

Abstract
An animal model most sensitive for measuring anticipatory anxiety is fear conditioning, which is expressed by an enduring increase in synaptic strength in the amygdala. A converse view predicts that agents that induce long-term depression (LTD) of synaptic efficacy in the amygdala may be useful in the amelioration of stress disorders. In the present study, we show that activation of group II metabotropic glutamate receptor (mGluR II) by (2S,3S,4S)-2-(carboxycyclopropyl) glycine (l-ccg) induces an LTD in the basolateral amygdala neurons. The effect was concentration-dependent with a maximal inhibition of ∼30%. The induction of l-CCG LTD required concurrent synaptic activity, required presynaptic but not postsynaptic Ca2+ increases, and was independent of NMDA receptors. l-CCG LTD was associated with an increase in the ratio of paired-pulse facilitation and was not occluded by low-frequency stimulation-induced LTD, suggesting that these two forms of LTD did not share a common underlying mechanism.
After eliciting LTD with l-CCG, application of isoproterenol increased the synaptic responses back to its original baseline, demonstrating that chemically depressed synapses could be potentiated by another chemical. A selective PKA inhibitor, KT 5720, by its own caused a depression of synaptic transmission and blockedl-CCG LTD, presumably by mimicking and thereby occluding any further depression. Together, these results suggest thatl-CCG LTD is induced by presynaptically mGluR II-mediated inhibition of Ca2+-sensitive adenylyl cyclase, resulting in a decrease in cAMP formation and PKA activation, which leads to a long-lasting decrease in transmitter release.
Keywords: amygdala, cAMP, PKA, synaptic plasticity, LTD, mGluR
The amygdala has been suggested to include an essential circuit for certain forms of neuroplasticity, such as emotional memory and epilepsy (Goddard et al., 1969; Davis et al., 1994; LeDoux 1994). One cellular model thought to store aversive emotional experiences in this structure is fear conditioning, which consists of freezing behavior evoked by the pairing of a conditioned stimulus (CS), such as a tone, with a noxious unconditioned footshock. The neural pathways mediating fear conditioning involve the transmission of sensory information about the CS to the lateral nucleus of amygdala (LA), which then projects to the basolateral nucleus (BLA) and other amygdala regions (LeDoux et al., 1990; Pitkanen et al., 1997;Maren, 1999). Several lines of evidence indicate that the BLA plays an important role in both acquisition and expression of conditioned fear. For example, reversible inactivation of the BLA neurons with lidocaine (Parent and McGaugh, 1994), as well as blockade of NMDA or glucocorticoid receptors in the BLA (Miserendino et al., 1990; Fanselow and Kim, 1994; Liang et al., 1994; Roozendaal and McGaugh, 1997), prevents the acquisition of conditioned fear. Thus, the BLA receives synaptic input from many primary sensory structures, and lesion of this structure yields deficits in Pavlovian fear conditioning.
Long-term potentiation (LTP) of synaptic responses, triggered by high-frequency stimulation (HFS) of excitatory afferents, is a leading cellular mechanism for learning and memory (Bliss and Collingridge, 1993). In view of the facts that fear conditioning induces increases in amygdala synaptic transmission that resemble LTP (McKernan and Shinnick-Gallagher, 1997; Rogan et al., 1997), it is hypothesized that they may share a common mechanism (Maren, 1999). This hypothesis receives convincing support from genetic studies of mice that lack RasGRE, a neuron-specific guanine nucleotide-releasing factor. These mice have profound deficits in amygdala LTP, as well as impairments in consolidation of long-term memories for fear conditioning to both contextual and acoustic stimuli (Brambilla et al., 1997). If this theory is correct, one may predict that drugs that induce the converse form of LTP, long-term depression (LTD), of synaptic efficacy in the amygdala is useful for the amelioration of conditioned fear.
Recently, we have demonstrated that homosynaptic LTD could be induced at the amygdala LA–BLA synapse by prolonged low-frequency stimulation (LFS) (Wang and Gean, 1999). The induction of LTD required activation of NMDA receptors, postsynaptic Ca2+increases, and phosphatase activity. Thus, the induction of LFS LTD is likely at the postsynaptic site. In the present study, we have investigated the role of group II metabotropic glutamate receptor (mGluR II) in the induction of LTD using its selective agonist (2S,3S,4S)-2-(carboxycyclopropyl) glycine (l-CCG). We found that chemical-induced LTD is NMDA receptor-independent. It requires synaptic activation and presynaptic, but not postsynaptic, Ca2+increases and is associated with an increase in paired-pulse facilitation (PPF). These results suggest that, in contrast to LFS LTD,l-CCG LTD is induced and expressed presynaptically.
MATERIALS AND METHODS
Slice preparation. Male Sprague Dawley 4- to 6-week-old rats were decapitated, and their brains were rapidly removed and placed in cold oxygenated artificial CSF (ACSF) solution. Subsequently, the brain was hemisected and cut transversely posterior to the first branch and anterior to the last branch of the superior cerebral vein. The resulting section was glued to the chuck of a Vibroslice tissue slicer (Campden Instruments, Silbey, UK). Transverse slices of 500 μm thickness were cut, and the appropriate slices were placed in a beaker of oxygenated ACSF at room temperature for at least 1 hr before recording. ACSF solution had the following composition (in mm): NaCl 117, KCl 4.7, CaCl2 2.5, MgCl2 1.2, NaHCO3 25, NaH2PO4 1.2, and glucose 11. The ACSF was bubbled continuously with 95%O2–5%CO2 and had the pH of 7.4.
Intracellular recordings. A single slice was transferred to the recording chamber in which it was held submerged between two nylon nets and maintained at 32 ± 1°C. The chamber consisted of a circular well of a low volume (1–2 ml) and was perfused constantly at a rate of 2–3ml/min. Intracellular recording microelectrodes were pulled from 1.0 mm microfiber capillary tubing on a Brown-Flaming electrode puller (Sutter Instruments, San Rafael, CA). The electrodes were filled with 4 m potassium acetate with resistance ranging from 70 to 130 MΩ. For chelating intracellular Ca2+, the electrodes were filled with 50 mm BAPTA in addition to 3 mpotassium acetate. When BAPTA-containing electrodes were used, loading of the cells with BAPTA was assayed by the blockade of Ca2+-activated afterhyperpolarization. The microelectrode tips were positioned into the BLA.
Monosynaptic EPSPs were evoked in BLA neurons by electrical stimulation of afferents from the lateral nucleus of amygdala with a concentric bipolar stimulating electrode (SNE-100; David Kopf Instruments, Bern, Germany). Electrical stimuli (150 μsec in duration) were delivered at a frequency of 0.05 Hz. To induce LTD, LFS protocol was used, which consists of 900 pulses, delivered at 1 Hz at the same stimulation intensity used for baseline. All data were expressed as mean ± SEM. Statistical analysis was performed using the Student'st test, and p < 0.05 was considered statistically significant.
Drug application. Drugs were applied directly to the ACSF using a continuous gravity-fed bath application, and the concentration of applied drug reached equilibrium within 2–3 min.l-CCG, BAPTA-AM, andd-2-amino-5-phosphonovalerate (d-APV) were obtained from Research Biochemicals (Natick, MA). 2S,1S′2S′-2-methyl-2-(2′-carboxycyclo-propyl)glycine (MCCG) and (2S)-α-ethylglutamic acid (EGLU) were obtained from Tocris Cookson. (Bristol, UK). KT 5720 was obtained from Calbiochem-Novabiochem International (San Diego, CA).
RESULTS
LTD induced by l-CCG
Figure 1 illustrates the effect of a selective mGluR II agonist l-CCG on EPSP in BLA neurons. Superfusion of l-CCG for 10–15 min caused a rapid depression of EPSP, which was maintained in the presence of L-CCG. After washout of l-CCG, the amplitude of EPSP was only partially recovered to a stable response that was depressed relative to the initial baseline value. This l-CCG-induced LTD typically was stable for at least 60 min; therefore, we used the magnitude of LTD at this time point for statistical comparison. The effect of l-CCG was concentration-dependent. One, 10, and 30 μm produced an initial depression measuring −17.1 ± 2.1% (n = 16), −55.4 ± 4.3% (n = 12) and −61.0 ± 3.6% (n = 8), respectively, and LTD measuring −18.8 ± 2.9% (n = 16), −31.5 ± 3.0% (n = 12), and −35.0 ± 3.9% (n = 8), respectively (Fig. 1B). Furthermore, the effect ofl-CCG was mimicked by another selective mGluR II agonist (2S,1′R,2′R,3′R)-(2′,3′-dicarboxycyclopropyl)glycine (DCG-IV). Superfusion of DCG-IV (2 μm) caused an initial depression of −46.2 ± 6.0% (n = 5) and was followed by an LTD with the magnitude of −29.3 ± 10.1% (n = 5). l-CCG (10 μm)-induced LTD was not attributable to an alteration of resting membrane potential (RMP) or neuronal input resistance (IR) of the BLA neurons (RMP and IR were −67.0 ± 1.6 mV and 45.5 ± 3.3 MΩ before, and −66.0 ± 1.3 mV and 45.6 ± 2.4 MΩ 60 min after the washout ofl-CCG) (cf. Neugebauer et al., 1997)
Fig. 1.
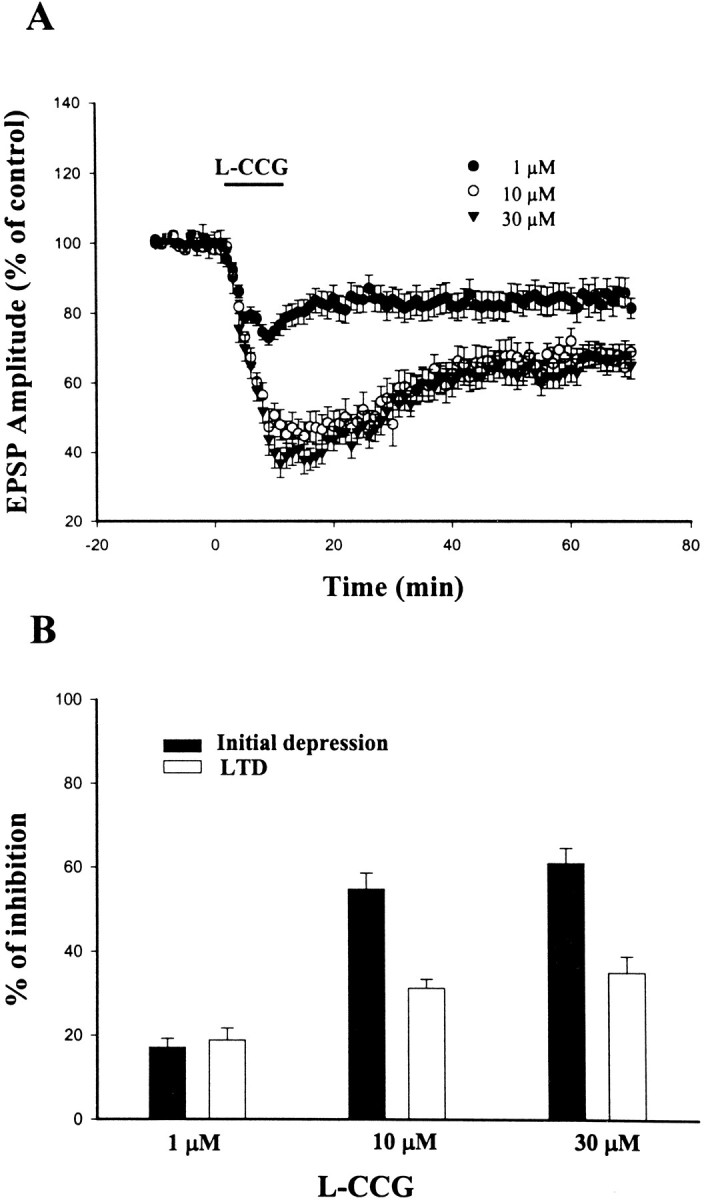
Concentration-dependent depression of EPSP byl-CCG. A, Application of l-CCG for 10 min resulted in an initial depression of EPSP, which was followed, upon washout of the l-CCG, by LTD.B, Concentration-dependent effects of l-CCG on the initial depression and LTD.
We have shown previously that LFS consistently induced LTD at the LA–BLA synapse but not at the ventral endopyriform nucleus (VEN)–BLA synapse (Wang and Gean, 1999). To test whetherl-CCG-induced LTD is also restricted at the LA–BLA synapse, we performed the experiments by placing the stimulating electrode on the VEN. In six neurons tested, l-CCG (10 μm) similarly induced an LTD at this pathway (−30.7 ± 3.6%, 60 min after the washout of l-CCG;n = 6).
Block of l-CCG-induced LTD by mGluR II antagonists
MCCG (100 μm), a selective antagonist for mGluR II, by itself did not affect EPSP significantly (97.8 ± 3.5% of control; n = 9; p > 0.1). However, as illustrated in Figure2A, it blocked the initial depression as well as the LTD induced byl-CCG. In the presence of MCCG, the initial depression and LTD induced by l-CCG were only −12.4 ± 4.4 (n = 9) and −9.6 ± 4.9% (n = 9), respectively, which were significantly less than that of without MCCG pretreatment (p < 0.001; unpaired t test for both initial depression and LTD).
Fig. 2.
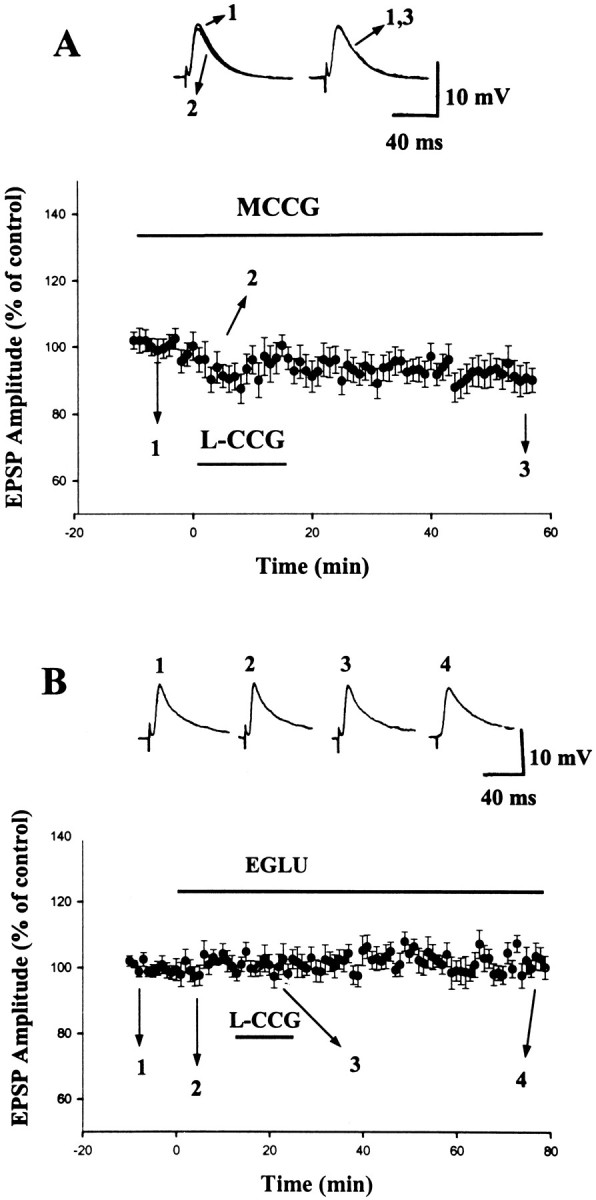
Blockade of l-CCG-induced LTD by mGluR II antagonists. A, Application ofl-CCG (10 μm) in the presence of MCCG (100 μm) failed to cause initial depression and LTD.Inset shows superimposed traces taken at the time points indicated. B, Application of EGLU (10 μm) did not affect the EPSP significantly but blocked the effect of l-CCG (10 μm). Insetshows the traces taken at the time points indicated.
Another mGluR II antagonist, EGLU (Jane et al., 1996), was also examined. Consistent with a previous report (Li et al., 1998), superfusion of EGLU (10 μm) did not affect the amplitude of EPSP (101 ± 1% of control; n = 7) but blocked the effect of l-CCG on EPSP (Fig.2B). In the presence of EGLU, the initial depression and LTD induced by l-CCG were 2.8 ± 4.1 (n = 7) and 0.1 ± 3.5% (n = 7), respectively, which were considerably less than in control neurons (p < 0.001 for both initial depression and LTD). Furthermore, the effect of EGLU was not mimicked by the selective mGluR I antagonist 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt). The initial depression and LTD induced byl-CCG (10 μm) in the presence of CPCCOEt (100 μm) were −39.1 ± 5.2 and −29.7 ± 8.8% (n = 3), respectively. Similarly, the group III mGluR antagonist (S)-2-methyl-2-amino-4-phosphonobutyrate (100 μm) itself affected neither synaptic transmission (107 ± 5%; n = 5) nor the LTD byl-CCG (10 μm) (−28.6 ± 5.0%; n = 5). These results confirm the mediation of LTD by group II mGluR.
l-CCG-induced LTD is independent of NMDA receptor activation
To determine whether NMDA receptor plays a role inl-CCG-induced LTD, NMDA receptor antagonistd-APV was applied before perfusion ofl-CCG. Figure 3Ashows that application of l-CCG (10 μm) in d-APV (50 μm) resulted in an initial depression of EPSP, which measured −58 ± 5% (n = 7), and was followed by an LTD with the magnitude of −31 ± 6% (n = 7). These values were indistinguishable from those observed in the absence of d-APV.
Fig. 3.
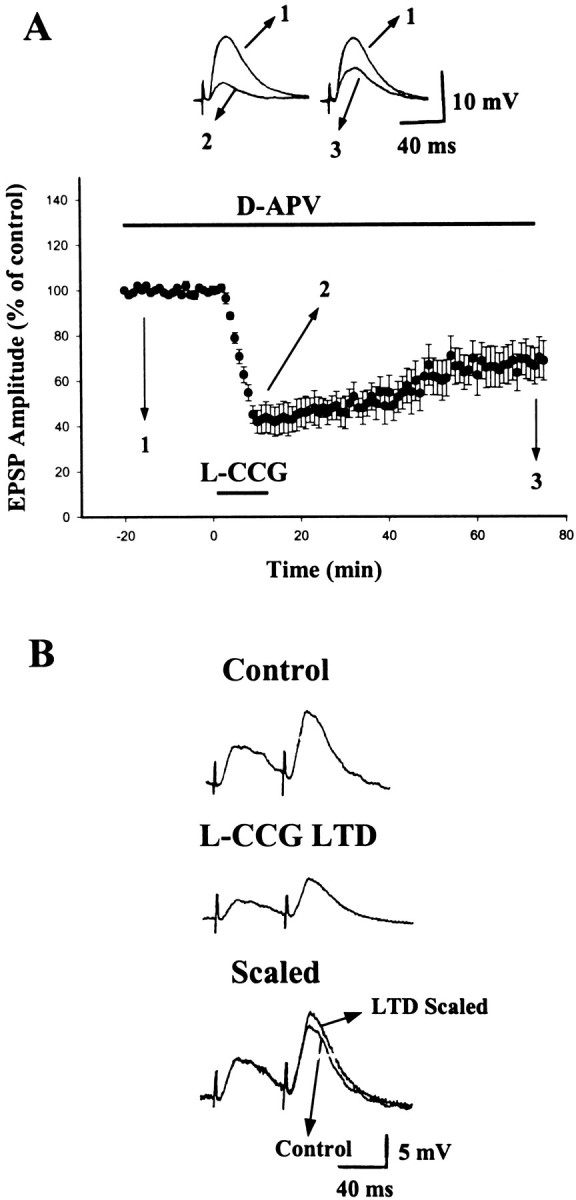
LTD induced by l-CCG is independent of NMDA receptor activation and is accompanied by an increase in PPF.A, Application of l-CCG (10 μm) in the presence of d-APV (50 μm) induced LTD that was not significantly different from control LTD. B, EPSPs evoked by paired stimuli in control and 60 min after the washout of l-CCG (10 μm). The control EPSP evoked by the first stimulus is scaled to that recorded 60 min after LTD induction. It is clear that there is a greater PPF after l-CCG LTD.
To localize the site of expression, we explored the effect ofl-CCG on PPF, because if l-CCG LTD involves presynaptic mechanisms, then it might be associated with an increase in PPF (Manabe et al., 1993; Schulz et al., 1994). A typical experiment is shown in Figure 3B in which a pair of synaptic responses was elicited with an interstimulus interval of 60 msec, and the ratio of PPF was compared between control and after LTD was induced. The result of seven experiments revealed that PPF was increased to 122 ± 6% (n = 7; p < 0.01) of baseline 60 min after LTD induction.
l-CCG-induced LTD and LFS-induced LTD are not mutually occluded
LTD induced by LFS requires NMDA receptor activation, whereasl-CCG LTD does not, suggesting different mechanisms of induction. We tested this prediction by performing occlusion experiments. In a first set of experiments, l-CCG LTD was saturated by giving a concentration of 10 μm, which induced the maximal LTD (Fig. 1). We then compared the magnitude of LTD induced under this condition with that induced withoutl-CCG pretreatment. A summary of nine experiments is shown in Figure 4A in whichl-CCG elicited an initial depression of −51.7 ± 4.8% and an LTD of −29.6 ± 2.9%. Subsequent LFS produced a further LTD (−14.3 ± 3.4%; n = 9;p < 0.01; paired t test). When the data were renormalized to the 10 min preceding application of LFS, responses were calculated to be depressed by 20.3 ± 3.4%. Thus, LFS was still able to elicit LTD, although its magnitude was less than that without previous application of l-CCG (−39.8 ± 8.5%; n = 16; p < 0.05; unpaired t test).
Fig. 4.

l-CCG-induced LTD and LFS-induced LTD are not mutually occluded. A, One application of 10 μm L-CCG for 15 min was given to saturate LTD. Sixty minutes after washout of l-CCG, 1 Hz stimulation (900 pulses, 15 min) was delivered. LFS produced a further depression.B, An example of experiments showing that, after LTD was induced to a maximal level by three periods of LFS, subsequent application of l-CCG (10 μm) induced a further LTD.
To further address this question, we performed the reverse occlusion experiment in which LTD was induced by LFS before l-CCG was applied. An example of these experiments is shown in Figure4B. LTD was induced to a maximal level (∼50%) by three epochs of LFS as indicated by a lack of further depression in response to the final epoch. Subsequent application ofl-CCG (10 μm) still induced a 12.5% LTD (25% if normalized to pre-l-CCG level) at 60 min after washout ofl-CCG. In nine neurons, under this condition,l-CCG (10 μm) elicited an average of 18.4 ± 3.1% (n = 8; p< 0.05; paired t test) of LTD, but its magnitude was less than that of untreated control slices (−31.5 ± 3.0%;n = 12; p < 0.05; unpairedt test).
Requirements of synaptic activity and presynaptic Ca2+ entry for l-CCG-induced LTD
To determine whether synaptic activation was required forl-CCG-induced LTD, experiments were performed in which synaptic stimulation was ceased both during the application ofl-CCG and 10 min after the washout, thereby avoiding Ca2+ entry into presynaptic boutons during application of l-CCG. As shown in Figure5, l-CCG did not induce LTD in the absence of synaptic stimulation. At 60 min after washout ofl-CCG, the EPSP amplitude measured 91.8 ± 7.4% (n = 8; p > 0.1).
Fig. 5.

Induction of l-CCG LTD is dependent on presynaptic activity. Summary of eight experiments showing thatl-CCG LTD was blocked when synaptic stimulation was stopped both during the application of l-CCG (10 μm) and 10 min after the washout.
To determine whether a rise in presynaptic Ca2+ is required for the action ofl-CCG, we buffered intraterminal Ca2+ using a membrane-permeant Ca2+ chelator, BAPTA-AM. Application of BAPTA-AM (100 μm) reduced the EPSP amplitude by 38.9 ± 4.6% (n = 8) and, in the presence of BAPTA-AM,l-CCG (10 μm) no longer induced initial depression (1.4 ± 1.2%; n = 8) or LTD (−0.8 ± 5.9%; n = 8) (Fig.6A). To differentiate presynaptic versus postsynaptic sites of action, we loaded the recorded postsynaptic neuron with BAPTA salt. After impalement, the cells were allowed to stabilize for at least 30 min to allow the cell to fill with BAPTA, which was manifested by blockade of slow afterhyperpolarization (Fig. 6B). Baseline responses were then obtained for an additional 10 min before superfusing l-CCG. As shown in Figure 6C, l-CCG still induced initial depression and LTD, measured −55.0 ± 3.5 and −25.0 ± 3.6% (n = 7), respectively. These values were not significantly different from those recorded with control electrodes. Collectively, these results suggest that an activity-mediated change in presynaptic but not postsynaptic Ca2+ level is required forl-CCG-induced LTD.
Fig. 6.

Presynaptic but not postsynaptic Ca2+ increase is required for l-CCG LTD.A, Summary of eight experiments showing that the membrane-permeable Ca2+ chelator BAPTA-AM (100 μm) suppressed synaptic transmission and blockedl-CCG LTD. B, Superimposedtraces taken immediately (1 min) and 30 min after the impalement of cell with a BAPTA-containing electrode to show the block of afterhyperpolarization. Electrode was filled with BAPTA (50 mm) as described in Materials and Methods.C, LFS LTD but not l-CCG LTD was blocked by intracellular BAPTA.
l-CCG-induced LTD can be reversed by isoproterenol-induced LTP
We sought to determine whether chemically depressed synapses could be potentiated by another chemical. After eliciting LTD withl-CCG, we applied isoproterenol (Iso). Iso was used because previous studies from this laboratory have shown that stimulation of β-adrenoceptors resulted in long-term enhancement of excitatory synaptic responses in the BLA neurons (Huang et al., 1996). As shown in Figure 7A, 20 min after LTD induction (−25.8 ± 4.6%; n = 7), application of Iso (15 μm) increased synaptic strength back to the original control level (107.7 ± 8.2%, measured at 60 min after the washout of Iso; n = 7). To further examine whether Iso-induced LTP could be depotentiated byl-CCG, we applied Iso andl-CCG in the reverse order. Figure 7Bshows a summary of eight experiments in which Iso (15 μm) induced LTP that stabilized at 138.4 ± 5.6% (n = 8) of baseline level (20 min after Iso;n = 8). l-CCG (10 μm) application 20 min after the washout of Iso caused a complete reversion of LTP. The EPSP amplitude was returned to 88.4 ± 6.5% (n = 8) of the initial control 60 min after l-CCG.
Fig. 7.
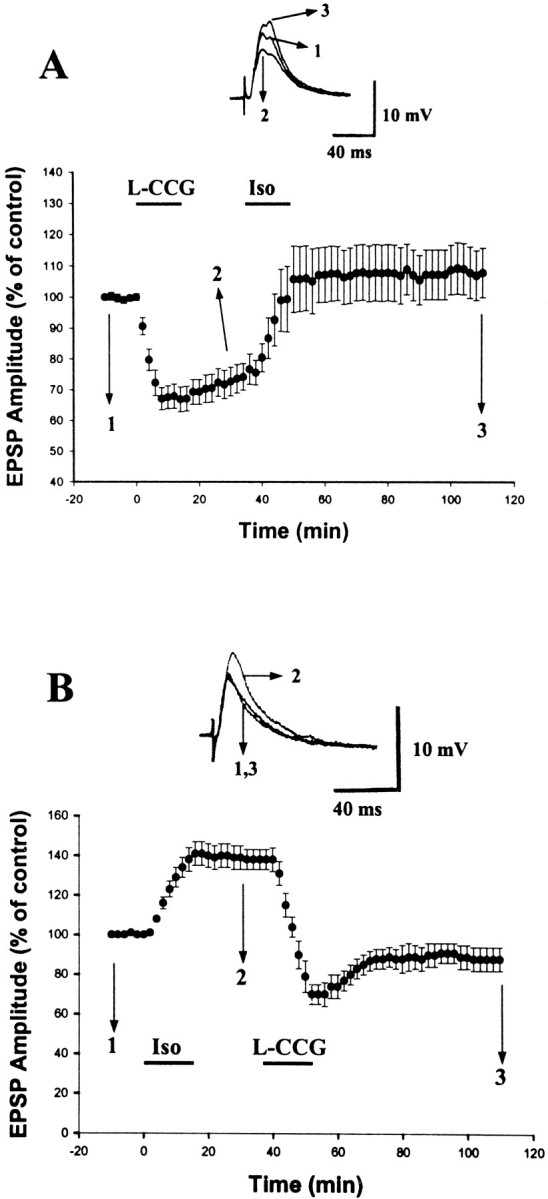
l-CCG LTD can be potentiated by Iso.A, Application of l-CCG (10 μm) induced an LTD. Twenty minutes after LTD stabilized, Iso (15 μm) was applied, which increased the synaptic strength back to the original level. B, Parallel experiments in which l-CCG and Iso were applied in the reverse order. Application of Iso (15 μm) induced an LTP.l-CCG (10 μm) application 20 min after the washout of Iso caused a complete reversal of LTP.
Involvement of protein kinase A inl-CCG-induced LTD
Mediating through G-proteins of Gi/Go classes, mGluR II is coupled negatively to adenylyl cyclase and thereby inhibits the formation of cAMP (Pin and Duvoisin, 1995). If mGluR-mediated decrease in cAMP level is necessary for triggering LTD, then pharmacological inhibition of cAMP and cAMP-dependent protein kinase A (PKA) should mimic the effect of l-CCG. We tested this possibility by application of KT5720, a PKA catalytic site antagonist. Application of KT5720 (1 μm) gradually reduced the amplitude of EPSP. The amplitudes of EPSP were 80.6 ± 3.3, 50.4 ± 2.9, and 50.5 ± 4.1% (n = 3) at 30, 60, and 90 min after the application of drug.
We next addressed the question of whether a decrease in PKA activity is important for l-CCG-induced LTD. To perform these experiments, slices were incubated in 1 μm KT 5720 for at least 1 hr before being transferred to the recording chamber in which the same concentration of KT 5720 was maintained. As shown in Figure8, l-CCG (10 μm) failed to induce LTD under this condition (100 ± 1% of control, measured at 60 min after the washout ofl-CCG; n = 6). It is noted that L-CCG still caused an initial depression when PKA activity was inhibited by KT 5720. These results suggest that the initial depression of transmitter release elicited by mGluR activation is not attributable to a decrease in cAMP. This conclusion was supported by the observation that inhibition of adenylyl cyclase with SQ 22536 (50 μm) completely abolished the induction of LTD (5.3 ± 4.3%; n = 6) without blocking the initial depression of EPSP by 10 μml-CCG (−38.2 ± 4.8%; n = 6).
Fig. 8.
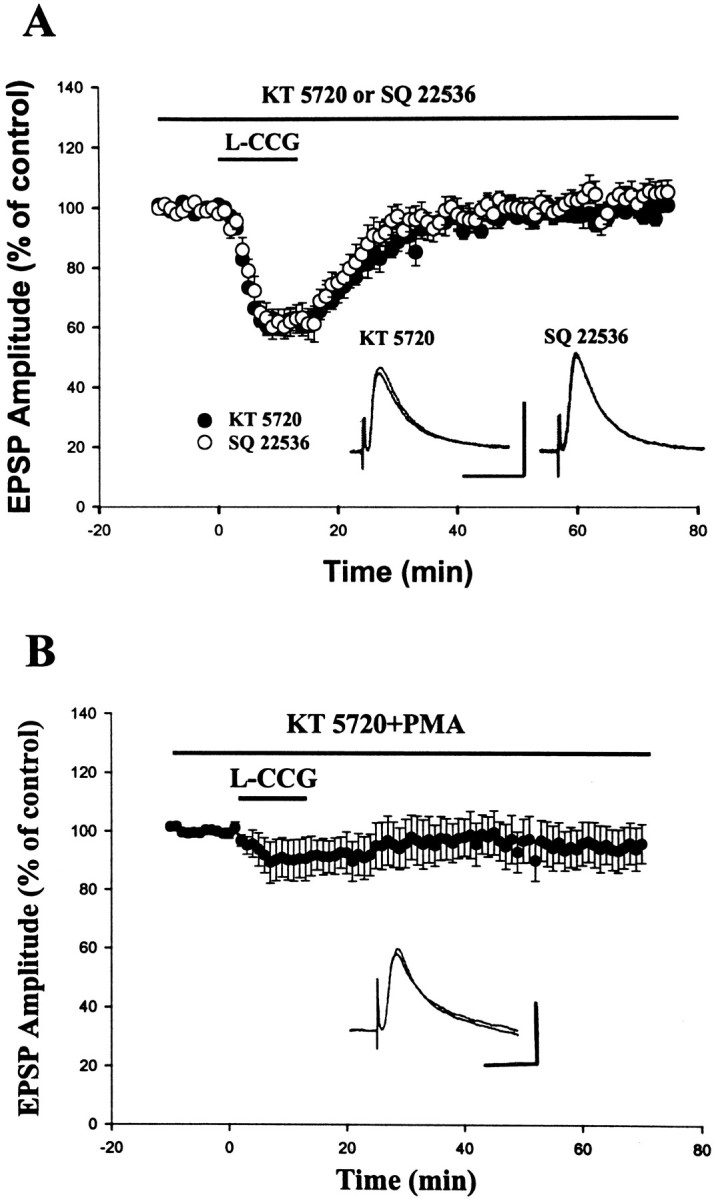
l-CCG LTD is blocked by adenylyl cyclase and PKA inhibitors. A, Slices were incubated for at least 1 hr in 1 μm KT 5720 or 20 min in 50 μm SQ 22536 before being transferred to the recording chamber in which the same concentration of drug was maintained.l-CCG LTD normally observed in control slices was blocked in the KT 5720- or SQ 22536-treated slices. Inset shows the representative traces taken before and 60 min after the washout of l-CCG. Calibration: 10 mV, 40 msec.B, Block of l-CCG-induced initial depression by PMA. Slices were incubated for at least 1 hr in KT 5720 (1 μm) plus PMA (1 μm). Insetshows the representative traces taken before and 60 min after the washout of l-CCG. Calibration: 10 mV, 40 msec.
Several G-protein-coupled receptors, such as mGluR II, GABAB, and adenosine A1receptors, exert inhibitory action on the synaptic transmission. Activation of these receptors also depresses voltage-dependent Ca2+ channels. Both G-protein-coupled receptor-mediated presynaptic inhibition and Ca2+ channel depression could be disrupted by phorbol esters (Swartz et al., 1993), the protein kinase C (PKC) activators, suggesting that PKC may phosphorylate and consequently inactivate some classes of G-protein mediating these actions. We therefore tested whether PKC activation can influence mGluR II-mediated synaptic depression. In these experiments, slices were incubated for at least 1 hr in 1 μm KT 5720 to eliminate l-CCG LTD. As illustrated in Figure 8B,l-CCG-induced transient depression was blocked by the pretreatment of slices with the PKC activator PMA (1 μm).
Endogenous activation of mGluR II
It has been suggested that neurotransmitters uptake into neurons and glial cells is the major mechanism by which synaptically released neurotransmitter is removed from the extracellular space. To investigate whether synaptically released glutamate is capable of diffusing to presynaptic terminals and inducing LTD by activating at mGluR II, glutamate release is increased by HFS (100 Hz, 1 sec) and by blocking glutamate uptake (Scanziani et al., 1997). In these experiments, the slices were perfused with d-APV (50 μm) to prevent NMDA receptor-dependent LTP and LTD. In the initial experiments, HFS elicited a post-tetanic potentiation, which was followed by an LTD in only 6 of 15 neurons tested. If mGluR activation depends on the spread of glutamate, the activation of these receptors should be enhanced by blocking the uptake of glutamate. Indeed, when the glutamate uptake blocker L-trans-pyrrolidine-2,4-dicarboxylic acid (trans-PDC) (100 μm) was applied, HFS induced an LTD in all seven neurons tested (−29.4 ± 3.6, 60 min after the stimulation; n = 7). We next tested whether mGluR II was involved in EPSP depression by applying MCCG (100 μm). In the presence of this mGluR II antagonist, HFS-induced LTD was blocked (−4.8 ± 2.9%, 60 min after the stimulation; n = 6), providing evidence that EPSP depression was caused by mGluR activation (Fig.9).
Fig. 9.

Glutamate release by brief titanic stimulation activates presynaptic mGluR II and induces LTD. In the presence ofd-APV (50 μm) and the glutamate uptake inhibitor trans-PDC (100 μm), brief titanic stimulation (100 Hz, 1 sec) evoked an LTD that was completely blocked by MCCG (100 μm). Inset shows the representative traces taken before and 60 min after the stimulation. Calibration: 10 mV, 40 msec.
DISCUSSION
The present results, in combination with previous work, clearly show that there are two forms of LTD that coexist at the same synapses of the rat amygdala. Both induction and expression mechanisms appear to be completely different; one induced by a mGluR II agonist requires Ca2+ influx into presynaptic terminals and is expressed by an enhanced transmitter release, and the other triggered by prolonged LFS is NMDA receptor-dependent and expressed postsynaptically. Importantly, like electrically induced synaptic plasticity, the same synapses that had undergone chemical-induced LTD could subsequently be potentiated by another chemical. Moreover, we have provided the first evidence that, when glutamate concentration is increased by high-frequency activity and by blocking glutamate uptake, mGluRs become activated, leading to an LTD of synaptic transmission. The use-dependent activation of presynaptic mGluRs that we describe here thus may have important physiological functions related to the amygdala, such as fear conditioning and kindling epileptogenesis.
Characteristics of l-CCG LTD
In this study, we have demonstrated that LTD can be induced in the basolateral nucleus of the rat amygdala by l-CCG, a selective mGluR II agonist. The effect of l-CCG is concentration-dependent with a maximal depression of ∼30%. The blockade of l-CCG-induced LTD by the selective mGluR II antagonists MCCG and EGLU confirms the mediation by mGluR II. LTD was not caused by a slow washout of l-CCG because application of MCCG during the washing period did not affect the magnitude of LTD (our unpublished observation). Furthermore, potentiation consistently did occur in response to Iso after induction of LTD, which argues strongly against excitotoxicity as a mechanism for the observed synaptic depression.
Group II mGluRs are located both presynaptically and postsynaptically, and their involvement in the induction of LTD has been described at the hippocampal mossy fiber–CA3 (Yokoi et al., 1996; Domenici et al., 1998; Tzounopoulos et al., 1998), medial perforant path–dentate gyrus (Huang et al., 1999), and the perirhinal cortex (Cho et al., 2000). In these studies, the locus of mGluR II responsible for LTD induction is not consistent; some studies favored a postsynaptic mechanism for mGluR II-mediated LTD (Huang et al., 1999; Cho et al., 2000), whereas others suggested a presynaptic mGluR II-mediated long-lasting depression of transmitter release (Tzounopoulos et al., 1998; Kobayashi et al., 1999). We have reported previously that LTD could be reliably induced in the amygdala by LFS, which requires activation of both NMDA and mGluRs, and depends on postsynaptic Ca2+increases and phosphatase activity. The precise mechanisms underlying the synergy between NMDA and mGluRs in the LTD induction remain to be identified. However, as described recently for LFS LTD in the perirhinal cortex (Cho et al., 2000), it is possible that mGluR II facilitates mGluR I-mediated increases in intracellular Ca2+. This facilitation plus NMDA receptor activation may be necessary for LTD induction at resting membrane potentials. Thus, in the amygdala, mGluR II may play an important role in both presynaptic and postsynaptic forms of LTD.
The lack of effect of loading postsynaptic cells with Ca2+ chelator to impede l-CCG LTD strongly suggests that this form of LTD is independent of postsynaptic Ca2+ influx. This finding, together with the blockade of LTD by extracellular perfusion of membrane-permeable Ca2+ chelator, indicates that an elevation of presynaptic Ca2+ is crucial forl-CCG-induced LTD in the amygdala. The results also show that l-CCG LTD is dependent on presynaptic activity but is not dependent on NMDA receptor activation. This reinforces the notion that postsynaptic mechanisms may not be involved because, in pathways in which postsynaptic mechanism is important, LTD induction is usually blocked by NMDA receptor antagonists (Dudek and Bear, 1992; Mulkey and Malenka, 1992) and is not dependent on presynaptic activity. In addition, an increase in PPF during l-CCG LTD provides further evidence that not only the induction but also the expression is presynaptic and caused by a decrease in transmitter release. The selective PKA inhibitor KT 5720 on its own caused a depression of synaptic transmission and blocked l-CCG LTD, presumably by mimicking and thereby occluding any further depression. Therefore, one potential mechanism by which presynaptic Ca2+ entry could be linked to LTD induction is mGluR II-mediated decrease in Ca2+-sensitive adenylyl cyclase activity, which leads to a decrease in cAMP formation and the activity of PKA, resulting in a long-lasting decrease in transmitter release. These data, combined with previous work on Iso-induced LTP (Huang et al., 1996), provides evidence that synaptic plasticity in the amygdala neurons is at least partially controlled by the presynaptic cAMP–PKA signaling pathway.
In the presence of KT 5720 or SQ 22536, l-CCG is no longer able to induce an LTD but is still capable of causing an initial depression of synaptic transmission. These results suggest thatl-CCG has effects on the initial depression and LTD may be mediated through different mechanisms. One type of inhibitory pathway coupling neurotransmitter receptors to voltage-dependent Ca2+ channels involves G-proteins and is membrane delimited (Hille, 1994). This inhibitory pathway is generally thought to be responsible for neurotransmitter-mediated inhibition of Ca2+ channels and synaptic transmission and could be disrupted by activation of PKC (Thompson and Gahwiler, 1992; Swartz et al., 1993; Kamiya and Yamamoto, 1997). In the present study, the l-CCG-induced initial depression is markedly reduced by PMA, implicating that it is likely attributable to a block of presynaptic voltage-dependent Ca2+channels. These results, coupled with the observation thatl-CCG LTD was absent in rab3A-deficient mice in the mossy fiber–CA3 synapse (Tzounopoulos et al., 1998), suggesting thatl-CCG-induced synaptic depression may have two components: the initial depression that involves modulation of presynaptic voltage-dependent Ca2+ channels and LTD, which may affect a rab3A-dependent vesicle exocytosis. Future experiments to simultaneously record nerve terminal Ca2+ currents and postsynaptic responses should be able to clarify this issue.
Comparison of two forms of LTD in the amygdala
Previously, we have reported that LTD could be reliably induced by LFS in the amygdala (Wang and Gean, 1999). Although l-CCG LTD described here is phenomenologically similar to that of LFS LTD, the underlying cellular mechanisms appear to be quite different. LFS LTD in the amygdala requires activation of the NMDA receptors, depends on postsynaptic Ca2+ increases and phosphatase activity (Wang and Gean, 1999), and is independent of the presynaptic phenomenon of paired-pulse facilitation. Therefore, the mechanism of LFS LTD in the amygdala is qualitatively similar to those of hippocampal CA1 neurons and several other synapses in the brain in which a dependence on postsynaptic Ca2+increases is evident (Dudek and Bear, 1992; Mulkey and Malenka, 1992).
In contrast, l-CCG LTD is NMDA receptor-independent and is not blocked by loading the postsynaptic cell with BAPTA salt. It is prevented by BAPTA-AM, suggesting that Ca2+ increases in the presynaptic terminals are necessary for its induction. Although LFS after induction of l-CCG LTD was unable to depress the responses to the same degree as in naïve synapses, depression consistently did occur. The inability to fully depress the synapses in these experiments is not surprising, giving the well documented “metaplastic” effects of previously reduced activity that would slide the modification threshold, θm, to a lower value, making it more difficult to further depress synapses (Abraham and Bear, 1996; Holland and Wagner, 1998). Thus, saturation of one form of LTD did not occlude the other. Paired-pulse experiments revealed that PPF ratio was increased in l-CCG LTD, suggesting that the maintenance is likely to occur at presynaptic sites. In these respects,l-CCG LTD in the amygdala closely resembles that recently described at hippocampal mossy fiber–CA3 synapse in which LTD requires presynaptic Ca2+ increases and is expressed at the presynaptic sites (Tzounopoulos et al., 1998;Kobayashi et al., 1999).
Functional implications
As stated in the introductory remarks, drugs that produce LTD in the amygdala may exhibit anxiolytic effect on patients with post-traumatic stress disorders. The other pathophysiology involving the amygdala is kindling phenomenon, which refers to the progressive development of partial and generalized seizures as a result of repeated subconvulsive stimuli (Goddard et al., 1969; McNamara, 1986). Kindling has been regarded as a chronic model of human temporal lobe epilepsy, and the amygdala is one of the most sensitive sites to induce kindling (Loscher et al., 1995). Electrophysiological recordings have demonstrated that kindling is expressed by a sustained enhancement of excitatory synaptic responses in the amygdala (Gean et al., 1989;Rainnie et al., 1992). In addition, clinical studies have reported that patients with complex partial seizures of temporal lobe origin may experience behavioral disorders, such as depressive and anxiety-related symptoms (Adamec, 1990). The finding of l-CCG LTD in the amygdala suggests that mGluR II agonists may be potential anticonvulsants in certain forms of seizure disorders. Indeed, group II mGluR agonists have been shown to attenuate epileptiform bursting observed in kindled rats (Neugebauer et al., 1997; Keele et al., 1999) and suppressed bicuculline-induced bursts in cortical neurons (Burke and Hablitz, 1995).
Footnotes
This study was supported by National Science Council Grant NSC89-2320-B006-011 and Academic Excellence Program of the Ministry of Education Grant 89-B-FA08-1-4 of Taiwan, Republic of China.
Correspondence should be addressed to Dr. Po-Wu Gean, Department of Pharmacology, College of Medicine, National Cheng-Kung University, Tainan, Taiwan 701. E-mail: powu@mail.ncku.edu.tw.
REFERENCES
- 1.Abraham WC, Bear MF. Metaplasticity: the plasticity of synaptic plasticity. Trends Neurosci. 1996;19:126–130. doi: 10.1016/s0166-2236(96)80018-x. [DOI] [PubMed] [Google Scholar]
- 2.Adamec R. Kindling, anxiety and limbic epilepsy: human and animal perspectives. In: Wada JA, editor. Kindling 4. Plenum; New York: 1990. pp. 329–341. [Google Scholar]
- 3.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 4.Brambilla R, Gnesutta N, Minichiello L, White G, Roylance AJ, Herron CE, Ramsey M, Wolfer DP, Cestari V, Rossi-Arnaud C, Grant SG, Chapman PF, Lipp HP, Sturani E, Klein R. A role for the Ras signaling pathway in synaptic transmission and long-term memory. Nature. 1997;390:281–286. doi: 10.1038/36849. [DOI] [PubMed] [Google Scholar]
- 5.Burke JP, Hablitz JJ. Modulation of epileptiform activity by metabotropic glutamate receptors in immature rat neocortex. J Neurophysiol. 1995;73:205–217. doi: 10.1152/jn.1995.73.1.205. [DOI] [PubMed] [Google Scholar]
- 6.Cho K, Kemp N, Noel J, Aggleton JP, Brown MW, Bashir ZI. A new form of long-term depression in the perirhinal cortex. Nat Neurosci. 2000;3:150–156. doi: 10.1038/72093. [DOI] [PubMed] [Google Scholar]
- 7.Davis M, Rainnie D, Cassell M. Neurotransmission in the rat amygdala related to fear and anxiety. Trends Neurosci. 1994;17:208–214. doi: 10.1016/0166-2236(94)90106-6. [DOI] [PubMed] [Google Scholar]
- 8.Domenici MR, Berretta N, Cherubini E. Two distinct forms of long-term depression coexist at the mossy fiber–CA3 synapse in the hippocampus during development. Proc Natl Acad Sci USA. 1998;95:8310–8315. doi: 10.1073/pnas.95.14.8310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudek SM, Bear MF. Homosynaptic long-term depression in area CA1 of hippocampus and effects of N-methyl-d-aspartate receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fanselow MS, Kim JJ. Acquisition of contextual Pavlovian fear conditioning is blocked by application of an NMDA receptor antagonist d,l-2-amino-5-phosphonovaleric acid to the basolateral amygdala. Behav Neurosci. 1994;108:210–212. doi: 10.1037//0735-7044.108.1.210. [DOI] [PubMed] [Google Scholar]
- 11.Gean PW, Shinnick-Gallagher P, Anderson AC. Spontaneous epileptiform activity and alteration of GABA- and of NMDA-mediated neurotransmission in amygdala neurons kindled in vivo. Brain Res. 1989;494:177–181. doi: 10.1016/0006-8993(89)90160-1. [DOI] [PubMed] [Google Scholar]
- 12.Goddard GV, McIntyre DC, Leech DEA. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. 1969;25:295–330. doi: 10.1016/0014-4886(69)90128-9. [DOI] [PubMed] [Google Scholar]
- 13.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 14.Holland LL, Wagner JJ. Primed facilitation of homosynaptic long-term depression and depotentiation in rat hippocampus. J Neurosci. 1998;18:887–894. doi: 10.1523/JNEUROSCI.18-03-00887.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang CC, Hsu KS, Gean PW. Isoproterenol potentiates synaptic transmission primarily by enhancing presynaptic calcium influx via P- and/or Q-type calcium channels in the rat amygdala. J Neurosci. 1996;16:1026–1033. doi: 10.1523/JNEUROSCI.16-03-01026.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang L, Killbride J, Rowan MJ, Anwyl R. Activation of mGluR II induces LTD via activation of protein kinase A and protein kinase C in the dentate gyrus of the hippocampus in vitro. Neuropharmacology. 1999;38:73–83. doi: 10.1016/s0028-3908(98)00168-3. [DOI] [PubMed] [Google Scholar]
- 17.Jane DE, Thomas NK, Tse HW, Watkins JC. Potent antagonists at the l-AP4- and (1S,3S)-ACPD-sensitive presynaptic metabotropic glutamate receptors in the neonatal rat spinal cord. Neuropharmacology. 1996;35:1029–1035. doi: 10.1016/s0028-3908(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 18.Kamiya H, Yamamoto C. Phorbol ester and forskolin suppress the presynaptic inhibitory action of group-II metabotropic glutamate receptor at rat hippocampal mossy fiber synapse. Neuroscience. 1997;80:89–94. doi: 10.1016/s0306-4522(97)00098-5. [DOI] [PubMed] [Google Scholar]
- 19.Keele NB, Neugebauer V, Shinnick-Gallagher P. Differential effects of metabotropic glutamate receptor antagonists on bursting activity in the amygdala. J Neurophysiol. 1999;81:2056–2065. doi: 10.1152/jn.1999.81.5.2056. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi K, Manabe T, Takahashi T. Calcium-dependent mechanisms involved in presynaptic long-term depression at the hippocampal mossy fiber-CA3 synapse. Eur J Neurosci. 1999;11:1633–1638. doi: 10.1046/j.1460-9568.1999.00578.x. [DOI] [PubMed] [Google Scholar]
- 21.LeDoux JE. The amygdala: contributions to fear and stress. Semin Neurosci. 1994;6:231–237. [Google Scholar]
- 22.LeDoux JE, Cicchetti P, Xagoraris A, Romanski LM. The lateral amygdaloid nucleus: sensory interface of the amygdala in fear conditioning. J Neurosci. 1990;10:1062–1069. doi: 10.1523/JNEUROSCI.10-04-01062.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Weiss SRB, Chuang DM, Post RM, Rogawski MA. Bidirectional synaptic plasticity in the rat basolateral amygdala: characterization of an activity-dependent switch sensitive to the presynaptic metabotropic glutamate receptor antagonist 2S-α-ethylglutamic acid. J Neurosci. 1998;18:1662–1670. doi: 10.1523/JNEUROSCI.18-05-01662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang KC, Hon W, Davis M. Pre- and posttraining infusion of N-methyl-d-aspartate receptor antagonists into the amygdala impair memory in an inhibitory avoidance task. Behav Neurosci. 1994;108:241–253. doi: 10.1037//0735-7044.108.2.241. [DOI] [PubMed] [Google Scholar]
- 25.Loscher W, Ebert U, Wahnschaffe U, Rundfeldt C. Susceptibility of different cell layers of the anterior and posterior part of the piriform cortex to electrical stimulation and kindling: comparison with the basolateral amygdala and “area tempestas.”. Neuroscience. 1995;66:265–276. doi: 10.1016/0306-4522(94)00614-b. [DOI] [PubMed] [Google Scholar]
- 26.Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired-pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- 27.Maren S. Long-term potentiation in the amygdala: a mechanism for emotional learning and memory. Trends Neurosci. 1999;22:561–567. doi: 10.1016/s0166-2236(99)01465-4. [DOI] [PubMed] [Google Scholar]
- 28.McKernan MG, Shinnick-Gallagher P. Fear conditioning induces a lasting potentiation of synaptic currents in vitro. Nature. 1997;390:607–610. doi: 10.1038/37605. [DOI] [PubMed] [Google Scholar]
- 29.McNamara JO. Kindling model of epilepsy. Adv Neurol. 1986;44:303–318. [PubMed] [Google Scholar]
- 30.Miserendino MJ, Sananes CB, Melia KR, Davis M. Blocking of acquisition but not expression on conditioned fear-potentiated startle by NMDA antagonists in the amygdala. Nature. 1990;345:716–718. doi: 10.1038/345716a0. [DOI] [PubMed] [Google Scholar]
- 31.Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 32.Neugebauer V, Keele NB, Shinnick-Gallagher P. Epileptogenesis in vivo enhances the sensitivity of inhibitory presynaptic metabotropic glutamate receptors in basolateral amygdala neurons in vitro. J Neurosci. 1997;17:983–995. doi: 10.1523/JNEUROSCI.17-03-00983.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parent MB, McGaugh JL. Posttraining infusion of lidocaine into the amygdala basolateral complex impairs retention of inhibitory avoidance training. Brain Res. 1994;661:97–103. doi: 10.1016/0006-8993(94)91186-x. [DOI] [PubMed] [Google Scholar]
- 34.Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- 35.Pitkanen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–522. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- 36.Rainnie DG, Asprodini EK, Shinnick-Gallagher P. Kindling-induced long-lasting changes in synaptic transmission in the basolateral amygdala. J Neurophysiol. 1992;67:443–454. doi: 10.1152/jn.1992.67.2.443. [DOI] [PubMed] [Google Scholar]
- 37.Rogan MT, Staubli UV, LeDoux JE. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 38.Roozendaal B, McGaugh JL. Glucocorticoid receptor agonist and antagonist administration into the basolateral but not central amygdala modulates memory storage. Neurobiol Learn Mem. 1997;67:176–179. doi: 10.1006/nlme.1996.3765. [DOI] [PubMed] [Google Scholar]
- 39.Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- 40.Schulz PE, Cook EP, Johnston D. Changes in paired-pulse facilitation suggest presynaptic involvement in long-term potentiation. J Neurosci. 1994;14:5325–5337. doi: 10.1523/JNEUROSCI.14-09-05325.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swartz KJ, Merritt A, Bean BP, Lovinger DM. Protein kinase C modulates glutamate receptor inhibition of Ca++ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- 42.Thompson SM, Gahwiler BH. Comparison of the action of baclofen at pre- and postsynaptic receptors in the rat hippocampus. J Physiol (Lond) 1992;451:329–345. doi: 10.1113/jphysiol.1992.sp019167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tzounopoulos T, Janz R, Sudhof TC, Nicoll RA, Malenka RC. A role for cAMP in long-term depression at hippocampal mossy fiber synapses. Neuron. 1998;21:837–845. doi: 10.1016/s0896-6273(00)80599-1. [DOI] [PubMed] [Google Scholar]
- 44.Wang SJ, Gean PW. Long-term depression of excitatory synaptic transmission in the rat amygdala. J Neurosci. 1999;19:10656–10663. doi: 10.1523/JNEUROSCI.19-24-10656.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yokoi M, Kobayashi K, Manabe T, Takahashi T, Sakaguchi I, Katsuura G, Shigemoto R, Ohishi H, Nomura S, Nakamura K, Nakao K. Impairment of hippocampal mossy fiber LTD in mice lacking mGluR2. Science. 1996;273:645–647. doi: 10.1126/science.273.5275.645. [DOI] [PubMed] [Google Scholar]