

Abstract
The transcription factor ΔFosB is induced in the hippocampus and other brain regions by repeated electroconvulsive seizures (ECS), an effective antidepressant treatment. The unusually high stability of this protein makes it an attractive candidate to mediate some of the long-lasting changes in the brain caused by ECS treatment. To understand how ΔFosB might alter brain function, we examined the gene expression profiles in the hippocampus of inducible transgenic mice that express ΔFosB in this brain region by the use of cDNA expression arrays that contain 588 genes. Of the 430 genes detected, 20 genes were consistently upregulated, and 14 genes were downregulated, by >50%. One of the upregulated genes is cyclin-dependent kinase 5 (cdk5). On the basis of its purported role in regulating neuronal structure, we studied directly whether cdk5 is a true target for ΔFosB. Upregulation of cdk5 immunoreactivity in the hippocampus was confirmed by Western blotting in the ΔFosB-expressing transgenic mice as well as in rats treated chronically with ECS. Chronic ECS treatment also increased, in the hippocampus, the phosphorylation state of tau, a microtubule-associated protein that is a known substrate for cdk5. A 1.6 kb fragment of the cdk5 promoter was cloned, and activity of the promoter was found to be increased after overexpression of ΔFosB in cell culture. Moreover, mutation of the single consensus activator protein-1 site contained within the cdk5 promoter fragment completely abolished activation of the promoter by ΔFosB. Together, these results suggest that cdk5 is one target by which ΔFosB produces some of its physiological effects in the hippocampus and thereby mediates certain long-term consequences of chronic ECS treatment.
Keywords: cdk5, ΔFosB, hippocampus, electroconvulsive seizures, transcription factors, antidepressant treatments, inducible transgenic mice, gene expression
Repeated administration of electroconvulsive seizures (ECS) is one of the most effective treatments for depression; yet the mechanisms by which it exerts its clinical effects remain incompletely understood. Regulation of gene expression by specific transcription factors may be one important mechanism involved, because the beneficial effects of ECS treatment can last long after the last ECS. Two transcription factors that are induced by chronic ECS in the hippocampus, a brain region implicated in depression and antidepressant treatments (Duman et al., 1999), are cAMP response element binding protein (Nibuya et al., 1996) and ΔFosB (Hope et al., 1994a; Chen et al., 1995, 1997). ΔFosB is of particular interest because of its unique temporal properties (Nestler et al., 1999). ΔFosB is induced in the hippocampus and in certain regions of cerebral cortex only after repeated ECS administration. Moreover, after being induced, it persists in the brain for relatively long periods of time (several weeks) because of its extraordinary stability. Thus, ΔFosB could be an important mediator of some of the long-lasting adaptive changes that chronic ECS treatment produces in the hippocampus.
ΔFosB is a truncated splice variant of the fosBgene. ΔFosB heterodimerizes with JunD and, to a lesser extent, JunB to form activator protein-1 (AP-1) complexes that bind to specific AP-1 sites contained within the 5′-promoter regions of certain genes (Chen et al., 1995). ΔFosB-containing AP-1 complexes are reported to act as both transcriptional repressors and activators in vitro, depending on the gene and cell type involved (Dobrazanski et al., 1991; Nakabeppu and Nathans, 1991; Yen et al., 1991; Chen et al., 1997). Identification of specific target genes via which ΔFosB produces its physiological effects in vivo is an important step in understanding the functional role played by this novel transcription factor. One approach to answer this question is to evaluate genes with known AP-1 sites as potential physiological targets for ΔFosB. This approach has succeeded recently in identifying two glutamate receptor subunits as possible targets for ΔFosB: the NR1 subunit of NMDA receptors (Hiroi et al., 1998) and the GluR2 subunit of AMPA receptors (Kelz et al., 1999).
A more open-ended and complementary approach is to use differential gene expression analysis to identify genes that are regulated by ΔFosB in vivo. To do this effectively, however, it must be possible to induce ΔFosB selectively within brain regions of interest in adult animals. We have accomplished this goal by use of the tetracycline-regulated gene expression system and have constructed bitransgenic mice that express ΔFosB in selected brain regions, including the hippocampus, in an inducible and brain region-specific manner (Chen et al., 1998; Kelz et al., 1999). Such inducible transgenic mice represent ideal tools with which to search for physiological targets for ΔFosB, because they avoid the developmental compensations that complicate many conventional transgenic and knock-out animals.
In the present study, we used cDNA arrays to analyze gene expression patterns in the hippocampus of these ΔFosB-expressing mice as a means of identifying novel targets for this transcription factor in vivo. One of the many genes found to be consistently regulated in the hippocampus after ΔFosB expression was that encoding cyclin-dependent kinase 5 (cdk5). We further pursued regulation of cdk5, because this protein appeared to be of particular interest as a putative target for ΔFosB in the hippocampus (see Discussion). In agreement with the DNA array finding, we show here that cdk5 does indeed appear to be a physiological target of ΔFosB, and for chronic ECS treatment, in the hippocampus. The results thus illustrate the usefulness of combining an open-ended DNA array-based approach with inducible, tissue-specific transgenic mice to identify novel targets for ΔFosB in the brain.
MATERIALS AND METHODS
ΔFosB-expressing mice. Bitransgenic 11A or 1A mice, which express ΔFosB in an inducible and brain region-specific manner (including the hippocampus), were used in these studies (Chen et al., 1998; Kelz et al., 1999). In initial experiments, 1A mice that contained both transgenes (NSE-tTA and TetOp-ΔFosB; see Fig. 1) were compared with littermates that contained only one transgene (NSE-tTA). In later experiments, 11A mice were used. The mice were conceived and raised on doxycycline (100 μg/ml) in the drinking water, which is known to suppress ΔFosB expression completely (Chen et al., 1998). Half of the littermates were removed from doxycycline at 3–4 weeks of age and were used 6 weeks later, at which time ΔFosB expression is known to be turned on. The hippocampus was removed from decapitated mice by gross dissection. 1A mice were used in initial studies, because this line expresses much higher levels of ΔFosB compared with the 11A mice, although the pattern of expression between the two lines appears to be equivalent (Chen et al., 1998; Kelz et al., 1999).
Fig. 1.
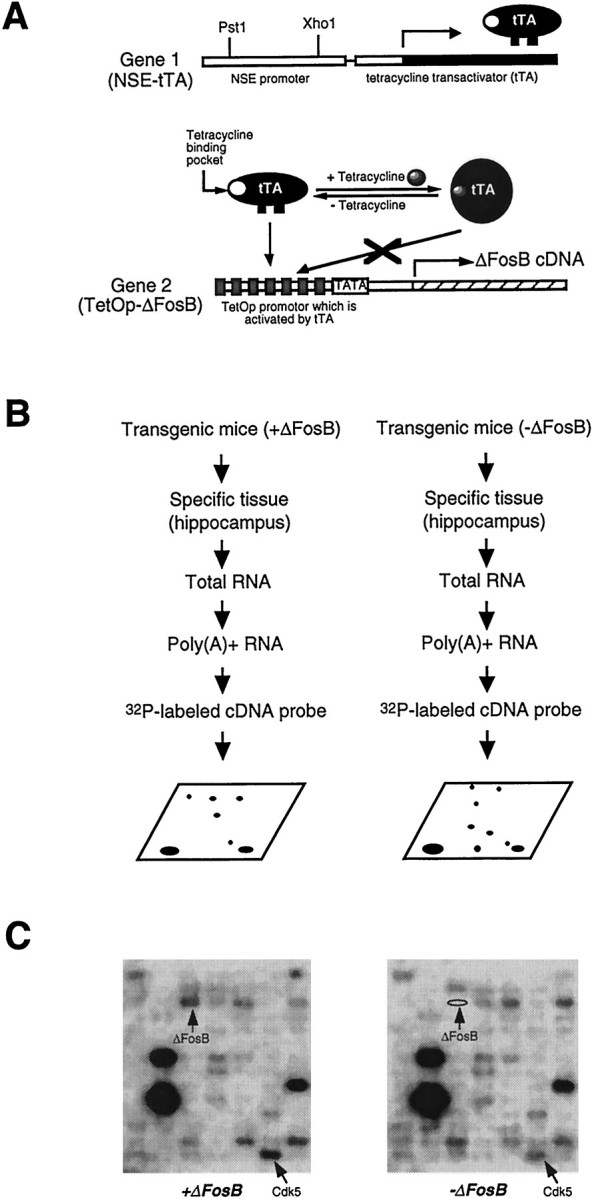
Identification of cdk5 as one of the downstream target genes for ΔFosB in the hippocampus of inducible bitransgenic mice using cDNA expression arrays. A, Schematic diagram of the tetracycline expression system used for the inducible tissue-specific expression of ΔFosB (Chen et al., 1998). Gene 1 encodes the tetracycline transactivator (tTA) under the control of the neuron-specific enolase (NSE) promoter. Gene 2 encodes ΔFosB under the control of the tetracycline-responsive promoter with seven tetracycline operators (TetOp). B, Strategy for searching downstream target genes for ΔFosB in the hippocampus of inducible ΔFosB-expressing bitransgenic mice. Total RNA was isolated from five bitransgenic mice, either expressing or not expressing ΔFosB, and pooled. Poly(A+) RNA was isolated from the pooled total RNA and used as a template for the synthesis of a32P-labeled cDNA probe. The cDNA probes were hybridized to the arrays, and the arrays were analyzed by the PhosphorImager.C, Gene expression profiles of the hippocampus of the bitransgenic mice, either expressing or not expressing ΔFosB, from a portion of the resulting cDNA expression arrays. Positions of the ΔFosB and cdk5 genes are indicated by arrows. The results are representative of three independent determinations.
cDNA expression arrays. Total RNA was isolated from the hippocampus of bitransgenic mice, one group maintained on doxycycline and the other group removed from doxycycline, by the use of an RNAqueous phenol-free total RNA isolation kit (Ambion, Austin, TX). Poly(A+) RNA was isolated from the total RNA by the use of the Oligotex mRNA isolation kit (Qiagen, Hilden, Germany). The poly(A+) RNA was used as a template for the synthesis of32P-labeled cDNA probes. The cDNA probes (1500 cpm/μl) were hybridized to Atlas mouse cDNA expression arrays (Clontech, Cambridge, UK). The arrays were then exposed to a phosphorimaging screen for 16–24 hr, and the hybridization signal was analyzed with a Bio-Rad (Hercules, CA) GS-363 PhosphorImager.
ECS treatments. Male Sprague Dawley rats (initial weight, ∼200 gm; Charles River Laboratories, Wilmington, MA) were used for all experiments. ECS was administered, as before (Hope et al., 1994a), via ear clip electrodes (45 mA; 0.3 sec). Chronically treated animals received a single ECS daily for 10 d and were used 18 hr later. Control animals received chronic sham treatment, in which electrodes were clipped onto the rats' ears but no current was applied. The hippocampus was obtained by gross dissection.
Gel shift assays. Gel shift assays were performed on the basis of published procedures (Hope et al., 1994a). Mouse or rat hippocampus was homogenized with Dounce homogenizers in 20 vol of electrophoretic mobility shift assay (EMSA) buffer: 20 mmHEPES, pH 7.9, 0.4 m NaCl, 20% glycerol, 5 mmMgCl2, 0.5 mm EDTA, 0.1 mm EGTA, 1% Nonidet P-40, 10 μg/ml leupeptin, 0.1 mmp-aminobenzamidine, 1 μg/ml pepstatin, 0.5 mm PMSF, and 5 mm DTT. The homogenates were incubated on ice for 30 min before centrifugation at 15,000 × g for 20 min at 4°C. Aliquots of supernatants (containing 20 μg of protein) were incubated at 20°C for 20 min with 1 μg of poly(dI–dC)·poly(dI–dC), 40 μg of bovine serum albumin, 10 mm Tris-HCl, pH 7.9, 10 mm KCl, 1 mm EDTA, 4% glycerol, and 1 ng of the radioactively labeled AP-1 probe. The samples were incubated for 20 min at 20°C and electrophoresed at 150 V for 2 hr in a nondenaturing 6% acrylamide and 0.16%N,N′-methylenebisacrylamide gel containing 25 mm Tris-borate buffer, pH 8.3, 1 mm EDTA, and 1.6% glycerol. The gels were dried and exposed to x-ray film. Levels of AP-1 binding were quantified by measuring the optical density of specific bands by the use of an image analysis system with NIH Image software, version 1.41.
Western blotting. Western blotting was performed as described previously (Hope et al., 1994b). Mouse or rat hippocampus was homogenized in Dounce homogenizers in 10 vol of EMSA buffer. Aliquots of these cellular extracts (containing 50 μg of protein) were then applied to a 10% acrylamide and 0.27%N,N′-methylenebisacrylamide resolving gel for SDS-PAGE overnight at 75 V and electrotransferred to nitrocellulose filters at 200 mA for 3 hr. The blots were incubated in blocking buffer, with four changes of 15 min each, containing 2% nonfat dry milk powder in PBS-Tween (10 mm sodium phosphate, pH 7.4, 150 mm NaCl, and 0.1% Tween 20) and incubated overnight at 4°C in a 1:200,000 dilution of anti-cdk5 antibody (Santa Cruz Biotechnology, Santa Cruz, CA), 1:10,000 dilution of anti-phospho-tau antibody (Roche Products, Mannheim, Germany), 1:2000 dilution of anti-p35 antibody (N-20; Santa Cruz Biotechnology), or 1:2000 dilution of anti-p35/p25 antibody (C-19; Santa Cruz Biotechnology) in blocking buffer with 0.05% sodium azide. The blots were washed four times for 15 min each in blocking buffer and incubated in a 1:4000 dilution of goat anti-rabbit antibody conjugated to horseradish peroxidase (Vector Laboratories, Burlingame, CA) in blocking buffer for 2 hr. The blots were washed eight times for 15 min each with PBS-Tween alone, developed with the enhanced chemiluminescence (ECL) system of Amersham (Arlington Heights, IL), and exposed to Hyperfilm-ECL (Amersham) for 5–60 sec. Levels of protein immunoreactivity were quantified by either measuring the optical density of specific bands using an image analysis system with NIH Image software, version 1.41, or measuring the light intensity using a Bio-Rad PhosphorImager.
Cloning cdk5 promoter. Genomic DNA from mouse tail was used as a template in PCR to clone a portion of the 5′-promoter region of the cdk5 gene (Ohshima et al., 1996). The accuracy of the PCR product was confirmed by DNA sequencing. Primers were designed on the basis of GenBank sequence information of the cdk5 gene: upstream primer, 5′-CCA GCA GCC AGA GGG GAC TCT-3′, and downstream primer, 5′-AGG TGC CTA GAG GAA GGT TG-3′. The PCR product (1.6 kb) was cloned into the pGL3-basic vector, which contains a luciferase reporter gene. The plasmid was designated pGL3-cdk5-luc.
Transfection. Cells (2.5 × 105) of an inducible ΔFosB-expressing C6 glioma cell line [described in Chen et al. (1997)] in 2 ml of DMEM containing 10% FBS were inoculated into each well of six-well Falcon plates. After 16 hr of incubation at 37°C in 5% CO2, the cells were transfected with 2.5 μg of pGL3-cdk5-luc DNA or pGL3-cdk5mut-luc DNA (see below) by the use of the lipofectamine reagent (Life Technologies, Gaithersburg, MD). Cells were harvested 24 hr later.
Site-directed mutagenesis. Primers containing mutations in the AP-1 site of the cdk5 promoter (mutant primer 1, 5′-GGG TGT TTG TCG ACT CCA GCG ACC TCC TGA CA-3′; mutant primer 2, GTC GCT GGA GTC GAC AAA CAC CCA ACC AGG TCA-3′) were paired with either upstream or downstream primers for the cdk5 promoter in PCR. The AP-1 site was replaced by use of the restriction enzyme SalI site. The PCR products were digested with SalI and then ligated by T4 DNA ligase. The ligated PCR product was used as a template and amplified by PCR using upstream and downstream cdk5 primers. The PCR product containing the mutated cdk5 promoter fragment was cloned into the pGL3-basic vector. The mutant plasmid was designated pGL3-cdk5mut-luc.
RESULTS
Analysis of gene expression profiles in ΔFosB-expressing mice
In previous studies, we used the tetracycline gene regulation system (Gossen and Bujard, 1992) to develop bitransgenic mice that support the inducible expression of ΔFosB in specific brain regions, including the hippocampus (Chen et al., 1998) (Fig.1A). Expression of ΔFosB is tightly regulated by doxycycline, an analog of tetracycline, in bitransgenic mice. To search for downstream targets for ΔFosB in the hippocampus, we analyzed RNA samples derived from this brain region of ΔFosB-expressing mice and of their littermates not expressing ΔFosB, by the use of commercially available cDNA expression arrays as shown in Figure 1, which contain 588 genes in each array. Levels of probe hybridization to each gene were quantified by PhosphorImager (see Materials and Methods). A portion of the resulting arrays are shown in Figure 1C. As would be expected, levels of hybridization tofosB DNA itself were ∼4.5-fold higher with probe derived from the hippocampus of ΔFosB-expressing mice than with that of doxycycline-suppressed controls (Table1). The magnitude of this increase corresponds to the degree of induction of ΔFosB observed previously in the bitransgenic mice (see Kelz et al., 1999), which in turn is similar to that obtained with chronic ECS treatment (Hope et al., 1994a). This result offers some validation of the ability to detect altered gene expression by use of the cDNA expression arrays.
Table 1.
Analysis of gene expression profiles in the hippocampus of inducible ΔFosB-expressing transgenic mice
| Gene | GenBank accession number | % Regulation |
|---|---|---|
| FosB | x14897 | 467% |
| ATP-dependent DNA helicase II | x66323 | 135% |
| Somatostatin receptor 2 | m181832 | 102% |
| Caspase-11 | u59463 | 92% |
| PCNA | x53068 | 87% |
| Relaxin | z27088 | 83% |
| Translin | x81464 | 83% |
| DNA ligase III | u66058 | 79% |
| MLH1 DNA mismatch repair protein | u59883 | 77% |
| Ung1 | x99018 | 68% |
| Oxidative stress-induced protein | u40930 | 67% |
| Glutathione S-transferase Pi 1 | d30687 | 67% |
| MHR23B | x92411 | 67% |
| Cdk5 | d29678 | 61% |
| DNA excision repair protein ERCC5 | d16306 | 61% |
| Ets-related Sap1A | z36885 | 58% |
| IL-10 receptor | l12120 | 55% |
| Bax | l22472 | 51% |
| RIP cell death protein | u25995 | 51% |
| Ubiquitin-conjugated enzyme | x96859 | 51% |
| PI3 kinase p110 | u03279 | −76% |
| PI3 kinase p85 | m60651 | −76% |
| Tie 2 proto-oncogene | s67051 | −77% |
| Cyclin C | u62638 | −77% |
| Myeloblastin, serine protease | u43525 | −77% |
| IL-6 receptor gp130 | m83336 | −78% |
| 5-HT 1b | z11597 | −79% |
| Leukocyte adhesion LFA-1 | x14951 | −79% |
| GABA-A transporter 3 | l04663 | −79% |
| PAX-8 | x57487 | −80% |
| Desmocolin 2 | l33779 | −81% |
| 5-HT 2 | s49542 | −82% |
| Interferon inducible protein 1 | u19119 | −84% |
| Kruppel-like factor | u25096 | −86% |
PhosphorImager analysis of gene expression profiles of the hippocampus of mice expressing ΔFosB or not expressing ΔFosB revealed 34 genes that were upregulated or downregulated by ΔFosB by >50%. The results are representative of three independent determinations.
IL, Interleukin; LFA, leukocyte function-associated antigen; PI3, phosphatidylinositol 3; PCNA, proliferating cell nuclear antigen; MHR23B, Rad23 UV excision repair protein homologue; ERCC5, DNA excision repair protein; PAX, paired box protein; MLH1, MLH1 DNA mismatch repair protein.
By comparing the gene expression profile from bitransgenic mice expressing ΔFosB with the profile from mice not expressing ΔFosB in three independent trials, we identified genes that are consistently upregulated or downregulated >50% after ΔFosB expression. Of the 430 genes detected on the resulting arrays, 20 genes were reliably upregulated by at least 50%, whereas 14 genes were reliably downregulated by at least 50%. These genes, which are listed in Table1, encode for a wide variety of proteins, including neurotransmitter receptors and transporters and intracellular signaling proteins. Because the sensitivity of these cDNA expression arrays is much lower than that of Northern blotting and the signals of many mRNAs detected by the arrays were close to background, the percentage of regulation was considered a semiquantitative measure of gene regulation.
Cdk5 is one of the downstream target genes for ΔFosB
One of the genes identified on the DNA arrays as upregulated by ΔFosB is that for cdk5 (Fig. 1C), which was increased by ∼61% in ΔFosB-expressing mice (Table 1). As a first step to confirm that this upregulation is not a false-positive result, levels of cdk5 immunoreactivity were measured by Western blotting in the hippocampus of an independent group of ΔFosB-expressing mice and their doxycycline-suppressed littermates. As shown in Figure2, levels of cdk5 were increased by close to 50% after ΔFosB expression.
Fig. 2.

Upregulation of cdk5 immunoreactivity in the hippocampus of inducible bitransgenic mice after ΔFosB expression.A, A representative immunoblot shows cdk5 levels in the hippocampus of bitransgenic mice expressing (+) or not expressing (−) ΔFosB. B, Levels of cdk5 immunoreactivity are given as arbitrary OD units and are expressed as the mean ± SEM (n = 5 animals in each treatment group). *p < 0.05 by Student's ttest.
To test whether ΔFosB regulation of cdk5 might be a direct effect of the transcription factor on the cdk5 gene, we cloned a 1.6 kb fragment of the 5′-promoter of the cdk5 gene by PCR and placed it into a reporter vector (pGL3-basic), which contains a luciferase reporter gene. This fragment of the cdk5 promoter, as published previously (Ohshima et al., 1996), contains several regulatory elements including individual AP-1, AP-2, CRE, NF-IL6, and SP1 sites (Fig.3A). The cdk5-luciferase construct was analyzed in a stable C6 glioma cell line in which ΔFosB expression is under the control of the tetracycline system (Chen et al., 1997). The cdk5 promoter exhibited strong activity in this cell line in the absence of ΔFosB, with 20-fold higher levels of luciferase seen as compared with the pGL3-basic control plasmid. Activity of the cdk5 promoter was increased by ∼75% when ΔFosB was induced by the removal of tetracycline (Fig. 3B). To test whether the AP-1 site in the promoter region is responsible for this regulation, we investigated the ability of ΔFosB to regulate a modified cdk5 promoter in which this site was altered by site-directed mutagenesis (Fig. 3A). As shown in Figure 3B, mutation of the AP-1 site completely abolished upregulation of cdk5 promoter activity by ΔFosB.
Fig. 3.

Induction of cdk5 promoter activity by ΔFosB.A, Schematic structure of a fragment of the 5′-promoter of the cdk5 gene is shown. Several putative response elements within the promoter region are indicated. The AP-1 site framedby a rectangular box and its adjacent sequences are shown. The AP-1 sequence in a mutated promoter (underlined sequence) is also shown. B, Luciferase activity was measured in a C6 glioma cell line that supports the inducible expression of ΔFosB (Chen et al., 1997) transfected with the wild-type (cdk5-luc) or mutated (mutcdk5-luc) cdk5 promoter in pGL3-basic. Data are expressed as the mean percent change in promoter activity in the presence of ΔFosB compared with that in the absence of ΔFosB (± SEM; n = 3). The results are representative of two independent replications. *p< 0.05 by Student's t test.
To confirm further the role of the AP-1 element in the regulation of cdk5 promoter activity by ΔFosB, we performed gel shift assays using an AP-1 oligonucleotide derived from the cdk5 promoter (Fig.4A). The results showed robust induction of AP-1-binding activity in the hippocampus after ΔFosB expression (Fig. 4B). This activity was caused by ΔFosB, because it was disrupted by including an anti-ΔFosB antibody in the assay mixture (data not shown).
Fig. 4.
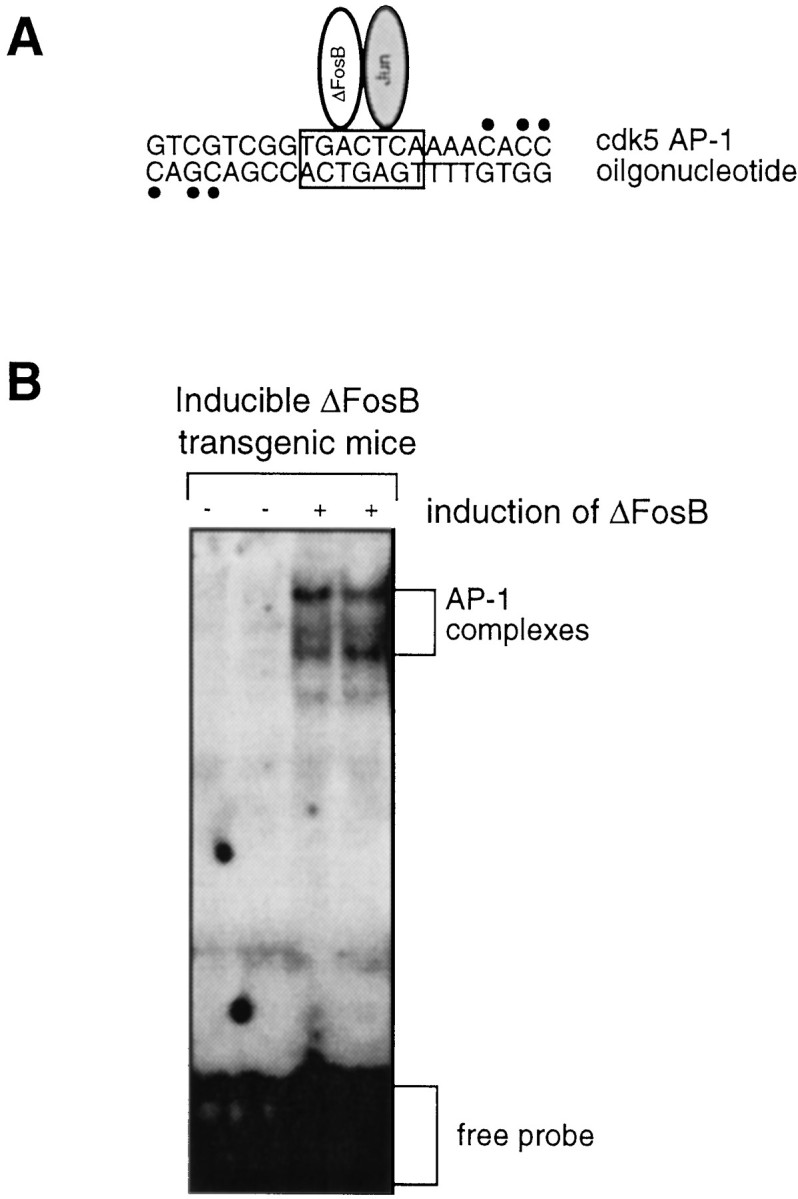
Upregulation of cdk5 AP-1-binding activity in the hippocampus of inducible bitransgenic mice after ΔFosB expression.A, The sequence of the cdk5 AP-1 oligonucleotide used as the probe is shown. The 32P-labeled nucleotides are indicated by dots. B, A representative autoradiogram shows the dramatic induction of cdk5 AP-1-binding activity after ΔFosB expression. The results are representative of three independent replications.
Upregulation of cdk5 in the hippocampus by chronic ECS treatment
Because chronic ECS treatment induces high level of ΔFosB in the hippocampus, it is hypothesized that chronic ECS should also upregulate cdk5 in this brain region. To test this hypothesis, we analyzed levels of cdk5 expression in the hippocampus of Sprague Dawley rats treated chronically with ECS. It was found that chronic ECS treatment increased levels of cdk5 immunoreactivity in the hippocampus by ∼50% (Fig.5A). Chronic ECS treatment was also found to increase levels of AP-1 binding, using the AP-1 element in the cdk5 promoter, in the hippocampus by more than twofold (Fig. 5B).
Fig. 5.
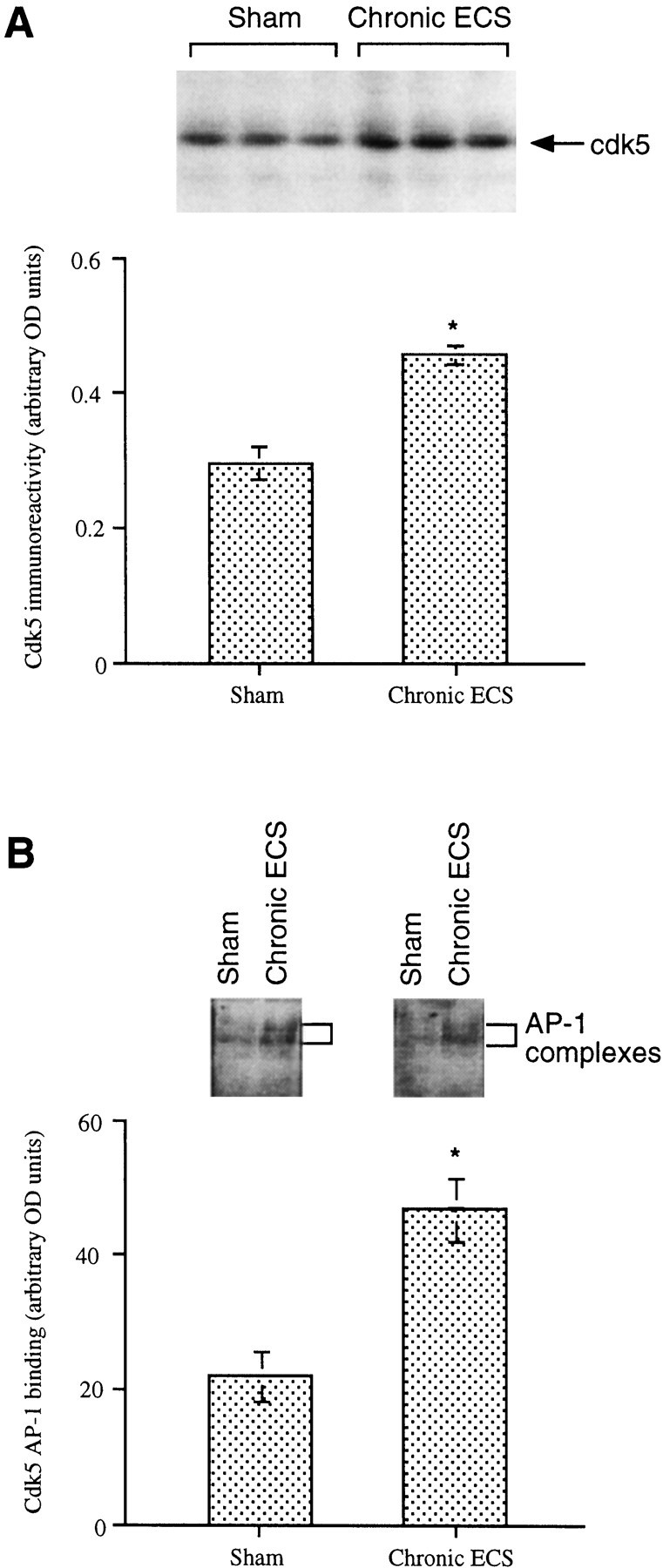
Upregulation of cdk5 immunoreactivity in rat hippocampus by chronic ECS treatment. A,Top, A representative immunoblot shows cdk5 levels in the hippocampus after sham or ECS treatment. Bottom, Levels of cdk5 immunoreactivity are given as arbitrary OD units and are expressed as the mean ± SEM (n = 8 animals in each treatment group). B, Top, A representative autoradiogram shows cdk5 AP-1-binding activity after sham or ECS treatment. Bottom, Levels of cdk5 AP-1-binding activity are given as arbitrary OD units and are expressed as the mean ± SEM (n = 8 animals in each treatment group). *p < 0.05 by Student'st test.
Evidence of increased cdk5 catalytic activity after chronic ECS treatment
Several substrates for cdk5 have been identified in brain. Prominent among these is the microtubule-associated protein tau (Patrick et al., 1999). To test whether upregulation of cdk5 immunoreactivity is associated with an increase in its catalytic activity, we analyzed levels of phosphorylated tau in the hippocampus of rats treated chronically with ECS. Five phosphorylated tau isoforms were detected by Western blotting. Each of these isoforms was upregulated by chronic ECS treatment (Fig.6).
Fig. 6.

Upregulation of tau phosphorylation in rat hippocampus by chronic ECS treatment. A, A representative immunoblot showing levels of phospho-tau proteins after sham or ECS treatment. B–F, Levels of phospho-tau, in arbitrary OD units, for each tau isoform ± SEM (n = 3). *p < 0.05 by Student's t test.
The catalytic activity of cdk5 depends on its cofactor, termed p35, that must be cleaved into a 25 kDa product, termed p25, to activate the kinase (Patrick et al., 1999). To determine whether upregulation of cdk5 immunoreactivity is associated with corresponding changes in the levels of p35 or its p25 fragment, levels of these proteins were analyzed by Western blotting in the hippocampus of rats after chronic ECS treatment. Chronic ECS failed to alter p35 levels in this brain region but did cause a significant (46%) increase in the levels of p25 immunoreactivity (Fig. 7).
Fig. 7.

Regulation of p35 and p25 immunoreactivity in rat hippocampus by chronic ECS treatment. A, A representative immunoblot shows p35 and p25 levels after sham or chronic ECS treatment. B, Levels of p35 and p25 immunoreactivity are given as arbitrary OD units and are expressed as the mean ± SEM (n = 8 animals in each treatment group). *p < 0.05 by Student'st test.
DISCUSSION
The results of the present study demonstrate, by use of DNA array technology, that cdk5 is one of the downstream target genes for ΔFosB. Cdk5 immunoreactivity is increased in the hippocampus of mice after the inducible expression of ΔFosB within this brain region. Binding at an AP-1 site present within the 5′-promoter region of the cdk5 gene is also increased after ΔFosB expression. ΔFosB expression also increases activity of the cdk5 promoter in vitro, and this increase is abolished after mutation of the AP-1 site contained within the promoter. Furthermore, chronic ECS treatment, which has been shown previously to induce ΔFosB dramatically in the hippocampus, also increases levels of cdk5 immunoreactivity, cdk5 AP-1-binding activity, and the state of phosphorylation of the microtubule-associated protein tau, a known cdk5 substrate, in this brain region.
The upregulation of cdk5 by chronic ECS treatment in the hippocampus is an interesting finding, based on evolving evidence that chronic ECS treatment may produce some of its clinically beneficial effects by promoting the growth and sprouting of several types of hippocampal neurons (Duman et al., 1997, 1999). Thus, chronic ECS increases the expression of the neurotrophic factor BDNF in the rat hippocampus (Nibuya et al., 1995). Chronic ECS also enhances the sprouting of granule cell neurons in the hippocampal dentate gyrus (Vaidya et al., 1999) and even increases the birth of new granule cell neurons (Madsen et al., 2000). Conversely, stress exerts the opposite effects in rodent models; it decreases BDNF expression, the sprouting of several neuronal cell types, and neurogenesis in the hippocampus (Smith et al., 1995;Sapolsky, 1996; Brown et al., 1999; Gould and Tanapat, 1999). These effects can be prevented by previous treatment with ECS. Several classes of chemical antidepressants can exert some, but not all, of the aforementioned effects. The relevance of these findings in animal models to psychiatric phenomena in humans is indicated by the observation of reduced hippocampal volume in patients with depression or other stress-related disorders (Sheline et al., 1996; Lupien et al., 1998; Bremner et al., 2000).
Cdk5 is a plausible mediator of some of these effects of chronic ECS administration, which is why, among all of the gene products identified on the arrays as putative targets for ΔFosB (Table 1), we focused first on cdk5. Cdk5 belongs to a family of cyclin-dependent kinases that are known to play an important role in the regulation of cell growth (Lee et al., 1997; Zheng et al., 1998). Among this kinase family, cdk5 is unique with respect to its enrichment in nervous tissue and, in particular, in the fully differentiated adult brain (Hellmich et al., 1992). Several neural proteins have been shown to be phosphorylated by cdk5 in recent years (Julien and Mushynski, 1998;Bibb et al., 1999; Ahlijanian et al., 2000). Prominent among these substrates are several proteins important for neuronal structure, including tau and neurofilament proteins (Patrick et al., 1999). Moreover, a dominant-negative mutant of ckd5 inhibits neurite outgrowth in primary neuronal cultures (Nikolic et al., 1996), and cdk5 knock-out mice show abnormal development of the hippocampus and cerebral cortex (Gilmore et al., 1998). Cdk5 also has been shown to enhance axonal growth in cultured neurons (Paglini et al., 1998). Although further work is needed to link causally ECS-induced upregulation of cdk5 to enhanced sprouting and growth of hippocampal neurons, the present results identify cdk5 as one candidate molecule that may mediate some of the long-term adaptive changes in the hippocampus induced by chronic ECS treatment.
The enzymatic activity of cdk5 in postmitotic neurons depends on a neuron-specific activator, p35 (Patrick et al., 1999). This effect of p35 requires its proteolytic cleavage into p25, which directly activates the enzyme. As a result, upregulation of cdk5 immunoreactivity in the hippocampus would be expected to result in increased cdk5 catalytic activity only if the amount of p35 (or p25) in this tissue is not limiting. The result that chronic ECS causes increased phosphorylation of tau is consistent with an increase in cdk5 catalytic activity. Further evidence of this interpretation is our finding that chronic ECS treatment also increases levels of p25 immunoreactivity in the hippocampus. The mechanism underlying this upregulation of p25, which occurs in the absence of a detectable change in the levels of p35, is unknown.
A novel aspect of this study is its application of DNA arrays to the analysis of mice that support the inducible and brain region-specific expression of a transcription factor, in this case ΔFosB. Cdk5 is one of 34 genes that were consistently upregulated or downregulated in the hippocampus after expression of ΔFosB. A major limitation of DNA array technology, as well as of several other methodologies used to analyze differential gene expression (such as differential display and subtraction hybridization), is the large number of false-positive results obtained. Although cdk5 proved to be a bona fide target for ΔFosB, it remains to be seen whether the other genes identified as putative ΔFosB targets (Table 1) are also true targets. Another limitation with DNA array technology, at least with the filter-based arrays used in the present study, is the relatively low sensitivity of detection. It is quite likely that additional targets for ΔFosB can be found by the use of more sensitive detection methods. Our results illustrate still another limitation of the use of DNA arrays, namely, that after putative genes are identified by the use of arrays, one is faced with studying the role of any individual gene by more conventional (and labor-intensive) methods. Thus, although we identified 34 putative targets for ΔFosB, we are still obligated to characterize the precise function of each gene one at a time.
Nevertheless, the results of the present study demonstrate the potential power of DNA array analyses in identifying novel targets for ΔFosB in the brain. On the basis of available models of ΔFosB action, we would not otherwise have thought of cdk5 as a potential mediator of this transcription factor. Cdk5 has been implicated previously in the hyperphosphorylation of tau seen in certain neurodegenerative disorders (see Baumann et al., 1993; Alvarez et al., 1999; Patrick et al., 1999). On the basis of our findings with DNA arrays, our results further implicate cdk5 in the neural plasticity that accompanies the treatment of depression with chronic ECS. Such observations could help explain the pathophysiology of depression as well as provide new leads to the development of more effective antidepressant treatments.
Footnotes
This work was supported by grants from the National Institute of Mental Health and the National Institute on Drug Abuse.
Correspondence should be addressed to Dr. Eric J. Nestler, Department of Psychiatry, The University of Texas Southwestern Medical Center, 5323 Harry Hines Boulevard, Dallas, TX 75390-9070. E-mail:eric.nestler@utsouthwestern.edu.
REFERENCES
- 1.Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvarez A, Toro R, Caceres A, Maccioni RB. Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett. 1999;459:421–426. doi: 10.1016/s0014-5793(99)01279-x. [DOI] [PubMed] [Google Scholar]
- 3.Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Lett. 1993;336:417–424. doi: 10.1016/0014-5793(93)80849-p. [DOI] [PubMed] [Google Scholar]
- 4.Bibb JA, Snyder GL, Nishi A, Yan Z, Meijer L, Fienberg AA, Tsai LH, Kwon YT, Girault JA, Czernik AJ, Huganir RL, Hemmings HC, Jr, Nairn AC, Greengard P. Phosphorylation of DARPP-32 by Cdk5 modulates dopamine signalling in neurons. Nature. 1999;402:669–671. doi: 10.1038/45251. [DOI] [PubMed] [Google Scholar]
- 5.Bremner JD, Narayan M, Anderson ER, Staib LH, Miller H, Charney DS. Smaller hippocampal volume in major depression. Am J Psychiatry. 2000;157:115–118. doi: 10.1176/ajp.157.1.115. [DOI] [PubMed] [Google Scholar]
- 6.Brown ES, Rush AJ, McEwen BS. Hippocampal remodeling and damage by corticosteroids: implications for mood disorders. Neuropsychopharmacology. 1999;21:474–484. doi: 10.1016/S0893-133X(99)00054-8. [DOI] [PubMed] [Google Scholar]
- 7.Chen J, Kelz MB, Hope BT, Nakabeppu Y, Nestler EJ. Chronic Fos-related antigens: stable variants of deltaFosB induced in brain by chronic treatments. J Neurosci. 1997;17:4933–4941. doi: 10.1523/JNEUROSCI.17-13-04933.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Kelz MB, Zeng G, Sakai N, Steffen C, Shockett PE, Picciotto MR, Duman RS, Nestler EJ. Transgenic animals with inducible, targeted gene expression in brain. Mol Pharmacol. 1998;54:495–503. doi: 10.1124/mol.54.3.495. [DOI] [PubMed] [Google Scholar]
- 9.Chen JS, Nye HE, Kelz MB, Hiroi N, Nakabeppu Y, Hope BT, Nestler EJ. Regulation of ΔFosB and FosB-like proteins by electroconvulsive seizure (ECS) and cocaine treatments. Mol Pharmacol. 1995;48:880–889. [PubMed] [Google Scholar]
- 10.Dobrazanski P, Noguchi T, Kovary K, Rizzo CA, Lazo PS, Bravo R. Both products of the fosB gene, FosB and its short form, FosB/SF, are transcriptional activators in fibroblasts. Mol Cell Biol. 1991;11:5470–5478. doi: 10.1128/mcb.11.11.5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular hypothesis of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 12.Duman RS, Malberg J, Thome J. Neural plasticity to stress and antidepressant treatment. Biol Psychiatry. 1999;46:1181–1191. doi: 10.1016/s0006-3223(99)00177-8. [DOI] [PubMed] [Google Scholar]
- 13.Gilmore EC, Ohshima T, Goffinet AM, Kulkarni AB, Herrup K. Cyclin-dependent kinase 5-deficient mice demonstrate novel developmental arrest in cerebral cortex. J Neurosci. 1998;18:6370–6377. doi: 10.1523/JNEUROSCI.18-16-06370.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gould E, Tanapat P. Stress and hippocampal neurogenesis. Biol Psychiatry. 1999;46:1472–1479. doi: 10.1016/s0006-3223(99)00247-4. [DOI] [PubMed] [Google Scholar]
- 16.Hellmich MR, Pant HC, Wada E, Battey JF. Neuronal cdc2-like kinase: a cdc2-related protein kinase with predominantly neuronal expression. Proc Natl Acad Sci USA. 1992;89:10867–10871. doi: 10.1073/pnas.89.22.10867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hiroi N, Marek GJ, Brown JR, Ye H, Saudou F, Vaidya VA, Duman RS, Greenberg ME, Nestler EJ. Essential role of the fosB gene in molecular, cellular, and behavioral actions of chronic electroconvulsive seizures. J Neurosci. 1998;18:6952–6962. doi: 10.1523/JNEUROSCI.18-17-06952.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hope BT, Kelz MB, Duman RS, Nestler EJ. Chronic electroconvulsive seizure (ECS) treatment results in expression of a long-lasting AP-1 complex in brain with altered composition and characteristics. J Neurosci. 1994a;14:4318–4328. doi: 10.1523/JNEUROSCI.14-07-04318.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hope BT, Nye HE, Kelz MB, Self DW, Iadarola MJ, Nakabeppu Y, Duman RS, Nestler EJ. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994b;13:1235–1244. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 20.Julien JP, Mushynski WE. Neurofilaments in health and disease. Prog Nucleic Acid Res Mol Biol. 1998;61:1–23. doi: 10.1016/s0079-6603(08)60823-5. [DOI] [PubMed] [Google Scholar]
- 21.Kelz MB, Chen JS, Carlezon WA, Jr, Whisler K, Gilden L, Beckmann AM, Steffen C, Zhang YJ, Marotti L, Self DW, Tkatch T, Baranauskas G, Surmeier DJ, Neve RL, Duman RS, Picciotto MR, Nestler EJ. Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature. 1999;401:272–276. doi: 10.1038/45790. [DOI] [PubMed] [Google Scholar]
- 22.Lee KY, QI Z, Yu YP, Wang JH. Neuronal Cdc2-like kinases: neuron-specific forms of Cdk5. Int J Biochem Cell Biol. 1997;29:951–958. doi: 10.1016/s1357-2725(97)00048-4. [DOI] [PubMed] [Google Scholar]
- 23.Lupien SJ, de Leon M, de Santi S, Convit A, Tarshish C, Nair NP, Thakur M, McEwen BS, Hauger RL, Meaney MJ. Cortisol levels during human aging predict hippocampal atrophy and memory deficits. Nat Neurosci. 1998;1:69–73. doi: 10.1038/271. [DOI] [PubMed] [Google Scholar]
- 24.Madsen TM, Treschow A, Bengzon J, Bolwig TG, Lindvall O, Tingstrom A. Increased neurogenesis in a model of electroconvulsive therapy. Biol Psychiatry. 2000;47:1043–1049. doi: 10.1016/s0006-3223(00)00228-6. [DOI] [PubMed] [Google Scholar]
- 25.Nakabeppu Y, Nathans D. A naturally occurring truncated form of FosB that inhibits Fos/Jun transcriptional activity. Cell. 1991;64:751–759. doi: 10.1016/0092-8674(91)90504-r. [DOI] [PubMed] [Google Scholar]
- 26.Nestler EJ, Kelz MB, Chen J. ΔFosB: a molecular mediator of long-term neural and behavioral plasticity. Brain Res. 1999;835:10–17. doi: 10.1016/s0006-8993(98)01191-3. [DOI] [PubMed] [Google Scholar]
- 27.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nibuya M, Nestler EJ, Duman RS. Chronic antidepressant administration increases the expression of cAMP response element binding protein (CREB) in rat hippocampus. J Neurosci. 1996;16:2365–2372. doi: 10.1523/JNEUROSCI.16-07-02365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 30.Ohshima T, Kozak CA, Nagle JW, Pant HC, Brady RO, Kulkarni AB. Molecular cloning and chromosomal mapping of the mouse gene encoding cyclin-dependent kinase 5 regulatory subunit p35. Genomics. 1996;35:372–375. doi: 10.1006/geno.1996.0370. [DOI] [PubMed] [Google Scholar]
- 31.Paglini G, Pigino G, Kunda P, Morfini G, Maccioni R, Quiroga S, Ferreira A, Caceres A. Evidence for the participation of the neuron-specific CDK5 activator P35 during laminin-enhanced axonal growth. J Neurosci. 1998;18:9858–9869. doi: 10.1523/JNEUROSCI.18-23-09858.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 33.Sapolsky RM. Glucocorticoids and atrophy of the human hippocampus. Science. 1996;273:749–750. [Google Scholar]
- 34.Sheline YI, Wany P, Gado MH, Cernansky JG, Vannier MW. Hippocampal atrophy in recurrent major depression. Proc Natl Acad Sci USA. 1996;93:3908–3913. doi: 10.1073/pnas.93.9.3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith MA, Makino S, Kvetnansky R, Post RM. Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J Neurosci. 1995;15:1768–1777. doi: 10.1523/JNEUROSCI.15-03-01768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vaidya VA, Siuciak JA, Du F, Duman RS. Hippocampal mossy fiber sprouting induced by chronic electroconvulsive seizures. Neuroscience. 1999;89:157–166. doi: 10.1016/s0306-4522(98)00289-9. [DOI] [PubMed] [Google Scholar]
- 37.Yen J, Wisdom RM, Tratner I, Verma IM. An alternative spliced form of FosB is a negative regulator of transcriptional activation and transformation by Fos proteins. Proc Natl Acad Sci USA. 1991;88:5077–5081. doi: 10.1073/pnas.88.12.5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng M, Leung CL, Liem RK. Region-specific expression of cyclin-dependent kinase 5 (cdk5) and its activators, p35 and p39, in the developing and adult rat central nervous system. J Neurobiol. 1998;35:141–159. doi: 10.1002/(sici)1097-4695(199805)35:2<141::aid-neu2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]