993a), as well as in naturally sleeping cats (Steriade et al., 1996;Amzica and Steriade, 1998a) and humans (Steriade et al., 1993a;Achermann and Borbély, 1997; Amzica and Steriade, 1997). Recently, the slow oscillation was described in cortical glial cells (Amzica and Steriade, 1998b; Amzica and Neckelmann, 1999). The pathological oscillations result from the paroxysmal development of the slow oscillation into cortical spike-wave (SW) seizures (Steriade and Amzica, 1994; Steriade and Contreras, 1995; Steriade et al., 1998).
In view of their electrophysiological and anatomical properties, as well as their activities during the above-mentioned states, we hypothesized that glial cells contribute to the shape, and undergo the influence, of field potentials. As a consequence, we assumed that intraglial potentials would be reflected by the extraglial potentials, and vice versa. We therefore aimed at disclosing the dynamic evolution of the neuron–glial relationship during SW seizures compared with normal slow sleep oscillations.
MATERIALS AND METHODS
Animal preparation and recordings. Sixty adult cats of either sex were used for these experiments. They were deeply anesthetized with a mixture of ketamine and xylazine (10–15 mg/kg and 2–3 mg/kg, respectively, i.m.). The surgical procedure started with intubation, muscle paralysis with gallamine triethiodide, and artificial ventilation (20–30 cycles min). The end-tidal CO2 concentration was maintained at ∼3.7% (±0.2) by adjusting the O2 concentration in the airflow of the ventilation. All pressure points to be incised were infiltrated with lidocaine. EEG and heart rate were continuously monitored to detect the slightest decrease in the level of anesthesia, and supplementary doses of the anesthetic were given at the first signs of activated EEG or accelerated pulse rate (>110 beats/min). The craniotomy exposed mainly the suprasylvian gyrus. Cisternal drainage, hip suspension, pneumothorax, and filling of the hole in the calvarium with a 4% solution of agar were used to enhance the stability of the intracellular recordings. To compensate for fluid loss, the cats were periodically injected intravenously with saline (20–30 ml per experiment).
Intracellular recordings from areas 5 and 7 (see Fig.1A) were obtained with glass micropipettes (tip diameter <0.5 μm) filled with potassium acetate (3m, impedance 30–40 MΩ). The same micropipettes were occasionally used to record d.c. (direct current) extracellular potentials. The signals recorded with glass pipettes were passed through a high-impedance amplifier with active bridge circuitry (bandpass: d.c. to 9 kHz). The rest of the field potentials [alternating current (a.c.) traces] were recorded with tungsten macroelectrodes (0.5–1 MΩ impedance) inserted in the depth of the cortex (∼1 mm). In some cats (n = 5) we used arrays of eight tungsten electrodes to record the depth profile of slow sleep and SW seizure activities. The distance between the tips of the electrodes in the coaxial array was 0.25 mm. The array was lowered under visual control, perpendicular to the cortical surface, such that the uppermost lead remained on the pial surface. The EEG potentials were bandpass-filtered between 0.3 and 1000 Hz. The macroelectrodes were also used for stimulation purposes (stimuli lasting for 0.1–0.2 msec, intensity of 0.05–0.8 mA). All signals were digitally converted (20 kHz sampling rate) and recorded on tape for off-line analysis.
Fig. 1.

Normal responsiveness of cortical neurons and glia to cortical stimulation. A, Top view of cat's brain with the localization of association areas 5 and 7 in the suprasylvian gyrus. B, The impalement of a glial cell is marked (open arrowhead) by a sudden voltage deflection from extracellular potential values (∼0 mV) to −80 mV. Intraglial potentials (slow depolarizations) are reversed with respect to the extracellular ones. C, Double intracellular (neuron–glia) and field potential recording in cortical area 5. Response to a single cortical shock (black triangle) delivered close to the field electrode. The recording sites correspond to those indicated in A. The neuronal response consisted of an initial depolarization crowned by action potentials, an inhibitory potential, and a rebound excitation. The corresponding responses in the glia were a sluggish depolarizing slope, a slow further depolarization, and a negative wave, respectively. This shape was reproduced in the depth-EEG recording, with the exception of the early response, which appeared as a negative potential.D, Average of 25 responses evoked by the cortical stimulation. The initial glial depolarization (a) is clearly separated from the following positive wave (b) by a change of the depolarizing slope. In this and the following figures, all potentials are presented with the positivity upward. Intracellular recordings are all at rest (zero current), unless expressly indicated, and the resting membrane potential is indicated at left.
Ten cats were used for experiments using K+-sensitive electrodes. The procedure was similar to the one used in other studies (Janigro et al., 1997; de Curtis et al., 1998). Recordings were made with double-barrel pipettes in which the K+-sensitive electrode was treated with dimethyldichlorosilane and dried at 120°C for 2 hr, and the tip was filled with the K+ ionophore I-cocktail A (Fluka, Buchs, Switzerland). The rest of the K+-sensitive barrel was filled with KCl (0.2 m). The reference barrel was backfilled with potassium fluoride (2 m). The composition of the calibration solution was (in mm): NaCl 126, KCl 2.3, NaHCO3 26, MgSO4 1.3, CaCl2 2.4, KH2PO4 1.2, glucose 15, HEPES 5, thiourea 0.4, and 3% dextran 70,000, pH 7.3. We then substituted KCl for the NaCl resulting in a final K+ concentration of 1–20 mm. The relationship between the measured voltage and the K+ concentration was derived in accordance with the Nicolsky-Eisenmann equation (Ammann, 1986).
Ideally, disclosing the relationship between neurons and glia and their respective contribution to the shape of the field potentials would require recordings of simultaneous pairs within the same pool of neurons. However, using the in vivo preparation renders this task impossible because visual control is limited and cannot penetrate the depths of the cortex. This is why we approached this issue by means of two indirect procedures. (1) We used a fixed electrode to control the changes of the state of the network and a mobile electrode that would, under close control of its movements, record neurons, glia, and EEG within a restricted area (<10 μm). (2) We also used various combinations of double recordings (neuron–glia, neuron–EEG, and glia–EEG) at a short distance between the electrodes (<1 mm, mostly 0.5 mm) to collect information about simultaneously occurring potentials.
SW seizures occurred spontaneously (63% of the seizures), were triggered by electric stimulation (15% of the seizures), or were induced with bicuculline (0.2 mm solution in saline). In the latter case, a syringe needle was inserted in the rostral part of the suprasylvian gyrus (>5 mm away from the closest recording electrode to avoid bicuculline spillage in the recording area), and small amounts (0.02–0.05 μl) of bicuculline leaked slowly into the cortex.
At the end of the experiments, cats received a lethal dose of intravenous pentobarbital sodium.
Analysis. To establish time relationships between glial, neuronal, and field potentials, we used wave-triggered averages (WTAs) and correlative analyses. The WTA procedure consisted of averaging stereotyped sweeps extracted around a reference point. In the case of the slow oscillation, it has been shown that a cycle, starting at the onset of the depolarization and most often followed by a brief spindle sequence, represents a K-complex (Amzica and Steriade, 1997,1998a). Thus, the reference point was considered to be the location of the steepest slope at the onset of the K-complex. The choice of this criterion is justified by the assumption that at the maximum slope point, most of the population cells discharge in synchrony. The identification of such points during a recording required the differentiation of one channel and the detection of one local maximum per oscillation cycle. This produced a table with time stamps, which served for the extraction, from all recorded channels, of equal-duration sweeps representing individual K-complexes. Finally, the averaged sweeps from each channel generated the average K-complex. A similar procedure was applied to the calculation of rhythmic SW complexes.
The time relation, as well as the degree of resemblance between pairs of potentials, was quantified by means of cross-correlations, as defined for time series (Bendat and Piersol, 1980). The sign (positive or negative) of the highest peak of a cross-correlation suggests the phase relationship between the two waves (in-phase or phase-opposed, respectively), whereas its amplitude suggests the degree of global resemblance of the two waves (on a normalized scale from 0 to 1). The abscissa of the highest peak indicates an eventual time lag between the two signals.
RESULTS
Database and cellular identification
A total of 279 cells (146 neurons and 133 glia), obtained from 60 cats, were used in this study. Of those 279 cells, 67 were double intracellular recordings (neuron–glia). Only neurons with stable membrane potential (Vm) at rest (without current) for >15 min and more negative than −60 mV, and with overshooting action potentials, were considered for analysis. The criteria for intraglial impalements were as follows. (1) Impalement was accompanied by a sudden drop of the Vmfrom 0 mV to a resting level more negative than − 70 mV (Fig.1B). (2) TheVm remained stable for at least 15 min and did not require the application of steady hyperpolarizing currents. (3) No action potentials were triggered, either spontaneously or by depolarizing pulses. We did not include in the present database glial recordings with resting Vm in a more depolarizing range (McKhann et al., 1997).
The resting Vm of neurons was measured during the troughs of the slow oscillation, in the absence of synaptic activity. At the level of the whole neuronal population, theVm was −72 ± 2.7 mV (mean ± SD), and the input resistance resulting from hyperpolarizing pulses of 1 nA was 23 ± 3.2 MΩ. Under similar conditions, the averageVm of glia was −88 ± 3.6 mV, and the input resistance was 12 ± 1.4 MΩ. All cells were recorded within cortical association suprasylvian areas 5 and 7 (Fig.1A), at a depth of <1.5 mm from the surface.
Neuronal and glial responses to cortical stimuli
Under normal conditions, with the cortex displaying the slow oscillation, cortical stimuli applied in the vicinity of the impaled cells elicited in neurons EPSP–IPSP sequences followed by a rebound excitation (Fig. 1C,D). In the case of simultaneously recorded glia, the initial neuronal EPSP corresponded to the onset of a sluggish depolarization (Fig. 1D,segment a). The subsequent neuronal IPSP was associated with an additional depolarization of the glia (Fig. 1D, segment b). The rebound excitation of neurons was reflected in the glia as a hyperpolarizing potential followed by a depolarization above the control level. The correlate of these cellular responses in the depth field potential was an initial biphasic negativity, a subsequent positivity, and a late negativity corresponding to the rebound excitation of the neurons. With the exception of the initial part of responses, the depth a.c. field potentials had a shape similar to those of glial d.c. activities (Fig.1C,D).
The average hyperpolarization of neurons (n = 50) at a resting Vm of −65 mV was 1.7 ± 0.2 mV, whereas the corresponding glial depolarization at a restingVm of −80 mV was 1.1 ± 0.1 mV. The amplitude of the postinhibitory rebound (K-complex) was 7 ± 0.6 mV (depolarizing) in neurons and much smaller, 0.3 ± 0.1 mV (hyperpolarizing), in glia. This voltage distribution among neurons and glia suggests that the reciprocal interaction between them is not the same during the various components of the cortically evoked response (see Discussion).
The anesthesia used in our cats (ketamine–xylazine) produced hypersynchronous oscillations that often triggered SW seizures, evolving gradually from the slow oscillation (Steriade et al., 1998). The responsiveness of cortical neurons during such seizures is dealt with in another paper (Steriade and Amzica, 1999). Briefly, the neuronal response evolved from the normal pattern (Fig. 1C), yielding a progressive increase of the excitatory components of the response and the suppression of the interposed inhibition until the response was transformed into a paroxysmal depolarizing shift (PDS). We present below the glial evoked potentials during seizures because of the presence of a peculiar component that was also found during spontaneous activities.
To disclose the relationship between intraglial and field potentials during SW seizures, we used d.c. recordings through pairs of micropipettes, and we stimulated the cortex close to one of them (Fig.2). At the moment of the stimulation, the brain was in a state in which seizures of the type depicted in Figure2A had already occurred. The depth field potential corresponding to a PDS is shown in Figure 2B. It consisted of a biphasic negativity lasting for 350–400 msec. Its amplitude was a function of the distance from the stimulation site (the closer the stimulation site, the larger the response). After testing the responses with both electrodes in an extracellular environment, we obtained intraglial recordings with one of them (the impalement was <10 μm away from the previous location of the recording electrode). The intraglial expression of the PDS started in all tested cells (n = 78) with a short-lasting negative potential followed by a large, long-lasting depolarization (Fig. 2C). The comparison between the extracellular and intracellular shapes of the PDS at the same recording site shows that the initial negative potential was present in both situations (Fig. 2, dotted vertical line joining the extracellular and intracellular negativities at recording Site 1). The extracellular negativity, calculated after eliminating the trend of the subsequent longer lasting negative potential, represented 56% of its intracellular amplitude. The subsequent intracellular depolarization was reflected in the depth-EEG by a larger negative potential. The comparison between intraglial and EEG potentials recorded at different moments is possible because the two successive extracellular responses at the site where the recording electrode did not move were identical, proving that the state of the cortical network did not change, from the recorded period shown in Figure 2B to the period depicted inC.
Fig. 2.

Glial responsiveness to cortical stimulation in a seizure-prone cortex. A, One of the SW seizures recurring periodically in this cat. The seizures started with isolated PDSs (left) and continued with periodic SW complexes at 2–4 Hz. Two d.c. recording pipettes (1 and2) and a stimulating electrode were placed in the depth of the suprasylvian gyrus, according to the brain scheme on theright. The intraglial recording during the seizure was recorded with pipette 1, whereas the depth-EEG activity was recorded with a macroelectrode placed close to the pipette. All recordings in B and C display averaged (n = 25) responses. The black arrowhead indicates the stimulation artifact. B, Both pipettes record extracellular activities. Note the higher amplitude response closer to the stimulation site. C, Responses at the same location, after impaling a glial cell at site1 (resting Vm at −100 mV) just below (2 μm) the recording shown in B. The glial response consists of an initial negative deflection, followed by a huge, round, positive wave. The vertical dotted linepoints to the simultaneous occurrence of a field negativity in both extracellular (B) and intraglial (C) recordings. The trace ingray represents the difference between the intracellular response and the field response at site1. The inset displays the expanded negativity of the glial response, before and after correction.
The possibility that recorded intraglial activities are contaminated by ephaptic transmission of extracellular fields was also envisaged. We therefore subtracted the extracellular field potential (Fig.2B, Site 1) from the intracellular response (Fig. 2C, Site 1). The corrected trace (displayed in gray in Fig. 2C, Site 1) still contains a negative indentation before the onset of the depolarization (Fig. 2C, inset). This observation, together with the fact that at the scale of all recorded glia the field negativity represented only 56% of the intracellular amplitude, suggests that at least part of the latter is generated by nonephaptic mechanisms.
Spontaneous activity patterns during slow-wave sleep and SW seizures
The ketamine–xylazine anesthesia used in this study produces an electrographic pattern very similar to the one present during natural slow-wave sleep (Steriade et al., 1996). A slow cortical oscillation (<1 Hz) dominates the activity of neurons (Steriade et al., 1993a,b) and glia (Amzica and Steriade, 1998b; Amzica and Neckelmann, 1999) and is reflected in the EEG (Contreras and Steriade, 1995). This pattern can be recognized in the periods preceding or following some of the SW seizures depicted in this paper (Figs. 3,7) and consists of alternating periods of depolarization and hyperpolarization.
Fig. 3.
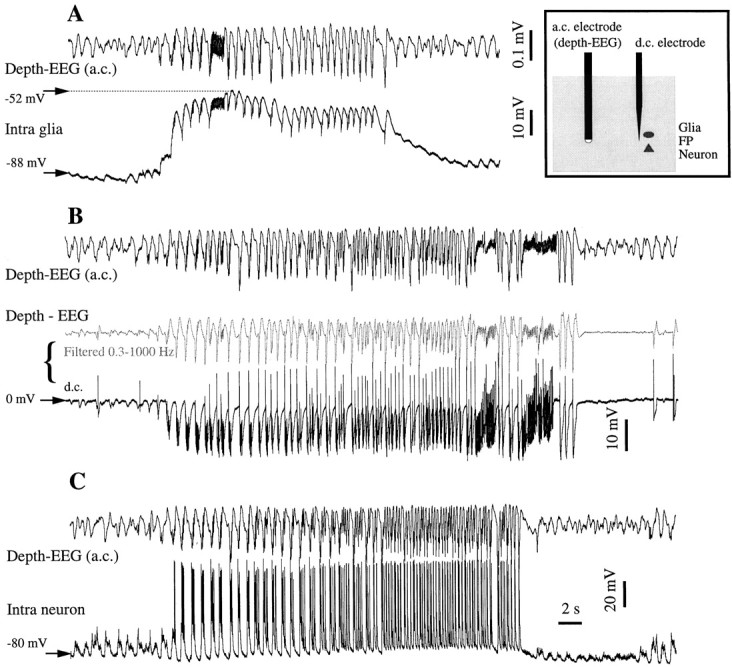
SW seizures recorded successively in various couple configurations. We kept one a.c. EEG electrode at a fixed depth, while penetrating with a pipette and recording first a glia (A), then d.c. extracellular field potentials (B), then a neuron (C). All seizures evolved from sleep-like, slowly oscillating patterns and produced recursive spike-and-wave complexes at 1.5–3 Hz. The seizure in the glia and neuron recordings is associated with a steady depolarization, which corresponds to a steady hyperpolarization in the d.c. extracellular recording. The vertical distance between the glia and the neuron is <10 μm, whereas the horizontal distance between the two electrodes is ∼0.5 mm. The EEG voltage calibration bar inA and the time calibration bar in C are common for all panels. In B, the depth-EEG recorded with the d.c. electrode (in black) was digitally filtered off-line between 0.3 and 1000 Hz (middle trace ingray) to emphasize that both d.c. and a.c. electrodes illustrate the same activity.
Fig. 7.

Glial transient negative potentials appear mostly during epileptiform activities. Double intracellular recording (neuron–glia) in association with area 5. A, Short SW seizure evolving from a slow oscillation pattern. The epileptic episode is accompanied in the glia recording by a persistent depolarization (above the horizontal dotted line). B, WTAs from the slow oscillation (1) and from the SW seizure (2) triggered with the steepest positive slope of the neuron (vertical dotted line). Note the additional depolarizing peak over the depolarization of the neuron in B2 and the corresponding negative potential superimposed on the glial potential. C, Superimposition of the neuronal and glial WTAs, from A andB, respectively. Traces marked with 1 are from the slow oscillation WTA; those with marked with 2are from the SW seizure. Vertical lines point to the excess of depolarization, and horizontal lines mark hyperpolarization during the seizure as compared with the slow oscillation activity.
SW seizures preferentially occur during natural resting sleep (Steriade, 1974; Kellaway, 1985). The progressive evolution from the slow oscillation toward rhythmic SW complexes at 2–4 Hz, often associated with fast runs (10–15 Hz), has been shown in cortical neurons (Steriade and Amzica, 1994; Steriade and Contreras, 1995;Steriade et al., 1998). Regardless of the recording configuration, spontaneous seizures developed from the slow oscillation by accelerating the pace of the oscillation to reach 2–3 Hz and by increasing the amplitude of the oscillatory complexes (Fig. 3). Occasionally, epochs with fast runs (∼10 Hz) were present during the seizure (Fig. 3A,B). In intraglial recordings, the oscillatory pattern was superimposed on a steady depolarization (Fig. 3A). This steady depolarization is in agreement with previous reports (Grossman and Hampton, 1968; Sypert and Ward, 1971). This steady depolarization was also present in neurons (see Fig. 5), although it was somewhat concealed by the firing of action potentials. It consisted of the progressively increased frequency of the intraneuronal PDSs (Fig. 3C). The depolarizing plateau of both neurons and glia was reflected in the extracellular d.c. field potentials as a sustained negative trend (Fig.3B, bottom trace). When this trace was digitally filtered in the frequency range used for AC recordings, its aspect was very similar to the one recorded directly nearby (Fig. 3B, compare middle and top traces).
Fig. 5.

Neuron–glia interaction during SW seizures. Continuous recording containing a double neuron–glia impalement (A), a neuron-field recording (B), and a double field d.c. recording (C) in cortical association area 7. The two electrodes are separated by <1 mm. The transition fromA to B is marked by the withdrawal of the pipette from the glia (oblique open arrowhead). During the neuron field recording (C), the second pipette is also withdrawn from the neuron (oblique open arrowhead at left), and a few seconds later it impales again, presumably the same neuron (oblique open arrowhead at right). Epochs within the squares are expanded above (A and B) or below (C) the respective panels. Note the recurrent sharp negative intraglial deflections associated with sharp neuronal depolarizing potentials (A).
To quantify the relationship between the main oscillatory components recorded in neurons, glia, and EEG belonging to the same pool of cells (separated by <10 μm), we performed WTAs (see Materials and Methods), separately, for the slow oscillation and the SW seizure (Fig.4). The a.c. electrode did not change its position, and thus we used its signal as a reference to perform the WTAs and the correlation analysis. WTAs were triggered with the steepest negative slope of the field potential at the start of each new oscillatory cycle (Fig. 4A, Slow oscillation; Fig. 4B, SW seizure). Each trace in Figure 4A1 (left) represents a K-complex (sharp depth–negative wave) during one of the three recording configurations depicted in the three panels of Figure 3. They are presented superposed and suggest the limited variability of their shape during the consecutive recording periods. Thereafter, we chose a 1 sec window (square) during which the resemblance of the three traces was >95% (see cross-correlations in the right panel). We also calculated the respective WTAs from the other recording electrode (Fig. 4A2,left). The cross-correlations derived from within the same window (Fig. 4A2, right) show a high central peak (∼88%). The negative sign of the peak is explained by the fact that the extracellular field potential is reversed with respect to both neuronal and glial potentials. The abscissa of the peak provides information about the time lag between the two activities contributing to the respective cross-correlation. In this case, and in relation to the focal depth-EEG activity, the neuronal depolarization preceded glia by 160 msec. It is worth mentioning that both peak amplitude and time lag reflect global properties of averaged activities and that slightly different time and resemblance relationships, as well as dynamics, may be obtained for limited segments of the potentials (e.g., the onset of the oscillatory cycle).
Fig. 4.

WTAs and correlative analyses for the activities presented in Figure 3. WTAs were triggered with the sharpest negative slope of the K-complex (A) and of the EEG spike (B) in the a.c. trace (Site 1, left panels). The triggering point is indicated by thevertical dotted lines. Two situations were analyzed: slow-wave sleep activities (A) and SW seizures (B). The three traces displayed in each left panel correspond to one of the three recording configurations described in the previous figure. The WTA traces recorded at Site 1 were superimposed to reveal a 1 sec epoch (in the square) in which their superimposition was optimal (see cross-correlations at right with correlative peaks >95%). The same 1 sec epoch was then used to calculate cross-correlations between the respective traces recorded atSite 2. These cross-correlations show roughly that during both slow oscillations and SW seizures, extracellular potentials reflect reverted intraglial and/or intraneuronal potentials. They also disclose time relationships between intracellular and extracellular potentials within cells separated by <10 μm.
A similar procedure was applied for the SW seizure (Fig.4B). It is obvious that, although thinner and ampler, the shape of the SW complexes (Fig. 4B1,left) is similar to the one of the K-complexes during sleep (Fig. 4A1, left), suggesting their common underlying mechanism. However, the neuron–EEG relationship (Fig.4B2, right) changed during seizure, compared with the one during sleep: the amplitude of the correlation peaks is higher (95%) and the time lag changed its sign (now the neuronal activity precedes the field by 50 msec). This is in line with previous findings that the relationship between pools of neurons evolves dynamically at the onset and during the SW seizures (Steriade and Amzica, 1994; Steriade and Contreras, 1995). The glia–EEG relationship remained constant, implying that the neuron–glia relationship changed during the SW seizure with respect to the control condition. In this case, the paroxysmal depolarization of the neuron preceded the glial depolarization by 170 msec.
The situation presented above (Figs. 3, 4) was representative for all recordings of this type (n = 24), namely, a fixed EEG macroelectrode and a moving microelectrode recording, successively, at least a cell and the adjacent field potential. Without exception, the global relationship between intracellular and d.c. extracellular potentials was one of reversal. The synchrony between cells and field potentials increased with the transition from slow oscillations to SW seizures. In 53% of the cases, the time relationship between cells and fields changed sign, meaning that for instance, if the cell was preceding the field at the beginning of the seizure, the opposite was detected later, during the seizure. In the rest of the cases the sign remained constant as the time relationship between the cell and its field remained constant throughout the recording. In 87% of the cases the time lag diminished during the transition, betraying an increase in the synchrony of the network. No systematic precursor time procession was observed for glia or neurons.
Negative intraglial potentials
Double intracellular recordings (neuron–glia) were performed from 67 pairs. In all cases, at least one of the electrodes recorded extracellular activities before or after the impalement (Fig.5). This configuration allowed only recordings of pairs of cells situated at some distance (generally 0.5–1 mm). Hence, we may assume that the respective cells did not belong to the same pool. However, the fact that these activities are the result of highly synchronized networks should yield pertinent conclusions for the voltage relationships between neurons, glia, and EEG.
Such recordings allowed us to shed light on an intriguing phenomenon that was noticed during intraglial recordings, especially those associated with paroxysmal activities. As already shown in Figure 2, the intraglial recordings displayed an early, short-lasting hyperpolarizing potential. This fact is surprising because all neurotransmitters tested up to now have only depolarizing actions on the glial membrane (see Discussion). Figure 5 contains a continuous double recording during recurrent SW seizures. Initially, during a simultaneous neuron–glia recording, both cells were steadily depolarized during a seizure (Fig. 5A). The expanded detail in Figure 5A shows two types of potentials: (1) neuronal fast transient depolarizing events (<50 msec) associated with fast negative deflections in the glia and (2) slower depolarizing potentials (>0.5 sec) in both neuron and glia. Although the former were reversed in the glia with respect to the neuron, the latter displayed similar time courses.
At the end of the seizure, the intraglial electrode was withdrawn (Fig.5B, oblique arrowhead) and a new seizure occurred. The expanded detail in Figure 5B shows that the general aspect of neuronal potentials remained the same as in the previous seizure. The extracellular d.c. field potentials recorded in the immediate vicinity of the previous glial cell displayed, in association with the excitatory neuronal events, exclusively negative potentials, which were larger and longer lasting than the ones recorded in the glia. They were superimposed on a persistent hyperpolarizing trend. A few seconds later, as a new seizure developed, the intraneuronal electrode was also withdrawn, and extracellular d.c. field potentials were recorded with both pipettes (Fig. 5C). The detail in the inset illustrates the resemblance between the EEGs at the two locations. The presence of negative intraglial potentials coincident with the onset of the PDSs on one hand, and their eventual concealing by depolarizing phasic potentials on the other hand, suggests that the intraglial activities recorded by our microelectrodes result from the summation of field effects and ionic currents (see Discussion).
To further test this hypothesis, we derived WTAs from the three above-mentioned recording configurations (Fig.6). Several stereotyped components are emphasized: during the double impalement (Fig. 6A), the neuronal SW complex contained an initial transient depolarization (NTD) superimposed on a steady depolarization (NSD). The equivalent waves in the intraglial recordings were a transient negativity (GTN) and a steady depolarization (GSD). These components were determined with reference (horizontal dotted lines) to theVm measured during the trough of the neuron (corresponding to the “wave” component of the EEG) preceding the onset of the phasic paroxysmal depolarization. It is worth mentioning that the aspect of the intraneuronal SW complex did not change from one seizure to the other (compare the respective traces in Fig. 6A,B), thus making possible the comparison between the intraglial and extraglial potentials. Moreover, the WTAs of the field potentials at the two recording sites (Fig. 6C) are almost identical (correlation factor >98%). The extracellular field potential produced after withdrawing the intraglial electrode (Fig. 6B) contains a transient negativity (FTN) and a steady negativity (FSN). Assuming that the GTN is caused exclusively by the ephaptic transmission of the FTN, the extraglial field potential (Fig. 6B) was subtracted from the intraglial WTA (Fig. 6A), producing a corrected trace (Fig. 6A in gray). This curve provides a maximal estimation (in the depolarizing sense) of the true intraglial potential. However, the GTN matches the reversed NTD (correlation factor −97%) much better than the FTN matches the NTD (correlation coefficient −86%). This difference may be attributable to the prolonged duration of the FTN. It seems reasonable therefore to hypothesize that the GTN is mainly caused by the reversed reflection of the NTD, whereas the FTN reflects superimposed reversed activities of both neurons and glia.
Fig. 6.

WTAs (n = 40) for the activities displayed in Figure 5. WTAs were triggered with the steepest slope at the onset of a SW complex as recorded with the second pipette (neuron pipette in the A and B and extracellular field recordings in C).A, Dual neuron–glia impalement. Two components were evident in the neuron: a transient depolarization (NTD) followed by a steady depolarization (NSD). The corresponding potentials in the glial recording were a transient negativity (GTN) and a steady depolarization (GSD). The gray trace resulted from the subtraction of the extraglial WTA (B) from the intraglial WTA (A). B, Recording with the first electrode withdrawn from the glia and the second electrode in the same neuron as in A. The same components were present in the neuronal SW complex. The NTD was associated in the extracellular field with a transient negative deflection (FTN), similar, to, although broader than, theGTN. The NSD corresponded to a steady negative potential (FSN).C, With both pipettes withdrawn from the respective cells, the WTAs at the two locations were identical. The calibration bar is the same for all panels.
This aspect was found, with certain variability, in all 67 pairs. The variability was caused by factors beyond our experimental control (e.g., in some pairs the SW complexes of the seizures had different frequencies or occurred with or without polyspikes, modifying the duration of the SW complex). To quantify the cell–field relationship regardless of these and other factors, we took into consideration only relative measures such as the increase in amplitude or duration from one state to the other.
Although the FTN and the GTN had a similar shape in all 52 recorded neuron–glia pairs, the amplitude of the FTN was higher than the amplitude of the GTN (20 ± 5%), and its duration, measured at half amplitude, was longer (38 ± 7%). The GTN could be reduced by the superimposition of the round depolarizing potential building up the GSD. Globally, the extracellular potential reflected the reversed intraneuronal potential (average correlation factor of 88%) better than the reversed intraglial potential (average correlation factor of 66%). Thus, the relationship between the glial and the field transient negativities (Fig. 6) suggests that the glial transient negativity reflects field potentials rather than intracellular hyperpolarizing potentials.
Several lines of evidence support this idea. On a few occasions (three neuron–glia pairs recorded intracellularly during 16 seizures), SW complexes in neurons consisted of the usual depolarizing–hyperpolarizing cycle of the slow oscillation overridden by paroxysmal depolarizations that had no clear relationship to the onset of the oscillatory cycle (Fig. 7). The seizures appeared spontaneously, evolving from slow oscillating patterns, were short in duration (<15 sec), and were not followed by postictal depression (Fig. 7A). This was in contrast to the majority of SW seizures, in which the PDSs resulted from the paroxysmal evolution of the slow oscillation and erupted from the very onset of the depolarization. In these cases, PDSs were superimposed over the depolarizing phase of the slow oscillation and kept a distinct shape and onset. We used these seizures to study relationships between these paroxysmal depolarizations and the intraglial negativities. WTAs of normal slow oscillation cycles (Fig. 7B1) yielded to the pattern associated with the K-complex. WTAs triggered with the same steep onset of the depolarization, but during the seizure, they generated a similar depolarizing pattern in the neuron on top of which the paroxysmal depolarization was evident (Fig. 7B2). This additional neuronal depolarization was reflected in the glial and field potential by a negative potential as well as an increased excitation. The superimposition of the neuronal WTAs resulting from the slow oscillation (1) and the seizure (2) (Fig.7C) shows the excess of depolarization in the neuron during the seizure (surface filled with vertical lines), compared with the slow oscillation. At the same time, the superimposition of the glial WTAs reveals a more negative potential for the initial part of the SW complex (surface marked with horizontal lines) and a subsequent excess of depolarization (surface filled with vertical lines). The increased glial depolarization after the increased neuronal depolarization during the seizure suggests that the increased negativity reflects field phenomena rather than a neurotransmitter-induced hyperpolarization.
Further support for the idea that glial negativities result from the reversal of the neuronal depolarization is shown in Figure8. It represents one of the cases in which the glial depolarization started before the neuronal one and in which the depolarizing trend of the glial PDS was interrupted by a negative potential. The calculation of the first derivative of each WTA (Fig. 8, gray traces) disclosed several key points (marked with black dots on the derivative curves) related to slope changes in the respective potentials. First, the moment where the derivative of the glial WTA became positive marks the onset of the glial depolarization (see black arrow pointed toward the intraglial WTA). At that moment, none of the other traces showed any sign of systematic variation. The next key point occurred in the neuronal WTA, at the moment where the onset of the neuronal depolarization reached its maximum slope. This event happened simultaneously with the maximum negative slope of the glial negativity and with a local maximum of the field potential (coincidence points are indicated by oblique empty arrows). Finally, the minimum of the depth-EEG is associated with a change in the slope of the glial depolarization and with the maximum of the neuronal PDS (however, the latter point is less reliable because of the abundant spiking of the neuron).
Fig. 8.

Evidence that the glial transient negativities reflect neuronal depolarizations. Shown are simultaneous intracellular recordings of neuronal and glial activities, together with d.c. extracellular field potentials. Black traces represent WTAs (n = 50) of rhythmic PDSs. The threegray traces are derivatives (d/dt) of the respective potentials. Several points (black dots) were marked on the three derivatives: the onset of the glial depolarization at the point where its derivative becomes positive, the maximum of the neuronal derivative coinciding with the maximum slope of the neuronal depolarization, and the minimum of the depth-EEG at the moment where the field derivative is zero. A black arrow and avertical dotted line mark the correspondence of these points with the associated potential in each trace. Open arrows point toward the coincidence with particular shapes: the maximum slope at the onset of the neuronal depolarization coincides with the maximum negative slope at the onset of the corresponding glial negativity (top arrow) and with a dicrotic swing in the EEG (bottom arrow). The EEG minimum is associated with the change in slope of the glial depolarization. Also note that the glial depolarization starts before the neuronal depolarization.
The analysis of these shape features suggests that the presence of the glial negativity interrupting the course of the depolarization reflects a gradient of the reversed intraneuronal PDS and not the direct field potential because (1) there is coincidence between the steepest slopes of the neuron and glia; (2) there is potential reversal; and (3) the time course of the field potential (measured in d.c. current and with similar electrodes as those used for the impalements) is different from the intraglial potential. Additionally, the coincidence between the minimum of the depth-EEG, the maximum of the neuronal PDS, and the reduction of rising slope in the glia further supports the idea that reversed neuronal potentials contribute to the shape of the glial intracellular activities.
To further test whether the intraglial negativities result from field effects or from membrane conductances, we established that these phenomena are dependent on the evolution of the seizure and not on the membrane polarization (Fig. 9). The seizure presented in Figure 9A was one of the numerous (130) recurrent seizures induced with bicuculline. They were used here because of the stereotyped pattern induced in glia (Amzica and Neckelmann, 1999, their Fig. 4) and for their periodic recurrence. Sweeps were extracted with reference to the steepest negative slope of the neighboring field potential (Fig. 9B, vertical dotted lines). They reflect the evolution from isolated PDSs (Fig.9B1), initial ictal PDSs (Fig. 9B2), and fast runs (Fig. 9B3) to rhythmic (∼2 Hz) ictal PDSs during the late stages of the SW seizure (Fig. 9B4,5). This evolution is accompanied by the appearance and progressive increase of negative glial potentials at the beginning of each SW complex. Because this development was also associated with the spontaneous depolarization of the glial membrane, we also recorded several seizures (n = 10) under steady depolarizing current. The comparison of isolated PDSs occurring, at rest and under current, before the onset of the seizure (Fig. 9C1) shows that no negative potentials appear at more depolarized membrane potentials. InC we superimpose the first PDS of the seizure inA, at resting Vm, and a first PDS recorded during the next seizure under steady depolarization (+1.5 nA). The Vm of the latter (Fig.9C1, −65 mV), measured before the PDS onset, is in the range in which negative potentials were apparent in the middle of the seizure (see the Vmwhere the negative potential occurs in Fig. 9C4, bottom trace). Conversely, by comparing SW complexes from the middle of seizures, and regardless of the Vm, one obtains similar amplitudes of the negative potentials (Fig.9C4). Thus, negative potentials are not generated by voltage-dependent mechanisms but rather by the imposition of external field potentials through the glial membrane.
Fig. 9.

Glial transient negative potentials are modulated by the evolution of the seizure and are not voltage dependent.A, Intraglial and field potential (a.c.) recording during a seizure starting with isolated PDSs (1) and continuing with recurrent SW complexes (2,4, 5) and fast runs (3). B, In each panel, superimposition of five sweeps extracted around the point of maximum negative slope of the field potential (dotted vertical line). The five panels correspond to the underlined epochs in A. Three of the sweeps in panel 1 were taken from a previous seizure recorded in the same glia. Note the absence of negative potentials in B1 and their progressive appearance from B2 to B5. C, Superimposition of two interictal (1) and two ictal (4) PDSs recorded without current (at rest) and when injecting +1.5 nA steady current into the glial cell. The sweeps belong to successive seizures and to periods similar to the ones indicated in A. Regardless of the amount of current injected and of the imposed membrane potential, there were no negative transient potentials at the beginning of the seizure (C1), and they had similar evolutions during seizures.
The dynamic evolution of various parameters defining the glial transient negative potentials is depicted in Figure10. Each glial negativity was characterized by its amplitude, duration, surface area, and theVm at which it occurred. Comparison between different seizures was made possible by normalizing the duration of these seizures (Fig. 10A–C,abscissae). The period from the onset of the first ictal PDS to the last PDS was divided into 10 equal windows, and for a given window we calculated the various parameters for each SW complex. Then, the average value for each parameter was plotted as a single point corresponding to the respective value. Only five representative seizures are depicted in Figure 10; however, the analysis was performed for a total of 50 seizures.
Fig. 10.

Dynamic evolution of the parameters of the glial negativity as a function of the seizure development. Each panel contains the results from five seizures and their average. Seizures, regardless of their duration, were divided into 10 equal windows. The surface area of glial negativities at the beginning of a SW complex (A), their duration (B), and their amplitude (C) were calculated for all SW complexes contained in each window and averaged. This average is plotted against the respective window ordinal, expressed as a percentage of the total time of the seizure. Therefore the abscissae inA–C represent the percentage of the total duration of the seizure. Each panel also contains the grand average for the five seizures depicted (thick line). D, The surface area of the glial negativities plotted against the membrane potential at which they occurred shows nonlinear dependence, suggesting that they are not a voltage-dependent phenomenon.
The first window generally produced very few and small negativities. They increased within the next two to three epochs to reach relatively stable values thereafter. The maximum average amplitude of the negativity was 4.4 mV (Fig. 10C), whereas the maximum average duration reached 147 msec (Fig. 10B). A small decrease of 5% in amplitude (also reflected in the surface area) (Fig.10A) was observed during the last window, before the arrest of the seizure. In each panel we also introduced data from seizures induced with bicuculline (traces with black points and asterisks), because in these seizures the location of the focus was known. From the two situations, thetrace with the black points stems from a glia recorded at a closer distance to the infusion site than the one depicted with asterisks. Thus, glial transient negativities were more ample closer to the presumed focus of the seizure. This finding was consistent in all of the 130 seizures induced with bicuculline. The analysis of surface–Vm relationships failed to disclose a linear relationship (Fig. 10D). Although there were linear segments within a graph, the nonlinear segments were such that they preclude the possibility that glial negativities are a voltage-dependent phenomenon. The linear segments in individual curves may result from the continuous increase of the glial negativity occurring simultaneously with the depolarization of the cell during the initial part of the seizure. After the sequence of values toward the end of the seizures, it appeared that the surface of the negative potentials remained constant, or even increased, as theVm started to hyperpolarize. One of the tested cells (depicted with black dots) showed nonlinear behavior at a Vm of −80 mV, whereas others showed it at −60–65 mV. Voltage-dependent conductances may be activated during SW seizures, but they cannot account for the above-mentioned behavior. This suggests that the glial negativity is not a voltage-dependent phenomenon.
We claim that negative intraglial potentials are not the sheer effect of ephaptic propagation of extracellular potentials into the glia. First, glial impalements were assessed by steep potential drops after penetration of the membrane (Fig. 1B). The leak conductance produced by impalement of cells is unlikely to be higher for glia than for neurons, so as to favor the leak of extracellular currents into the glia, and a resting membrane potential of −88 mV cannot betray a leaky membrane. Second, glial activities showed clear sustained depolarizations in association with negative extracellular d.c. potentials (Figs. 1B, 5, 6,11). Third, the time response of the intracellular electrodes used in this study is much faster than the time course of intraglial events, and their impedance is high enough to prevent them from recording distal fields. Thus, during impalements, they record only intracellular activity.
Fig. 11.

Evolution of glial transient negative potentials evoked by cortical stimulation during SW seizures. A, SW seizure induced by cortical stimulation close to the recording site (area 5) of a glia and d.c. field potentials (left) and expansion of three sweeps (marked with asterisks) to show the evolution of the intracellular negativity as a function of the progression of the seizure (right). B, Evoked potentials (n = 50). Stimuli were delivered at 10 Hz and continued beyond the period depicted atleft. The glial evoked response displays an initial transient negative potential followed by a steady depolarization. The transient negativity was practically identical to that recorded in the d.c. field potential, whereas the depolarization appeared reversed in the depth-EEG.
Similar results were obtained from seizures induced by electrical stimulation (88 seizures). Simultaneous intraglial and d.c. field potential recordings show that the average response to the cortical stimulus (Fig. 11B) starts with a negative potential of comparable amplitude in both electrodes. However, the averaging procedure obscures the dynamic evolution of the response. This is shown in Figure 11A, right, where progressively larger negative potentials develop with the advancement of the seizure. In such cases, again, the amplitude of the glial negative potentials was not related to the Vm (data not shown). It is equally noteworthy that the rest of the evoked response consists of a steady depolarization in the intraglial response (Fig.11B, vertical lines), which corresponds to the negative extracellular potential (Fig. 11B,horizontal lines).
Extracellular K+ measurements in relation to glial activities
Glial cells are known as reliable K+detectors (Nicholls and Kuffler, 1964; Kuffler et al., 1966). However, there is no mention of the extracellular K+ activity associated with the slow sleep oscillation. We measured the [K+]o with ion-sensitive electrodes in 10 animals. During the slow oscillation, the variations of the K+ activity followed those of the EEG (Fig.12A). Recordings were made with double-barrel electrodes, which allowed an estimate of the amount of field potential activity recorded through the ion-sensitive electrode. After correction, the amplitude of the K+ oscillations was in the range of 1–2 mm. The average amplitude of the K+ increase during a cycle of the slow oscillation, calculated from 10 episodes with slow oscillations (each episode containing 60 periods), was 1.8 ± 0.4 mm.
Fig. 12.

Potassium activities related to the slow oscillations and SW seizures. A, Depth-EEG and K+ variations during a period with slow (<1 Hz) oscillations. The right panel depicts WTAs (n = 40) from the two leads. The WTAs were triggered with the steepest descending slope of the field potential (vertical dotted line). B, SW seizure recorded with a double-barrel pipette (Depth-EEG andPotassium) and with an intracellular microelectrode (Intra glia). In this panel, field potentials were not subtracted from the K+-sensitive or intraglial potentials because field and glial activities were time-lagged and because negative K+ potentials were more ample than their field equivalents. The three underlined epochs are expanded below and display two interictal PDSs (1), ictal PDSs during the initial part (2), and the middle of the seizure (3).
SW seizures were associated, as already reported (Fertziger and Ranck, 1970; Futamachi et al., 1974; Moody et al., 1974), with a steady increase of the [K+]o, which matched the intracellular voltage variations of glial cells (Fig.12B). Additionally, phasic increases of K+ were seen for both interictal and ictal PDSs. The amplitude of [K+]o during isolated interictal PDSs was in the order of 3–4 mm (after eliminating field contributions), whereas the same measurement during ictal PDSs was less reliable because of the superimposition of the steady accumulation of K+ during the seizure. It becomes clear from the expanded details (Fig. 12B1–3) that the intraglial negativities are not caused exclusively by ephaptic transmission of extracellular field spikes because the latter have smaller amplitudes and a slower time course than the former. However, they may affect the potentials recorded by the ion-sensitive electrode.
Relationship of intraglial potentials with the depth profile of the SW complexes
The results presented above show the coherent activities of neurons and glial cells. It is known from numerous studies that cortical activities display a dipolar behavior. In the particular case of epileptic discharges of the type recorded in this study, a depth-to-surface potential reversal has been identified for the SW complexes (Kostopoulos et al., 1982; Petsche et al., 1984; Kandel and Buzsáki, 1997; Steriade et al., 1998). This brings into discussion the behavior of glial cells located in the surface of the cortex in association with closely recorded field potentials.
We performed simultaneous intraglial and depth profile recordings. For the latter we used multiple electrodes aligned vertically (Fig.13) (fields are filtered between 0.3 Hz and 1 kHz). Impaling superficial glia proved to be a difficult task, and we recorded only three cells within the first 300 μm of cortical depth. However, in all recorded glia, regardless of their depth, the intracellular potentials displayed consistent patterns, similar to the ones depicted in the present paper for deeply situated cells. The general pattern of the seizure was unaffected by the position of the impaled glia and consisted of round phasic depolarizing potentials becoming rhythmic and synchronized during SW seizures. They were exclusively depolarizing during the interictal spikes preceding the onset of the paroxysm (Fig. 13B1) and started to display small negative potentials at the beginning of the seizure (Fig.13B2), which gradually increased toward the end of the seizure (Fig. 13B3,4). WTAs of the SW complexes (Fig. 13C) further emphasize the evolution of these negative potentials with the seizure.
Fig. 13.
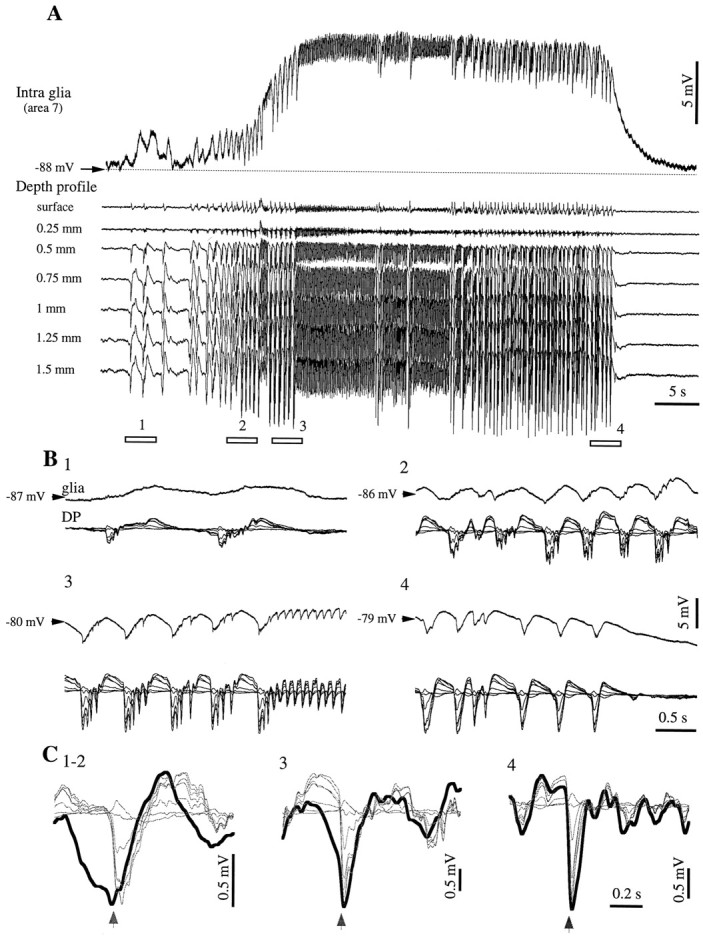
Relationship of intraglial potentials with the depth profile of SW seizures. A, Intraglial recording together with field potentials recorded at seven equidistant depths of the cortex. The distance between the pipette and the multiple electrodes was ∼1 mm, and the glia was recorded at a depth of ∼1.5 mm. The seizure starts with a few isolated PDSs and continues with rhythmic PDSs interrupted by sequences of fast runs. The four underlined periods are expanded in B. Small negative intraglial potentials appear toward the end of panel 2. C, WTAs triggered with the most negative slope at the onset of the EEG spike in the deepest lead. Averages were made with 10 sweeps taken within the beginning of the seizure (1–2), the middle of the seizure (3; with the exclusion of the fast runs), and the end of the seizure (4). The arrows belowindicate the triggering moment of the WTA. The glial WTA is drawn with a thick line. Note the increasing resemblance between intraglial and depth field potentials and the reversal of the latter in the surface of the cortex.
The laminar profile of the seizure shows that the EEG SW complexes were made of depth-negative potentials followed by slower positive waves. The amplitude of these components increased with the depth, and they were reversed in the surface. The reversal occurred at a depth ∼0.3 mm. Comparison between intraglial potentials and the depth profile shows that, at least for the initial epoch, the repolarization of the glial cell at the end of a SW cycle had a shape that was distinct from the one recorded in the depth of the cortex. However, as the seizure evolved, intraglial potentials became more similar to those of the depth field potentials, and the amplitude of the intraglial negativity increased (Fig. 13C3,4). Still, the superficial components of the field were not related to the intraglial components. This further reinforces the idea that the intraglial negativity is not induced primarily by ephaptic transmission. It also suggests that the genesis of superficial local field potentials is caused mainly by sink currents reentering the apical dendrites of neurons, whereas local cells do not have a major contribution.
DISCUSSION
We have shown that neurons and glia respond coherently to cortical stimuli and that they oscillate together during slow sleep oscillations and SW seizures. The neuronal potentials during the onset of the PDS are reflected in intraglial activities as negative potentials, and we propose that transient negative intraglial potentials are a field effect resulting from the reversal of neuronal hypersynchronous phasic events. This finding is based on dual glia–neuron recordings, as well as on consecutive glia–neuron impalements, and on the recording of d.c. extracellular field potentials. It suggests that the genesis of extracellular field potentials involves reciprocal and dynamic interactions between neurons and glia. Secondly, we show that cortical glial cells may contribute to the excitability of cortical neurons.
Contribution of glia to cortical evoked activities
Cortical responses during double intracellular recordings (Fig. 1) indicate that glial cells respond to cortical stimuli. Under our experimental condition it was not possible to discern whether the glial responses were induced (1) by the glial uptake of ions and/or neurotransmitters released by the neurons or (2) by neuronal synapses contacting the glial membrane. Some evidence suggests that both cases could be at work. Through their anatomic position, some glial cells sheathe synapses and increase the efficiency of synaptic transmission (Pfrieger and Barres, 1997). These glial cells would be directly subject to the influence of neurotransmitters because their membrane is endowed with receptors for glutamate (Sontheimer et al., 1988;Steinhäuser and Gallo, 1996) and GABA (Bormann and Kettenmann, 1988; MacVicar et al., 1989; Rosier et al., 1993). In addition, glia contain, synthesize, and release neuroactive compounds, including amino acid transmitters such as glutamate and aspartate (Martin et al., 1990;Levi and Patrizio, 1992, Araque et al., 1999) as well as GABA (Levi and Gallo, 1995), which could contribute to the closing of the neuron–glia feedback loop.
The fact that the initial barrage of neuronal EPSPs betraying excitation is associated with glial depolarization (Fig.1C,D) suggests the implication of neuronal K+ released during the EPSP or a direct glutamatergic action of neurons. The subsequent neuronal IPSP is produced by the activation of GABAA and GABAB receptors (Connors et al., 1988), although the contribution of the latter is less important in vivo(Contreras et al., 1997). It was also suggested that the late part of the inhibitory response might be attributable to disfacilitation during which K+ currents, responsible for the resting Vm, dominate the membrane behavior (Contreras et al., 1997). In any event, glia are expected to become depolarized by either uptake of K+released by neurons after their activation through GABAB receptors or opening of Cl− channels by GABAA action (Kettenmann and Schachner, 1985). This was indeed observed, and the neuronal IPSP is reflected in the glia by a depolarizing potential (Fig. 1C,D). The rebound excitation after the neuronal IPSP reflects a synchronous event known as the cortical K-complex (Amzica and Steriade, 1998). The glia became hyperpolarized at the beginning and slowly depolarized thereafter. This phenomenon is rather surprising because no hyperpolarizing actions have been previously described in glial cells. Indeed, the active or passive uptake of K+, as well as the higher concentration of intraglial Cl− ions, would only depolarize glia. Thus, the hyperpolarization depicted in Figure1C,D could be either the result of a yet unknown mechanism actively hyperpolarizing glial cells or, as we postulate, the field reflection of the intracellular potentials of nearby neurons.
The fact that the extracellular K-complex and the preceding depth-positive wave have shapes similar to intraglial potentials indicates that both intraglial and extracellular electrodes pick up the reversed potentials of the intraneuronal activities. This reasoning fails in the case of the initial excitation, probably because it reflects a different phenomenon. For instance, it could be envisaged that the initial synaptic excitation is produced only by a signal traveling exclusively through axons, possibly directly contacting glia, which would result in negligible field effects. In contrast, the subsequent inhibitory–rebound sequences would reflect more generalized discharges invading the somas and the dendrites of neurons. On the other hand, in epileptic tissue, the initial negativities present at the onset of the glial response (Fig. 2C) may be explained by the hypersynchronous nature of the directly triggered PDSs. The ample neuronal depolarization is reflected, reversed, in the extracellular space and could undergo the same process across glial membranes that are brought closer to the neuronal membranes by the glial swelling (Amzica and Neckelmann, 1999). The subsequent glial-positive potential may be the superimposition of a membrane depolarization over a field negativity and thus may have underevaluated amplitude (Fig. 2C).
Contribution of glia to cortical spontaneous activities
Spontaneous activities, such as the ones recorded in this study, are characterized by the synchrony between neuronal elements (Steriade and Amzica, 1994; Amzica and Steriade, 1995; Contreras and Steriade, 1995; Colder et al., 1996; Neckelmann et al., 1998). The slow oscillation is composed of alternative depolarizing and hyperpolarizing levels of the neuronal Vm (Fig. 8). The depolarizing phase of the slow oscillation is made of EPSPs and GABAA IPSPs (Steriade et al., 1993a), whereas the hyperpolarizing phase is associated with reduced synaptic activity in the cortical network caused by disfacilitation and with relatively increased resistance of the neuronal membrane (Contreras et al., 1996). Although the slow oscillation evolves synchronously in neurons and glia, each cycle of the slow oscillation has different potential shapes in the two cell categories. Only very small negative intraglial potentials could be recorded at the onset of the excitation during the slow oscillation (Fig. 7B1). Because each cycle of the slow oscillation represents a K-complex (Amzica and Steriade, 1997, 1998a), it is not surprising that they had amplitudes similar to those evoked by cortical stimuli (see above).
For the rest of the potentials contributing to the slow oscillation, it is reasonable to assume that the glialVm reflects the variations in [K+]o attributable to neuronal activity (Figs. 5, 12A). From the present results it may already be anticipated that glial cells, beyond their passive role in regulating [K+]o (Newman, 1995), may also play an active role in setting the pace of the slow oscillation. During one cycle of the slow oscillation, the maximal glial depolarization is reached much later than the end of the neuronal depolarization (Fig. 7B1), and the glialVm returns to control values at precisely the end of the cycle. This behavior could result from a close interaction between neurons and glia, which may control the pace of the oscillation as well as the ionic equilibrium in the extracellular space. Besides the neurotransmitter-based dialogue between neurons and glia (Araque et al., 1998, 1999), an indirect communication may be supported by the [K+]o, which is known to modulate neuronal excitability. For instance, it has been shown in cerebellar neurons that increasing the [K+]o up to 4.5 mm leads to increases in firing rate and synaptic responsiveness; however, further elevations induce a drop in excitability (Kocsis et al., 1983) and would thus end the neuronal firing and produce a progressive disfacilitation of the network. Here we report that the [K+]o amplitude during the slow oscillation reached 1–2 mm (Fig.12A), which when added to the physiological values of resting concentrations (∼3 mm) (Futamachi et al., 1974) brings the excitability of cortical neurons to oscillate between hyperexcitability and hypoexcitability.
The development of SW seizures from the slow oscillation and the common mechanisms underlying the genesis of SW complexes and K-complexes makes the discussion of SW seizures similar to the one of the slow oscillation. The differences mainly concern the larger amount of depolarization expressed by all recorded cells and thus the increase in [K+]o (Fig.12B) and the prominent glial negative potential at the onset of SW complexes (Figs. 4B2,6A, 7B2, 8, 9, 11-13). Several lines of evidence support the view that the latter component is related to field effects. First, these negative potentials reflect reverted neuronal potentials. Second, ephaptic potentials have been mentioned mostly during paroxysmal events (Taylor and Dudek 1982; Dudek et al., 1986;Hochman et al., 1995). Third, the intraglial negative transients are not voltage dependent (Figs. 9-10), and finally, this phenomenon is potentiated by the shrinkage of the extracellular space, which is known to occur during SW seizures (Dietzel et al., 1980). Intraglial negative potentials appear gradually with the development of the seizure (Fig.9). This observation corroborates a recent in vivo study, under similar experimental conditions, showing that the shrinkage of the extracellular space during SW seizures is caused by the progressive swelling of glial cells (Amzica and Neckelmann, 1999).
Relationships between neuronal, glial, and field potentials
The extracellular field potential is generated as a result of current flow within cells. The way these currents make up the field potentials depends on the time course of the intracellular currents. These phenomena are well documented separately either for neurons, particularly the occurrence of action potentials (Terzuolo and Araki, 1961), or for glia, especially for persistent potentials (Somjen, 1973), but they were not investigated in neocortical neuron–glia pairs. The contribution of glia to the genesis of faster events may be envisaged, because these cells are at least as numerous as neurons (Streit, 1995) and their membrane resistance is lower than that of neurons (Trachtenberg and Pollen, 1970), thus favoring the extracellular reflection of intraglial potentials.
We propose that the genesis of field potentials results from complex interactions between neurons and glia and that the state of the cortical networks modulates this interaction (Fig.14). During the slow oscillation, which dominates the electric activity of the cortex during quiet sleep, the extracellular milieu undergoes the influence of neuronal and glial potentials (Fig. 14, left). The transmembrane currents of both types of cells summate in the interstitial space, and the ephaptic field effects play a reduced role, if any. Paroxysmal oscillations induce glial swelling and the reduction of the extracellular space (Fig. 14, right), and besides the aforementioned mechanisms, field effects across the touching membranes may be superimposed on the intracellular activities. Large neuronal potentials are expected to be reflected reversed in glia. A reciprocal action would also be justified; however, the slower time course of intraglial events makes them less likely to be recognized within intraneuronal potentials, where they might be included in the neuronal phasic events. A more quantitative study is required to establish the dynamic weights of each type of potential in the resulting EEG.
Fig. 14.
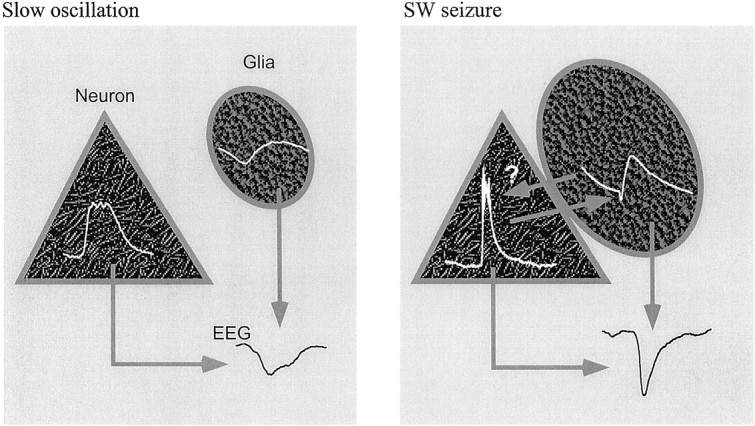
Schematic diagram of the mechanisms generating field potentials during slow oscillations (left) and SW seizures (right). An averaged cycle is drawn in each cell (white traces). During the slow oscillation, reversed neuronal and glial potentials contribute to the genesis of the extracellular field potential (EEG). SW seizures are accompanied by glial swelling, which may bring patches of cellular membranes into contact, allowing intraneuronal potentials to appear reversed, as field potentials, in the glial cells (arrowfrom neuron to glia points toward the glial negativity). The reverse pathway might also be at work (arrow from glia to neuron). Both intraneuronal and intraglial activities contribute to the shape of the extracellular field potential.
Footnotes
This study was supported by the Medical Research Council of Canada (Grants MT-15681 to F.A. and MT-3689 to M.S.). F.A. is a Scholar of Fonds de la Recherche en Santé de Québec. We thank D. Drolet and P. Giguère for technical assistance. We are also indebted to A. Manfridi for his assistance during experiments with potassium-sensitive electrodes.
Correspondence should be addressed to Florin Amzica, Laboratoire de Neurophysiologie, Faculté de Médecine, UniversitéLaval, Québec, Canada G1K 7P4. E-mail:florin.amzica@phs.ulaval.ca.
REFERENCES
- 1.Achermann P, Borbély AA. Low-frequency (<1 Hz) oscillations in the human sleep EEG. Neuroscience. 1997;81:213–222. doi: 10.1016/s0306-4522(97)00186-3. [DOI] [PubMed] [Google Scholar]
- 2.Ammann D. Ion selective microelectrodes. Springer; Berlin: 1986. [Google Scholar]
- 3.Amzica F, Neckelmann D. Membrane capacitance of cortical neurons and glia during sleep oscillations and spike-wave seizures. J Neurophysiol. 1999;82:2731–2746. doi: 10.1152/jn.1999.82.5.2731. [DOI] [PubMed] [Google Scholar]
- 4.Amzica F, Steriade M. Short- and long-range neuronal synchronization of the slow (<1 Hz) cortical oscillation. J Neurophysiol. 1995;75:20–38. doi: 10.1152/jn.1995.73.1.20. [DOI] [PubMed] [Google Scholar]
- 5.Amzica F, Steriade M. The K-complex: Its slow (<1 Hz) rhythmicity and relation to delta waves. Neurology. 1997;49:952–959. doi: 10.1212/wnl.49.4.952. [DOI] [PubMed] [Google Scholar]
- 6.Amzica F, Steriade M. Cellular substrates and laminar profile of sleep K-complex. Neuroscience. 1998a;82:671–686. doi: 10.1016/s0306-4522(97)00319-9. [DOI] [PubMed] [Google Scholar]
- 7.Amzica F, Steriade M. Electrphysiological correlates of sleep delta waves. Electroencephalogr Clin Neurophysiol. 1998b;107:69–83. doi: 10.1016/s0013-4694(98)00051-0. [DOI] [PubMed] [Google Scholar]
- 8.Araque A, Sanzgiri RP, Parpura V, Haydon PG. Calcium elevation in astrocytes causes an NMDA receptor-dependent increase in the frequency of miniature synaptic currents in cultured hippocampal neurons. J Neurosci. 1998;18:6822–6829. doi: 10.1523/JNEUROSCI.18-17-06822.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. doi: 10.1016/s0166-2236(98)01349-6. [DOI] [PubMed] [Google Scholar]
- 10.Bendat JS, Piersol AG. Engineering applications of correlation and spectral analysis. Wiley; New York: 1980. [Google Scholar]
- 11.Bormann J, Kettenmann H. Patch clamp study of GABA receptor Cl− channels in cultured astrocytes. Proc Natl Acad Sci USA. 1988;85:8336–8340. doi: 10.1073/pnas.85.23.9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colder BW, Wilson CL, Frysinger RC, Chao LC, Harper RM, Engel J., Jr Neuronal synchrony in relation to burst discharge in epileptic human temporal lobes. J Neurophysiol. 1996;75:2496–2508. doi: 10.1152/jn.1996.75.6.2496. [DOI] [PubMed] [Google Scholar]
- 13.Connors BW, Malenka RC, Silva LR. Two inhibitory postsynaptic potentials, and GABAA and GABAB receptor-mediated responses in neocortex of rat and cat. J Physiol (Lond) 1988;406:443–468. doi: 10.1113/jphysiol.1988.sp017390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Contreras D, Steriade M. Cellular basis of EEG slow rhythms: a study of dynamic corticothalamic relationships. J Neurosci. 1995;15:604–622. doi: 10.1523/JNEUROSCI.15-01-00604.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Contreras D, Timofeev I, Steriade M. Mechanisms of long-lasting hyperpolarizations underlying slow sleep oscillations in cat corticothalamic networks. J Physiol (Lond) 1996;494:251–264. doi: 10.1113/jphysiol.1996.sp021488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Contreras D, Durmüller N, Steriade M. Absence of prevalent laminar distribution of IPSPs in association cortical neurons of cat. J Neurophysiol. 1997;78:2742–2753. doi: 10.1152/jn.1997.78.5.2742. [DOI] [PubMed] [Google Scholar]
- 17.de Curtis M, Manfridi A, Biella G. Activity-dependent pH shifts and periodic recurrence of spontaneous interictal spikes in a model of focal epileptogenesis. J Neurosci. 1998;18:7543–7551. doi: 10.1523/JNEUROSCI.18-18-07543.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dichter MA, Herman CJ, Selzer M. Silent cells during interictal discharges and seizures in hippocampal penicillin foci. Evidence for the role of extracellular K+ in the transition from the interictal state to seizures. Brain Res. 1972;48:173–183. doi: 10.1016/0006-8993(72)90177-1. [DOI] [PubMed] [Google Scholar]
- 19.Dietzel I, Heinemann U, Hofmeier G, Lux HD. Transient changes in the size of the extracellular space in the sensorimotor cortex of cats in relation to stimulus-induced changes in potassium concentration. Exp Brain Res. 1980;40:432–439. doi: 10.1007/BF00236151. [DOI] [PubMed] [Google Scholar]
- 20.Dudek FE, Snow RW, Taylor CP. Role of electrical interactions in synchronization of epileptiform bursts. Adv Neurol. 1986;44:593–617. [PubMed] [Google Scholar]
- 21.Duffy S, Fraser DD, MacVicar BA. Potassium channels. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 185–201. [Google Scholar]
- 22.Fertziger AP, Ranck JB., Jr Potassium accumulation in interstitial space during epileptiform seizures. Exp Neurol. 1970;26:571–585. doi: 10.1016/0014-4886(70)90150-0. [DOI] [PubMed] [Google Scholar]
- 23.Futamachi KJ, Mutani R, Prince DA. Potassium activity in rabbit cortex. Brain Res. 1974;75:5–25. doi: 10.1016/0006-8993(74)90767-7. [DOI] [PubMed] [Google Scholar]
- 24.Grossman RG, Hampton T. Depolarization of cortical glial cells during electrical activity. Brain Res. 1968;11:316–324. doi: 10.1016/0006-8993(68)90027-9. [DOI] [PubMed] [Google Scholar]
- 25.Hochman DW, Baraban SC, Owens JW, Schwartzkroin PA. Dissociation of synchronization and excitability in furosemide blockade of epileptiform activity. Science. 1995;270:99–102. doi: 10.1126/science.270.5233.99. [DOI] [PubMed] [Google Scholar]
- 26.Hubbard JI, Llinás R, Quastel DMJ. Electrophysiological analysis of synaptic transmission. Arnold; London: 1969. [Google Scholar]
- 27.Janigro D, Gasparini S, D'Ambrosio R, McKhann G, II, DiFrancesco D. Reduction of K+ uptake in glia prevents long-term depression maintenance and causes epileptiform activity. J Neurosci. 1997;17:2813–2824. doi: 10.1523/JNEUROSCI.17-08-02813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kandel A, Buzsáki G. Cellular-synaptic generation of sleep spindles, spike-and-wave discharges, and evoked thalamocortical responses in the cortex of the rat. J Neurosci. 1997;17:6783–6797. doi: 10.1523/JNEUROSCI.17-17-06783.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kellaway P. Sleep and epilepsy. Epilepsia. 1985;26 [Suppl] 1:15–30. doi: 10.1111/j.1528-1157.1985.tb05720.x. [DOI] [PubMed] [Google Scholar]
- 30.Kettenmann H, Schachner M. Pharmacological properties of γ-aminobutyric acid-, glutamate-, and aspartate-induced depolarizations in cultured astrocytes. J Neurosci. 1985;5:3295–3301. doi: 10.1523/JNEUROSCI.05-12-03295.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kocsis JD, Malenka RC, Waxman SG. Effects of extracellular potassium concentration on the excitability of the parallel fibres of the rat cerebellum. J Physiol (Lond) 1983;334:225–244. doi: 10.1113/jphysiol.1983.sp014491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kostopoulos G, Avoli M, Pellegrini A, Gloor P. Laminar analysis of spindles and spikes of the spike and wave discharge of feline generalized penicillin epilepsy. Electroencephalogr Clin Neurophysiol. 1982;53:1–13. doi: 10.1016/0013-4694(82)90101-8. [DOI] [PubMed] [Google Scholar]
- 33.Kuffler SW, Nicholls JG, Orkand RD. Physiological properties of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:768–787. doi: 10.1152/jn.1966.29.4.768. [DOI] [PubMed] [Google Scholar]
- 34.Levi G, Gallo V. Release of neuroactive amino acids from glia. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 815–826. [Google Scholar]
- 35.Levi G, Patrizio M. Astrocyte heterogeneity: endogenous amino acids levels and release evoked by non-N-methyl-d-aspartate receptor agonists and by potassium-induced swelling in type-1 and type-2 astrocytes. J Neurochem. 1992;58:1943–1952. doi: 10.1111/j.1471-4159.1992.tb10073.x. [DOI] [PubMed] [Google Scholar]
- 36.Lux HD, Neher E. The equilibrium time course of [K+]o in cat cortex. Exp Brain Res. 1973;17:190–205. doi: 10.1007/BF00235028. [DOI] [PubMed] [Google Scholar]
- 37.MacVicar BA, Tse FW, Crichton SA, Kettenmann H. GABA-activated Cl− channels in astrocytes of hippocampal slices. J Neurosci. 1989;9:3577–3583. doi: 10.1523/JNEUROSCI.09-10-03577.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin DL, Madelian V, Seligmann B, Shain W. The role of osmotic pressure and membrane potential in K+-taurine release from cultured astrocytes and LRM55 cells. J Neurosci. 1990;10:571–577. doi: 10.1523/JNEUROSCI.10-02-00571.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKhann GM, D'Ambrosio R, Janigro D. Heterogeneity of astrocyte resting membrane potentials and intracellular coupling revealed by whole-cell and gramicidin-perforated patch recordings from cultured neocortical and hippocampal slice astrocytes. J Neurosci. 1997;17:6850–6863. doi: 10.1523/JNEUROSCI.17-18-06850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moody WJ, Jr, Futamachi KJ, Prince DA. Extracellular potassium activity during epileptogenesis. Exp Neurol. 1974;42:248–263. doi: 10.1016/0014-4886(74)90023-5. [DOI] [PubMed] [Google Scholar]
- 41.Neckelmann D, Amzica F, Steriade M. Spike-wave complexes and fast components of cortically generated seizures. III. Synchronizing mechanisms. J Neurophysiol. 1998;80:1480–1494. doi: 10.1152/jn.1998.80.3.1480. [DOI] [PubMed] [Google Scholar]
- 42.Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 43.Newman EA. Glial cell regulation of extracellular potassium. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 717–731. [Google Scholar]
- 44.Nicholls JG, Kuffler SW. Extracellular space as a pathway for exchange between blood and neurons in the central nervous system of the leech: ionic composition of glial cells and neurons. J Neurophysiol. 1964;27:645–671. doi: 10.1152/jn.1964.27.4.645. [DOI] [PubMed] [Google Scholar]
- 45.Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- 46.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 47.Pasti L, Volterra A, Pozzan T, Carmignoto C. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pedley TA, Traub RD. Physiological basis of the EEG. In: Daly DD, Pedley TA, editors. Current practice of clinical electroencephalography. Raven; New York: 1990. pp. 107–137. [Google Scholar]
- 49.Petsche H, Pockberger H, Rappelsberger P. On the search for the sources of the electroencephalogram. Neuroscience. 1984;11:1–27. doi: 10.1016/0306-4522(84)90212-4. [DOI] [PubMed] [Google Scholar]
- 50.Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- 51.Rosier A, Arckens L, Orban GA, Vandesande F. Immunocytochemical detection of astrocyte GABAA receptors in cat visual cortex. J Histochem Cytochem. 1993;41:685–692. doi: 10.1177/41.5.8385682. [DOI] [PubMed] [Google Scholar]
- 52.Somjen GG. Electrogenesis of sustained potentials. Prog Neurobiol. 1973;1:201–237. [PubMed] [Google Scholar]
- 53.Somjen GG. Electrophysiology of mammalian glial cells in situ. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 319–331. [Google Scholar]
- 54.Somjen GG, Trachtenberg M. Neuroglial as generator of extracellular current. In: Speckmann EJ, Caspers H, editors. Origin of cerebral field potentials. Thieme; Stuttgart: 1979. pp. 21–32. [Google Scholar]
- 55.Sontheimer H, Ritchie JM. Voltage-gated sodium and calcium channels. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 202–220. [Google Scholar]
- 56.Sontheimer H, Kettenmann H, Backus KH, Schachner M. Glutamate opens Na+/K+ channels in cultured astrocytes. Glia. 1988;1:328–336. doi: 10.1002/glia.440010505. [DOI] [PubMed] [Google Scholar]
- 57.Speckmann EJ, Caspers H. Origin of cerebral field potentials. Thieme; Stuttgart: 1979. [Google Scholar]
- 58.Speckmann EJ, Elger CE. Introduction to neurophysiological basis of the EEG and DC potentials. In: Niedermeyer E, Lopes da Silva F, editors. Electroencephalography. Basic principles, clinical applications, and related fields. Williams & Wilkins; Baltimore: 1999. pp. 15–27. [Google Scholar]
- 59.Steinhäuser C, Gallo V. News on glutamate receptors in glial cells. Trends Neurosci. 1996;19:339–345. doi: 10.1016/0166-2236(96)10043-6. [DOI] [PubMed] [Google Scholar]
- 60.Steriade M. Interneuronal epileptic discharges related to spike-and-wave cortical seizures in behaving monkeys. Electroencephalogr Clin Neurophysiol. 1974;37:247–263. doi: 10.1016/0013-4694(74)90028-5. [DOI] [PubMed] [Google Scholar]
- 61.Steriade M, Amzica F. Dynamic coupling among neocortical neurons during evoked and spontaneous spike-wave seizure activity. J Neurophysiol. 1994;72:2051–2069. doi: 10.1152/jn.1994.72.5.2051. [DOI] [PubMed] [Google Scholar]
- 62.Steriade M, Amzica F. An intracellular study of excitability in the seizure-prone neocortex in vivo. J Neurophysiol. 1999;82:3108–3122. doi: 10.1152/jn.1999.82.6.3108. [DOI] [PubMed] [Google Scholar]
- 63.Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci. 1995;15:623–642. doi: 10.1523/JNEUROSCI.15-01-00623.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Steriade M, Nuñez A, Amzica F. A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993a;13:3252–3265. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Steriade M, Nuñez A, Amzica F. Intracellular analysis of relations between the slow (<1 Hz) neocortical oscillation and other sleep rhythms of the electroencephalogram. J Neurosci. 1993b;13:3266–3283. doi: 10.1523/JNEUROSCI.13-08-03266.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Steriade M, Amzica F, Contreras D. Synchronization of fast (30–40 Hz) spontaneous cortical rhythms during brain activation. J Neurosci. 1996;16:392–417. doi: 10.1523/JNEUROSCI.16-01-00392.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Steriade M, Amzica F, Neckelmann D, Timofeev I. Spike-wave complexes and fast components of cortically generated seizures. II. Extra- and intracellular patterns. J Neurophysiol. 1998;80:1456–1479. doi: 10.1152/jn.1998.80.3.1456. [DOI] [PubMed] [Google Scholar]
- 68.Streit W. Microglial cells. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 85–96. [Google Scholar]
- 69.Sypert GW, Ward AA., Jr Unidentified neuroglia potentials during propagated seizures in neocortex. Exp Neurol. 1971;33:239–255. doi: 10.1016/0014-4886(71)90018-5. [DOI] [PubMed] [Google Scholar]
- 70.Taylor CP, Dudek FE. Synchronous neural afterdischarges in rat hippocampal slices without active chemical synapses. Science. 1982;218:810–812. doi: 10.1126/science.7134978. [DOI] [PubMed] [Google Scholar]
- 71.Terzuolo CA, Araki T. An analysis of intra-versus extracellular potential changes associated with activity of single spinal motoneurons. Ann NY Acad Sci. 1961;94:547–558. doi: 10.1111/j.1749-6632.1961.tb35558.x. [DOI] [PubMed] [Google Scholar]
- 72.Trachtenberg MC, Pollen DA. Neuroglia: biophysical properties and physiologic function. Science. 1970;167:1248–1252. doi: 10.1126/science.167.3922.1248. [DOI] [PubMed] [Google Scholar]
- 73.Walz W. Distribution and transport of chloride and bicarbonate ions across membranes. In: Kettenmann H, Ransom BR, editors. Neuroglia. Oxford UP; New York: 1995. pp. 221–229. [Google Scholar]
- 74.Zuckermann EC, Glaser GH. Hippocampal epileptic activity induced by localized ventricular perfusion with high-potassium cerebrospinal fluid. Exp Neurol. 1968;20:87–110. doi: 10.1016/0014-4886(68)90126-x. [DOI] [PubMed] [Google Scholar]