

Abstract
Acoustic overstimulation is one of the major causes of hearing loss. Glutamate is the most likely candidate neurotransmitter for afferent synapses in the peripheral auditory system, so it was proposed that glutamate excitotoxicity may be involved in noise trauma. However, there has been no direct evidence that noise trauma is caused by excessive release of glutamate from the inner hair cells (IHCs) during sound exposure because studies have been hampered by powerful glutamate uptake systems in the cochlea. GLAST is a glutamate transporter highly expressed in the cochlea. Here we show that after acoustic overstimulation, GLAST-deficient mice show increased accumulation of glutamate in perilymphs, resulting in exacerbation of hearing loss. These results suggest that GLAST plays an important role in keeping the concentration of glutamate in the perilymph at a nontoxic level during acoustic overstimulation. These findings also provide further support for the hypothesis that IHCs use glutamate as a neurotransmitter.
Keywords: glutamate transporter, excitotoxicity, noise trauma, inner hair cell, knock-out mouse, supporting cell
Acoustic trauma is one of the major causes of hearing loss. Exposure to an intense, loud noise causes afferent dendrite swelling below inner hair cells (IHCs) as well as mechanical damage to outer hair cells (OHCs) (Robertson, 1983; Saunders et al., 1985). Because glutamate is the most likely candidate neurotransmitter for IHC–auditory nerve synapses (Ottersen et al., 1998) and the noise-induced dendrite damage is very similar to that seen after exposure of the cochlea to glutamate receptor agonists (Puel et al., 1991; Puel, 1995), it has been suggested that noise-induced hearing loss may be caused, in part, by glutamate excitotoxicity. However, no previous studies directly examined the excess release of glutamate during sound exposure and its correlation with the functional impairment because powerful glutamate uptake systems rapidly remove synaptically released glutamate from the extracellular space. To date, five subtypes of Na+-dependent glutamate transporters, abbreviated as GLT-1, GLAST, EAAC1, EAAT4, and EAAT5, have been cloned (Arriza et al., 1997; Kanai, 1997; Tanaka, 2000). Previous studies demonstrate that GLAST is present in the rat and guinea pig cochlea (Li et al., 1994; Furness and Lehre, 1997). Thus, GLAST-deficient mice provide an in vivo model for studying an excitotoxic mechanism producing noise-induced hearing loss (Harada et al., 1998; Watase et al., 1998; Watanabe et al., 1999). We now report that after noise overstimulation, GLAST-deficient mice exhibit increased accumulation of glutamate in perilymphs, resulting in exacerbation of hearing loss.
MATERIALS AND METHODS
Immunohistochemistry. The animals were deeply anesthetized with sodium pentobarbital (50 mg/kg, i.p.). Before systemic transcardial perfusion with 4% paraformaldehyde in 0.1m phosphate buffer, pH 7.25, the same fixative was injected through the tympanic membrane. The temporal bones were removed immediately after perfusion and post-fixed with 4% paraformaldehyde at 4°C for 3 hr. They were then incubated in 7% EDTA (in H2O) for decalcification for 2 weeks. Serial cryostat sections (15 μm thick) were cut and placed on silane-coated slides. Immunocytochemical staining of GLAST was performed by an indirect immunofluorescence method. The specimens were incubated with the following solutions: (1) rabbit polyclonal anti-GLAST antibody (4 μg/ml, kindly provided by Dr. N. C. Danbolt, University of Oslo, Norway, in PBS with 0.3% Triton X-100), overnight at 4°C; (2) biotinylated anti-rabbit IgG (Vector Laboratories, Burlingame, CA), overnight at 4°C; and (3) streptavidin-fluorescein isothiocyanate (FITC) (Amersham, Arlington Heights, IL), 2 hr at room temperature. For double staining of GLAST and glutamine synthetase, the specimens were incubated with the following solutions: (1) a mixture of rabbit polyclonal anti-GLAST antibody (A522, raised against a synthetic peptide corresponding to amino acids 522–541) (Lehre et al., 1995), and mouse monoclonal anti-glutamine synthetase (MAB302; Chemicon, Temecula, CA) diluted with 0.3% Triton X-100, overnight at 4°C; (2) a mixture of the secondary antibodies, biotinylated anti-rabbit IgG (Amersham) and FITC-conjugated goat anti-mouse IgG (Amersham), overnight at 4°C; and (3) streptavidin Texas Red (Amersham), 2 hr at room temperature. The antibody dilutions were 4 μg/ml (A522), 1:1000 (MAB302), or 1:100 (secondary antibodies). The specimens were examined with a confocal scanning microscope (Olympus Optical, Tokyo, Japan) equipped with the appropriate filter combinations. Specificity of anti-GLAST antibody has been verified previously (Lehre et al., 1995). Four adults (12–16 weeks old) of each genotype were examined.
Microdialysis. Mice were anesthetized with a mixture of nitrous oxide/oxygen (1:1) gas and 3% halothane. During the experiment, body temperature was kept at 36–37°C by a heating lamp. In the right ear, the otic bulla was exposed by a retroauricular incision. A small hole, 0.3 mm in diameter, was drilled into the basal turn of the cochlea, and a probe with a dialysis membrane (Eicom, Kyoto, Japan) 1 mm in length and 0.22 mm in diameter was positioned in the tympanic scala through the hole using a micromanipulator. The probe was sealed and fixed with dental cement. The microdialysis system was perfused with Ringer's solution at a flow rate of 0.6 μl/min using a microinfusion pump (BRC, Nagoya, Japan). The samples of the dialysate, 3 μl each, were sequentially collected every 5 min in sampling tubes in an ice bath: 4 samples before noise exposure, 6 samples during noise exposure, and 24 samples after noise exposure. The concentration of glutamate was measured by an enzymatic cycling procedure, as reported by Mitani et al. (1994). Eight adults (12–16 weeks old) of each genotype were examined.
Auditory brainstem responses. Auditory brainstem responses (ABRs) were obtained from mice anesthetized with a mixture of nitrous oxide/oxygen (1:1) gas and 3% halothane. Responses were differentially recorded between subcutaneous stainless steel electrodes at the vertex (active) and mastoid (reference), and the lower back served as ground. Testing was performed in a sound-attenuated box. The ABRs, in response to the sound of clicks, were recorded using a signal processor (NEC Synax 1200, Tokyo, Japan). The average of 300 responses was taken. Threshold is defined as the intensity level at which an ABR wave I with an amplitude of 0.05 μV was seen in two averaged runs. Eight adults (12–16 weeks old) of each genotype were examined.
Sound overexposure. The acoustically traumatizing stimulus [4 kHz continuous pure tone at 105 dB sound pressure level (SPL) for 30 min] was produced by a National VP-7421A signal generator (Matsushita Electric, Tokyo, Japan) and amplified by a Kenwood KA-990-D amplifier (Kenwood, Tokyo, Japan). Intense sound was delivered from a Fostex FT96H loudspeaker (Fostex) in front of the head of the anesthetized animal in a sound exposure box (40 × 15 × 15 cm) under anesthesia. One animal was exposed at a time.
Histological analysis. The cochleae were perfused (10 min)in situ with 2.5% glutaraldehyde in 0.1m phosphate buffer. The animals were then killed, and the cochleae were quickly removed and immersed in the same fixative for 8 hr at 4°C. They were post-fixed in 2% OsO4 (2 hr), dehydrated in ethanol, and embedded in epoxy resin. Ultrathin sections were made from the basal turn and then counterstained with uranyl acetate and lead citrate before being observed under a transmission electron microscope (Hitachi H800).
We quantified the loud sound-induced damage to fibers by counting the number of swollen dendritic terminals in contact with the IHCs at presound exposure and at 5 and 120 min after sound exposure. The criteria for swollen dendritic terminals were as follows: (1) the appearance of clear cytosol and the presence of a rounded, regular membrane. (2) The contrast of the intracellular content was weaker than that of IHCs. For the size, we measured the length along the long axis of dendritic terminals in four control cochleae with no treatment. The three largest oval-shaped dendritic terminals were then chosen from each section. The average length along the long axis of these dendritic terminals was 1.2 μm. (3) In cochleae after sound exposure, dendritic terminals with a length of <1.2 μm were not counted as swollen. Four adults (12–16 weeks old) of each genotype were examined.
RESULTS
Localization of a glutamate transporter, GLAST, in the mouse cochlea
GLAST was found in the region of the IHCs, forming flask-shaped outlines with the shape of the IHC body and around the spiral ganglion cells (Fig. 1A). There was no significant staining in the region of the OHCs, in agreement with previous results (Furness and Lehre, 1997). GLAST was also found in the fibrocytes in the limbus and the spiral ligament (Fig.1A). In GLAST-deficient mice, however, there is no significant immunoreactivity, despite morphologically normal appearance (Fig. 1B). From the immunocytochemistry result in Figure 1A, it is difficult to determine the cellular origin of the GLAST immunoreactivity. To address this question, we performed a double-labeling experiment using GLAST antibody together with glutamine synthetase (GS) antibody that has been shown to be a specific marker for the supporting cells (Takumi et al., 1997; Ottersen et al., 1998). Superimposed images of GLAST and GS show that green areas (indicating GS) were found evenly in the supporting cells, whereas orange areas (indicative of colocalization) and red areas (indicating GLAST) occurred along the margins of these cells (Fig.1C,D), indicating that GLAST is localized in the supporting cells around the IHCs.
Fig. 1.

Images by confocal laser scanning microscopy.A, B, Immunofluorescence for GLAST in the cochlea in wild-type (A) and GLAST-deficient (B) mice. The interference image is superimposed on the immunofluorescent image. C, Double immunofluorescence for GLAST (red, rabbit antibody) and glutamine synthetase (green, mouse) in the cochlea in wild-type mice. Green areas (indicating single labeling for glutamine synthetase) occur in the central parts of the supporting cells, whereas yellow areas (indicative of colocalization) occur along the margins of these cells.D, A bright field of C.IHC, Inner hair cell; OHC, outer hair cell; LIM, limbus; SG, spiral ganglion;TM, tectorial membrane; BM, basilar membrane. Scale bars: A, B, 100 μm;C, D, 50 μm.
Monitoring of glutamate concentration in noise-stimulated perilymph
To examine the role of GLAST in glutamate clearance, we measured the basal concentration and time course of free glutamate in perilymphs during and after noise exposure using a microdialysis technique (Hakuba et al., 1997). Before noise exposure, the basal glutamate level in the mutant mice [0.90 ± 0.06 pmol/μl (mean ± SEM);n = 8] was significantly elevated in comparison with the wild-type mice (0.58 ± 0.07 pmol/μl; n = 8) (p < 0.01, t test) (Fig.2). Moreover, a significant increase in glutamate level was observed during and after exposure in the mutant mice (p < 0.05), whereas no significant increase in glutamate level was observed in the wild-type mice (Fig.2). These results indicate that GLAST is an important determinant of glutamate clearance in the cochlea and that glutamate is the most likely candidate neurotransmitter for the afferent hair cell synapses.
Fig. 2.

Changes in glutamate concentration in the perilymph before, during, and after 105 dB sound exposure in wild-type (n = 8, open circles) and GLAST-deficient mice (n = 8, closed circles). Data are mean ± SEM (bars) values.
The effects of noise trauma in GLAST-deficient mice
To test the excitotoxic nature of acoustic trauma, we compared functional and structural damage after exposure to a 4 kHz pure tone at a 105 dB SPL for 30 min in wild-type and mutant mice. Functional damage was estimated using ABRs. The ABR thresholds of mutant mice (33.75 ± 2.46 dB SPL; n = 8) were slightly but significantly higher than thresholds of wild-type mice (23.75 ± 1.57 dB SPL; n = 8) (p < 0.005,t test) before exposure. Amplitudes of the five principal waves of mutant mice were decreased, although no change in latencies of all five waves could be detected (data not shown). Thus, although GLAST is expressed in astrocytes of brainstem structures (the cochlear nucleus and inferior colliculus), it is likely that the ABR threshold elevation of mutant mice is of peripheral origin. After sound exposure, the homozygous mutants displayed a significant rise in ABR thresholds and a significant delay in the recovery of the ABR thresholds (Fig.3). Histologically, we examined sections from the basal turn of the cochlea, corresponding to a frequency of 4 kHz (Greenwood, 1990). No excitotoxic damage (i.e., the afferent dendrite swelling below IHCs) was observed in mutant mice at presound exposure (Fig. 4B). Noise exposure acutely produced afferent nerve terminal swelling at the IHC in both wild-type and mutant mice (Fig. 4, Table1). Two hours after exposure, the morphological appearance of the IHC and dendrite region had returned to almost normal in wild-type mice (Fig. 4E), whereas the swelling was still observed in mutant mice (Fig.4F). Figure 4 shows cross-sections of IHCs taken from typical animals from the two groups (wild-type and mutant). All eight cochleae from two groups of four mice each used for histological analysis showed almost similar results. These results indicate that GLAST-deficient mice show a significant increase in noise-induced hearing loss because of the exacerbation of dendrite damage, confirming the excitotoxic nature of noise-induced hearing loss.
Fig. 3.

Shifts of the ABR thresholds after 105 dB sound exposure in wild-type (n = 8, open circles) and GLAST-deficient (n = 8,closed circles) mice. Data are mean ± SEM (bars) values. (* p < 0.005; **p < 0.05; data were analyzed by ttest).
Fig. 4.
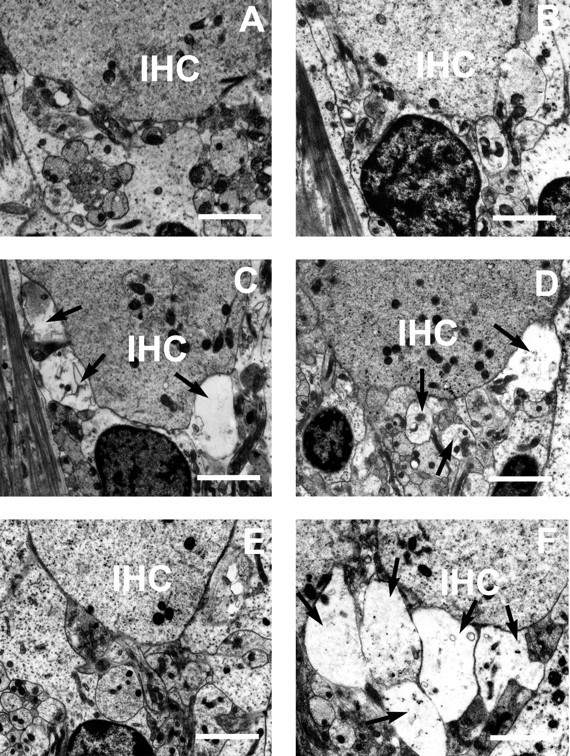
Effect of 105 dB sound exposure on the auditory dendrites below the IHCs in wild-type (A,C, E) and GLAST-deficient (B, D, F) mice. Typical transmission electron micrographs showing IHC region, presound exposure (A, B), 5 min (C, D), and 120 min (E, F) after sound exposure.Arrows indicate massive swelling of structures in dendrite region. All micrographs are taken from the basal turn of the cochlea. Scale bars, 2 μm.
Table 1.
Means and SEs of the number of swollen dendritic terminals before and after sound exposure in wild-type and GLAST-deficient mice
| Time | Wild type | Mutant | Wild vs mutant |
|---|---|---|---|
| Pre (n= 8) | 0 ± 0 | 0 ± 0 | NS |
| 5 min (n = 8) | 2.50 ± 0.40 | 2.88 ± 0.33 | NS |
| 120 min (n = 8) | 0.88 ± 0.30 | 2.75 ± 0.37 | p < 0.05 |
Student's t test was used to estimate the significance of the difference in the numbers of swollen dendritic profiles between the wild-type and mutant mice. n, Number of cochleae observed; NS, not significant. All data are expressed as mean ± SEM.
DISCUSSION
Our results suggest that GLAST plays a neuroprotective role against noise-induced hearing loss. Glutamate neurotoxicity is implicated in a number of pathological states, such as ischemia, aminoglycoside antibiotics-induced hearing loss, neural presbycusis, and certain forms of peripheral tinnitus (Puel, 1995; Basile et al., 1996). Pujol et al. (1993) reported a protective effect of piribedil, a D2 dopamine receptor agonist, against the radial dendritic swelling induced by transient ischemia (Pujol et al., 1993). Accordingly, bromocriptine may serve as a prototype for the development of new therapeutic agents for these pathological conditions because bromocriptine is a glutamate transporter activator as well as a D2 dopamine receptor agonist (Yamashita et al., 1995, 1998).
GLAST deficiency led to a slightly increased level of the basal glutamate in perilymph before noise exposure and the slight (∼10 dB) ABR threshold elevations in untreated mutant mice. However, no obvious abnormality at hair cell or dendrite levels could be observed before noise exposure in mutant mice. Previous studies show that, when hearing losses after noise trauma are not >40 dB, only light mechanical and/or electrical dysfunction occurs (Puel et al., 1998). Thus it is likely that the increased glutamate level in untreated mutant mice is sufficient to elevate the ABR threshold, but insufficient to produce structural damage, although we cannot rule out a possibility that some other yet undetected defect in mutant mice leads to the baseline ABR threshold elevation. In the present study, we could not detect increased amounts of glutamate in noise-stimulated perilymphs in the wild-type mice, consistent with previous studies (Eybalin, 1993). However, we succeeded in demonstrating an increase of the perilymphatic concentration of glutamate during and after sound stimulations in GLAST-deficient mice. These results indicate that GLAST is a powerful glutamate uptake system in the cochlea and that GLAST hampers the reliable detection of increased amounts of glutamate in noise-stimulated perilymphs by previous works. Our data also provide direct evidence for the hypothesis that glutamate is a neurotransmitter of synapses between IHC and the auditory nerve. In our data, there are two discrepancies between glutamate levels and the effects of glutamate on the ABR thresholds (Figs. 2, 3). First, GLAST mutant mice show a partial recovery of the ABR threshold, whereas glutamate levels in perilymphs remain high even at 120 min after sound exposure. The mechanism of such discrepancy is unknown, but a reasonable explanation is that a decrease of the cochlea blood flow and damage to hair-cell stereocilia induced by acoustic overstimulation could recover within 120 min after sound exposure. Further work will be required to unravel the detailed mechanisms of recovery. Second, noise causes an ABR threshold elevation but no glutamate rise in the wild-type mice. This discrepancy can be explained by the inherent problem in a microdialysis approach. Because a microdialysis technique cannot directly measure the concentration of glutamate in the synaptic cleft but monitors the concentration of glutamate that diffuses out of the synaptic cleft into perilymph, and because the wild-type mice have a powerful glutamate uptake system (GLAST) in the cochlea, we cannot detect an increase of the perilymphatic concentrations of glutamate during and after sound stimulation in the wild-type mice, although the concentration of glutamate in the synaptic cleft really increases.
GLAST mutant mice will provide a model system for the investigation of the contribution of excitotoxic mechanisms to various cochlear diseases.
Footnotes
This work was supported in part by research grants from the Ministry of Education, Science, and Culture of Japan, the Ministry of Health and Welfare of Japan, Uehara Memorial Foundation, Brain Science Foundation, Yamada Science Foundation, Toyota Physical and Chemical Research Institute, The Karoji Memorial Fund, and the Japan Spina Bifida and Hydrocephalus Research Foundation. We thank Yutaka Takumi, Naoya Iijima, and Katsuhiko Kobatashi for their help with immunocytochemical techniques.
Correspondence should be addressed to Kohichi Tanaka, Department of Molecular Neuroscience, Medical Research Institute, Tokyo Medical and Dental University, 1-5-45 Yushima, Bunkyo-ku, Tokyo 113-8519, Japan. E-mail: tanaka.aud@mri.tmd.ac.jp.
REFERENCES
- 1.Arriza JL, Eliasof S, Kavanaugh MP, Amara SG. Excitatory amino acid transporter 5, a retinal glutamate transporter coupled to a chloride conductance. Proc Natl Acad Sci USA. 1997;94:4155–4160. doi: 10.1073/pnas.94.8.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Basile AS, Huang JM, Xie C, Webster D, Berlin C, Skolnick P. N-methyl-d-aspartate antagonists limit aminoglycoside antibiotic-induced hearing loss. Nat Med. 1996;2:1338–1343. doi: 10.1038/nm1296-1338. [DOI] [PubMed] [Google Scholar]
- 3.Eybalin M. Neurotransmitters and neuromodulators of the mammalian cochlea. Physiol Rev. 1993;73:309–373. doi: 10.1152/physrev.1993.73.2.309. [DOI] [PubMed] [Google Scholar]
- 4.Furness DN, Lehre KP. Immunocytochemical localization of a high-affinity glutamate-aspartate transporter, GLAST, in the rat and guinea-pig cochlea. Eur J Neurosci. 1997;9:1961–1969. doi: 10.1111/j.1460-9568.1997.tb00763.x. [DOI] [PubMed] [Google Scholar]
- 5.Greenwood DD. A cochlear frequency-position function for several species—29 years later. J Acoust Soc Am. 1990;87:2592–2605. doi: 10.1121/1.399052. [DOI] [PubMed] [Google Scholar]
- 6.Hakuba N, Gyo K, Yanagihara N, Mitani A, Kataoka K. Efflux of glutamate into the perilymph of the cochlea following transient ischemia in the gerbil. Neurosci Lett. 1997;230:69–71. doi: 10.1016/s0304-3940(97)00462-x. [DOI] [PubMed] [Google Scholar]
- 7.Harada T, Harada C, Watanabe M, Inoue Y, Sakagawa T, Nakayama N, Sasaki S, Okuyama S, Watase K, Wada K, Tanaka K. Functions of the two glutamate transporters GLAST and GLT-1 in the retina. Proc Natl Acad Sci USA. 1998;95:4663–4666. doi: 10.1073/pnas.95.8.4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanai Y. Family of neutral and acidic amino acid transporters: molecular biology, physiology, and medical implications. Curr Opin Cell Biol. 1997;9:565–572. doi: 10.1016/s0955-0674(97)80035-x. [DOI] [PubMed] [Google Scholar]
- 9.Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li HS, Niedzielski AS, Beisel KW, Hiel H, Wenthold RJ, Morley BJ. Identification of a glutamate/aspartate transporter in the rat cochlea. Hear Res. 1994;78:235–242. doi: 10.1016/0378-5955(94)90029-9. [DOI] [PubMed] [Google Scholar]
- 11.Mitani A, Andou Y, Matsuda S, Arai T, Sakanaka M, Kataoka K. Origin of ischemia-induced glutamate efflux in the CA1 field of the gerbil hippocampus: an in vivo brain microdialysis study. J Neurochem. 1994;63:2152–2164. doi: 10.1046/j.1471-4159.1994.63062152.x. [DOI] [PubMed] [Google Scholar]
- 12.Ottersen OP, Takumi Y, Matsubara A, Landsend AS, Laake JH, Usami S. Molecular organization of a type of peripheral glutamate synapse: the afferent synapses of hair cells in the inner ear. Prog Neurobiol. 1998;54:127–148. doi: 10.1016/s0301-0082(97)00054-3. [DOI] [PubMed] [Google Scholar]
- 13.Puel JL. Chemical synaptic transmission in the cochlea. Prog Neurobiol. 1995;47:449–476. doi: 10.1016/0301-0082(95)00028-3. [DOI] [PubMed] [Google Scholar]
- 14.Puel JL, Pujol R, Ladrech S, Eybalin M. Alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid electrophysiological and neurotoxic effects in the guinea-pig cochlea. Neuroscience. 1991;45:63–72. doi: 10.1016/0306-4522(91)90103-u. [DOI] [PubMed] [Google Scholar]
- 15.Puel JL, Ruel J, d'Aldin CG, Pujol R. Excitotoxicity and repair of cochlear synapses after noise-trauma induced hearing loss. NeuroReport. 1998;9:2109–2114. doi: 10.1097/00001756-199806220-00037. [DOI] [PubMed] [Google Scholar]
- 16.Pujol R, Puel JL, Gervais d'Aldin C, Eybalin M. Pathophysiology of the glutamatergic synapses in the cochlea. Acta Otolaryngol (Stockh) 1993;113:330–334. doi: 10.3109/00016489309135819. [DOI] [PubMed] [Google Scholar]
- 17.Robertson D. Functional significance of dendritic swelling after loud sounds in the guinea pig cochlea. Hear Res. 1983;9:263–278. doi: 10.1016/0378-5955(83)90031-x. [DOI] [PubMed] [Google Scholar]
- 18.Saunders JC, Dear SP, Schneider ME. The anatomical consequences of acoustic injury: a review and tutorial. J Acoust Soc Am. 1985;78:833–860. doi: 10.1121/1.392915. [DOI] [PubMed] [Google Scholar]
- 19.Takumi Y, Matsubara A, Danbolt NC, Laake JH, Storm-Mathisen J, Usami S, Shinkawa H, Ottersen OP. Discrete cellular and subcellular localization of glutamine synthetase and the glutamate transporter GLAST in the rat vestibular end organ. Neuroscience. 1997;79:1137–1144. doi: 10.1016/s0306-4522(97)00025-0. [DOI] [PubMed] [Google Scholar]
- 20.Tanaka K. Functions of glutamate transporters in the brain. Neurosci Res. 2000;37:15–19. doi: 10.1016/s0168-0102(00)00104-8. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe T, Morimoto K, Hirano T, Suwaki H, Watase K, Tanaka K. Amygdala-kindled and pentylenetetrazole-induced seizures in glutamate transporter GLAST-deficient mice. Brain Res. 1999;845:92–96. doi: 10.1016/s0006-8993(99)01945-9. [DOI] [PubMed] [Google Scholar]
- 22.Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita H, Kawakami H, Zhang YX, Tanaka K, Nakamura S. Neuroprotective mechanism of bromocriptine. Lancet. 1995;346:1305. doi: 10.1016/s0140-6736(95)91911-2. [DOI] [PubMed] [Google Scholar]
- 24.Yamashita H, Kawakami H, Zhang YX, Tanaka K, Nakamura S. Effect of amino acid ergot alkaloids on glutamate transport via human glutamate transporter hGluT-1. J Neurol Sci. 1998;155:31–36. doi: 10.1016/s0022-510x(97)00266-9. [DOI] [PubMed] [Google Scholar]