

Abstract
The development of cortical neurons in vivo andin vitro is accompanied by alterations in NMDA receptor subunit expression and concomitant modifications in the pharmacological profile of NMDA-activated ionic currents. For example, we observed that with decreasing NR2B/NR2A subunit expression ratio, the block of NMDA receptor-mediated whole-cell responses by the NR2B-selective antagonist haloperidol was also decreased. In mature cultures (>22 d in vitro), however, NMDA responses obtained from excised nucleated macropatches, which comprised a large portion of the soma, remained strongly antagonized by haloperidol. These results suggest that in more mature neurons NR1/NR2B receptors appear to be preferentially expressed in the cell body. As predicted from the whole-cell recording pharmacological profile, NMDA-induced toxicity was largely unaffected by haloperidol in mature cultures. However, haloperidol effectively blocked glutamate toxicity in the same cultures, suggesting that the neurotoxic actions of this amino acid were mostly due to the activation of somatic NMDA receptors. In experiments in which the potency of glutamate toxicity was increased by the transport inhibitorl-trans-pyrrolidine-2,4-dicarboxylic acid, the neuroprotective effects of haloperidol were significantly diminished. This was likely because of the fact that glutamate, now toxic at much lower concentrations, was able to reach and activate dendritic receptors under these conditions. These results strongly argue that exogenous glutamate and NMDA normally induce excitotoxicity at distinct cellular locations in mature mixed neuronal cultures and that NR1/NR2B receptors remain an important component in the expression of glutamate, but not NMDA-induced excitotoxicity.
Keywords: haloperidol, development, excitotoxicity, NMDA receptors, glutamate, NR2B subunit, glutamate transport, cortical neurons, tissue culture
Neurons and glia grown in tissue culture have been important experimental models for the study of the cellular and molecular mechanisms underlying excitotoxicity (Rothman, 1983; Choi et al., 1987). Observations obtained with these in vitro systems have had direct applications to the study of neurodegenerative processes in vivo (Doble, 1999). An important component of excitotoxicity that has been studied in detail in culture systems is the glutamate uptake transport system and its role in regulating NMDA receptor-mediated responses, including neuronal injury (Garthwaite, 1985; Rosenberg and Aizenman, 1989; Sugiyama et al., 1989; Rosenberg et al., 1992). Indeed, deficits in glutamate uptake have been implicated in human neurological disease (Rothstein et al., 1992; Lin et al., 1998). We previously determined that a powerful glutamate uptake system in mixed cultures of astrocytes and neurons influenced the potency of transported agonists in mediating cell death and had a dramatic effect in altering the neuroprotective properties of competitive NMDA receptor antagonists (Speliotes et al., 1994). The avid uptake system present in these cultures, combined with the fact that most synapses are buried beneath an astrocyte layer (Harris and Rosenberg, 1993), led us to hypothesize that the action of exogenous glutamate on neurons was restricted to the cell bodies. We further proposed that nontransported agonists such as NMDA could penetrate through the interstices of the tissue to reach receptors on dendrites. In essence, we suggested that glutamate and NMDA induced NMDA receptor-mediated toxicity at different cellular locations and possibly through different mechanisms (Speliotes et al., 1994).
Recently published results by Tovar and Westbrook (1999) and Rumbaugh and Vicini (1999) led us to revisit and test this hypothesis. Using the NR1/NR2B subunit-specific blockers ifenprodil (Williams, 1993) and CP 101,606 (Boeckman and Aizenman, 1996), these investigators reported that NR1/NR2B receptors segregate to extrasynaptic regions as hippocampal and cerebellar neurons mature developmentally both in tissue culture and in vivo. Importantly, through the use of excised “nucleated” macropatches, Rumbaugh and Vicini (1999)observed that the NR1/NR2B NMDA receptor configuration was selectively expressed in the soma of cerebellar granule neurons at late developmental stages. In the present study, we have used haloperidol, another antagonist with a high selectivity for the NR1/NR2B subunit configuration (Ilyin et al., 1996; Lynch and Gallagher, 1996), to confirm that NR1/NR2B receptors also segregate to the cell body in mature rat cortical neurons in culture. The somatic segregation of these receptors and their distinctive pharmacology allowed us to evaluate whether glutamate toxicity was restricted to the cell body at advanced developmental stages.
MATERIALS AND METHODS
Materials. All drugs and tissue culture reagents were obtained from Sigma (St. Louis, MO) with the exception of Fe-supplemented bovine calf serum (HyClone, Logan, UT), tetrodotoxin (Calbiochem, La Jolla, CA),l-trans-pyrrolidine-2,4-dicarboxylic acid (PDC; Tocris Neuramin, Bristol, England), CP 101,606 (a gift from Dr. W. F. White, Pfizer Central Research, Groton, CT), and CGS-19755 (a gift from Dr. P. Ornstein, Eli Lilly, Indianapolis, IN). RNA extraction from cortical cultures was achieved by using a commercially available kit (RNeasy; Qiagen, Santa Clarita, CA). Components for the RNase protection assay were purchased from Promega (Madison, WI) and Ambion (Austin, TX). NMDA subunit probes were generous gifts from Drs. Jie Zhong and Perry Molinoff (Bristol-Myers, Squibb, Wallingford, CT). Probes were labeled using [32P]UTP from DuPont–New England Nuclear (Boston, MA).
Astrocyte-rich cortical cultures. Cerebral cortices were obtained from embryonic day 16 (E16) Sprague Dawley rats and dissociated by previously described methods (Hartnett et al., 1997). Cells were plated onto poly-l-lysine-coated glass coverslips in either 6-well or 24-well culture dishes at a density of 300,000–500,000 cells/ml of growth media (v/v mixture of 80% DMEM, 10% Ham's F12, 10% heat-inactivated iron-supplemented bovine calf serum, 25 mm HEPES, 24 U/ml penicillin, 24 μg/ml streptomycin, and 2 mml-glutamine) and maintained at 37°C in 5% CO2. A mitotic inhibitor (cytosine arabinoside; 2 μm) was added once at 15 DIV after which the growth medium contained no F-12 and only 2% serum. Media was partially replaced with fresh growth medium 3 times per week. At 3–5 weeks in vitro these cultures contain ∼10–20% neurons (Rosenberg and Aizenman, 1989; Rosenberg, 1991).
RNase protection assay. RNA was harvested from the cultures and tested for degradation with an agarose-formaldehyde gel; the 18 S and 28 S rRNA bands were visualized with ethidium bromide. The concentration of total RNA was measured for every preparation, with a yield between 30 and 50 μg of RNA per two to three 6-well plates. One microgram of linearized NR subunit plasmid was added to a 20 μl solution containing [32-P]UTP (800 Ci/mmol; 10 mCi/ml), nucleotides (ATP, GTP, and CTP; 10 mmeach), unlabeled UTP (100 μm), SP6 or T7 RNA polymerase (20 U), transcription buffer, and RNasin RNase Inhibitor (1 U). The mixture was incubated for 1.5 hr at 30°C. DNA template was then degraded with 5 U of DNase I. The probes were removed from unincorporated nucleotides using phenol chloroform extraction and pelleted by ethanol precipitation. Total RNA samples (10 μg) were incubated with labeled probes (5 × 105 cpm) and hybridized overnight at 50°C. The solution was digested with RNase A and RNase T1 at 30°C for 1 hr followed by a proteinase K-1% SDS incubation for 30 min at 37°C. The hybridized RNA species was extracted using phenol chloroform and pelleted by ethanol precipitation. The protected RNA species were separated by electrophoresis (8 m urea, 5% acrylamide gel). Bands were visualized and quantified using a PhosphorImager (Storm; Molecular Dynamics, Sunnyvale, CA).
Electrophysiology. Electrophysiological recordings were conducted using the whole-cell configurations of the patch-clamp technique as described earlier (Tang and Aizenman, 1993; Brimecombe et al., 1998). The extracellular recording solution contained (in mm): NaCl, 150; KCl, 2.8; CaCl2, 1.0; HEPES, 10, 0.3 μm tetrodotoxin, 10 μmglycine; pH adjusted to 7.2 with 0.3 m NaOH. The intracellular (pipette) solution contained (in mm): CsF, 140; EGTA, 10; CaCl2, 1.0; pH adjusted to 7.2 with CsOH. Recordings were performed with 2–3 MΩ electrodes. Partial compensation for series resistance (80%) was performed in some experiments. Currents were filtered at 0.5–1 kHz and digitized at 1–2 kHz. Drugs were dissolved in external solution and applied to cells using a multibarrel fast perfusion system (Warner Instruments, Hamden, CT). The current amplitude measurement to calculate haloperidol block was performed on the portion of the trace immediately before the removal of this drug. The time course of haloperidol application used was chosen from preliminary experiments with longer applications in which we noted the time required to reach steady state and selected the briefest drug application that was near this level.
Toxicity assays. Cell survival was assessed on the cortical cells plated or transferred to 24-well culture plates. Cells were exposed to either control solution [minimum essential medium (MEM) plus 0.01% BSA and 25 mm HEPES, with 10 μmglycine], NMDA or glutamate for 30 min at 37°C and 5% CO2 after which the treatment was terminated by two complete rinses with MEM (a 200:1 dilution). Cells were maintained for 18–20 hr after the exposure and the amount of lactate dehydrogenase released into the medium was measured spectrophotometrically using a previously described protocol (Hartnett et al., 1997). Neuroprotection provided by the various agents tested was derived from the following equation:
RESULTS
Changes in NMDA receptor subunit expression in cultured cortical neurons with development
Total RNA was isolated from the cortical cultures on a weekly basis for up to 4 weeks. Using an RNase protection assay, we determined the levels of expression of the NR1, NR2A, and NR2B subunits (Fig.1A). NR1 message was present at week one, and its levels increased throughout the maturation of the cultures. NR2B message was also reliably detected starting at week one, and its expression remained relatively constant. In contrast, NR2A message was very low relative to NR1 and NR2B during the first 2 weeks in vitro. However, expression of NR2A nearly doubled by the third week, and doubled again by week four. We observed that the ratio of NR2B to NR2A message, averaged across three independent experiments, decreased significantly (p < 0.05, ANOVA) from ∼6.5 at 8 d in vitro (DIV) to nearly 2 at 29 DIV (Fig. 1B). These data essentially reproduce earlier in vitro and in vivo findings that show that NR2A expression occurs later in cerebral cortex development when compared with NR1 and NR2B (Monyer et al., 1994; Zhong et al., 1994,1995; Portera-Cailliau et al., 1996; Wenzel et al., 1997).
Fig. 1.
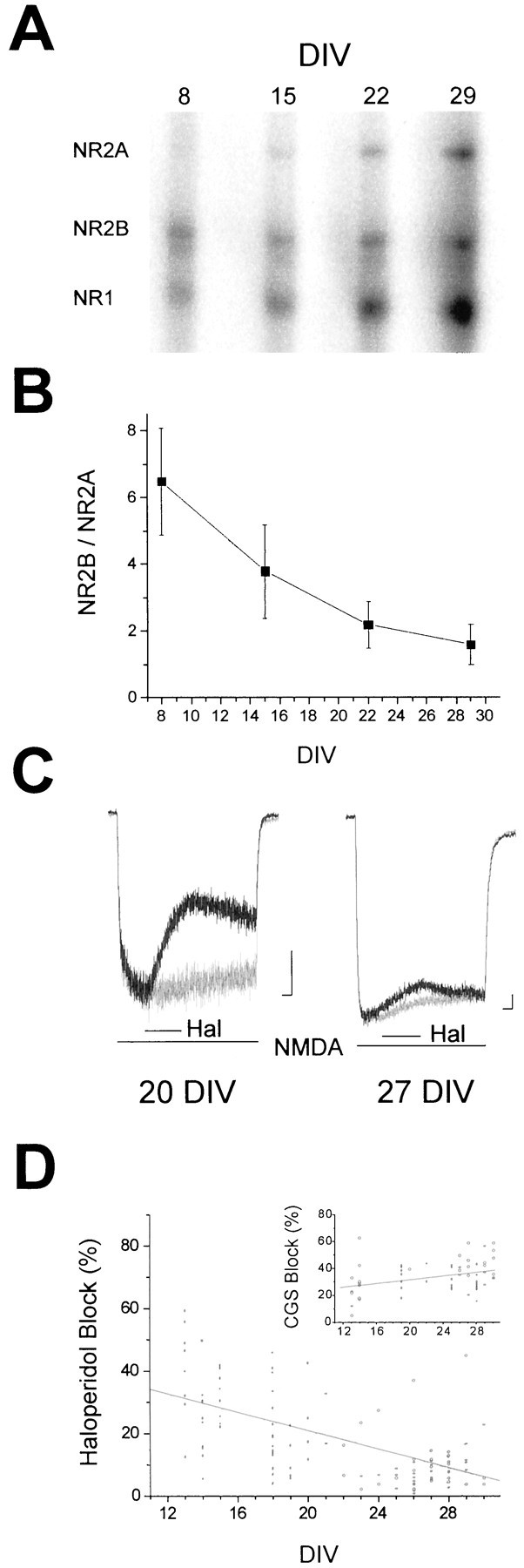
Onset of NMDA receptor subunit expression in rat cortex in vitro. A, Representative gel from an RNase protection assay demonstrating the time course of NMDA receptor subunit expression in cortical neurons grown in astrocyte-rich cultures for up to 4 weeks. The expression of NMDA receptor subunits was determined by using 32P-lableled probes specific for NR1, NR2A, and NR2B.B, Quantification of the ratio of NR2B to NR2A band densities during the maturation of the cultures. Values represent the mean ± SEM of three separate experiments performed on three independent cultures. The decrease in band density ratio during development was statistically significant (p< 0.05; ANOVA). C, Whole-cell voltage-clamped (−60 mV) currents obtained from cortical neurons in astrocyte-rich cultures at two different developmental stages invitro. Currents were recorded during fast application of 30 μm NMDA in the absence or presence of 10 μm haloperidol (Hal). Thelinesbelow the traces indicate drug applications. Calibration: 150 pA, 1 sec. D, Responses such as those shown in C were used to measure the steady-state block by haloperidol of NMDA-elicited currents at various developmental time points. The measurements of current inhibition were corrected for the desensitization observed when agonist was applied alone (C, light traces). Points represent individual cells (n = 118); the line represents a regression through the points (r = −0.55). The decrease in haloperidol block with development was highly significant (p < 0.0001; ANOVA). Insetrepresents the degree of block produced by 0.7 μmCGS-19755 of 30 μm NMDA-induced responses recorded under similar circumstances (n = 69);r = 0.35. The increase in block with development was statistically significant (p < 0.05; ANOVA).
To confirm that the observed changes in the NR2B to NR2A expression ratio reflected a functional change in NMDA receptor properties, we performed electrophysiological recordings. Whole-cell currents evoked by 30 μm NMDA in the absence or presence of 10 μm haloperidol were elicited in a total of 118 neurons from developmental ages ranging from 13 to 30 DIV (Fig. 1C). Haloperidol and other noncompetitive ifenprodil-like drugs have been shown to be highly selective antagonists of NR1/NR2B receptors, having a much lower affinity for receptors assembled with the NR1/NR2A subunit configuration (Ilyin et al., 1996; Lynch and Gallagher, 1996) or on putative NR1/NR2A/NR2B-containing receptors (Boeckman and Aizenman, 1996; Brimecombe et al., 1997; Vicini et al., 1998; Tovar and Westbrook, 1999). The concentration of haloperidol chosen for these studies (10 μm) is near the EC50 reported for recombinant NR1/NR2B receptors and less than the EC5 for the NR1/NR2A receptor configuration (Ilyin et al., 1996; Lynch and Gallagher, 1996). We observed a highly significant (p < 0.0001, ANOVA) decrease in the sensitivity to haloperidol with NMDA-induced responses in neurons at later developmental stages (Fig.1D), similar to what had previously been described (Ilyin et al., 1996; Lynch and Gallagher, 1996). This result indicates that the decrease in NR2B relative to NR2A during development can be functionally detected.
In contrast to this finding, the block induced by 0.7 μmCGS-19755, a competitive antagonist at the glutamate recognition site of the NMDA receptor (Murphy et al., 1988; Aizenman and Hartnett, 1992), was observed to increase slightly but significantly (p < 0.01, ANOVA) with development in a total of 69 additional neurons tested at 13–30 DIV (Fig.1D, inset). This increase in block could be reflective of the fact that CGS-19755 has a slightly higher affinity for NR1/NR2A receptors when compared with NR1/NR2B (Laurie and Seeburg, 1994; Boeckman and Aizenman, 1996).
Developmental changes in NMDA receptor-mediated toxicity
We next evaluated whether the changes in NMDA receptor subunits during development influenced the pharmacological profile of excitotoxicity in our cultures. Sister cultures at two developmental stages (15–18 and 25 DIV) were exposed for 30 min to 100 and 300 μm NMDA and assayed for neuronal cell death a day later. NMDA receptor-mediated injury was normalized to the nearly complete neuronal death produced by an overnight exposure to 1 mm kainate (Koh and Choi, 1988). As seen previously in our culture system (Sinor et al., 1997) and in others (Choi, 1985), younger cultures were substantially less sensitive to NMDA toxicity than mature cultures, although a considerable amount of cell death could be produced with the higher NMDA concentration in the immature neurons (Fig. 2A). The extent of NMDA toxicity in these young cultures was of sufficient magnitude to allow us to test the neuroprotective effects of haloperidol (10 μm) at this developmental stage. This antagonist rescued ∼50% of 13–19 DIV neurons after exposure to 300 μm NMDA. In contrast, haloperidol was much less effective in protecting 25–32 DIV cells (∼15% survival; Fig.2B). This observation suggests that the decrease in NR2B expression relative to NR2A during the maturation of the cultures influences the effectiveness of haloperidol in blocking NMDA-induced neurotoxicity. By comparison, 10 μm CGS-19755 was equally effective in protecting young and mature cells exposed to 300 μm NMDA (Fig. 2B). Hence, the modest increase in CGS-19755 block observed with the electrophysiological measurements during development (Fig.1D, inset) may not have been of sufficient magnitude to be detected with the toxicity assay. A series of comparative experiments were also performed using ifenprodil (1 μm), the prototypical NR1/NR2B-selective antagonist (Williams, 1993), to ensure that the neuroprotective effects of haloperidol were indeed because of NMDA receptor block. Results from representative experiments performed on young (18 DIV) and mature (27 DIV) cultures showing typical LDH values obtained are shown (Fig.2C,D). The effects of ifenprodil were virtually indistinguishable from those of haloperidol.
Fig. 2.
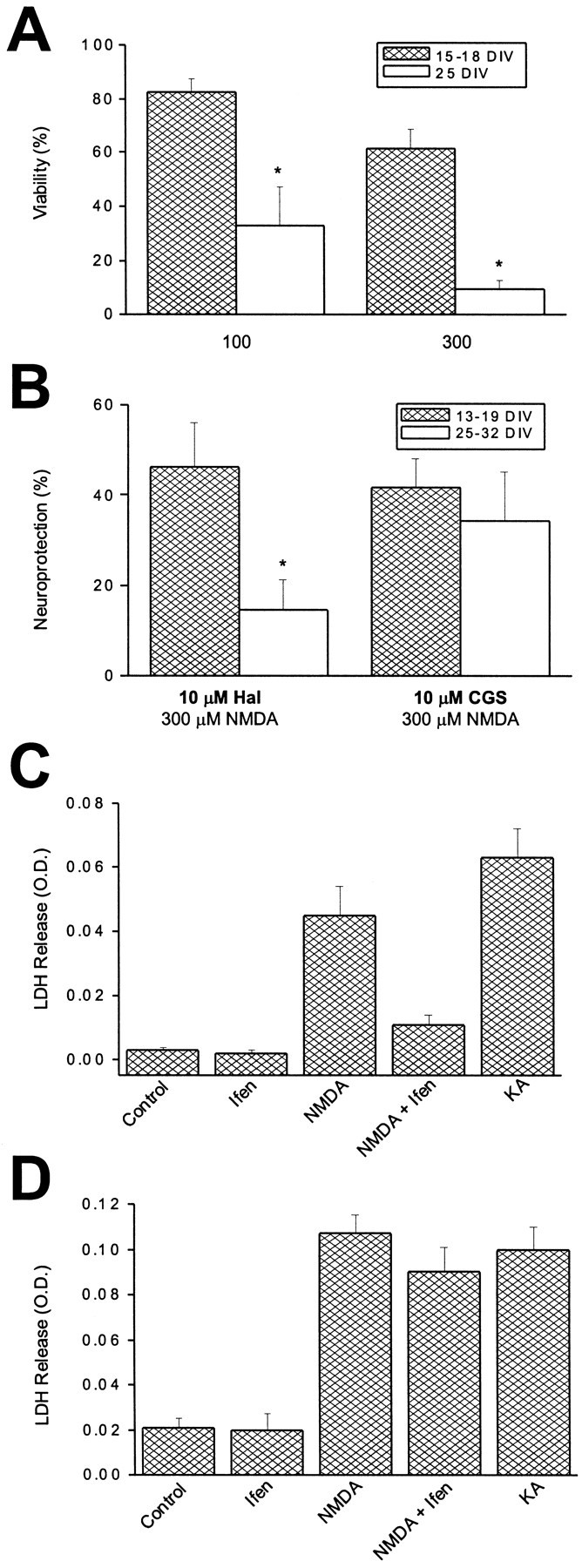
Developmental changes in NMDA toxicity.A, Neuronal viability of cortical neurons in vitro 24 hr after a 30 min exposure to either 100 or 300 μm NMDA. Experiments were performed on sister cultures at two developmental ages. Results were normalized to the death produced by an overnight exposure to 1 mm kainate.Asterisks denote significant differences in viability between the two developmental ages (p < 0.005; paired t test). Values represent the mean ± SEM of seven (15–18 DIV) and four (25 DIV) independent experiments, each performed in quadruplicate. B, Degree of neuroprotection afforded by either haloperidol (10 μm) or CGS- 19755 (10 μm) against 300 μmNMDA (30 min exposure) in cortical neurons at two developmental periodsin vitro. Haloperidol was significantly less effective in protecting mature neurons compared with younger cells (p < 0.05; unpaired t test). CGS-19755 protected neurons equally regardless of developmental age. Values represent the mean ± SEM of five independent experiments performed in quadruplicate. C, A representative experiment performed on a young culture (18 DIV) using 1 μm ifenprodil (Ifen) for comparison purposes. This figure shows LDH release values obtained; note the substantial neuroprotection against 300 μm NMDA produced by the NR1/NR2B-selective blocker. Results are the mean ± SD of a single experiment performed in quadruplicate. A total of four independent experiments were performed on 18–20 DIV cultures with essentially identical results. KA represents an overnight exposure to 1 mm kainate. D, A similar experiment performed on a mature culture (27 DIV). Note the relative lack of neuroprotection produced by ifenprodil (1 μm). Results are the mean ± SD of a single experiment performed in quadruplicate. A total of four independent experiments were performed on 26–32 DIV cultures with essentially identical results.
NMDA receptor subunits are functionally segregated in mature neurons
As indicated earlier, NMDA-evoked whole-cell currents at latein vitro stages were poorly antagonized by 10 μm haloperidol (Fig. 1C,D). In fact, the average inhibition produced by haloperidol in cells 22 DIV or older was only 9.6 ± 1.1% (n = 57). This is the same period of development that we had previously showed to closely coincide with the full expression of sensitivity to NMDA-induced toxicity (Fig.2A; Sinor et al., 1997). Hence, the NR1/NR2B component of the total receptor population in cortical neurons appears to diminish at a time when NMDA receptor-mediated excitotoxicity becomes fully expressed. Interestingly, mice deficient in the NR2A subunit are more resistant to stroke-induced CNS damage when compared with wild-type controls (Morikawa et al., 1998).
Recent reports have suggested that any remaining NR1/NR2B receptors in mature hippocampal or cerebellar neurons are, for the most part, restricted to extrasynaptic or somatic regions (Rumbaugh and Vicini, 1999; Tovar and Westbrook, 1999). We confirmed that this also occurred in cortical neurons at 22–30 DIV by using a similar strategy to that described by Rumbaugh and Vicini (1999). The degree of haloperidol (10 μm) block of 30 μm NMDA-induced currents was compared between whole-cell recordings, and responses obtained after excision of nucleated patches from the same neurons. These macrovesicles essentially represent the excised soma, or a large portion of it. Haloperidol blocked the whole-cell NMDA response by an average of 5.9 ± 2.5% in four of these neurons, which is a value very similar to that obtained earlier for cells older than 22 DIV. In contrast, the block was significantly greater (p< 0.005, paired t test) in the nucleated patches obtained from these cells, averaging 51.5 ± 5.9% (Fig.3A,C). In five other cells we measured the block of NMDA-induced currents by 0.7 μm CGS-19755, which was not significantly different (p > 0.05, paired t test) in the intact cells (34.9 ± 5.5%) and in nucleated patches (45.2 ± 10.8%; Fig. 3B,C). These results confirm that NR1/NR2B receptors are present in mature cortical neurons, and that the majority of them segregate to the cell body. Conversely, the density of NR1/NR2A or putative NR1/NR2A/NR2B appears to be equally distributed throughout somatic and extrasomatic regions at late developmental stages.
Fig. 3.
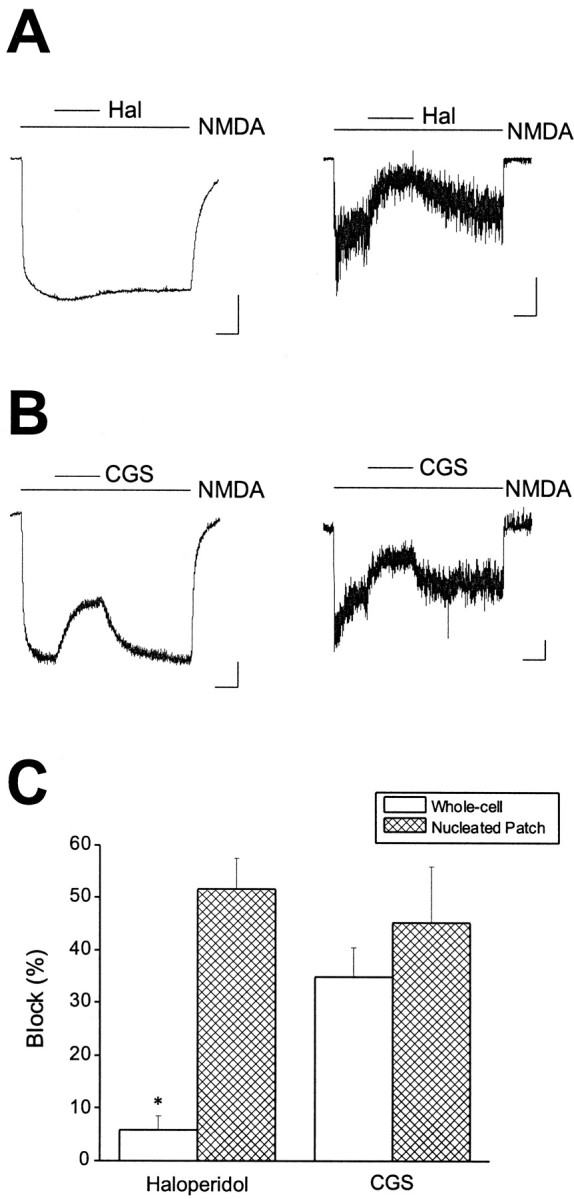
Haloperidol block reveals segregation of NR1/NR2B receptors to the soma in mature cortical neurons. A,NMDA (30 μm)-induced responses obtained from a mature cortical neuron (25 DIV) before (left trace) or after excision of a nucleated patch (right trace). Recordings were obtained in the absence and presence of 10 μmhaloperidol as indicated by the linesabove the traces. A more substantial block was observed in the nucleated patch when compared with the whole cell. Similar results were obtained in a total of four cells. B, NMDA (30 μm)-induced responses obtained from a second cell (28 DIV) before (left trace) or after excision of a nucleated patch (right trace). Recordings were obtained in the absence and presence of 0.7 μm CGS-19755, as indicated by the lines above the traces. A similar degree of block was observed in both recording configurations. Calibration: 500 pA whole-cell, 50 pA patch, 2 sec. Similar results were obtained in a total of five cells. C, Pooled block data showing a significant difference in haloperidol block between whole-cell recordings and nucleated patches (p < 0.005; paired ttest).
NMDA and glutamate induce excitotoxicity at different cellular locations
In previous work we proposed that glutamate and NMDA induce NMDA receptor-mediated excitotoxicity at different cellular locations (Speliotes et al., 1994). We suggested that glutamate injures neurons by acting directly on the cell body, whereas NMDA killed by stimulating receptors on cell processes. The observed segregation of NR1/NR2B receptors to the soma at late developmental stages provided us with the opportunity to test this hypothesis. We exposed mature sister cultures (>25 DIV) to either 300 μm NMDA or 300 μmglutamate in the absence or presence of 10 μmhaloperidol. Glutamate toxicity in these cultures at late developmental stages is mediated solely by NMDA receptors activation (Aizenman and Hartnett, 1992). Once again, haloperidol was marginally effective in blocking NMDA-mediated damage in these cells (Figs. 4,5A,C). Strikingly, glutamate-mediated toxicity was significantly antagonized (56.1 ± 9.6% neuroprotection; n = 7; p < 0.05; paired t test) by the NR1/NR2B receptor blocker (Figs.4, 5A,C). In accordance with this finding, whole-cell currents generated by 30 μm glutamate in 25 DIV cells were inhibited nearly 40% by 10 μmhaloperidol (38.2 ± 3.2% block; n = 5). This block is not significantly different (p > 0.05, unpaired t test) from the inhibition induced by haloperidol of NMDA responses in the nucleated patches. Furthermore, the difference between the block of glutamate toxicity and glutamate-induced currents by haloperidol was not statistically significantly (p > 0.05, unpaired t test). These results are consistent with our hypothesis. Similar results were obtained using 1 μm ifenprodil as the antagonist. In 26–32 DIV cultures, this drug induced 51.6 ± 14.7% neuroprotection against glutamate and only 20.6 ± 7.9% neuroprotection versus NMDA (p < 0.05, pairedt test; n = 4).
Fig. 4.
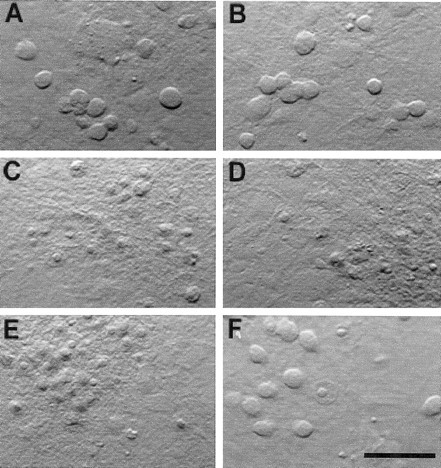
Haloperidol block distinguishes between glutamate and NMDA toxicity. Photomicrographs obtained from cortical cultures 24 hr after a 30 min exposure to vehicle (MEM; A), 10 μm haloperidol (B), 300 μm NMDA (C), NMDA plus haloperidol (D), 300 μm glutamate (E), or glutamate plus haloperidol (F). Note the relative lack of neuroprotection by haloperidol against NMDA toxicity compared with glutamate. Scale bar, 100 μm. Please see Figure 5 for quantification of the results.
Fig. 5.
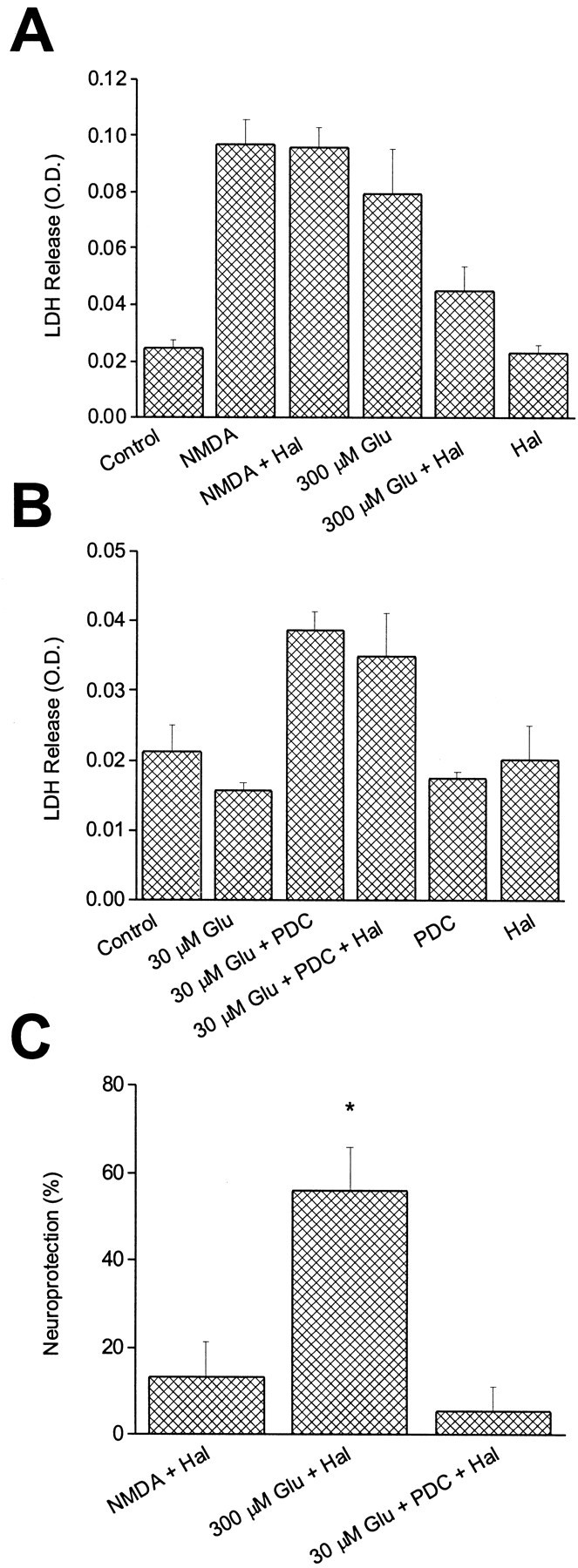
NMDA and glutamate toxicity have distinct pharmacological properties. A, Single representative experiment that shows contrast between the effect of haloperidol against NMDA toxicity and glutamate toxicity in mature neurons (25–27 DIV). Values represent the mean ± SD of LDH released into the medium by cortical cultures 24 hr after a 30 min exposure to vehicle (Control), 10 μm haloperidol, 300 μm NMDA, NMDA plus haloperidol, 300 μmglutamate, glutamate plus haloperidol, or haloperidol alone. The experiment was performed in quadruplicate. B, Addition of the glutamate transport inhibitor PDC (100 μm) increased the potency of glutamate in mature cultures (25–27 DIV). Values represent the mean ± SD of LDH released into the medium by cultures 24 hr after exposure to vehicle (Con), 30 μm glutamate, glutamate plus PDC, glutamate plus PDC and 10 μm haloperidol, and PDC or haloperidol alone. Note that under these conditions, haloperidol no longer saves against glutamate toxicity. This single representative experiment was performed in quadruplicate. C, Data such as those shown inA and B were normalized and averaged across four to seven independent experiments performed in quadruplicate to illustrate the degree of neuroprotection afforded by 10 μm haloperidol against 300 μm NMDA, 300 μm glutamate, or 30 μm glutamate in the presence of 100 μm PDC. Asterisk denotes significant difference between Glu + Hal group and the other two experimental groups (p < 0.01; ANOVA with Tukey–Kramer multiple comparisons test). Values represent the mean ± SEM (n = 5 for NMDA + Hal;n = 7 for Glu + Hal; n = 4 for Glu + PDC + Hal).
In accordance with our model, increasing the potency of glutamate by using a glutamate transporter blocker (Robinson et al., 1993; Dugan et al., 1995) should be the direct consequence of glutamate gaining access to NMDA receptors on dendrites. Under these conditions, haloperidol should lose its neuroprotective properties if our hypothesis is correct. We first confirmed that the glutamate transport inhibitor PDC did in fact shift the EC50 for glutamate toxicity without altering the NMDA concentration–response relationship. As reported by others (Robinson et al., 1993; Dugan et al., 1995), we observed that the EC50 for glutamate in inducing cell death shifted approximately eightfold (p < 0.05; paired t test) in the presence of 100 μm PDC (a concentration of the transport inhibitor that is minimally toxic; Blitzblau et al., 1996), without affecting the NMDA EC50 in mature neurons (>25 DIV; Table 1). Hence, concentrations of glutamate that were marginally or nontoxic under normal conditions now became significantly lethal. Under these circumstances, the neuroprotective effects of haloperidol (10 μm) against glutamate disappeared (Figs. 5B,C). These results provide conclusive evidence that the model predicted by our previous investigations (Speliotes et al., 1994) was correct.
Table 1.
Effects of glutamate transport inhibition on excitotoxicity
| EC50(μm) | |||
|---|---|---|---|
| NMDA | NMDA + PDC | Glutamate | Glutamate + PDC |
| 44.1 ± 17.5 | 28.2 ± 7.9 | 320.2 ± 42.1 | 42.9 ± 9.2* |
Significantly different from glutamate EC50 (p < 0.05; paired t test). PDC, l-trans-pyrrolidine-2,4-dicarboxylic acid (100 μm). EC50 values represent the mean ± SEM of three independent concentration–response determinations, each performed in quadruplicate.
DISCUSSION
Mixed cultures of rat cerebral cortex contain various non-neuronal cells types, including astrocytes, oligodendrocytes, microglia, and ependymal cells (Dichter, 1978). These cells, especially astrocytes, represent the largest component of the cultures, contributing up to ∼80–90% of the total cellular mass at 3–5 weeks in vitro (Rosenberg, 1991). Previous work by our group and by other investigators had suggested that the presence of a glutamate transport system in glia strongly influenced the ability of glutamate to kill neurons in culture (Garthwaite, 1985; Rosenberg and Aizenman, 1989;Sugiyama et al., 1989; Rosenberg et al., 1992). We also reported that the presence of the transport system could distort the pharmacological profile of competitive antagonists in protecting against the neurotoxic actions of transported agonists such as glutamate, but not against nontransported agonists like NMDA (Speliotes et al., 1994). These observations, coupled with the fact that dendrites are shielded from the external milieu by an astrocyte layer (Harris and Rosenberg, 1993), led us to propose that exogenous glutamate never reaches the sites that are accessible to NMDA to injure neurons. We hypothesized that exogenous glutamate is toxic neurons under these circumstances by activating somatic receptors exclusively (Speliotes et al., 1994). In the present study, we provide experimental evidence that directly supports this hypothesis. The fact NR1/NR2B receptors appear to segregate to the cell body as neurons mature in mixed cultures allowed us to demonstrate that haloperidol could effectively block glutamate-induced neurotoxicity without substantially altering NMDA-mediated cell death. Furthermore, a glutamate transport inhibitor was used to allow glutamate to reach dendritic receptors and become a more potent toxin. Under these circumstances, haloperidol became ineffective in blocking neuronal death, similar to what was observed when NMDA was used as the stimulus.
An alternative scenario, which may result in the receptor segregation we observe in the mature cultures, is that NR1/NR2B-containing neurons simply perish during development. Although we normally do not see widespread natural neuronal cell death even after 6 weeks in culture, and NR2B message does not decline with development, we cannot totally rule out this possibility. Nonetheless, whatever population of neurons becomes prominent at later stages in development, it still has NR1/NR2B receptors segregated to the soma, which allows us to test our hypothesis.
Glutamate-induced toxicity triggered by activation of somatic receptors requires a high concentration of agonist, and therefore, protection by competitive antagonists like d-APV or CGS-19755 can only be achieved by very high concentrations of these drugs (Speliotes et al., 1994). The high concentration of glutamate required to produce toxicity via somatic receptors suggests that the mechanisms by which glutamate and NMDA injure neurons in mixed cultures may be quite different. Results from several studies have strongly argued that the compartmentalization and association of NMDA receptors with other proteins or subcellular organelles may be critical for triggering excitotoxicity (Furukawa et al., 1997; Peng and Greenamyre, 1998;Sattler et al., 1998, 1999). As such, it is possible that the coupling between the NMDA receptor and the intracellular processes necessary to trigger cell death vary between the dendrites and the cell body. The high concentration of glutamate that is required to kill neurons in mixed cultures cannot be accounted for by differences in agonist affinity between NMDA receptor subtypes (Laurie and Seeburg, 1994;Boeckman and Aizenman, 1996), and is reminiscent of the levels of glutamate needed to elicit oxidative stress and cell death via inhibition of cystine transport in cell lines, neurons and oligodendrocytes (Murphy et al., 1989,, 1990; Oka et al., 1993; Ratan et al., 1994; Li et al., 1997). We currently favor the hypothesis that glutamate toxicity in mature astrocyte-rich cortical cultures requires not only NMDA receptor activation, but also at least a second injurious process, such as oxidative stress (Coyle and Puttfarcken, 1993). Future experiments will be designed to test this hypothesis.
Glutamate-induced neurotoxicity in mixed cultures is a commonly used model of neuronal injury despite the fact that the NMDA receptors that cluster in dendritic spines (O'Brien et al., 1998) are likely to be critical in mediating more relevant pathophysiological processes, such as those that are initiated during ischemia (Sattler et al., 2000). Indeed, synaptically released glutamate appears to mediate a large component of the excitotoxic damage under these circumstances (Rothman, 1983, 1984; Monyer et al., 1992; Sattler et al., 2000). Hence, the demonstration that NMDA and glutamate act at different cellular locations presented in this study warrants the reassessment of previous work focusing on glutamate-mediated intracellular alterations that are limited to nonsynaptic regions, including imaging studies at the soma.
Footnotes
This work was funded in part by grants from the National Association for Research in Schizophrenia and Depression (E.A.), Ron Shapiro Charitable Foundation (P.A.R.), and the Muscular Dystrophy Association (P.A.R.) and by National Institutes of Health Grants NS29365 (E.A.) and NS38475 (P.A.R.). E.A. and D.N.L. are supported by the American Heart Association. We thank B. A. McLaughlin, K. Hartnett, K. Kandler, and G. A. Herin for helpful discussions and suggestions, and Koichi Takimoto for advice with the RNase protection assay.
Correspondence should be addressed to Dr. Elias Aizenman, Department of Neurobiology, University of Pittsburgh School of Medicine, E-1456 BST, Pittsburgh, PA 15261. E-mail: redox+@pitt.edu.
REFERENCES
- 1.Aizenman E, Hartnett KA. The action of CGS-19755 on the redox enhancement of NMDA toxicity in rat cortical neurons in vitro. Brain Res. 1992;585:28–34. doi: 10.1016/0006-8993(92)91187-j. [DOI] [PubMed] [Google Scholar]
- 2.Blitzblau R, Gupta S, Djali S, Robinson MB, Rosenberg PA. The glutamate transport inhibitor l-trans-pyrrolidine-2,4-dicarboxylate indirectly evokes NMDA receptor mediated neurotoxicity in rat cortical cultures. Eur J Neurosci. 1996;8:1840–1852. doi: 10.1111/j.1460-9568.1996.tb01328.x. [DOI] [PubMed] [Google Scholar]
- 3.Boeckman FA, Aizenman E. Pharmacological properties of acquired excitotoxicity in Chinese hamster ovary cells transfected with N-methyl-d-aspartate receptor subunits. J Pharmacol Exp Ther. 1996;279:515–523. [PubMed] [Google Scholar]
- 4.Brimecombe JC, Boeckman FA, Aizenman E. Functional consequences of NR2 subunit composition in single recombinant N-methyl-d-aspartate receptors. Proc Natl Acad Sci USA. 1997;94:11019–11024. doi: 10.1073/pnas.94.20.11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brimecombe JC, Gallagher MJ, Lynch DR, Aizenman E. An NR2B point mutation affecting haloperidol and CP101,606 sensitivity of single recombinant N-methyl-d-aspartate receptors. J Pharmacol Exp Ther. 1998;286:627–634. [PubMed] [Google Scholar]
- 6.Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293–297. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- 7.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 9.Dichter MA. Rat cortical neurons in cell culture: culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res. 1978;149:279–293. doi: 10.1016/0006-8993(78)90476-6. [DOI] [PubMed] [Google Scholar]
- 10.Doble A. The role of excitotoxicity in neurodegenerative diseases: implications for therapy. J Pharmacol Exp Ther. 1999;81:163–221. doi: 10.1016/s0163-7258(98)00042-4. [DOI] [PubMed] [Google Scholar]
- 11.Dugan LL, Bruno VMG, Amagasu SM, Giffard RG. Glia modulate the response of murine cortical neurons to excitotoxicity: glia exacerbate AMPA neurotoxicity. J Neurosci. 1995;15:4545–4555. doi: 10.1523/JNEUROSCI.15-06-04545.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furukawa K, Fu W, Li Y, Witke W, Kwiatkowski DJ, Mattson MP. The actin-severing protein gelsolin modulates calcium channel and NMDA receptor activities and vulnerability to excitotoxicity in hippocampal neurons. J Neurosci. 1997;17:8178–8186. doi: 10.1523/JNEUROSCI.17-21-08178.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garthwaite J. Cellular uptake disguises action of l-glutamate on N-methyl-d-aspartate receptors. Br J Pharmacol. 1985;85:297–307. doi: 10.1111/j.1476-5381.1985.tb08860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harris KM, Rosenberg PA. Localization of synapses in rat cerebral cortex in dissociated cell culture. Neuroscience. 1993;53:495–508. doi: 10.1016/0306-4522(93)90214-z. [DOI] [PubMed] [Google Scholar]
- 15.Hartnett KA, Stout AK, Rajdev S, Rosenberg PA, Reynolds IJ, Aizenman E. NMDA receptor-mediated neurotoxicity: a paradoxical requirement for extracellular Mg2+ in Na+/Ca2+-free solutions in rat cortical neurons in vitro. J Neurochem. 1997;68:1836–1845. doi: 10.1046/j.1471-4159.1997.68051836.x. [DOI] [PubMed] [Google Scholar]
- 16.Ilyin VI, Whittemore ER, Guastella J, Weber E, Woodward RM. Subtype-selective inhibition of N-methyl-d-aspartate receptors by haloperidol. Mol Pharmacol. 1996;50:1541–1550. [PubMed] [Google Scholar]
- 17.Koh JY, Choi DW. Vulnerability of cultured cortical neurons to damage by excitotoxins: differential susceptibility of neurons containing NADPH-diaphorase. J Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laurie DJ, Seeburg PH. Ligand affinities at recombinant N-methyl-d-aspartate receptors depend on subunit composition. Eur J Pharmacol Mol Pharmacol Sec. 1994;268:335–345. doi: 10.1016/0922-4106(94)90058-2. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 20.Lin CL, Bristol LA, Jin L, Dykes-Hoberg M, Crawford T, Clawson L, Rothstein JD. Aberrant RNA processing in a neurodegenerative disease: the cause for absent EAAT2, a glutamate transporter, in amyotrophic lateral sclerosis. Neuron. 1998;20:589–602. doi: 10.1016/s0896-6273(00)80997-6. [DOI] [PubMed] [Google Scholar]
- 21.Lynch DR, Gallagher MJ. Inhibition of N-methyl-d-aspartate receptors by haloperidol: developmental and pharmacological characterization in native and recombinant receptors. J Pharmacol Exp Ther. 1996;279:154–161. [PubMed] [Google Scholar]
- 22.Monyer H, Giffard RG, Hartley DM, Dugan LL, Goldberg MP, Choi DW. Oxygen or glucose deprivation-induced neuronal injury in cortical cell cultures is reduced by tetanus toxin. Neuron. 1992;8:967–973. doi: 10.1016/0896-6273(92)90211-u. [DOI] [PubMed] [Google Scholar]
- 23.Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 24.Morikawa E, Mori H, Kiyama Y, Mishina M, Asano T, Kirino T. Attenuation of focal ischemic brain injury in mice deficient in the epsilon1 (NR2A) subunit of NMDA receptor. J Neurosci. 1998;18:9727–9732. doi: 10.1523/JNEUROSCI.18-23-09727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- 26.Murphy DE, Hutchinson AJ, Hurt SD, Williams M, Sills MA. Characterization of the binding of [3H]CGS-19755: a novel N-methyl-d-aspartate antagonist with nanomolar affinity in rat brain. Br J Pharmacol. 1988;95:932–938. doi: 10.1111/j.1476-5381.1988.tb11723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 28.O'Brien R, Lau LF, Huganir RL. Molecular mechanisms of glutamate receptor clustering at excitatory synapses. Curr Opin Neurobiol. 1998;8:364–369. doi: 10.1016/s0959-4388(98)80062-7. [DOI] [PubMed] [Google Scholar]
- 29.Oka A, Belliveau MJ, Rosenberg PA, Volpe JJ. Vulnerability of oligodendroglia to glutamate: pharmacology, mechanisms, and prevention. J Neurosci. 1993;13:1441–1453. doi: 10.1523/JNEUROSCI.13-04-01441.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peng TI, Greenamyre JT. Privileged access to mitochondria of calcium influx through N-methyl-d-aspartate receptors. Mol Pharmacol. 1998;53:974–980. [PubMed] [Google Scholar]
- 31.Portera-Cailliau C, Price DL, Martin LJ. N-methyl-d-aspartate receptor proteins NR2A and NR2B are differentially distributed in the developing rat central nervous system as revealed by subunit-specific antibodies. J Neurochem. 1996;66:692–700. doi: 10.1046/j.1471-4159.1996.66020692.x. [DOI] [PubMed] [Google Scholar]
- 32.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 33.Robinson MB, Djali S, Buchhalter JR. Inhibition of glutamate uptake with l-trans-pyrrolidine-2,4-dicarboxylate potentiates glutamate toxicity in primary hippocampal cultures. J Neurochem. 1993;61:2099–2103. doi: 10.1111/j.1471-4159.1993.tb07447.x. [DOI] [PubMed] [Google Scholar]
- 34.Rosenberg PA. Accumulation of extracellular glutamate and neuronal death in astrocyte-poor cortical cultures exposed to glutamine. Glia. 1991;4:91–100. doi: 10.1002/glia.440040111. [DOI] [PubMed] [Google Scholar]
- 35.Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg PA, Amin S, Leitner M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J Neurosci. 1992;12:56–61. doi: 10.1523/JNEUROSCI.12-01-00056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rothman SM. Synaptic activity mediates death of hypoxic neurons. Science. 1983;220:536–537. doi: 10.1126/science.6836300. [DOI] [PubMed] [Google Scholar]
- 38.Rothman SM. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci. 1984;4:1884–1891. doi: 10.1523/JNEUROSCI.04-07-01884.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 40.Rumbaugh G, Vicini S. Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J Neurosci. 1999;19:10603–10610. doi: 10.1523/JNEUROSCI.19-24-10603.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71:2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x. [DOI] [PubMed] [Google Scholar]
- 42.Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- 43.Sattler R, Xiong Z, Lu W-Y, MacDonald JF, Tymianski M. Distinct roles of synaptic and extrasynaptic NMDA receptors in excitotoxicity. J Neurosci. 2000;20:22–33. doi: 10.1523/JNEUROSCI.20-01-00022.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sinor JD, Boeckman FA, Aizenman E. Intrinsic redox properties of N-methyl-d-aspartate receptor can determine the developmental expression of excitotoxicity in rat cortical neurons in vitro. Brain Res. 1997;747:297–303. doi: 10.1016/s0006-8993(96)01237-1. [DOI] [PubMed] [Google Scholar]
- 45.Speliotes EK, Hartnett KA, Blitzblau RC, Aizenman E, Rosenberg PA. Comparison of the potency of competitive NMDA antagonists against the neurotoxicity of glutamate and NMDA. J Neurochem. 1994;63:879–885. doi: 10.1046/j.1471-4159.1994.63030879.x. [DOI] [PubMed] [Google Scholar]
- 46.Sugiyama K, Brunori A, Mayer ML. Glial uptake of excitatory amino acids influences neuronal survival in cultures of mouse hippocampus. Neuroscience. 1989;32:779–791. doi: 10.1016/0306-4522(89)90298-4. [DOI] [PubMed] [Google Scholar]
- 47.Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- 49.Wenzel A, Fritschy JM, Mohler H, Benke D. NMDA receptor heterogeneity during postnatal development of the rat brain: differential expression of the NR2A, NR2B, and NR2C subunit proteins. J Neurochem. 1997;68:469–478. doi: 10.1046/j.1471-4159.1997.68020469.x. [DOI] [PubMed] [Google Scholar]
- 50.Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- 51.Zhong J, Russell SL, Pritchett DB, Molinoff PB, Williams K. Expression of mRNAs encoding subunits of the N-methyl-d-aspartate receptor in cultured cortical neurons. Mol Pharmacol. 1994;45:846–853. [PubMed] [Google Scholar]
- 52.Zhong J, Carrozza DP, Williams K, Pritchett DB, Molinoff PB. Expression of mRNAs encoding subunits of the NMDA receptor in developing rat brain. J Neurochem. 1995;64:531–539. doi: 10.1046/j.1471-4159.1995.64020531.x. [DOI] [PubMed] [Google Scholar]