

Abstract
Behavioral studies indicate that the ability to acquire long-term memories is severely impaired during sleep. It is unclear, however, why the highly synchronous discharge of neurons during sleep should not be followed by the induction of enduring plastic changes. Here we show that the expression of phosphorylated CRE-binding protein, Arc, andBDNF, three genes whose induction is often associated with synaptic plasticity, is high during waking and low during sleep. We also show that the induction of these genes during waking depends on the activity of the noradrenergic system, which is high in waking and low in sleep. These molecular results complement behavioral evidence and provide a mechanism for the impairment of long-term memory acquisition during sleep.
Keywords: Arc, BDNF, neural plasticity, memory, norepinephrine, P-CREB, sleep, sleep deprivation, waking
Behavioral studies have shown that sleep severely impairs the ability to acquire long-term memories (Tononi and Cirelli, 2000). For example, a large number of studies have failed to demonstrate the transfer of any learning from sleep to consecutive waking (Simon and Emmons, 1956). The retention of new information is possible only when the subject is awake during the presentation of the stimulus or when this presentation induces activation in the electroencephalogram (EEG) (Portnoff et al., 1966;Koukkou and Lehmann, 1968). Animal studies have shown that the impairment in the acquisition of long-term memories during sleep is not merely caused by a reduction of brain activity or by the reduced response to sensory inputs. In non-rapid eye movement (non-REM) sleep, neurons in the cortex and thalamus fire in synchronous bursts at overall rates only slightly lower than those in waking (Steriade, 1999). Furthermore, even if the reduced response to external stimuli is bypassed by high-frequency stimulation, hippocampal long-term potentiation (LTP) can be produced during waking but not during non-REM sleep (Leonard et al., 1987; Bramham and Srebro, 1989).
From an evolutionary perspective, the suppression of long-term memory acquisition during sleep would seem to serve an adaptive purpose. In general, plastic changes leading to the acquisition of new information should occur when neural activity is related to the environment, but not when the brain is active off-line (Tononi and Cirelli, 2000). However, the molecular correlates of the impairment of long-term memory acquisition during sleep are unclear. For example, despite the behavioral evidence against sleep learning, it has been suggested that synchronous activity bursts during non-REM sleep may be associated with massive influx of calcium and may constitute an ideal trigger for the induction of plasticity-related genes (Buzsáki, 1998). Thus, it is of interest to establish whether genes thought to be associated with the occurrence of plastic changes are preferentially induced during waking or during sleep and, if so, what the underlying neural mechanisms might be.
In this study, we examined the brain expression in sleep and waking of three molecular markers whose induction has been linked to the acquisition of long-term memories: phosphorylated CRE-binding protein (P-CREB) (Silva et al., 1998), Arc (Link et al., 1995; Lyford et al., 1995), and BDNF (McAllister et al., 1999). We found that P-CREB, Arc, and BDNF are expressed at high levels in the cerebral cortex and hippocampus of rats who have been awake for a few hours, whereas their levels are low in rats who have been asleep, independently of circadian time. Noradrenergic neurons, which fire during waking in response to salient events, decrease their firing rate or cease firing altogether during sleep (Aston-Jones and Bloom, 1981a,b; Rasmussen et al., 1986). We found that, if cortical noradrenergic innervation is destroyed, the expression of P-CREB, Arc, and BDNF in the cerebral cortex during waking is significantly reduced, despite the presence of a normal waking EEG. Altogether, these results provide a molecular correlate for the impairment of the acquisition of long-term memories during sleep and point to a gating function of the noradrenergic system.
MATERIALS AND METHODS
Surgery and sleep recording
Adult male WKY rats (∼300 gm) were implanted with chronic electrodes for EEG and with neck muscle electrodes for electromyography. EEG electrodes were located over frontal cortex (2 mm anterior to the bregma and 2 mm lateral to the midline) and over occipital cortex (4 mm posterior to the bregma and 3.8 mm lateral to the midline). Rats were individually housed in a recording cage (12/12 hr light/dark cycle; lights on at 10 A.M.; ∼150 lux; 25 ± 1°C) and polygraphically recorded until the percentages and distribution of sleep states were regular. Each day from 09:30–10:00 A.M. the rats were allowed to play with a new object to familiarize them with the sleep-deprivation procedure. Sleeping rats were killed during the light hours, at the end of a long period of sleep (>45 min; interrupted by periods of wakefulness of <2 min) and after spending at least 75% of the previous 3 or 8 hr asleep. Sleep-deprived rats were also killed during the light period, after being kept awake for 1–9 hr by introducing novel objects in their recording cage. Spontaneously awake rats were killed during the dark phase, after a long period of continuous wakefulness (>1.5 hr; interrupted by periods of sleep of <5 min) and after spending at least 70% of the previous 3–8 hr awake. All animals were anesthetized (<2 min) with isoflurane in their own cage and then killed by decapitation or perfused with 4% paraformaldehyde. Animal protocols followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by The Neurosciences Institute.
EEG signals were low-pass filtered (−3 dB at 30 Hz; 24 dB/octave), analog-to-digital converted (sampling rate, 128 Hz), and subjected to spectral analysis. EEG power density values were computed for successive 4 sec epochs (24 hr of recording for each animal) in the frequency range from 0.25 to 25 Hz (collapsed into 0.5 Hz bins between 0.25 and 5 Hz and into 1 Hz bins between 5.25 and 25 Hz).
Lesions of the noradrenergic system
N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine. Rats were pretreated with the selective serotonin uptake inhibitor fluoxetine (10 mg/kg, i.p.; Sigma, St. Louis, MO) to prevent possible effects on serotoninergic terminals and 30 min later injected intraperitoneally withN-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4; 50 mg/kg; n = 12; Research Biochemicals, Natick, MA). Rats fully recovered from DSP-4 injection within 24–48 hr. Rats were recorded for 1–2 weeks before and after the injection.
6-Hydroxydopamine. Under pentobarbital anesthesia (60–75 mg/kg, i.p.), rats were implanted with electrodes for EEG recording as described above. During the same surgical session, rats were infused with 6-hydroxydopamine (6-OHDA; Research Biochemicals) unilaterally into the left or right locus coeruleus by way of a 24 gauge stainless steel needle connected to a 5 μl Hamilton syringe. The stereotaxic coordinates according to the atlas of Paxinos and Watson (1986) were 0.74 mm posterior to the interaural line, 7.5 mm below the dura, and 1.2 mm lateral to the midline. Rats were pretreated with fluoxetine (10 mg/kg, i.p.), to prevent possible effects of 6-OHDA on serotoninergic terminals. The volume of 6-OHDA injected was 0.5–1 μl of a solution of 2.5 μg/μl 6-OHDA in saline containing 1 mg/ml ascorbic acid and was delivered over 5 min. The needle was left in place an additional 5 min to avoid back-diffusion. After recovery from anesthesia, rats were recorded (both right and left hemispheres) continuously for 2–3 weeks. Noradrenergic cell bodies and fibers were identified by incubating free-floating sections of the entire brain with a monoclonal antibody against tyrosine hydroxylase (anti-TH; 1:1000; Boehringer Mannheim, Indianapolis, IN). Only animals that showed an extensive (>80%) decrease in cortical TH immunostaining after DSP-4 or 6-OHDA treatment were used. Cortical TH staining was evaluated by densitometry using an Image-1/Metamorph imaging system (Universal Imaging Corporation, West Chester, PA).
Ribonuclease protection assay, in situ hybridization, and cDNA microarrays
After the rats were killed, the head was cooled in liquid nitrogen, and the whole brain was removed. The right cerebral cortex and hippocampus were dissected, whereas the rest of the brain was left intact for in situ hybridization experiments. Samples were immediately frozen on dry ice and stored at −80°C. Total RNA was isolated from the right cerebral cortex by using Trizol (Life Technologies, Gaithersburg, MD) according to the manufacturer's instructions.
Ribonuclease protection assays. Antisense RNA probes complementary to the coding region of BDNF [nucleotides 333–740 (Maisonpierre et al., 1991)] and Arc [nucleotides 777–1055 (Lyford et al., 1995)] were synthesized by runoff transcription from a linearized DNA template using the MAXIscriptin vitro transcription kit (Ambion, Austin, TX) and [α-32P]UTP (DuPont NEN, Boston, MA). Ribonuclease protection assays (RPAs) were performed using the RPAII kit (Ambion).
In situ hybridization. The protocol for in situ hybridization was as described in Pompeiano et al. (1994). Slides were scanned with a PhosphorImager (Molecular Dynamics, Sunnyvale, CA) and exposed to Biomax film (Eastman Kodak, New Haven, CT).
cDNA microarrays. Total pooled RNA from the right cerebral cortex was converted into 32P-labeled first-strand cDNA and used to hybridize cDNA microarrays (rat atlas cDNA array 1.0 and 1.2; Clontech, Palo Alto, CA) according to the manufacturer's instructions.
Immunocytochemistry and ELISA
Immunocytochemistry was performed on frontal sections (50 μm) of the entire brain using the following antibodies: polyclonal anti-rat P-CREB (1:1000; Upstate Biotechnology, Lake Placid, NY), polyclonal anti-rat Arc (1:10,000; gift of Dr. Dietmar Kuhl, Max-Planck-Institut, Heidelberg, Germany) (Guthrie et al., 2000), and polyclonal anti-rat Arc (1:1000; Transduction Laboratories, Lexington, KY). The P-CREB antibody recognizes phosphorylated but not dephosphorylated CREB, and that fact has been used to demonstrate the activity dependence of nuclear CREB phosphorylation (Ginty et al., 1993; Deisseroth et al., 1996). The two different antibodies specific for Arc gave similar results. For double-labeling experiments, antibodies against glial fibrillary acidic protein (1:1000; Sigma) and microtubule-associated protein 2 (1:250; Sigma) or parvalbumin (Parv-19; 1:1000; Sigma) were also used on a subset of sections. Cell counting was performed with the Image-1/Metamorph imaging system by observers blind to the origin of the sections. The evaluation was based on at least three sections per region per animal. The background level was set such that only unequivocally positive, darkly stained cells were counted. The nonparametric Mann–Whitney U test was used for the statistical analysis of the results.
ELISA was performed with a BDNF Emax Immunoassay System kit (Promega, Madison, WI) according to the manufacturer's instructions.
RESULTS
The level of P-CREB in the cerebral cortex is high during waking and low during sleep, irrespective of circadian time
Rats kept on a 12/12 hr light/dark cycle are asleep for most of the light period and awake for most of the dark period. We selected rats that had been asleep for the first 3 or 8 hr of the light period, rats that had been spontaneously awake for the first 3 or 8 hr of the dark period, and rats that had been sleep-deprived during the light period for 3 or 8 hr. The use of these different groups of animals allowed us to distinguish between changes in gene expression related to sleep and waking per se as opposed to circadian time or to the sleep-deprivation procedure (Fig.1A).
Fig. 1.

CREB phosphorylation in the cerebral cortex after sleep and waking. A, The experimental conditions chosen to distinguish changes associated with sleep (S), waking (W), circadian factors, and the sleep-deprivation procedure (SD). A 12 hr light/dark cycle is indicated by the horizontal bar. Three hours (3h) and 8 hr (8h) of S,SD, and W are shown.B, Anti-P-CREB staining in coronal sections of parietal cortex (layers II–VI) from a rat that slept for 3 hr (S) and a rat that was sleep-deprived for 3 hr (SD). Scale bar, 100 μm. C, Mean number (±SEM) of P-CREB-immunoreactive neurons in entorhinal (ent), parietal (par), temporal (te), and occipital (occ) cortex after 3 or 8 hr of S (n = 4/group), 3 or 8 hr of SD (n = 4/group), and 3 or 8 hr of spontaneous W (n = 3 after 3 hr; n = 2 after 8 hr). The sampled area was 500 μm wide across all cortical layers. The number of P-CREB-immunoreactive neurons was significantly higher inSD and W with respect to S(Mann–Whitney U test, *p < 0.01). No differences were found between SD andW. For each condition, there were no differences between data from the 3 and 8 hr groups, which were therefore pooled.
We first investigated whether CREB is differentially phosphorylated depending on the behavioral state of the animals. CREB phosphorylation (P-CREB) at Ser133 follows increases in the intracellular concentration of Ca2+ or cAMP or the activation of Ras-dependent protein kinases (De Cesare et al., 1999). P-CREB is induced by stimuli that produce either potentiation or depression of synaptic strength but not by high-frequency firing alone (Deisseroth et al., 1996). P-CREB and the activation of CREB-dependent transcription play a crucial role in the acquisition of different forms of long-term memory in the hippocampus and the cerebral cortex. For example, CREB-regulated transcription participates in long-term facilitation in Aplysia and in long-term memory in Drosophila. Mice lacking CREB show deficits in the late phase of LTP and in the acquisition of long-term memory. Finally, antisense oligonucleotides directed against CREB mRNA can block the conversion of short-term into long-term memory [see references in Silva et al. (1998)].
As shown in Figure 1B, P-CREB immunolabeling was low in rats killed after either 3 or 8 hr of sleep. The pattern of staining varied from one animal to another. In some rats, weakly stained nuclei were seen in all cortical layers, whereas in others they were present only in layers II-III and V or in layer V only (Fig.1B). By contrast, after 3 hr of sleep deprivation or spontaneous waking, P-CREB immunostaining was consistently high in all cortical layers (Fig. 1B) and in all cortical areas (Fig. 1C). No further increase was seen after 8 hr of waking. On the basis of neutral red and methyl green counterstaining, it appeared that >95% of all cortical cells of awake rats showed a strong, dark P-CREB staining. P-CREB staining did not colocalize with glial fibrillary acidic protein staining and was present in both parvalbumin-negative (excitatory) and parvalbumin-positive (inhibitory) neurons (data not shown).
The expression of Arc in the cerebral cortex is high during waking and low during sleep and changes as a function of the duration of waking
Arc is unique among activity-regulated genes in that, after induction, its mRNA is selectively targeted near activated postsynaptic sites, where it may play a role in activity-dependent synaptic plasticity requiring protein synthesis (Steward et al., 1998). A recent study, using antisense oligonucleotides, has shown that Arc protein expression is required for the late phase of LTP in the rat hippocampus. In addition, interfering with Arc protein expression impairs long-term memory for a spatial task, whereas task acquisition and short-term memory are not affected (Guzowski et al., 2000).
We found that Arc mRNA levels were low after either 3 or 8 hr of sleep and increased twofold after 3 hr of either spontaneous waking or sleep deprivation. This increase was maintained after 8 hr of waking (Fig. 2A,B).In situ hybridization experiments indicated thatArc induction during waking, although particularly evident in the cerebral cortex, was not limited to this brain region (Fig.2C).
Fig. 2.

Arc expression in the cerebral cortex after sleep and waking. A, cDNA microarrays showing cortical Arc mRNA levels (arrowheads) after 3 hr (3h) or 8 hr (8h) of sleep (S; n = 7), sleep deprivation (SD; n = 7), and waking (W; n = 6).B, Densitometric analysis performed by scanning the microarrays with a PhosphorImager. The y-axis values refer to signal intensity (arbitrary units). C,In situ hybridization for Arc mRNA in brain sections of a representative rat killed after 8 hr ofS and of a rat killed at the same circadian time after 8 hr of SD. Scale bar, 1.5 mm. D, Arc levels measured with immunocytochemistry in the parietal cortex of rats after 3 hr of S and 3 hr of W. Scale bar, 100 μm. E, Mean number (±SEM) of Arc-immunoreactive neurons in parietal cortex after 3 hr ofS (n = 8), SD(n = 8), and W(n = 5). The sampled area was a 500-μm-wide cortical column spanning all layers (Mann–Whitney Utest, *p < 0.01). F, Double labeling in the parietal cortex of a rat that was sleep deprived for 3 hr. Arc immunoreactivity (black cells;left) does not colocalize with parvalbumin immunoreactivity (white cells; right). Scale bar, 50 μm.
The ability of Arc mRNA to move toward dendritic sites of recent synaptic activity, as well as the subsequent local translation of Arc, has been demonstrated so far only in the hippocampus (Steward et al., 1998). In the cerebral cortex, Arc mRNA moves from nucleus to cytoplasm after seizures or after the exploration of a novel environment (Guzowski et al., 1999). However, it is unknown whether or not Arc also localizes to the dendritic arbor after a physiological stimulus. It is also unclear whether Arc is selectively expressed by specific subpopulations of cortical cells.
To determine whether Arc levels were higher in waking with respect to sleep, another group of animals was perfused after 3 hr of sleep, sleep deprivation, or spontaneous waking, and the brains were processed for immunocytochemistry. In agreement with mRNA levels, after 3 hr of sleep, Arc levels were found to be extremely low in most brain regions, including the cerebral cortex (Fig. 2D). In a few cases, however, a faint cytoplasmic staining was seen in large pyramidal cells of layer V, notably in parietal cortex, and vertically oriented, weakly stained fibers were observed in layers II-III and V. By contrast, after 3 hr of spontaneous waking or sleep deprivation, high levels of Arc were found in most regions of both isocortex and allocortex (Fig. 2D,E). Darkly stained cells were present in layers II-VI, but within each layer the intensity of the staining varied from one cell to another. In most cells the staining could be seen to extend from the cell body to the dendritic arbor, notably to the apical dendrites. We concluded that Arc was selectively expressed in neurons because Arc-positive cells never stained with an antibody for glial fibrillary acidic protein (data not shown). When sections were simultaneously reacted with antibodies against Arc and parvalbumin, no double-labeled neurons were seen in any cortical regions, suggesting that Arc is localized in glutamatergic but not in GABAergic neurons (Fig. 2F).
We then determined the time course of Arc expression by examining, in addition to the rats killed after 3 hr of waking (dark period), other animals killed after 1, 6, and 9 hr of spontaneous waking (dark period) or sleep deprivation (light period; Fig.3, n = 2 for each time point). After 1 hr of waking, strong Arc staining was present in the cytoplasm of large pyramidal cells in layer V, and vertically oriented labeled fibers were clearly present throughout layers II-V (Fig.3A). The most noticeable difference with respect to 3 hr of waking was the lower number of darkly stained cells in layers II-IV (Fig. 3A) and VI (Fig. 3B). After 6 hr of waking, the pattern of Arc expression was very similar to that observed after 3 hr of waking. After 9 hr of waking, by contrast, few darkly stained cells were present in layers II-IV (Fig. 3A) and VI (Fig.3B). Only faint cytoplasmic staining was observed in layer V. However, long, darkly stained fibers were present in most neocortical areas, notably in layers II-IV (Fig. 3A).
Fig. 3.
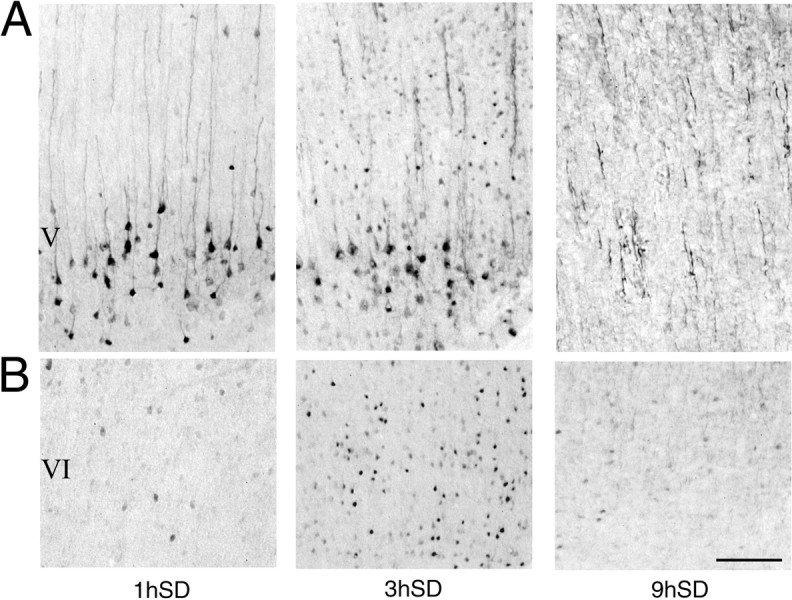
Arc expression in the parietal cortex as a function of the duration of waking. A, B, Arc levels measured with immunocytochemistry in cortical layers V (A) and VI (B) after 1 hr (1h), 3 hr (3h), and 9 hr (9h) of sleep deprivation (SD). Scale bar, 200 μm.
Taken together, these results show that spontaneous waking, but not sleep, is associated with the induction of Arc mRNA and the translation of Arc in excitatory cortical neurons. Arc immunostaining is both cytoplasmic and dendritic after short periods of waking and is mainly dendritic after longer periods.
The expression of BDNF in the cerebral cortex is high during waking and low during sleep, irrespective of circadian time
BDNF is thought to modulate activity-dependent plasticity on the basis of three main lines of evidence (Thoenen, 1995; McAllister et al., 1999). (1) BDNF expression is strongly modulated by neuronal activity, (2) BDNF potentiates both spontaneous and evoked synaptic transmission, and (3) BDNF is able to alter dendritic morphology. Mice lacking BDNF show a deficit in the induction of LTP, which can be rescued with recombinant BDNF or reexpression of the BDNF gene (Korte et al., 1996; Patterson et al., 1996). Conversely, dendrites fromBDNF-overexpressing neurons undergo massive sprouting (Wilson Horch et al., 1999).
It was shown recently that BDNF mRNA levels are higher during the dark period relative to the light period (Bova et al., 1998;Berchtold et al., 1999). However, it was unclear whether the fluctuations in BNDF expression were caused by circadian or other factors. We measured BDNF mRNA levels by using RPA. No differences inBDNF mRNA levels in the cerebral cortex were observed after 3 hr of sleep, spontaneous waking, or sleep deprivation (data not shown). However, cortical BDNF expression was considerably higher after 8 hr of waking relative to 8 hr of sleep, irrespective of whether waking occurred spontaneously during the dark period or via sleep deprivation during the light period (Fig.4A,B). Moreover,in situ hybridization experiments showed that the induction of BDNF during waking was not limited to the cerebral cortex but could be observed also in other brain regions, including the hippocampus and the thalamus (Fig. 4C). These results indicate that behavioral state (waking), rather than circadian time (dark period), is responsible for the induction of BDNF.
Fig. 4.

BDNF expression in the cerebral cortex after sleep and waking. A, RPA showing corticalBDNF mRNA levels after 8 hr of sleep (S;n = 7), sleep deprivation (SD;n = 7), and waking (W;n = 6). A β-actinantisense riboprobe was used to normalize the amount of sample RNA.Lane 1 (from left), Molecular weight markers. Lane 2, BDNF andβ-actin riboprobes hybridized with 10 μg of yeast RNA, incubated without RNase mixture. Most of the signal is the full-length BDNF (arrow) andβ-actin (arrowhead) probes. Lanes 3–8, BDNF andβ-actin probes hybridized under conditions of excess probe with 2 μg of pooled RNA. S,lanes 3 and 4; SD, lanes 5and 6; W, lanes 7 and 8. The protected fragments are 407 bp for BDNF and 250 bp for β-actin. B, Densitometric analysis performed by scanning the RPA gel with a PhosphorImager. The y-axis values refer to signal intensity (arbitrary units). Relative to S,BDNF mRNA levels were higher in both SDand W rats (t test, p= 0.021 and 0.028, respectively). C, In situ hybridization for BDNF mRNA in brain sections of representative rats killed after 8 hr of Sand 8 hr of SD. Scale bar, 1.5 mm. D, BDNF levels (picograms per milligram of protein) measured with ELISA in the cerebral cortex of rats after 8 hr of S,SD, and W. Each columnrepresents an individual rat (mean of 3 measurements ± SEM). Animals were divided into two groups on the basis of their age: 8–9 weeks (left) and 15–16 weeks (right). BDNF levels for all rats were measured simultaneously on the same assay. BDNF levels were higher inSD and W rats than in Srats (F = 21.11; p < 0.01).E, Densitometric analysis showing changes inTrkB mRNA levels, as measured by RPA, after 8 hr ofS (n = 7), SD(n = 7), and W(n = 6). The y-axis values refer to signal intensity (arbitrary units). Relative to S, cortical TrkB mRNA levels were significantly higher after 8 hr of SD (t test,p = 0.032) but not after 8 hr of W(t test, p = 0.058).
We then measured BDNF protein levels in the cerebral cortex after 8 hr of sleep, spontaneous waking, and sleep deprivation using an ELISA immunoassay. We used two batches of rats, ∼8 and ∼16 weeks old. ANOVA showed that, irrespective of age, cortical BDNF levels were higher in waking than in sleeping rats (Fig. 4D;F = 21.11; p < 0.01). There was also a significant effect of age (F = 44.01; p< 0.01) but no interaction between age and behavioral state (F = 2.53). Therefore, both mRNA and protein levels ofBDNF are increased in the cerebral cortex during waking with respect to sleep, independently of circadian factors or stress.
BDNF acts via the specific tyrosine kinase receptor TrkB. Electrical or pharmacological stimulation increases TrkB mRNA and protein levels (Nibuya et al., 1995). Moreover, BDNF and TrkB play an essential role in hippocampus-mediated learning (Minichiello et al., 1999). To determine whether TrkB expression is also induced by waking, TrkB mRNA levels were measured with RPA.TrkB expression did not change after 3 hr of sleep, sleep deprivation, or waking (data not shown). By contrast, relative to the levels with 8 hr of sleep, TrkB mRNA levels in the cerebral cortex were increased by 31% after 8 hr of sleep deprivation (t test, p = 0.032) and by 13% after 8 hr of spontaneous waking (t test, p = 0.058; Fig. 4E).
The expression of plasticity-related genes during waking depends on the noradrenergic system
Certain neuromodulatory systems with diffuse projections, such as the noradrenergic and serotoninergic systems, are plausible candidates for enabling the expression of P-CREB, Arc, andBDNF during waking, when they fire both tonically and phasically, but not during sleep, when their firing is reduced or ceases altogether (McGinty and Harper, 1976; Aston-Jones and Bloom, 1981a). It was shown in a previous study that, after lesions of the noradrenergic system, the expression of Fos, NGFI-A, and P-CREB in the cerebral cortex during waking is reduced and becomes as low as it normally is during sleep (Cirelli et al., 1996). Whether the release of norepinephrine can also regulate the expression of BNDFand Arc was not known.
The majority of noradrenergic neurons are located in the two symmetric brainstem nuclei of the locus coeruleus, which send projections to the entire brain. We used two different approaches to lesion the noradrenergic system selectively. The neurotoxin DSP-4, which can be administered systemically, destroys fibers originating in the locus coeruleus by accumulating within the noradrenergic nerve endings and depleting them of catecholamines (Fritschy and Grzanna, 1989). A different neurotoxin, 6-OHDA, also causes a rapid, long-lasting, and selective depletion of brain norepinephrine but acts with a different mechanism (Bloom, 1971). 6-OHDA does not cross the blood–brain barrier and must be directly infused into the locus coeruleus. Because noradrenergic projections to the cerebral cortex are almost exclusively ipsilateral, when injected in the locus coeruleus of one side, 6-OHDA completely destroys the noradrenergic fibers of the corresponding cerebral hemisphere but spares the other side of the brain, thereby permitting an intra-animal comparison.
We first established that sleep-waking patterns had not been modified as a result of noradrenergic lesions. In agreement with previous results (Cirelli et al., 1996), percentages of waking and sleep did not significantly differ before and 8–10 d after the injection (light hours, waking, 35 ± 1%; non-REM sleep, 51 ± 4%; REM sleep, 14 ± 2%; dark hours, waking, 65 ± 2%; non-REM sleep, 30 ± 4%; REM sleep, 5 ± 1%) and were not significantly different from those of age-matched controls. Moreover, we found that raw EEG signals and the EEG power density spectra were similar before and 8–14 d after the injection of DSP-4, as described previously (Cirelli et al., 1996). Similarly, we did not detect any difference between the raw EEG on the intact and lesioned side in rats treated with 6-OHDA.
We measured cortical Arc and BDNF mRNA levels after 8 hr of sleep deprivation in rats in which the cortical noradrenergic innervation had been destroyed 2 weeks earlier by DSP-4 and in control rats that had received a saline injection at the same time. The expression of Arc and of BDNF in the cerebral cortex was 50% lower in DSP-4-treated rats than in control rats (Fig. 5A,B).
Fig. 5.

BDNF and Arcexpression in the cerebral cortex after lesion of the noradrenergic system of the locus coeruleus. A, RPA showing corticalBDNF and Arc mRNA levels after 8 hr of sleep deprivation in control rats who received a saline injection (C; n = 4; lanes 1–3from left) and in rats treated with DSP-4 to destroy the noradrenergic innervation of the cerebral cortex (n= 4; lanes 4–6). Aβ-actin antisense riboprobe was used to normalize the amount of sample RNA. B, Densitometric analysis performed by scanning the RPA gel with a PhosphorImager. They-axis values refer to signal intensity (arbitrary units). BDNF and Arc mRNA levels were higher in C than in DSP-4 rats (t test,p = 0.030 and 0.00, respectively). C, Left, Coronal brainstem section of a representative rat in which the left locus coeruleus was destroyed by a local injection of 6-OHDA (the arrow indicates the right locus coeruleus). Tyrosine hydroxylase immunostaining, used to identify noradrenergic neurons and fibers in the locus coeruleus, is abolished on the side of the lesion, whereas it is intact on the other side. Scale bar, 1.2 mm.Right, A rostral coronal brain section immunostained against tyrosine hydroxylase from the same animal showing the almost complete depletion of noradrenergic fibers in the left cerebral cortex and hippocampus 2 weeks after the lesion. The noradrenergic innervation on the right side of the brain is intact. Scale bar, 2 mm. D, Arc immunostaining in a coronal section of parietal cortex adjacent to that shown in C. Arc expression is high on the intact side (right) after 3 hr of sleep deprivation, but it is as low as in sleep on the side where the noradrenergic innervation had been destroyed (left). Scale bar, 100 μm.
We also examined rats that had received 6-OHDA injections in the locus coeruleus of one side. As expected, after 1–2 weeks the noradrenergic neurons of the locus coeruleus of that side were completely destroyed (Fig. 5C), as was the case for the noradrenergic fibers in the ispilateral (but not contralateral) cerebral hemisphere and hippocampus (Fig. 5C). In rats that had been awake for 3 hr before being killed, Arc immunostaining was high in the cerebral cortex of the intact hemisphere, as was expected during waking, but low in the hemisphere in which noradrenergic fibers had been destroyed (Fig.5D).
Thus, in the absence of an intact noradrenergic system, waking behavior accompanied by normal low-voltage fast activity patterns is not sufficient for the induction of P-CREB (Cirelli et al., 1996),Arc, and BDNF that is normally seen in this behavioral state.
DISCUSSION
Sleep, waking, and memory acquisition
It is well established that the acquisition of long-term memories requires the activation of gene expression in the nucleus as well as protein synthesis, which may lead to structural changes in neural circuits (Squire and Kandel, 1999). Several experimental paradigms, including spatial learning tasks, high-frequency stimulation of the perforant path, experimental seizures, and kainate injections, have been used to identify genes whose induction may accompany synaptic plasticity (Nedivi et al., 1993; Lanahan and Worley, 1998; Matsuo et al., 2000). Among such genes are those coding for transcription factors (e.g., P-CREB, c-fos, and NGFI-A), chaperones and heat-shock proteins (e.g., BiP, Hsp70, and Hsc70), membrane, cytoskeleton, and vesicle-related proteins (e.g., clathrin, synaptotagmin IV, Arc, and Homer), growth factors (BDNF), and enzymes (e.g., cyclooxygenase and tissue plasminogen activator). Although the induction of most of these genes may be associated with the initiation of long-term plastic changes without being either necessary or sufficient for such changes to occur, a causal link has convincingly been established for at least P-CREB and Arc. Interfering with the induction of P-CREB and Arc is, indeed, sufficient to cause a selective deficit in long-term memory (Silva et al., 1998; Guzowski et al., 2000).
The present results demonstrate that levels of P-CREB, Arc, and BDNF in most cortical areas are much higher after waking than after sleep. The expression of Fos and NGFI-A, two transcription factors that have been variously associated with the induction of plastic changes, is also higher during waking relative to sleep (Cirelli and Tononi, 2000). Moreover, other genes that are frequently upregulated under conditions leading to plastic changes (e.g., those encoding BiP, synaptotagmin IV, calmodulin, and several neurotransmitter receptors) are also expressed at higher levels during waking than during sleep (C. Cirelli and G. Tononi, unpublished results). Thus, all available data indicate that molecular changes associated with the establishment of long-term changes take place during waking and much less or not at all during sleep.
These molecular findings go together with the behavioral and electrophysiological evidence indicating that the long-term acquisition of information is severely dampened if not blocked during sleep (Tononi and Cirelli, 2000), whereas it is facilitated by an increase in arousal levels (Cahill and McGaugh, 1996). Conversely, the present findings contradict the suggestion that sleep, notably non-REM sleep, may be a time during which the expression of plasticity-related genes may be preferentially induced via a massive influx of calcium during burst firing (Buzsáki, 1998). This suggestion is also put in question by the recent demonstration that Ca2+-induced Ca2+ release, an important link in the induction of gene expression, occurs predominantly in association with Ca2+ entry through L-type channels during the tonic firing of waking but not in association with Ca2+ entry through T-type channels during the burst firing of non-REM sleep (Budde et al., 2000).
The role of neuromodulatory systems in enabling memory acquisition in different behavioral states
During sleep, locus coeruleus neurons fire regularly at very low rates, whereas during waking they fire regularly at higher rates and emit phasic, short bursts of action potentials in response to salient events (Aston-Jones and Bloom, 1981a,b; Rasmussen et al., 1986). Norepinephrine released diffusely by these neurons over large portions of the brain enhances information transmission (Foote et al., 1991;McCormick et al., 1991; Kitchigina et al., 1997) and promotes attentive processes by increasing γ activity in the EEG [see Cape and Jones (1998) and references therein]. Studies on cortical and hippocampal slices indicate that norepinephrine can enable various forms of activity-dependent synaptic plasticity, including LTP and long-term depression (Stanton and Sarvey, 1987; Brocher et al., 1992;Kato, 1993; Huang and Kandel, 1996; Thomas et al., 1996; Katsuki et al., 1997; Kirkwood et al., 1999). Studies with cell cultures (Lin et al., 1998) and transgenic mice (Thome et al., 2000) show that norepinephrine can stimulate gene transcription by increasing the phosphorylation of CREB via activation of the cAMP cascade and protein kinase A (β-adrenoceptors) and/or via activation of calcium-dependent protein kinases (α1-adrenoceptors).
As shown here in the freely behaving animal, the expression of Arc and BDNF is high during waking, when locus coeruleus firing is high, and low during sleep, when locus coeruleus firing is reduced or absent. Most important, if the noradrenergic system is lesioned, waking behavior associated with a normal waking EEG (low-voltage fast activity) is not accompanied by the induction of these molecular markers of plasticity. Thus, the activation of the EEG can be completely dissociated from the activation of gene expression. These results are consistent with previous evidence indicating that the activity of the noradrenergic system is responsible for the differential expression of P-CREB, Fos, and NGFI-A (Cirelli et al., 1996). Thus, although several factors may contribute to the induction of such molecular markers during waking and not during sleep, including the reduced sensory input and the burst-pause mode of activity that characterize non-REM sleep, the activity of the noradrenergic system appears to play an important enabling function (cf. Flicker et al., 1981).
These observations prompt the question whether differences in the expression of plasticity-related genes similar to those observed between waking and sleep also occur between other conditions associated respectively with high locus coeruleus activity, such as orienting to novel stimuli, associative learning, and exploratory behavior, and with low locus coeruleus activity, such as grooming and consummatory behavior (Aston-Jones and Bloom, 1981a,b; Rasmussen et al., 1986). If this were the case, it would suggest that the firing of locus coeruleus neurons may play a key role in determining whether or not neural activity is accompanied by long-term neural plasticity. In view of the present study, it would be important to establish such a role under physiological conditions. To date, pharmacological studies assessing the role of norepinephrine in the acquisition of new memories have yielded mixed results, and mice lacking dopamine β-hydroxylase, which are unable to synthesize norepinephrine, are impaired in some learning paradigms but not in others [see Thomas and Palmiter (1997) and references therein]. The use of unilateral lesions of the noradrenergic system combined with experimental paradigms leading to lateralized learning may help clarify this issue.
Serotoninergic neurons also fire at higher levels during waking and decrease firing during sleep (McGinty and Harper, 1976). However, in sharp contrast to noradrenergic neurons, serotoninergic neurons are inactivated during orientation to salient stimuli and are activated instead during repetitive motor activity such as locomoting, grooming, or feeding (Jacobs and Fornal, 1999). Concurrently, information processing in various sensory pathways is inhibited (Jacobs and Fornal, 1999), and γ activity is reduced (Cape and Jones, 1998). Interestingly, lesions of the serotoninergic system do not affect the level of expression of P-CREB, Arc, or BDNF during waking and sleep (P. J. Shaw and G. Tononi, unpublished results). The histaminergic system also discharges at higher levels during waking than during sleep, but no data are available concerning the effects of histamine on the expression of molecular markers of plasticity.
Finally, acetylcholine release in the cerebral cortex and the activity of cholinergic neurons are higher during waking than during non-REM sleep, but they are also often higher in REM sleep than in non-REM sleep (Jasper and Tessier, 1971; Steriade at al., 1990). Although acetylcholine may, under certain conditions, play a gating role similar to that demonstrated here for norepinephrine, the short duration of REM sleep episodes in the rat (∼2 min) may be insufficient for triggering the transcription and translation of BDNF andArc. On the other hand, it should be emphasized that, if synaptic activation is strong enough or if other neuromodulatory systems are strongly activated for sufficient periods of time, the enabling function of the noradrenergic system may be bypassed. This may be the case, for example, with tetanic stimulation or after the administration of dopaminergic or cholinergic agonists (Zafra et al., 1991; Fosnaugh et al., 1995). Interestingly, in animals that experience a sustained rebound of REM sleep (3–4 hr) as a result of prolonged sleep deprivation, c-fos expression is induced in many limbic areas that receive strong cholinergic innervation (Cirelli and Tononi, unpublished results). Whether the expression of P-CREB,Arc, and BDNF is also increased remains to be determined.
Conclusions
The present results demonstrate that several molecular markers of neural plasticity, such as P-CREB, Arc, and BDNF, are induced during waking but not during sleep. They also indicate that the inactivity of the noradrenergic system is a key factor that prevents the induction of these genes during sleep, leading to an impairment of long-term memory acquisition. Although these results provide a molecular basis for the inability to acquire long-term memories during sleep, they do not exclude a contribution of sleep to other aspects of learning and memory, such as memory consolidation. Other molecular mechanisms, such as dendritic or somatic protein synthesis, receptor insertion and clustering, and synaptic capture, may occur preferentially during sleep, although at present no direct evidence is available. A better understanding of the molecular mechanisms of memory consolidation should provide an opportunity to determine whether sleep may play a specific and positive role in some aspect of memory.
Footnotes
The Neurosciences Institute is supported by the Neurosciences Research Foundation, which receives major support from Novartis. We thank G. A. Davis, M. C. Gallina, and D. F. Robinson for their expert contribution and Dr. D. Kuhl for the kind gift of the anti-Arc antibody.
Correspondence should be addressed to Dr. Chiara Cirelli, The Neurosciences Institute, 10640 John J. Hopkins Drive, San Diego, CA 92121. E-mail: cirelli@nsi.edu.
REFERENCES
- 1.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep–waking cycle. J Neurosci. 1981a;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aston-Jones G, Bloom FE. Norepinephrine-containing locus coeruleus neurons in behaving rats exhibit pronounced responses to non-noxious environmental stimuli. J Neurosci. 1981b;1:887–900. doi: 10.1523/JNEUROSCI.01-08-00887.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berchtold NC, Oliff HS, Isackson P, Cotman CW. Hippocampal BDNF mRNA shows a diurnal regulation, primarily in the exon III transcript. Mol Brain Res. 1999;71:11–22. doi: 10.1016/s0169-328x(99)00137-0. [DOI] [PubMed] [Google Scholar]
- 4.Bloom FE. Fine structural changes in rat brain after intracisternal injection of 6-hydroxydopamine. In: Malmfors T, Thoenen H, editors. 6-Hydroxydopamine and catecholamine neurons. North-Holland; Amsterdam: 1971. pp. 135–150. [Google Scholar]
- 5.Bova R, Micheli MR, Qualadrucci P, Grassi-Zucconi G. BDNF and trkB mRNAs oscillate in rat brain during the light-dark cycle. Mol Brain Res. 1998;57:321–324. doi: 10.1016/s0169-328x(98)00092-8. [DOI] [PubMed] [Google Scholar]
- 6.Bramham CR, Srebro B. Synaptic plasticity in the hippocampus is modulated by behavioral state. Brain Res. 1989;493:74–86. doi: 10.1016/0006-8993(89)91001-9. [DOI] [PubMed] [Google Scholar]
- 7.Brocher S, Artola A, Singer W. Agonists of cholinergic and noradrenergic receptors facilitate synergistically the induction of long-term potentiation in slices of rat visual cortex. Brain Res. 1992;573:27–36. doi: 10.1016/0006-8993(92)90110-u. [DOI] [PubMed] [Google Scholar]
- 8.Budde T, Sieg F, Braunewell K-H, Gundelfinger ED, Pape H-C. Ca2+-induced Ca2+ release supports the relay mode of activity in thalamocortical cells. Neuron. 2000;26:483–492. doi: 10.1016/s0896-6273(00)81180-0. [DOI] [PubMed] [Google Scholar]
- 9.Buzsáki G. Memory consolidation during sleep: a neurophysiological perspective. J Sleep Res. 1998;7[Suppl 1]:17–23. doi: 10.1046/j.1365-2869.7.s1.3.x. [DOI] [PubMed] [Google Scholar]
- 10.Cahill L, McGaugh JL. Modulation of memory storage. Curr Opin Neurobiol. 1996;6:237–242. doi: 10.1016/s0959-4388(96)80078-x. [DOI] [PubMed] [Google Scholar]
- 11.Cape EG, Jones BE. Differential modulation of high-frequency γ-electroencephalogram activity and sleep–wake state by noradrenaline and serotonin microinjections into the region of cholinergic basalis neurons. J Neurosci. 1998;18:2653–2666. doi: 10.1523/JNEUROSCI.18-07-02653.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cirelli C, Tononi G. On the functional significance of c-fos induction during the sleep/waking cycle. Sleep. 2000;23:453–469. [PubMed] [Google Scholar]
- 13.Cirelli C, Pompeiano M, Tononi G. Neuronal gene expression in the waking state: a role for the locus coeruleus. Science. 1996;274:1211–1215. doi: 10.1126/science.274.5290.1211. [DOI] [PubMed] [Google Scholar]
- 14.De Cesare D, Fimia GM, Sassone-Corsi P. Signaling routes to CREM and CREB: plasticity in transcriptional activation. Trends Biochem Sci. 1999;24:281–285. doi: 10.1016/s0968-0004(99)01414-0. [DOI] [PubMed] [Google Scholar]
- 15.Deisseroth K, Bito H, Tsien RW. Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron. 1996;16:89–101. doi: 10.1016/s0896-6273(00)80026-4. [DOI] [PubMed] [Google Scholar]
- 16.Flicker C, McCarley RW, Hobson JA. Aminergic neurons: state control and plasticity in three model systems. Cell Mol Neurobiol. 1981;1:123–166. doi: 10.1007/BF00710716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Foote SL, Berridge CW, Adams LM, Pineda JA. Electrophysiological evidence for the involvement of the locus coeruleus in alerting, orienting, and attending. In: Barnes CD, Pompeiano O, editors. Neurobiology of the locus coeruleus. Elsevier; Amsterdam: 1991. pp. 521–532. [DOI] [PubMed] [Google Scholar]
- 18.Fosnaugh JS, Bhat RV, Yamagata K, Worley PF, Baraban JM. Activation of arc, a putative “effector” immediate early gene, by cocaine in rat brain. J Neurochem. 1995;64:2377–2380. doi: 10.1046/j.1471-4159.1995.64052377.x. [DOI] [PubMed] [Google Scholar]
- 19.Fritschy J-M, Grzanna R. Immunohistochemical analysis of the neurotoxic effects of DSP-4 identifies two populations of noradrenergic axon terminals. Neuroscience. 1989;30:181–197. doi: 10.1016/0306-4522(89)90364-3. [DOI] [PubMed] [Google Scholar]
- 20.Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME. Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science. 1993;260:238–241. doi: 10.1126/science.8097062. [DOI] [PubMed] [Google Scholar]
- 21.Guthrie K, Rayhanabad J, Kuhl D, Gall C. Odors regulate Arc expression in neuronal ensembles engaged in odor processing. NeuroReport. 2000;11:1809–1813. doi: 10.1097/00001756-200006260-00003. [DOI] [PubMed] [Google Scholar]
- 22.Guzowski JF, McNaughton BL, Barnes CA, Worley PF. Environment-specific expression of the immediate early gene Arc in hippocampal neuronal ensembles. Nat Neurosci. 1999;2:1120–1124. doi: 10.1038/16046. [DOI] [PubMed] [Google Scholar]
- 23.Guzowski JF, Lyford GL, Stevenson GD, Houston FP, McGaugh JL, Worley PF, Barnes CA. Inhibition of activity-dependent Arc protein expression in the rat hippocampus impairs the maintenance of long-term potentiation and the consolidation of long-term memory. J Neurosci. 2000;20:3993–4001. doi: 10.1523/JNEUROSCI.20-11-03993.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang YY, Kandel ER. Modulation of both the early and the late phase of mossy fiber LTP by the activation of β-adrenergic receptors. Neuron. 1996;16:611–617. doi: 10.1016/s0896-6273(00)80080-x. [DOI] [PubMed] [Google Scholar]
- 25.Jacobs BL, Fornal CA. Activity of serotonergic neurons in behaving animals. Neuropsychopharmacology. 1999;21:9S–15S. doi: 10.1016/S0893-133X(99)00012-3. [DOI] [PubMed] [Google Scholar]
- 26.Jasper HH, Tessier J. Acetylcholine liberation from cerebral cortex during paradoxical (REM) sleep. Science. 1971;172:601–602. doi: 10.1126/science.172.3983.601. [DOI] [PubMed] [Google Scholar]
- 27.Kato N. Mechanisms of beta-adrenergic facilitation in rat visual cortex. NeuroReport. 1993;4:1087–1090. [PubMed] [Google Scholar]
- 28.Katsuki H, Izumu Y, Zorumski CF. Noradrenergic regulation of synaptic plasticity in the hippocampal CA1 region. J Neurophysiol. 1997;77:3013–3020. doi: 10.1152/jn.1997.77.6.3013. [DOI] [PubMed] [Google Scholar]
- 29.Kirkwood A, Rozas C, Kirkwood J, Perez F, Bear MF. Modulation of long-term synaptic depression in visual cortex by acetylcholine and norepinephrine. J Neurosci. 1999;19:1599–1609. doi: 10.1523/JNEUROSCI.19-05-01599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitchigina V, Vankov A, Harley C, Sara SJ. Novelty-elicited, noradrenaline-dependent enhancement of excitability in the dentate gyrus. Eur J Neurosci. 1997;9:41–47. doi: 10.1111/j.1460-9568.1997.tb01351.x. [DOI] [PubMed] [Google Scholar]
- 31.Korte M, Griesbeck O, Gravel C, Carroll P, Staiger V, Thoenen H, Bonhoeffer T. Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc Natl Acad Sci USA. 1996;93:12547–12552. doi: 10.1073/pnas.93.22.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koukkou M, Lehmann D. EEG and memory storage in sleep experiments with humans. Electroencephalogr Clin Neurophysiol. 1968;25:455–462. doi: 10.1016/0013-4694(68)90155-7. [DOI] [PubMed] [Google Scholar]
- 33.Lanahan A, Worley P. Immediate-early genes and synaptic function. Neurobiol Learn Mem. 1998;70:37–43. doi: 10.1006/nlme.1998.3836. [DOI] [PubMed] [Google Scholar]
- 34.Leonard BJ, McNaughton BL, Barnes CA. Suppression of hippocampal synaptic plasticity during slow-wave sleep. Brain Res. 1987;425:174–177. doi: 10.1016/0006-8993(87)90496-3. [DOI] [PubMed] [Google Scholar]
- 35.Lin RZ, Chen J, Hu ZW, Hoffman BB. Phosphorylation of the cAMP response element binding protein and activation of transcription by alpha1 adrenergic receptors. J Biol Chem. 1998;273:30033–30038. doi: 10.1074/jbc.273.45.30033. [DOI] [PubMed] [Google Scholar]
- 36.Link W, Konietzko U, Kauselmann G, Krug M, Schwanke B, Frey U, Kuhl D. Somatodendritic expression of an immediate early gene is regulated by synaptic activity. Proc Natl Acad Sci USA. 1995;92:5734–5738. doi: 10.1073/pnas.92.12.5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyford GL, Yamagata K, Kaufmann WE, Barnes CA, Sanders LK, Copeland NG, Gilbert DJ, Jenkins NA, Lanahan AA, Worley PF. Arc, a growth factor and activity-regulated gene, encodes a novel cytoskeleton-associated protein that is enriched in neuronal dendrites. Neuron. 1995;14:433–445. doi: 10.1016/0896-6273(95)90299-6. [DOI] [PubMed] [Google Scholar]
- 38.Maisonpierre PC, Le Beau MM, Espinosa R, Ip NY, Belluscio L, de la Monte SM, Squinto S, Furth ME, Yancopoulos GD. Human and rat brain-derived neurotrophic factor and neurotrophin-3: gene structures, distributions, and chromosomal localizations. Genomics. 1991;10:558–568. doi: 10.1016/0888-7543(91)90436-i. [DOI] [PubMed] [Google Scholar]
- 39.Matsuo R, Murayama A, Saitoh Y, Sakaki Y, Inokuchi K. Identification and cataloging of genes induced by long-lasting long-term potentiation in awake rats. J Neurochem. 2000;74:2239–2249. doi: 10.1046/j.1471-4159.2000.0742239.x. [DOI] [PubMed] [Google Scholar]
- 40.McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annu Rev Neurosci. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- 41.McCormick DA, Pape H-C, Williamson A. Actions of norepinephrine in the cerebral cortex and thalamus: implications for function of the central noradrenergic system. In: Barnes CD, Pompeiano O, editors. Neurobiology of the locus coeruleus. Elsevier; Amsterdam: 1991. pp. 293–305. [DOI] [PubMed] [Google Scholar]
- 42.McGinty DJ, Harper RM. Dorsal raphe neurons: depression of firing during sleep in cats. Brain Res. 1976;101:569–575. doi: 10.1016/0006-8993(76)90480-7. [DOI] [PubMed] [Google Scholar]
- 43.Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp H-P, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 44.Nedivi E, Hevroni D, Naot D, Israeli D, Citri Y. Numerous candidate plasticity-related genes revealed by differential cDNA cloning. Nature. 1993;363:718–721. doi: 10.1038/363718a0. [DOI] [PubMed] [Google Scholar]
- 45.Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J Neurosci. 1995;15:7539–7547. doi: 10.1523/JNEUROSCI.15-11-07539.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 47.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; Sydney: 1986. [DOI] [PubMed] [Google Scholar]
- 48.Pompeiano M, Cirelli C, Tononi G. Immediate-early genes in spontaneous wakefulness and sleep: expression of c-fos and NGFI-A mRNA and protein. J Sleep Res. 1994;3:80–96. doi: 10.1111/j.1365-2869.1994.tb00111.x. [DOI] [PubMed] [Google Scholar]
- 49.Portnoff G, Baekeland F, Goodenough DR, Karacan I, Shapiro A. Retention of verbal materials perceived immediately prior to onset of non-REM sleep. Percept Mot Skills. 1966;22:751–758. doi: 10.2466/pms.1966.22.3.751. [DOI] [PubMed] [Google Scholar]
- 50.Rasmussen K, Morilak DA, Jacobs BL. Single unit activity of locus coeruleus neurons in the freely moving cat. I. During naturalistic behavior and in response to simple and complex stimuli. Brain Res. 1986;371:324–334. doi: 10.1016/0006-8993(86)90370-7. [DOI] [PubMed] [Google Scholar]
- 51.Silva AJ, Kogan JH, Frankland PW, Kida S. CREB and memory. Annu Rev Neurosci. 1998;21:127–148. doi: 10.1146/annurev.neuro.21.1.127. [DOI] [PubMed] [Google Scholar]
- 52.Simon C, Emmons W. Responses to material presented during various levels of sleep. J Exp Psychol. 1956;51:89–97. doi: 10.1037/h0043637. [DOI] [PubMed] [Google Scholar]
- 53.Squire LR, Kandel ER. Memory: from mind to molecules. Scientific American; New York: 1999. [Google Scholar]
- 54.Stanton PK, Sarvey JM. Norepinephrine regulates long-term potentiation of both the population spike and dendritic EPSP in hippocampal dentate gyrus. Brain Res Bull. 1987;18:115–119. doi: 10.1016/0361-9230(87)90039-6. [DOI] [PubMed] [Google Scholar]
- 55.Steriade M. Coherent oscillations and short-term plasticity in corticothalamic networks. Trends Neurosci. 1999;22:337–345. doi: 10.1016/s0166-2236(99)01407-1. [DOI] [PubMed] [Google Scholar]
- 56.Steriade M, Datta S, Paré D, Oakson G, Dossi C. Neuronal activities in brain-stem cholinergic nuclei related to tonic activation processes in thalamocortical systems. J Neurosci. 1990;10:2541–2559. doi: 10.1523/JNEUROSCI.10-08-02541.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Steward O, Wallace CS, Lyford GL, Worley PF. Synaptic activation causes the mRNA for the IEG Arc to localize selectively near activated postsynaptic sites on dendrites. Neuron. 1998;21:741–751. doi: 10.1016/s0896-6273(00)80591-7. [DOI] [PubMed] [Google Scholar]
- 58.Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- 59.Thomas MJ, Moody TD, Makhinson M, O'Dell TJ. Activity-dependent β-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- 60.Thomas SA, Palmiter RD. Disruption of the dopamine beta-hydroxylase gene in mice suggests roles for norepinephrine in motor function, learning, and memory. Behav Neurosci. 1997;111:579–589. doi: 10.1037//0735-7044.111.3.579. [DOI] [PubMed] [Google Scholar]
- 61.Thome J, Sakai N, Shin K-H, Steffen C, Zhang Y-J, Impey S, Storm D, Duman RS. cAMP response element-mediated gene transcription is upregulated by chronic antidepressant treatment. J Neurosci. 2000;20:4030–4036. doi: 10.1523/JNEUROSCI.20-11-04030.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tononi G, Cirelli C (2000) Some consideration on sleep and neural plasticity. Arch Ital Biol, in press. [PubMed]
- 63.Wilson Horch H, Krüttgen A, Portbury SD, Katz LC. Destabilization of cortical dendrites and spines by BDNF. Neuron. 1999;23:353–364. doi: 10.1016/s0896-6273(00)80785-0. [DOI] [PubMed] [Google Scholar]
- 64.Zafra F, Castren E, Thoenen H, Lindholm D. Interplay between glutamate and γ-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc Natl Acad Sci USA. 1991;88:10037–10041. doi: 10.1073/pnas.88.22.10037. [DOI] [PMC free article] [PubMed] [Google Scholar]