

Abstract
Several biochemical changes have been described in noradrenergic neurons of the locus coeruleus (LC) after chronic morphine treatment. Changes in neurochemical expression in opioid afferent projections to the LC may be equally important in modulating noradrenergic neurons during chronic opiate exposure. To test the hypothesis that opioid peptides in LC afferents are altered after chronic opiate administration, we exposed adult male rats to either morphine or placebo pellets for 5 d. Tissue sections through the LC were processed for peroxidase or gold–silver labeling of methionine5-enkephalin (met-ENK) and analyzed using light or electron microscopy, respectively. Light level densitometry and ultrastructural analysis showed that there was a significant decrease in immunolabeling for ENK in LC-afferent terminals of morphine-treated rats. Western immunoblot analysis confirmed that protein levels for both leucine5- and methionine5-ENK were significantly decreased in tissue samples containing the LC after chronic morphine treatment. To test whether decreases in ENK protein expression were mirrored by decreases in gene expression, Northern blot analysis of preproenkephalin (PPE) mRNA was conducted in tissue samples obtained through the medulla, a brainstem area that contains the major opioid afferents to the LC. PPE mRNA was reduced in samples obtained from morphine-treated rats. Finally, in situ hybridization experiments confirmed significant decreases in PPE mRNA expression in the nucleus paragigantocellularis, a region known to provide a robust opioid input to the LC. These data suggest that there is a decrease in the synthesis of the opioid peptide mRNA and protein in the medullo-coerulear pathway after chronic exposure to morphine. Such alterations in opioid peptide levels during opiate dependence may contribute to the observed hyperactivity of LC neurons during opiate withdrawal.
Keywords: opioid, noradrenaline, morphine, arousal, enkephalin, opiate withdrawal
Locus coeruleus (LC) neurons are inhibited by acute exposure to exogenous opiates, a mechanism mediated by activation of an inward rectifying K+current through coupling with the Gi/o family of G-proteins (North et al., 1987; Alreja and Aghajanian, 1993). After chronic morphine administration, LC neurons develop cellular forms of tolerance and dependence where they gradually recover from the acute inhibitory effects of the drug (Andrade et al., 1983; Christie et al., 1987). After withdrawal of the exogenous opiates, LC neurons exhibit an augmented activation of their discharge activity (Aghajanian, 1982;Redmond and Huang, 1982; Rasmussen et al., 1990; Akaoka and Aston-Jones, 1991). This alteration in neuronal activity suggested that the α2-adrenergic agonist, clonidine, was therapeutically efficacious because of its known ability to inhibit the neuronal firing rate of noradrenergic LC neurons (Gold et al., 1981; Grant et al., 1988;Hayward et al., 1990; Rasmussen et al., 1990, 1996; Akaoka and Aston-Jones, 1991; Guitart et al., 1992, 1993; Maldonado and Koob, 1993; Aghajanian et al., 1994; Chieng and Christie, 1995; Kogan and Aghajanian, 1995; Maldonado et al., 1995; Rasmussen, 1995; Krystal et al., 1996). Several hypotheses have been presented to explain this increase in activity. Upregulation of cAMP in LC neurons may account for elevated basal firing rates (Kogan et al., 1992). In addition, the brainstem circuit linking the nucleus paragigantocellularis (PGi) and the LC has been implicated in contributing to the hyperactivity of LC neurons during opiate withdrawal because of increased glutamatergic transmission in this pathway (Rasmussen and Aghajanian, 1989; Akaoka and Aston-Jones, 1991; Tokuyama et al., 1998) during opiate withdrawal. Changes in neurochemical expression in afferent projections to the LC may be equally important in modulating noradrenergic neurons during chronic opiate exposure. The PGi not only provides the major source of glutamate (Ennis and Aston-Jones, 1988) to the LC but also contributes the major opioid innervation to the LC (Drolet et al., 1992). We have also shown that the excitatory transmitter, glutamate, coexists with the endogenous opioid peptide, enkephalin (ENK), in a subset of axon terminals in the LC (Van Bockstaele et al., 2000). Thus, alterations in ENK levels in medullary neurons projecting to the LC after chronic opiate exposure may contribute to the observed hyperactivity of LC neurons during opiate withdrawal by removing an inhibitory influence on LC neurons (Williams et al., 1987; Abercrombie and Jacobs, 1988) and causing coexisting excitatory transmitters, such as glutamate, to hyperactivate LC neurons.
Several lines of evidence indicate that, in some neuronal circuits, chronic administration of morphine decreases the expression of some endogenous opioid peptides, methionine5(met)-ENK and leucine5 (leu)-ENK (Gudehithlu et al., 1991; Tejwani et al., 1994), whereas others have not observed such changes (Childers et al., 1977; Fratta et al., 1977;Wesche et al., 1977). Although differences in the literature may be attributed to variability in routes of drug administration, it is also likely that there are regionally specific changes in ENK expression and protein levels after exposure to opiates. For example, Tejwani and Rattan (1997) have shown a 70% reduction in ENK levels in medullary regions that include the PGi. In the present study, immunocytochemistry and Western blot analysis were conducted after chronic morphine administration to examine levels of endogenous opioid peptide proteins in the LC of rat brain. These techniques showed a decrease in ENK immunoreactivity and protein levels in the LC. This correlated with a decrease in ENK gene expression as shown by Northern blot analysis andin situ hybridization experiments of preproenkephalin (PPE) mRNA in regions known to provide opioid inputs to the LC, specifically the PGi. A persistent diminution in ENK levels in opioid afferents to the LC during withdrawal from opiates would most likely contribute to the observed hyperactivity of these neurons in the withdrawn state.
MATERIALS AND METHODS
Drug administration. Fifty adult male Sprague Dawley rats (Harlan, Indianapolis, IN; 225–275 gm) were used in this study. Rats were subcutaneously implanted with two morphine pellets each (National Institute Drug Abuse: 75 mg morphine base) for a period of 5 d before transcardial perfusion or decapitation. Fresh tissue samples included micropunches of either the LC/peri-LC area or medullary regions. Control rats received placebo pellets. This dosage has been shown to yield physical dependence to the drug (Koob et al., 1992). These procedures have been approved by the Institutional Animal Care and Use Committee at Jefferson Medical College of Thomas Jefferson University and conform to National Institutes of Health guidelines.
Immunocytochemistry. After a 5 d period, rats were deeply anesthetized with sodium pentobarbital (60 mg/kg) and perfused transcardially through the ascending aorta with 50 ml of 3.8% acrolein (Electron Microscopy Sciences, Fort Washington, PA) and 200 ml of 2% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4.Vibratome sections (40 μm thick) containing the LC were placed for 30 min in 1% sodium borohydride in 0.1 m PB to remove reactive aldehydes, rinsed extensively in 0.1 m PB, and incubated for 30 min in 0.5% bovine serum albumin in 0.1 mPB before the primary antibody incubation. Tissue sections containing the LC were incubated at room temperature for 15–18 hr in one of the following: a mouse monoclonal anti-leucine5-ENK antibody (1:100; Fitzgerald Laboratories, Concord, MA) or a rabbit anti-methionine5-ENK (1:4000; Diasorin Laboratories, Stillwater, MN) (Van Bockstaele et al., 1996a; Van Bockstaele and Chan, 1997). Two antisera, a polyclonal antiserum (directed against met-ENK) that recognizes several opioid derivatives of the preproenkephalin A and B precursor, and a monoclonal antiserum (leu-ENK) that selectively recognizes derivatives of the preproenkephalin A precursor, were selected in this study to examine the distribution of ENK using both immunocytochemistry and Western blot analysis in the LC. The leu-ENK antibody displays ∼40% cross-reactivity with the C-terminal-extended met-ENK hexapeptides and 7% cross-reactivity with the extended heptapeptide (-Arg-Phe-OH) but does not recognize β-endorphin or dynorphin. The antibody directed against met-ENK was used in the light microscopy analysis. Both met- and leu-ENK antisera were used for the Western blot analysis. Sections were treated with 0.3% Triton X-100 when immunostaining for light microscopic analysis and were not treated with Triton X-100 for immunoelectron microscopic analysis. For light microscopic experiments, antigens were visualized using a standard immunoperoxidase method. Images of immunostained sections were captured using a Dage CCD C72 camera and control unit (using preset gain and black levels), and NIH Image 1.60 software. Uneven illumination across the field was compensated for by subtracting blank fields taken from a slide without tissue, and average pixel densities (out of 256 gray levels) were determined. To compensate for background staining levels, the average pixel density for five small regions that contained nonspecific staining was determined, and this value was subtracted from all density measurements made on that image. Data collected from each region were compared within groups to allow for variations in staining intensities that occur across different immunocytochemical processing runs. Data are reported as means ± SD. Significant decreases between the control and experimental groups were statistically assessed by the Mann–Whitney rank sum test.
For electron microscopy, antigens were visualized using a standard immunogold–silver method (Chan et al., 1990). Silver intensification of the gold particles was achieved using a silver enhancement kit (Amersham, Piscataway, NJ). Sections were rinsed in 0.1 mPB and incubated in 2% osmium tetroxide in 0.1 m PB for 1 hr, washed in 0.1 m PB, dehydrated, and flat-embedded in Epon 812. Thin sections of ∼55–65 nm were cut from the outer surface of the tissue with a diamond knife (Diatome, Fort Washington, PA). Portions of the LC were excised, mounted on Epon chucks, trimmed to trapezoidal blocks, and thin-sectioned (70 nm thickness) on a Leica ultramicrotome. Thin sections were collected on copper grids, counterstained with uranyl acetate and Reynolds' lead citrate, and examined with a Hitachi electron microscope. Captured images of selected thin sections were compared with captured light microscopic images of the blockface before sectioning, and boundaries of the LC were directly mapped onto images of thin sections selected for analysis (Fig. 3). The plastic tissue interface was determined on sections examined from placebo and experimental groups such that comparable depths could be evaluated from the surface of ultrathin sections and direct comparisons could be made between them (Fig. 3A). A profile containing one or two gold particles that was unlabeled in adjacent thin sections was designated as lacking detectable immunoreactivity. As observed in low-magnification electron micrographs, cellular compartments containing at least two or three gold particles were considered to be immunoreactive.
Fig. 3.
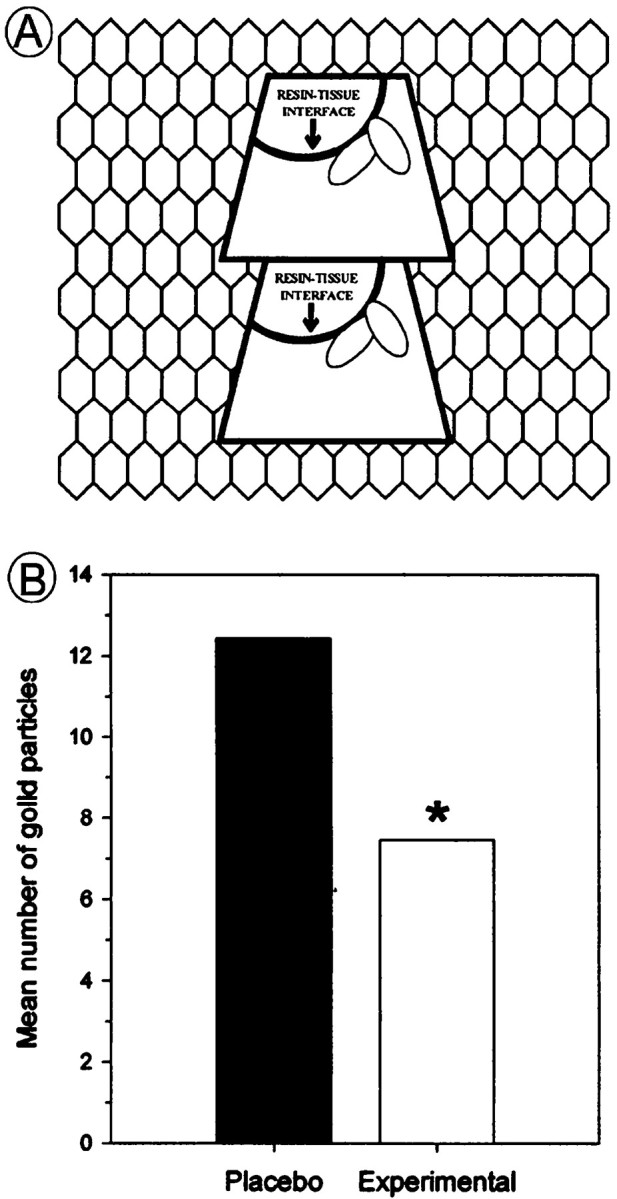
Schematic illustration showing results obtained from quantitative analysis of electron microscopic data.A, Schematic depiction of a copper grid containing a ribbon of sections obtained from 70-nm-thick ultrathin sections.Trapezoids included the LC/peri-LC areas from placebo- and morphine-treated rats. Sections were collected in serial order from the face of the block. Only sections containing the tissue/plastic (resin) interface were included in the analysis so that the results were not thwarted by variations in immunocytochemical labeling based on distances from the surface of the blockface. B, Bar graph showing that ENK-labeled axon terminals from the LC of morphine-treated rats exhibited fewer gold–silver particles as compared with ENK-labeled axon terminals analyzed from placebo-treated rats.
Western immunoblots. For Western blot analysis, samples were collected through the LC area of placebo- or morphine-treated rats and extracted in RIPA buffer containing the following inhibitor mixture: 3 mm sodium pyrophosphate, 50 mm sodium fluoride, 50 μg/ml aprotinin, 5 μg/ml leupeptin, 1 mm sodium vanadate, and 0.5 mmphenylmethylsulfonyl fluoride. Protein levels were quantitated from each sample, and soluble fractions were loaded at equal protein concentrations (37 μg per lane). The proteins were separated in a 4–20% Tris-glycine gel (Novex, San Diego, CA) and then electrophoretically transferred to Immobilon-P membrane (Millipore, Bedford, MA) at 290 mA for 3.5 hr. The blots were incubated in a TBS buffer containing 5% Blotto (nonfat dry milk) and 0.1% Tween 20 for 1 hr to block nonspecific binding sites and then incubated overnight at 4°C in primary antibody (1:1000). Blots were washed with TBS containing 0.1% Tween 20 and then incubated with horseradish peroxidase-conjugated antibodies from Amersham Life Sciences (Arlington Heights, IL) at a dilution of 1:7000 for 1 hr, followed by additional washes before developing with ECL reagents from Amersham Life Sciences. Blots were exposed to X-OMAT AR film from Kodak (Rochester, NY) for different lengths of time to optimize exposures. Bands on film were acquired for analysis using a TWAIN compliant scanner (Hewlett Packard Scan Jet 3C). Variations in the optical density of bands were compared using Kodak Digital Science ID.
RNA probe preparation. Two plasmids containing the entire coding region of rat PPE cDNA were a generous gift of Dr. S. Sabol (National Institutes of Health, Bethesda, MD). They were prepared by ligation of the 935 bp SacI–SmaI fragment of plasmid pRPE2 (Yoshikawa et al., 1984) into the plasmids pSP64/pSP65. For Northern blot analysis, pYSEC1 yielded transcripts that contained the coding sequence of PPE, whereas pYSEA1 transcripts served as controls.
Radioactive antisense cRNA copies were synthesized by incubation of 250 ng of linearized plasmid in 6 mmMgCl2, 30–40 mm Tris, pH 7.9, 2 mm spermidine, 10 mm NaCl, 10 mmdithiothreitol, 0.2 mm ATP/GTP/CTP, (α-35S)UTP, 40 U of RNAsin, and 20 U of SP6 RNA polymerase for 60 min at 37°C. Unincorporated nucleotides were removed using the ammonium acetate method; 100 μl of DNase solution (1 μl of DNase, 5 μl of 5 mg/ml tRNA, 94 μl of 10 mm Tris/10 mm MgCl2) was added, and 10 min later an extraction was accomplished using a phenol chloroform solution. The cRNA was precipitated with 80 μl of 5m ammonium acetate and 500 μl of 100% ethanol for 20 min on dry ice. The pellet was washed with 500 μl of ethanol, dried, and resuspended in 100 μl of 10 mm Tris/1 mmEDTA, pH 8.0. A concentration of 107cpm probe was mixed into 1 ml of hybridization solution (500 μl of formamide, 60 μl of 5 m NaCl, 10 μl of 1m Tris, pH 8.0, 2 μl of 0.5 m EDTA, pH 8.0, 20 μl of 50× Denhardt's solution, 200 μl of 50% dextran sulfate, 50 μl of 10 mg/ml tRNA, 10 μl of 1 m DTT, 118 μl of DEPC water minus volume of probe used). This solution was mixed and heated for 5 min at 65°C before being spotted on slides. Radioactive sense (control) cRNA copies were also prepared to verify the specificity of each probe. Hybridization with these probes did not reveal any positive signal.
RNA isolation and Northern blot analysis. Total cellular RNA (tcRNA) was isolated from 1 to 2 gm of each specific rat tissue according to the methods of Chirgwin et al. (1979). The tissues examined included the medulla, striatum, and heart of adult Sprague Dawley rats treated with either placebo or morphine pellets. tcRNA of each tissue was quantitated spectrophotometrically, and these values were confirmed by agarose/formaldehyde gel electrophoresis in the presence of 100 μg/ml ethidium bromide. Northern blot (RNA blot) analysis was performed with 10 μg of total cellular RNA using 2% agarose/formaldehyde gels and Biotrans nylon membranes (ICN Pharmaceuticals, Costa Mesa, CA). The Northern blots were probed with the pYSEA1- and pYSEC1-specific (950 bp) fragments in accordance with the prehybridization, hybridization, and wash conditions outlined below. The blots were prehybridized in 6× SSC (1× SSC is 0.15m NaCl plus 0.015 m sodium citrate), 5× Denhardt's, 1% SDS, 100 mm sodium phosphate, pH 6.5, 250 μg of salmon sperm DNA per ml at 65°C for 2 hr. Denatured probe was added directly to this mixture, and the membrane was hybridized overnight at 65°C. The hybridization solution was discarded, and the membranes were washed three times for 5 min each and one time for 20 min at room temperature in 2× SSC–0.2% SDS. The membranes were then washed once for 20 min at room temperature and once for 1 hr at 65°C with vigorous agitation in 0.2× SSC–0.2% SDS. Later, these same Northern blots were stripped and reprobed with glyceraldehyde phosphate dehydrogenase (GAPDH) for the purpose of normalization.
Northern blots were exposed to Kodak X-OMAT AR film using an intensifying screen, and multiple exposures were quantitated in the linear range using an LKB Ultrascan XL Enhanced Laser Densitometer and the Gelscan XL program (Pharmacia LKB Biotechnology, Piscataway, NJ). Quantitation values represent an average of multiple scannings of multiple blots to eliminate blot/band inconsistencies. Quantitation values were normalized to a standard specific activity, exposure time, and GAPDH expression. The resulting quantitation values were reported as a specific fold increase or decrease over the baseline value.
In situ hybridization histochemistry. Protocols for riboprobe synthesis, hybridization, and autoradiographic localization of mRNA signal were adapted from Simmons et al. (1989). All solutions were prepared in RNase-free conditions and sterilized to prevent RNA degradation. Tissue sections mounted onto poly-l-lysine-coated slides were desiccated under vacuum overnight, fixed in 4% paraformaldehyde for 20 min, and digested by proteinase K (20 mg/ml in 100 mm Tris HCl, pH 8.0, and 50 mm EDTA at 37°C for 25 min). Thereafter, the brain sections were rinsed in sterile DEPC water followed by a solution of 100 mm triethanolamine (TEA), pH 8.0, acetylated in 0.25% acetic anhydride in 100 mmTEA, and dehydrated through graded concentrations of alcohol (50, 70, 95, and 100%). After being dried under vacuum for a minimum of 2 hr, 90 μl of hybridization mixture (107cpm/ml) was spotted on each slide, sealed under a coverslip, and incubated at 60°C overnight (∼15–20 hr) in a slide warmer. Coverslips were then removed, and the slides were rinsed in 4× SSC at room temperature. Sections were digested by RNase A (10 mg/ml, 37°C, 30 min), rinsed in decreasing concentrations of SSC (2×, 1×, 0.5× SSC), washed in 0.1× SSC for 30 min at 60°C, pH 7.0, and dehydrated through graded concentrations of alcohol. After being dried for 2 hr under vacuum, the sections were exposed at room temperature to x-ray film (MR-1 Biomax, Kodak) for 18 hr, defatted in xylene, and coated with NTB2 nuclear emulsion (Kodak; diluted 1:1 with distilled water). Slides were exposed for ∼6 d, developed in D19 developer (Kodak) for 3.5 min at 14–15°C, rinsed in distilled water, and fixed in rapid fixer (Kodak) for 5 min. Thereafter, tissues were rinsed in running distilled water for 1–2 hr, dehydrated through graded concentrations of alcohol, cleared in xylene, and coverslipped with DPX neutral mounting medium (Aldrich, Milwaukee, WI).
Blind quantitative analysis of hybridization signal for the ENK mRNA signal in the PGi was performed on x-ray film (MR-1, Kodak). Transmittance values of the hybridization signal were measured using a Northern Light Desktop Illuminator (Imaging Research, St. Catharine, Ontario, Canada) with a Micro-Nikkor 60 mm mounted Sony camera coupled to a Power MacIntosh 8600/300. The signal was analyzed with NIH Image software, version 1.61 (W. Rasband, National Institutes of Health, Bethesda, MD). Densitometric analysis yielding measures of optical density was performed according to a standard scale that was established using 14C standard slides (American Radiolabeled Chemicals, St. Louis, MO).
RESULTS
Light microscope and electron microscope analysis of ENK in LC
Alternate sets of sections were processed in parallel for either peroxidase localization of ENK immunoreactivity for light microscopic analysis (Fig. 1A,B) or immunogold–silver localization of ENK for ultrastructural analysis (Fig. 1C, D). In sections obtained from placebo-treated rat brains, peroxidase labeling for ENK (Fig. 1A) was moderately distributed within the portion of the LC containing noradrenergic cell bodies (nuclear LC “core”) and was densely distributed in peri-LC areas including the dorsolateral peri-LC immediately ventral to the superior cerebellar peduncle as previously described (Van Bockstaele et al., 1995). Immunolabeling of ENK in sections through the LC obtained from morphine-treated rats (Fig. 1B) was decreased in both LC and peri-LC areas using brightfield microscopy.
Fig. 1.
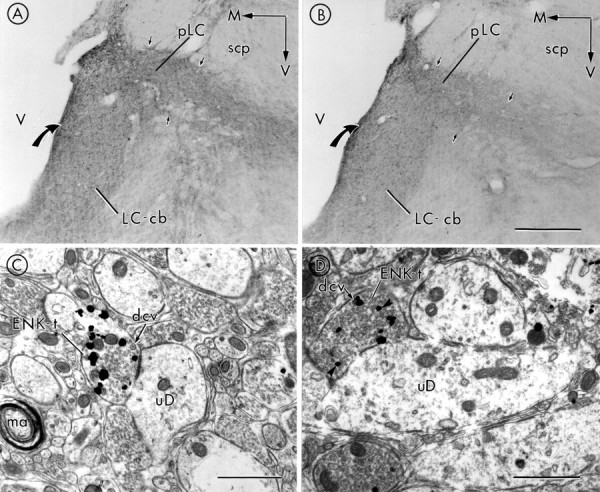
Photomicrographs showing peroxidase labeling for ENK in the caudal aspect of the LC cell body region (cb) (also defined as nuclear or core of the LC) and peri-LC (pLC) areas (region containing noradrenergic dendrites of LC neurons) of placebo- and morphine-treated rats.A, Peroxidase labeling for ENK can be detected in varicose processes in LC and peri-LC areas in a placebo-treated rat. Varicose processes are distributed throughout the region of the LC containing noradrenergic cell bodies (LC-cb) as well as in peri-LC areas (small black arrows), which are known to contain noradrenergic dendrites of LC neurons.B, There is a decrease in peroxidase immunoreactivity for ENK in the LC-cb region as well as in the peri-LC ventral to the superior cerebellar peduncle (scp;small black arrows) in morphine-treated animals. For both Aand B, a large black curved arrow points to the medial aspect of the LC immediately adjacent to the ventricle (V). Arrows point medially (M) and ventrally (V). C, D, Electron micrographs showing immunogold–silver labeling for ENK in axon terminals in the LC (ENK-t) of a placebo-treated rat (C) and a morphine-treated rat (D). Note that in C there are more gold–silver particles indicative of ENK immunolabeling in an axon terminal apposed to an unlabeled dendrite (uD), as compared with the ENK-t in the opiate-dependent rat (D). ENK-t in both C andD contains numerous small, clear vesicles as well as large, dense-core vesicles (dcv). Scale bars:A, B, 250 μm; C, 0.94 μm; D, 0.75 μm. ma, Myelinated axon.
To better quantify decreases in ENK immunoreactivity, subregions of the LC exhibiting peroxidase labeling for ENK were analyzed using brightfield microscopy densitometry throughout the rostrocaudal extent of the dorsal pons. As shown in Figure 2,A and B, the LC was subdivided according to the location of noradrenergic cell bodies as well as the location of noradrenergic dendrites known to extend from LC cell bodies (dorsolateral peri-LC, pLCdL; rostromedial, pLCrm). In the caudal and rostral subdivisions, varicose processes exhibiting peroxidase labeling for ENK were distributed in both LC and peri-LC areas. Light level densitometric analysis confirmed that immunoperoxidase labeling for ENK was decreased in sections obtained from morphine-treated rats (Fig. 2C). A statistically significant decrease could be discerned between the rostral and caudal LC + pLCrm, whereas the caudal and rostral pLCdL did not exhibit statistically different amounts of ENK immunoreactivity (Fig. 2D). These data suggest that the core of the LC + pLCrm exhibits a greater decrease in ENK immunoreactivity as compared with the dorsolateral peri-LC area.
Fig. 2.
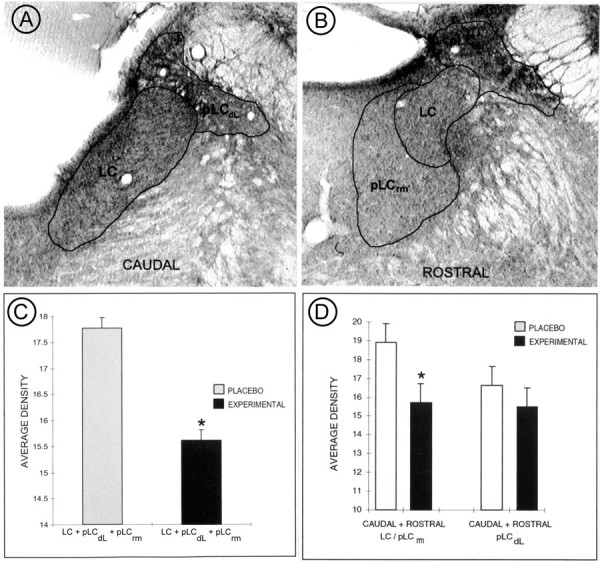
Light level densitometry measurements of coronal tissue sections taken from placebo- and morphine-treated rats that were processed for immunoperoxidase localization of ENK. A,B, Representative sampling of regions defined as LC (which comprises the core of the LC) and peri-LC (which comprises the dorsolateral aspect, pLCdL, as well as the rostromedial, pLCrm) at both caudal and rostral levels of the dorsal pons of control rats. The nuclear LC consists of portions of the dorsal pontine tegmentum containing noradrenergic cell bodies, whereas the peri-LC areas contain noradrenergic dendrites. Because the ENK labeling is extensive in peri-LC areas and extends into the medial parabrachial, sampling was restricted to portions of the neuropil known to contain noradrenergic dendrites. C,D, Bar graphs illustrating quantification of pixel values obtained from LC and peri-LC areas shown in A andB. In C, the values for the LC and pLC were combined, whereas in D the samples are shown for caudal + rostral LC/pLCrm and caudal + rostral pLCdL areas. Note that there is a statistically significant decrease in peroxidase immunolabeling in samples obtained from morphine-treated rats. There is also a statistically significant decrease in the caudal + rostral LC/pLCrm obtained from morphine-treated rats.
For electron microscopy, the core of the LC was sampled, and immunogold–silver detection was used to visualize ENK immunoreactivity. Maps were made of the ultrathin, serially cut sections containing ENK-labeled terminals (Fig.3A), and the plastic/tissue interface was determined. At the ultrastructural level, ENK immunoreactivity appeared as punctate black particles within axon terminals in the LC and peri-LC areas (Fig. 1C,D). Gold–silver labeling for ENK was distributed throughout the axoplasm in axon terminals that contained small clear and large dense-core vesicles (Fig. 1D). Random axonal profiles containing gold–silver labeling for ENK at the plastic/Epon interface were tallied, and the number of gold particles per axon terminal was counted. Approximately 35 profiles were examined from either placebo- or morphine-treated rats. The Mann–Whitney rank sum test indicated that there was a significant decrease in the number of gold particles within axon terminals sampled from the morphine-treated rat group (Fig. 3B) as compared with the placebo-treated rats.
Western blot analysis
To determine whether chronic exposure to morphine resulted in quantitative decreases in either met-ENK or leu-ENK levels, microsamples were collected through the LC and processed for Western blot analysis. Both opioid peptides were expressed in the LC and peri-LC areas, as previously described, using light level immunocytochemistry (Van Bockstaele et al., 1995, 1996a; Van Bockstaele and Chan, 1997). Several bands representing fragments of opioid precursor derivatives could be discerned in both immunoblots. The met-ENK antiserum recognizes fragments from both preproenkephalin A and B. However, the leu-ENK antibody only recognizes derivatives of the preproenkephalin A precursor and does not recognize dynorphin peptides or β-endorphin. Samples obtained from the placebo and experimental rats showed differential expression of opioid peptides. Bands located above the 33.9 kDa marker, which most likely represents the proenkephalin-like opioid peptide, exhibited a reduction in morphine-treated tissue samples. Specifically, for leu-ENK-labeled Western blots, the reduction ranged from 71 to 80%. Decreases for met-ENK ranged from 31 to 60%. These data suggest that chronic exposure to morphine decreases the amount of endogenous opioid peptides in the LC/peri-LC area (Fig. 4).
Fig. 4.

Western blot analysis of samples obtained from the LC of placebo-treated (P) and experimental (E) morphine-treated rats. Expression of the primary antibody directed against met- or leu-ENK was readily detected in microsamples obtained through the LC region. Multiple bands were detected on the gel; however, proteins that migrated above the 33.9 kDa marker most likely represent the migration of proenkephalin-like opioid peptides. Expression was reduced in samples of the LC obtained from morphine-dependent rats (E). The bar graph represents the mean net intensity observed from Western blots (n = 6) processed for met-ENK from placebo- and morphine-treated rats.
Northern blot analysis
Northern blot analysis of a probe recognizing an mRNA transcript for PPE, which was conducted on samples obtained from the medulla, striatum, and heart (control sample) of rats receiving placebo or morphine pellets for 5 d, identified a band of ∼950 bp (Fig.5). Higher levels of PPE mRNA were expressed in the striatum (Fig. 5D), as compared with PPE mRNA levels of samples obtained from the medulla (Fig. 5B) or heart (Fig. 5E), in placebo-treated rats. In samples obtained from morphine-treated rats, PPE mRNA was similar to that of samples obtained from placebo-treated rats. In contrast, medullary regions obtained from morphine-treated rats exhibited a twofold decrease in PPE mRNA expression (Fig. 5A), as compared with morphine-treated rats (Fig. 5B). Ethidium bromide staining and GAPDH expression demonstrated that similar quantities of total cellular RNA were added to all lanes (data not shown). These results indicate that the effects of morphine on ENK levels in the LC are the result of changes in gene expression of PPE mRNA.
Fig. 5.

Northern blot showing a 950 bp band indicative of PPE mRNA in three different tissue specimens: medulla oblongata containing the nucleus paragigantocellularis (PGi) from morphine-treated (lane A) and placebo-treated (lane B) rats, striatum from morphine-treated (lane C) and placebo-treated (lane D) rats, and heart (lane E, used as a positive control for PPE mRNA). Plasmids were successfully transformed into anEscherichia coli strain, and the insert DNA was isolated from the vector by restriction digestion using SacI andSmaI enzymes. Note that the level of PPE mRNA is reduced in lane A, representing medullary regions containing the PGi of morphine-treated rats as compared with a similar sample obtained from placebo-treated rats (lane B).
In situ hybridization analysis
In situ hybridization for PPE mRNA revealed a relatively high positive signal within PGi neurons of saline-treated rats (Fig.6B,C). Morphine treatment significantly reduced the expression of PPE mRNA in the PGi of morphine-treated rats (optical density: saline 2.93 ± 0.08, morphine 2.21 ± 0.11 arbitrary units, p < 0.0009) (Fig. 6B,C).
Fig. 6.
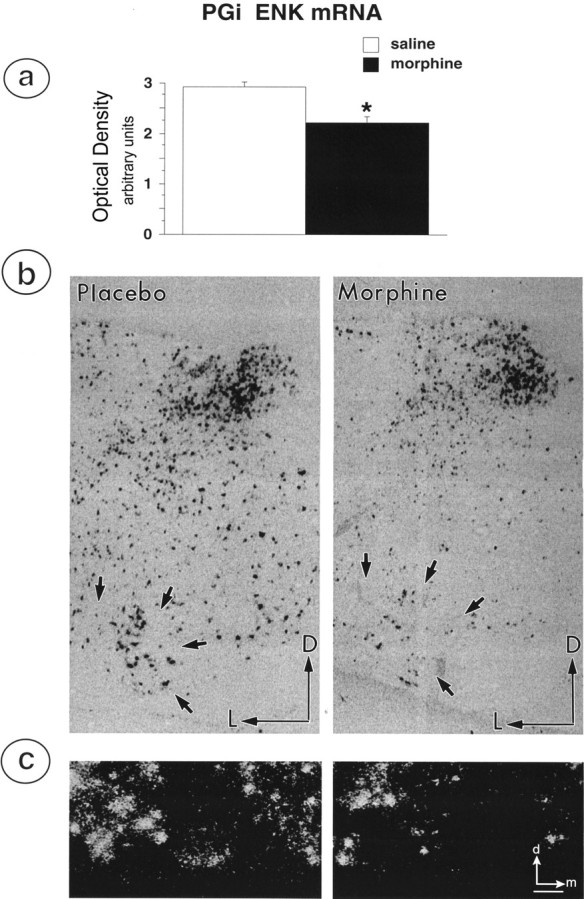
Brain sections through the medulla oblongata of morphine- and placebo-treated rats were processed for in situ hybridization of mRNA for PPE using a plasmid generously provided by Dr. S. Sabol (National Institutes of Health).a, Graphic illustration depicting the effects of morphine treatment on average optical density for ENK mRNA hybridization signal in the PGi. Chronic morphine treatment caused a statistically significant decrease in PPE mRNA expression in PGi neurons. b, Representative examples of coronal sections of medullary sections containing the PGi and processed for in situ hybridization of ENK mRNA expression in placebo- and morphine-treated rats. Straight black arrows indicate location of PGi neurons in the ventral medulla. Arrowspoint dorsally (D) and laterally (L). c, High-magnification image of in situ hybridization labeling of PGi neurons from a placebo-treated (left) and morphine-treated (right) rat. Arrows point dorsally (d) and medially (m). Expression of mRNA for PPE is reduced in samples obtained from morphine-treated rats. Scale bar, 100 μm.
DISCUSSION
This study provides convergent lines of evidence showing a decrease in opioid peptides in afferents to LC and peri-LC areas of opiate-dependent rats. In addition, the decrease in mRNA for PPE in medullary regions, such as the PGi, known to provide opioid input to the LC suggests that opioid peptide gene expression is diminished after chronic morphine exposure. Decreases in endogenous opioids in LC afferents are likely to influence the physiological activity of LC neurons. We have previously provided ultrastructural evidence that ENK coexists with glutamate in a subset of afferent terminals in the LC (Van Bockstaele et al., 2000). A persistent diminution in the presence of ENK in glutamatergic terminals in the LC during opiate withdrawal would suggest that there is a removal of an endogenous inhibitory transmitter that would normally hyperpolarize LC neurons. Thus, diminution in opioid-mediated synaptic transmission may contribute to the observed hyperactivity seen in the LC after withdrawal from opiates.
ENK is decreased after chronic morphine treatment in LC afferents
The present set of studies adds to the existing literature by showing decreases in endogenous opioid peptides in the medullo-coerulear pathway after chronic morphine administration. Decreases in the release of inhibitory-type neurotransmitters from LC afferents during opiate dependence may contribute, along with the reported alterations in signal transduction elements (Nestler and Aghajanian, 1997), to the altered phenotype observed after withdrawal from morphine. Specific changes in endogenous opioid peptide systems occur as a result of chronic opiate administration. For instance, chronic administration of morphine inhibits the release and biosynthesis of β-endorphin by exerting a negative feedback on pro-opiomelanocortin neurons (Bronstein et al., 1993). In other paradigms, the induction of limbic seizures by administration of kainic acid (Hong et al., 1988) has been shown to affect opioid peptide expression in a temporal fashion. Specifically, seizures initially release ENK from mossy fibers and subsequently increase the synthetic rate of opioid peptide-containing neurons in the hippocampus (Hong et al., 1993).
The results of this study suggest transmitter-specific neuroadaptations that occur in the medullo-coerulear circuit after chronic morphine treatment. In opiate-dependent rats, there is a marked increase in the discharge rate of noradrenergic LC neurons during antagonist-induced withdrawal (Aghajanian et al., 1994), which may be related to upregulation of cAMP in LC neurons (Kogan et al., 1992; Nestler et al., 1993). In vivo studies have also suggested that an increase in the release of excitatory amino acids from extrinsic afferents, such as from the PGi of the rostralventral medulla, occurs in the LC during withdrawal from opiates (Aghajanian et al., 1994; Zhang et al., 1994). However, additional brain nuclei are also likely be involved in mediating the somatic signs of opiate withdrawal (Christie et al., 1997; Caille et al., 1999; Delfs et al., 2000) because chemical lesions of the LC before induction of dependence do not attenuate all somatic signs precipitated by systemic naloxone (Chieng and Christie, 1995;Caille et al., 1999; Delfs et al., 2000).
Topographic decreases in ENK immunoreactivity within LC and peri-LC areas may be attributed to differences in the opioid innervation of these subregions by different opioid-containing afferents. We have previously identified the nucleus prepositus hypoglossi and the PGi as endogenous sources of ENK to the “nuclear” or core portion of the LC (Drolet et al., 1992). However, we now have evidence that additional afferents from opioid-enriched regions such as the central nucleus of the amygdala and the nucleus of the solitary tract (NTS) (Van Bockstaele et al., 1996b, 1998, 1999a) target peri-LC areas that contain noradrenergic dendrites of LC neurons. The present study supports alterations in opioid innervation to the nuclear or core aspect of the LC because this region exhibited a significant decrease in ENK innervation as compared with peri-LC areas. Interestingly, the core of the LC was shown to be the region containing most of the dually labeled synaptic endings containing both ENK and glutamate (Van Bockstaele et al., 2000). Taken with the decrease in PPE mRNA in medullary regions known to provide ENK innervation to the LC, our data suggest that ENK projections to the core aspect of the LC are selectively decreased. Opioid-containing afferents may also target specific subsets of LC somata and dendrites that may have distinct postsynaptic targets. It is known that noradrenergic neurons in the ventral aspect of the LC target medullary and spinal cord sites, whereas dorsal noradrenergic neurons project primarily to forebrain sites (Loughlin et al., 1986). Thus, ENK levels within projections from the medulla to the LC are decreased after chronic morphine treatment.
Expression of opioid peptide mRNA in medullary neurons
The present data using Northern blot analysis and in situ hybridization experiments reveal that decreases in ENK peptides in the LC are most likely attributed to decreases in the synthesis of PPE mRNA in neurons afferent to the LC, such as the PGi. Interestingly, others (Tejwani et al., 1994) have shown a 70% decrease in ENK levels in medullary regions of morphine-tolerant animals using radioimmunoassay experiments. Several lines of evidence point to alterations in PGi neurons during withdrawal. Increased c-fos expression has been reported in the PGi after chronic morphine treatment (Stornetta et al., 1993). Increased neuronal activity of presumed adrenergic neurons of the C1 cell group has also been reported during naloxone-precipitated withdrawal (Baraban et al., 1995). Lesions of the PGi have been shown to attenuate the hyperactivity seen after precipitated opiate withdrawal (Rasmussen and Aghajanian, 1989). We have recently described how PGi neurons that project to the LC exhibit prominent immunolabeling for μ opioid receptors (μORs) (Van Bockstaele et al., 1999b), suggesting the hypothesis that alterations in μORs in afferent neurons to the LC may affect the physiological activity of these afferent neurons and, subsequently, neurotransmitter release in LC. Taken together, these data suggest a critical role for circuits originating from the PGi. Because we have previously shown that the ventral medulla provides a significant opioid innervation to the LC, our results extend these observations by showing that decreases in opioid peptide mRNA most likely translate to decreased levels of ENK at postsynaptic targets such as the LC.
Decreases in ENK immunoreactivity in LC afferents after chronic morphine administration may be surprising in light of reports indicating increased levels of cAMP in a host of neurons. Chronic morphine has been shown to increase total levels of cAMP response element binding activity, which in turn should increase expression of the proenkephalin gene and raise ENK levels (Comb et al., 1986; Van Nguyen et al., 1990). However, in certain brain areas (such as the cortex), it has been shown that ENK levels are decreased in the presence of increased cAMP (Sheu et al., 1995). In the medullo-coerulear pathway, it is believed that cAMP levels are altered in LC neurons during chronic exposure to morphine; however, the relationship between cAMP and PPE mRNA expression in medullary projections to the LC has not been investigated. Therefore, although ENK and cAMP are altered during morphine tolerance and dependence, it is still not clear how cAMP affects ENK levels and whether the changes in the levels of cAMP and ENK are interrelated, which may affect the development of morphine tolerance and abstinence (Konradi et al., 1995).
It is tempting to speculate that neurons in the PGi may be poised for coactivating several brain sites that may participate in opiate withdrawal. We previously demonstrated that PGi neurons send axonal collaterals to both the LC and the NTS (Van Bockstaele and Aston-Jones, 1992). The concept that collaterals of PGi neurons may be important in activating additional brainstem circuits is supported by the recent description of the involvement of projections from the NTS to the bed nucleus of the stria terminalis (BNST) in mediating opiate withdrawal (Delfs et al., 2000). Local microinjections of the β-receptor antagonist, propranolol, attenuated withdrawal-induced aversion and also reduced teeth chatters, eye twitches, and wet-dog shakes (Aston-Jones et al., 1999). c-fos expression in the BNST, which is normally increased after withdrawal from opiates, was also shown to be significantly reduced in rats pretreated with propranolol (Aston-Jones et al., 1999).
Functional implications
Assigning a specific neuronal pathway to the mediation of the opiate withdrawal syndrome is not possible. However, it is feasible to dissect changes in defined groups of neurons once alterations in these pathways are known. The benefit of studying the medullocoerulear pathway is that there is substantial information available regarding its physiology and anatomy, and it is clear that this pathway undergoes changes after precipitated withdrawal. Our results indicate that there are decreases in ENK immunoreactivity in the LC after chronic opiate administration. Taken together with our recent description that ENK and glutamate are cotransmitters in a subset of axon terminals in the LC (Van Bockstaele et al., 2000), we postulate that during chronic opiate treatment, there is an imbalance in the levels of ENK (possibly in glutamatergic afferents) in axon terminals innervating LC neurons during chronic opiate dependence that may contribute to hyperactivity of these neurons during withdrawal.
Footnotes
This work was supported by an Established Investigator Award from the American Heart Association and National Institute of Drug Abuse, National Institutes of Health Grant DA10450 to E.V.B. G.D. held a scholarship from Le Fonds de la Recherche en Sante du Quebec. We gratefully acknowledge the gift of plasmids provided by Dr. S. Sabol (National Institutes of Health). We also appreciate the technical advice provided by Dr. Joseph Pierce on the light level densitometry aspect of this study, the technical assistance of Patti Telegan and Vijayalakshmi Gabetta, and comments provided by Dr. Fernando Garcia-Hernandez.
Correspondence should be addressed to Dr. Elisabeth Van Bockstaele, Department of Pathology, Anatomy and Cell Biology, Thomas Jefferson University, 1020 Locust Street, Suite 520, Philadelphia, PA 19107. E-mail:elisabeth.vanbockstaele@mail.tju.edu.
REFERENCES
- 1.Abercrombie ED, Jacobs BL. Systemic naloxone administration potentiates locus coeruleus noradrenergic neuronal activity under stressful but not non-stressful conditions. Brain Res. 1988;441:362–366. doi: 10.1016/0006-8993(88)91415-1. [DOI] [PubMed] [Google Scholar]
- 2.Aghajanian GK. Central noradrenergic neurons: a locus for the functional interplay between alpha-2 adrenoceptors and opiate receptors. J Clin Psychiatry. 1982;43:20–24. [PubMed] [Google Scholar]
- 3.Aghajanian GK, Kogan JH, Moghaddam B. Opiate withdrawal increases glutamate and aspartate efflux in the locus coeruleus: an in vivo microdialysis study. Brain Res. 1994;636:126–130. doi: 10.1016/0006-8993(94)90186-4. [DOI] [PubMed] [Google Scholar]
- 4.Akaoka H, Aston-Jones G. Opiate withdrawal-induced hyperactivity of locus coeruleus neurons is substantially mediated by augmented excitatory amino acid input. J Neurosci. 1991;11:3830–3839. doi: 10.1523/JNEUROSCI.11-12-03830.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alreja M, Aghajanian GK. Opiates suppress a resting sodium-dependent inward current and activate an outward potassium current in locus coeruleus neurons. J Neurosci. 1993;13:3525–3532. doi: 10.1523/JNEUROSCI.13-08-03525.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andrade R, Vandermaelen CP, Aghajanian GK. Morphine tolerance and dependence in the locus coeruleus: single cell studies in brain slices. Eur J Pharmacol. 1983;91:161–169. doi: 10.1016/0014-2999(83)90461-2. [DOI] [PubMed] [Google Scholar]
- 7.Aston-Jones G, Delfs JM, Druhan J, Zhu Y. The bed nucleus of the stria terminalis: a target site for noradrenergic actions in opiate withdrawal. Ann N Y Acad Sci. 1999;877:486–498. doi: 10.1111/j.1749-6632.1999.tb09284.x. [DOI] [PubMed] [Google Scholar]
- 8.Baraban SC, Stornetta RL, Guyenet PG. Effects of morphine and morphine withdrawal on adrenergic neurons of the rat rostral ventrolateral medulla. Brain Res. 1995;676:245–257. doi: 10.1016/0006-8993(95)00097-a. [DOI] [PubMed] [Google Scholar]
- 9.Bronstein DM, Day NC, Gutstein HB, Trujillo KA, Akil H. Pre- and posttranslational regulation of beta-endorphin biosynthesis in the CNS: effects of chronic naltrexone treatment. J Neurochem. 1993;60:40–49. doi: 10.1111/j.1471-4159.1993.tb05820.x. [DOI] [PubMed] [Google Scholar]
- 10.Caille S, Espejo EF, Reneric JP, Cador M, Koob GF, Stinus L. Total neurochemical lesion of noradrenergic neurons of the locus coeruleus does not alter either naloxone-precipitated or spontaneous opiate withdrawal nor does it influence ability of clonidine to reverse opiate withdrawal. J Pharmacol Exp Ther. 1999;290:881–892. [PubMed] [Google Scholar]
- 11.Chan J, Aoki C, Pickel VM. Optimization of differential immunogold–silver and peroxidase labeling with maintenance of ultrastructure in brain sections before plastic embedding. J Neurosci Methods. 1990;33:113–127. doi: 10.1016/0165-0270(90)90015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chieng B, Christie MJ. Lesions to terminals of noradrenergic locus coeruleus neurones do not inhibit opiate withdrawal behaviour in rats. Neurosci Lett. 1995;186:37–40. doi: 10.1016/0304-3940(95)11276-3. [DOI] [PubMed] [Google Scholar]
- 13.Childers SR, Simantov R, Snyder SH. Enkephalin: radioimmunoassay and radioreceptor assay in morphine dependent rats. Eur J Pharmacol. 1977;46:289–293. doi: 10.1016/0014-2999(77)90346-6. [DOI] [PubMed] [Google Scholar]
- 14.Chirgwin DM, Przybyla AE, MacDonald RJ, Rutter WJ. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry. 1979;18:5294–5299. doi: 10.1021/bi00591a005. [DOI] [PubMed] [Google Scholar]
- 15.Christie MJ, Williams JT, North RA. Cellular mechanisms of opioid tolerance: studies in single brain neurons. Mol Pharmacol. 1987;32:633–638. [PubMed] [Google Scholar]
- 16.Christie MJ, Williams JT, Osborne PB, Bellchambers CE. Where is the locus in opioid withdrawal? Trends Pharmacol Sci. 1997;18:134–140. doi: 10.1016/s0165-6147(97)01045-6. [DOI] [PubMed] [Google Scholar]
- 17.Comb M, Birnberg NC, Seasholtz A, Herbert E, Goodman HM. A cyclic AMP- and phorbol ester-inducible DNA element. Nature. 1986;323:353–356. doi: 10.1038/323353a0. [DOI] [PubMed] [Google Scholar]
- 18.Delfs J, Zhu Y, Druhan J, Aston-Jones G. Noradrenaline in the ventral forebrain is critical for opiate withdrawal-induced aversion. Nature. 2000;403:430–434. doi: 10.1038/35000212. [DOI] [PubMed] [Google Scholar]
- 19.Drolet G, Van Bockstaele EJ, Aston-Jones G. Robust enkephalin innervation of the locus coeruleus from the rostral medulla. J Neurosci. 1992;12:3162–3174. doi: 10.1523/JNEUROSCI.12-08-03162.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ennis M, Aston-Jones G. Activation of locus coeruleus from nucleus paragigantocellularis: a new excitatory amino acid pathway in brain. J Neurosci. 1988;8:3644–3657. doi: 10.1523/JNEUROSCI.08-10-03644.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fratta W, Yang HY, Hong J, Costa E. Stability of Met-enkephalin content in brain structures of morphine-dependent or foot shock-stressed rats. Nature. 1977;268:452–453. doi: 10.1038/268452a0. [DOI] [PubMed] [Google Scholar]
- 22.Gold MS, Pottash AC, Extein IL, Kleber HD. Neuroanatomical sites of action of clonidine in opiate withdrawal: the locus coeruleus connection. Prog Clin Biol Res. 1981;71:285–298. [PubMed] [Google Scholar]
- 23.Grant SJ, Huang YH, Redmond DE., Jr Behavior of monkeys during opiate withdrawal and locus coeruleus stimulation. Pharmacol Biochem Behav. 1988;30:13–19. doi: 10.1016/0091-3057(88)90419-4. [DOI] [PubMed] [Google Scholar]
- 24.Gudehithlu KP, Tejwani GA, Bhargava HN. Beta-endorphin and methionine-enkephalin levels in discrete brain regions, spinal cord, pituitary gland and plasma of morphine tolerant, dependent, and abstinent rats. Brain Res. 1991;553:284–290. doi: 10.1016/0006-8993(91)90836-k. [DOI] [PubMed] [Google Scholar]
- 25.Guitart X, Thompson MA, Mirante CK, Greenberg ME, Nestler EJ. Regulation of cyclic AMP response element-binding protein (CREB) phosphorylation by acute and chronic morphine in the rat locus coeruleus. J Neurochem. 1992;58:1168–1171. doi: 10.1111/j.1471-4159.1992.tb09377.x. [DOI] [PubMed] [Google Scholar]
- 26.Guitart X, Kogan JH, Berhow M, Terwilliger RZ, Aghajanian GK, Nestler EJ. Lewis and Fischer rat strains display differences in biochemical, electrophysiological and behavioral parameters: studies in the nucleus accumbens and locus coeruleus of drug naive and morphine-treated animals. Brain Res. 1993;611:7–17. doi: 10.1016/0006-8993(93)91770-s. [DOI] [PubMed] [Google Scholar]
- 27.Hayward MD, Duman RS, Nestler EJ. Induction of the c-fos proto-oncogene during opiate withdrawal in the locus coeruleus and other regions of rat brain. Brain Res. 1990;525:256–266. doi: 10.1016/0006-8993(90)90872-9. [DOI] [PubMed] [Google Scholar]
- 28.Hong JS, McGinty JF, Grimes L, Kanamatsu T, Obie J, Mitchell CL. Seizure-induced alterations in the metabolism of hippocampal opioid peptides suggest opioid modulation of seizure-related behaviors. NIDA Res Monogr. 1988;82:48–66. [PubMed] [Google Scholar]
- 29.Hong JS, McGinty JF, Lee PH, Xie CW, Mitchell CL. Relationship between hippocampal opioid peptides and seizures. Prog Neurobiol. 1993;40:507–528. doi: 10.1016/0301-0082(93)90020-s. [DOI] [PubMed] [Google Scholar]
- 30.Kogan JH, Aghajanian GK. Long-term glutamate desensitization in locus coeruleus neurons and its role in opiate withdrawal. Brain Res. 1995;689:111–121. doi: 10.1016/0006-8993(95)00545-2. [DOI] [PubMed] [Google Scholar]
- 31.Kogan JH, Nestler EJ, Aghajanian GK. Elevated basal firing rates and enhanced responses to 8-Br-cAMP in locus coeruleus neurons in brain slices from opiate-dependent rats. Eur J Pharmacol. 1992;211:47–53. doi: 10.1016/0014-2999(92)90261-2. [DOI] [PubMed] [Google Scholar]
- 32.Konradi C, Cole RL, Green D, Senatus P, Leveque JC, Pollack AE, Grossbard SJ, Hyman SE. Analysis of the proenkephalin second messenger-inducible enhancer in rat striatal cultures. J Neurochem. 1995;65:1007–1015. doi: 10.1046/j.1471-4159.1995.65031007.x. [DOI] [PubMed] [Google Scholar]
- 33.Koob GF, Maldonado R, Stinus L. Neural substrates of opiate withdrawal. Trends Neurosci. 1992;15:186–191. doi: 10.1016/0166-2236(92)90171-4. [DOI] [PubMed] [Google Scholar]
- 34.Krystal JH, Compere S, Nestler EJ, Rasmussen K. Nimodipine reduction of naltrexone-precipitated locus coeruleus activation and abstinence behavior in morphine-dependent rats. Physiol Behav. 1996;59:863–866. doi: 10.1016/0031-9384(95)02206-6. [DOI] [PubMed] [Google Scholar]
- 35.Loughlin SE, Foote SL, Bloom FE. Efferent projections of nucleus locus coeruleus: topographic organization of cells of origin demonstrated by three-dimensional reconstruction. Neuroscience. 1986;18:291–306. doi: 10.1016/0306-4522(86)90155-7. [DOI] [PubMed] [Google Scholar]
- 36.Maldonado R, Koob GF. Destruction of the locus coeruleus decreases physical signs of opiate withdrawal. Brain Res. 1993;605:128–138. doi: 10.1016/0006-8993(93)91364-x. [DOI] [PubMed] [Google Scholar]
- 37.Maldonado R, Valverde O, Garbay C, Roques BP. Protein kinases in the locus coeruleus and periaqueductal gray matter are involved in the expression of opiate withdrawal. Naunyn Schmiedebergs Arch Pharmacol. 1995;352:565–575. doi: 10.1007/BF00169392. [DOI] [PubMed] [Google Scholar]
- 38.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 39.Nestler EJ, Hope BT, Widnell KL. Drug addiction: a model for the molecular basis of neural plasticity. Neuron. 1993;11:995–1006. doi: 10.1016/0896-6273(93)90213-b. [DOI] [PubMed] [Google Scholar]
- 40.North RA, Williams JT, Surprenant A, Christie MJ. Mu and delta receptors belong to a family of receptors that are coupled to potassium channels. Proc Natl Acad Sci USA. 1987;84:5487–5491. doi: 10.1073/pnas.84.15.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rasmussen K. The role of the locus coeruleus and N-methyl-d-aspartic acid (NMDA) and AMPA receptors in opiate withdrawal. Neuropsychopharmacology. 1995;13:295–300. doi: 10.1016/0893-133X(95)00082-O. [DOI] [PubMed] [Google Scholar]
- 42.Rasmussen K, Aghajanian GK. Withdrawal-induced activation of locus coeruleus neurons in opiate-dependent rats: attenuation by lesions of the nucleus paragigantocellularis. Brain Res. 1989;505:346–350. doi: 10.1016/0006-8993(89)91466-2. [DOI] [PubMed] [Google Scholar]
- 43.Rasmussen K, Beitner-Johnson DB, Krystal JH, Aghajanian GK, Nestler EJ. Opiate withdrawal and the rat locus coeruleus: behavioral, electrophysiological, and biochemical correlates. J Neurosci. 1990;10:2308–2317. doi: 10.1523/JNEUROSCI.10-07-02308.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rasmussen K, Kendrick WT, Kogan JH, Aghajanian GK. A selective AMPA antagonist, LY293558, suppresses morphine withdrawal-induced activation of locus coeruleus neurons and behavioral signs of morphine withdrawal. Neuropsychopharmacology. 1996;15:497–505. doi: 10.1016/S0893-133X(96)00094-2. [DOI] [PubMed] [Google Scholar]
- 45.Redmond DE, Jr, Huang YH. The primate locus coeruleus and effects of clonidine on opiate withdrawal. J Clin Psychiatry. 1982;43:25–29. [PubMed] [Google Scholar]
- 46.Sheu MJ, Sribanditmongkol P, Santosa DN, Tejwani GA. Inhibition of morphine tolerance and dependence by diazepam and its relation to cyclic AMP levels in discrete rat brain regions and spinal cord. Brain Res. 1995;675:31–37. doi: 10.1016/0006-8993(95)00036-p. [DOI] [PubMed] [Google Scholar]
- 47.Simmons DM, Arriza JL, Swanson LW. A complete protocol for in situ hybridization of messenger RNAs in brain and other tissues with radiolabeled single stranded RNA probes. J Histotechnol. 1989;12:169–181. [Google Scholar]
- 48.Stornetta RL, Norton FE, Guyenet PG. Autonomic areas of rat brain exhibit increased Fos-like immunoreactivity during opiate withdrawal in rats. Brain Res. 1993;624:19–28. doi: 10.1016/0006-8993(93)90055-r. [DOI] [PubMed] [Google Scholar]
- 49.Tejwani GA, Rattan AK. Met-enkephalin alteration in the rat during chronic injection of morphine and/or midazolam. Brain Res. 1997;775:119–126. doi: 10.1016/s0006-8993(97)00875-5. [DOI] [PubMed] [Google Scholar]
- 50.Tejwani GA, Rattan AK, Koo KL, Matwyshyn GA, Bhargava HN. Methionine-enkephalin concentrations in discrete brain regions, spinal cord, pituitary gland and peripheral tissues of U-50,488H-tolerant and abstinent rats. Pharmacology. 1994;48:216–225. doi: 10.1159/000139183. [DOI] [PubMed] [Google Scholar]
- 51.Tokuyama S, Zhu H, Wakabayashi H, Feng YZ, Ho IK. The role of glutamate in the locus coeruleus during opioid withdrawal and effects of H-7, a protein kinase inhibitor, on the action of glutamate in rats. J Biomed Sci. 1998;5:45–53. doi: 10.1007/BF02253355. [DOI] [PubMed] [Google Scholar]
- 52.Van Bockstaele EJ, Aston-Jones G. Collateralized projections from neurons in the rostral medulla to the nucleus locus coeruleus, the nucleus of the solitary tract and the periaqueductal gray. Neuroscience. 1992;49:653–668. doi: 10.1016/0306-4522(92)90234-s. [DOI] [PubMed] [Google Scholar]
- 53.Van Bockstaele EJ, Chan J. Electron microscopic evidence for coexistence of leucine5-enkephalin and gamma-aminobutyric acid in a subpopulation of axon terminals in the rat locus coeruleus region. Brain Res. 1997;746:171–182. doi: 10.1016/s0006-8993(96)01194-8. [DOI] [PubMed] [Google Scholar]
- 54.Van Bockstaele EJ, Branchereau P, Pickel M. Morphologically heterogeneous met-enkephalin terminals form synapses with tyrosine hydroxylase-containing dendrites in the rat nucleus locus coeruleus. J Comp Neurol. 1995;363:423–438. doi: 10.1002/cne.903630307. [DOI] [PubMed] [Google Scholar]
- 55.Van Bockstaele EJ, Chan J, Biswas A. Ultrastructural evidence for convergence of enkephalin and adrenaline-containing axon terminals on common targets and their presynaptic associations in the rat nucleus locus coeruleus. Brain Res. 1996a;718:61–75. doi: 10.1016/0006-8993(96)00004-2. [DOI] [PubMed] [Google Scholar]
- 56.Van Bockstaele EJ, Chan J, Pickel VM. Input from central nucleus of the amygdala efferents to pericoerulear dendrites, some of which contain tyrosine hydroxylase immunoreactivity. J Neurosci Res. 1996b;45:289–302. doi: 10.1002/(SICI)1097-4547(19960801)45:3<289::AID-JNR11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 57.Van Bockstaele EJ, Colago EEO, Valentino RJ. Amygdaloid corticotropin-releasing factor targets locus coeruleus dendrites: substrate for the coordination of emotional and cognitive limbs of the stress response. J Neuroendocrinol. 1998;10:743–757. doi: 10.1046/j.1365-2826.1998.00254.x. [DOI] [PubMed] [Google Scholar]
- 58.Van Bockstaele EJ, Peoples J, Telegan P. Efferent projections of the nucleus of the solitary tract to peri-locus coeruleus dendrites in rat brain: evidence for a monosynaptic pathway. J Comp Neurol. 1999a;412:410–428. doi: 10.1002/(sici)1096-9861(19990927)412:3<410::aid-cne3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 59.Van Bockstaele EJ, Saunders A, Telegan P, Page ME. Localization of mu-opioid receptors to locus coeruleus-projecting neurons in the rostral medulla: morphological substrates and synaptic organization. Synapse. 1999b;34:154–167. doi: 10.1002/(SICI)1098-2396(199911)34:2<154::AID-SYN8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 60.Van Bockstaele EJ, Saunders A, Commons KG, Liu XB, Peoples J. Evidence for coexistence of enkephalin and glutamate in axon terminals and cellular sites for functional interactions of their receptors in the rat locus coeruleus. J Comp Neurol. 2000;417:103–114. doi: 10.1002/(sici)1096-9861(20000131)417:1<103::aid-cne8>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 61.Van Nguyen T, Kobierski L, Comb M, Hyman SE. The effect of depolarization on expression of the human proenkephalin gene is synergistic with cAMP and dependent upon a cAMP-inducible enhancer. J Neurosci. 1990;10:2825–2833. doi: 10.1523/JNEUROSCI.10-08-02825.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wesche D, Hollt V, Herz A. Radioimmunoassay of enkephalins. Regional distribution in rat brain after morphine treatment and hypophysectomy. Naunyn Schmiedebergs Arch Pharmacol. 1977;301:79–82. doi: 10.1007/BF00501267. [DOI] [PubMed] [Google Scholar]
- 63.Williams JT, Christie MJ, North RA, Roques BP. Potentiation of enkephalin action by peptidase inhibitors in rat locus ceruleus in vitro. J Pharmacol Exp Ther. 1987;243:397–401. [PubMed] [Google Scholar]
- 64.Yoshikawa K, Williams C, Sabol SL. Rat brain preproenkephalin mRNA. cDNA cloning, primary structure, and distribution in the central nervous system. J Biol Chem. 1984;259:14301–14308. [PubMed] [Google Scholar]
- 65.Zhang T, Feng Y, Rockhold RW, Ho IK. Naloxone-precipitated morphine withdrawal increases pontine glutamate levels in the rat. Life Sci. 1994;55:PL25–31. doi: 10.1016/0024-3205(94)90108-2. [DOI] [PubMed] [Google Scholar]