

Abstract
A central feature of drugs of abuse is to induce gene expression in discrete brain structures that are critically involved in behavioral responses related to addictive processes. Although extracellular signal-regulated kinase (ERK) has been implicated in several neurobiological processes, including neuronal plasticity, its role in drug addiction remains poorly understood. This study was designed to analyze the activation of ERK by cocaine, its involvement in cocaine-induced early and long-term behavioral effects, as well as in gene expression. We show, by immunocytochemistry, that acute cocaine administration activates ERK throughout the striatum, rapidly but transiently. This activation was blocked when SCH 23390 [a specific dopamine (DA)-D1 antagonist] but not raclopride (a DA-D2 antagonist) was injected before cocaine. Glutamate receptors of NMDA subtypes also participated in ERK activation, as shown after injection of the NMDA receptor antagonist MK 801. The systemic injection of SL327, a selective inhibitor of the ERK kinase MEK, before cocaine, abolished the cocaine-induced ERK activation and decreased cocaine-induced hyperlocomotion, indicating a role of this pathway in events underlying early behavioral responses. Moreover, the rewarding effects of cocaine were abolished by SL327 in the place-conditioning paradigm. Because SL327 antagonized cocaine-induced c-fos expression and Elk-1 hyperphosphorylation, we suggest that the ERK intracellular signaling cascade is also involved in the prime burst of gene expression underlying long-term behavioral changes induced by cocaine. Altogether, these results reveal a new mechanism to explain behavioral responses of cocaine related to its addictive properties.
Keywords: cocaine, ERK, Elk-1, c-fos expression, striatum, dopamine receptors, reward
A central feature of psychostimulants is to produce, in rodents, a release of dopamine (DA) in the mesolimbic system (Di Chiara and Imperato, 1988) and to induce gene expression in discrete brain structures (Hope et al., 1992;Moratalla et al., 1993). These molecular events are thought to be crucial for behavioral responses related to addictive processes (for review, see Nestler and Aghajanian, 1997; Berke and Hyman, 2000). In this way, acute psychostimulant administration has been reported to induce immediate early gene (IEG) expression in the striatum, a major cerebral target of DA inputs (Graybiel et al., 1990; Young et al., 1991; Hope et al., 1992; Moratalla et al., 1993; Konradi et al., 1994;Berke et al., 1998). Although their induction is transient because most mRNAs return to baseline within a few hours to a day, some of them are thought to form the basis of initial neural plasticity as they encode transcription factors (c-fos, JunB). Chronic exposure desensitizes the capability of psychostimulants to induce these proteins and results instead in a gradual long-term accumulation of novel Fos-related proteins, termed chronic FRAs (Hope et al., 1994), which are associated with long-lasting changes in synaptic efficacy and structural changes (Fitzgerald et al., 1996; Kelz et al., 1999).
The role of DA-D1 receptor subtype in transient bursts of altered gene expression and behavioral responses is now well established because both are blocked by D1 antagonists (Cabib et al., 1991; Young et al., 1991) and by the deletion of D1 receptors in knock-out mice (Xu et al., 1994a,b; Moratalla et al., 1996). By elevating intracellular cAMP levels, and thereby activating PKA, DA-D1 stimulation leads to cAMP response element-binding protein (CREB) phosphorylation, which in turn controls transcriptional expression via the cAMP and calcium response element (CRE) present in the promoter of various IEGs (such as c-fos, junB, or zif268) (for review, see Herdegen and Leah, 1998). However, recent evidences in PC12 and neuronal cells show that cAMP can also stimulate extracellular signal-regulated kinase (ERK) (Vossler et al., 1997; Impey et al., 1998; Vincent et al., 1998;Yao et al., 1998; York et al., 1998; Roberson et al., 1999), which is thought to play an important role in neuronal adaptive responses (English and Sweatt, 1996, 1997; Martin et al., 1997; Davis et al., 2000) as well as memory formation (Atkins et al., 1998). After activation, ERK proteins can translocate to the nucleus (Chen et al., 1992), where they phosphorylate the ternary complex factor Elk-1 (Gille et al., 1992, 1995) and thereby control c-fos transcription via the serum response element (SRE) (for review, see Hill et al., 1993; Marais et al., 1993; Hipskind et al., 1994a,b; Janknecht et al., 1994;Treisman, 1995; Zinck et al., 1995).
The aim of this study was to investigate whether acute cocaine administration induced ERK activation in the striatum. We report a rapid and transient activation of ERK in vivo, in different striatal subregions. This activation is linked to D1 receptor stimulation, with a partial contribution of D2 and NMDA receptors. Using a specific inhibitor of MEK, the dual-specific protein kinase that phosphorylates ERK, we partially antagonized cocaine-induced locomotor response and totally reversed cocaine-rewarding effects on the place preference paradigm. Furthermore, cocaine-induced c-fos expression and Elk-1 hyperphosphorylation were also blocked by the MEK inhibitor. This suggests that the control exerted by the ERK–Elk-1 cascade on the prime burst of gene expression in striatum plays a crucial role in some cocaine behavioral response, such as rewarding effects.
MATERIALS AND METHODS
Animals and drug. Male CD-1 mice (Charles River, France) weighing 22–24 gm were housed 10 per cage and acclimatized to the laboratory conditions (12 hr light/dark cycle, 21 ± 1°C room temperature) 1 week before the experiment. Food and water were available ad libitum. Behavioral tests and animal care were conducted in accordance with the standard ethical guidelines (National Institutes of Health, publication number 85–23, revised 1985; European Community Guidelines on the Care and Use of Laboratory Animals) and approved by the local ethics committee. All drugs were administrated by intraperitoneal injection. Cocaine (Sigma, Arbresle, France), R(+)-SCH 23390 (Sigma),S(−)-raclopride (Sigma), and (+)-MK 801 (Sigma) were dissolved in saline 0.9%. The MEK inhibitor SL327 was dissolved in DMSO and then diluted twice in sterile water.
Treatments. The same experimental conditions and doses were used for immunocytochemistry and behavioral assay. R(+)-SCH 23390 (0.1 mg/kg) and S(−)-raclopride (0.3 mg/kg) were injected 15 min before cocaine injection (20 mg/kg). (+)-MK 801 (0.15 mg/kg) was administered 30 min before cocaine treatment. SL327 (50 mg/kg) was injected 1 hr before cocaine administration. For the conditioned place preference experiment, SL327 and/or cocaine were administered every drug-conditioning day. For chronic treatments, cocaine (20 mg/kg) was injected once daily for 6 d. On day seven, a challenge of cocaine was performed at the same dose.
Tissue preparation for immunocytochemistry. Mice brains were fixed by intracardiac perfusion of 4% paraformaldehyde (PFA) in 0.1mNa2HPO4–NaH2PO4buffer, pH 7.5 (phosphate buffer), delivered with a peristaltic pump at 20 ml/min for 5 min. Brains were removed and post-fixed overnight in the same fixative solution. Sections (30 μm) were cut with a vibratome (Leica, Nussloch, Germany) and then kept in a solution containing 30% ethylene glycol, 30% glycerol, 0.1 mphosphate buffer, and 0.1% diethylpyrocarbonate (DEPC; Sigma, Deisenhofen, Germany) at −20°C until they were processed for immunocytochemistry.
Antibodies. The anti-active ERK antibody was a polyclonal antibody raised against the dually phosphorylated Thr/Glu/Tyr region within the catalytic core of the active form of p44–ERK1 and p42–ERK2 (anti-phospho Thr183-Tyr185 ERKs; New England Biolabs, Beverly, MA). The anti-active Elk-1 antibody was a monoclonal antibody directed against a phospho-Ser383 peptide corresponding to residues 379–392 of Elk-1 (Santa Cruz Biotechnology, Santa Cruz, CA). The c-fos antibody was a polyclonal antibody directed against residues 3–16 of human c-fos (Santa Cruz). The dilutions used for immunostaining were 1:400 for p-ERK antiserum; 1:250 for p-Elk-1 antiserum, and 1:1000 for c-fos.
Immunocytochemistry. The immunohistochemical procedure was adapted from protocols previously described (Sgambato et al., 1998) except that for the detection of phosphorylated proteins, 0.1 mm NaF was included in all buffers and incubation solutions. Day 1: Free-floating sections were rinsed in Tris-buffered saline (TBS; 0.25 m Tris and 0.5 m NaCl, pH 7.5), incubated for 5 min in TBS containing 3% H2O2 and 10% methanol, and then rinsed three rimes for 10 min each in TBS. After a 15 min incubation in 0.2% Triton X-100 in TBS, the sections were rinsed three times in TBS. These were incubated with the primary antibody for 72 hr (cFos) or overnight (p-ERK, p-Elk-1) at 4°C. Day 2: After three rinses in TBS, the sections were incubated for 2 hr at room temperature with the secondary biotinylated antibody (anti-IgG), using a dilution twice that of the first antibody in TBS. After being washed, the sections were incubated for 90 min in avidin–biotin–peroxidase complex (ABC) solution (final dilution, 1:50; Vector Laboratories, Peterborough, UK). The sections were then washed in TBS and twice in TB (0.25 m Tris, pH 7.5) for 10 min each, placed in a solution of TB containing 0.1% 3,3′-diaminobenzidine (DAB; 50 mg/100 ml), and developed by H2O2 addition (0.02%). After processing, the tissue sections were mounted onto gelatin-coated slides and dehydrated through alcohol to xylene for light microscopic examination.
P-ERK positive neurons were plotted at 10× magnification using a computerized image analyzer (Biocom). Cell counts were performed for each mouse in the whole striatum divided in dorsomedial (DM), dorsolateral (DL), core, and shell. In each region, the total amount of P-ERK-positive neurons (evaluated on the basis of a cytoplasmic and nuclear staining) was counted.
Adenylyl cyclase assays. Mouse brains were sectioned in 300-μm-thick slices in Ca2+-free artificial CSF (in mm: NaCl 125, KCl 2.4, KH2PO4 0.5, Na2SO4 0.5, MgCl2 1.93, NaHCO3 27, and glucose 10) using Vibroslice apparatus (Campden Instruments, Leicester, UK). Tissue microdisks were punched out from caudate putamen using a stainless steel cylinder and homogenized at 1 mg of protein per milliliter in a buffer containing 2 mm Tris-maleate, pH 7.2, 2 mm EGTA, and 300 mm sucrose using a Potter-Elvehjem apparatus. Adenylyl cyclase activity was measured by the conversion of α-(32P)-ATP into cyclic (32P)-AMP as described previously (Bockaert et al., 1977). Adenylyl cyclase activities were measured in the presence of vehicle or dopamine 10−4m and various concentrations of SL327 (0.3, 3, 10, 30, 100, and 300 μm). The cyclic (32P)-AMP formed was isolated according toSalomon et al. (1974), and dopamine response on adenylyl cyclase activity was calculated in picomoles of cAMP produced per minute and per milligram of protein and expressed as percentage increase.
Locomotor assay. Locomotor activity was measured by using locomotor activity boxes consisted in individual plastic rectangular area (9 × 20 × 11 cm; Imetronic). The boxes contained a line of photocells 2 cm above the floor to measure horizontal movements and another line located 6 cm above the floor to measure vertical activity (rearings). Mice were individually placed in the locomotor boxes 5 min after injection of drug or saline, and locomotion was recorded for 15 min in a low luminosity environment (20–25 lux).
Conditioned place preference. Cocaine-rewarding effects were evaluated in the conditioned place preference paradigm by using an unbiased procedure, as previously described (Maldonado et al., 1997). Briefly, the protocol consists of three different phases: preconditioning, conditioning, and postconditioning phases. During preconditioning phase, mice were allowed ad libitum access for 18 min to the two chambers that were distinguished by different patterns on floors and walls. During conditioning phase, mice received pairing of saline or cocaine (20 mg/kg) once a day for 6 d in separate compartments using a counterbalanced design. After injection, animals were confined to a given chamber for a period of 25 min. Each mouse received three pairings each in one chamber with cocaine and other with saline. The postconditioning phase began 24 hr after last conditioning session, when animals were permitted ad libitumaccess to both chambers. Place preference was quantified in terms of time spent in drug paired side. A score was calculated for each mouse as the difference between postconditioning and preconditioning time spent in drug-paired compartment.
Statistical analysis. Data were analyzed using one-way ANOVA between subjects for immunocytochemistry, locomotion, and score values of conditioned place preference. Post hoccomparisons were made using the Newman–Keuls test. Preconditioning and postconditioning times spent in the drug-paired compartment were analyzed using two-way ANOVA with between (treatment) and within (phase) groups factors, followed by corresponding one-way ANOVA andpost hoc comparisons (Newman–Keuls test). In all cases, significance was set at p < 0.05.
RESULTS
Acute cocaine treatment induces ERK activation throughout the striatum
After the administration of cocaine (20 mg/kg), the temporal pattern of ERK activation was analyzed in vivo, into the striatum, by immunocytochemistry. This activation was detected with an antiserum specifically recognizing the phosphorylated form (anti-phospho-Thr183 and Tyr185) of ERK proteins, as tested by Western blot (Sgambato et al., 1998; Davis et al., 2000). In saline-treated mice, no P-ERK immunoreactivity was found in the nucleus accumbens (NA), and only a low level was detectable in the dorsal striatum (Fig.1A,B). By contrast, 5 and 10 min after cocaine administration, immunostaining for P-ERK increased markedly throughout the dorsal striatum and the NA (Fig.1A,B). As exemplified in Figure 1B, P-ERK immunostaining corresponded to both P-ERK-positive neurons and neuropil. In neuronal cells, the immunolabeling was mainly present in cytoplasmic compartments (soma and dendrites) 5 min after cocaine administration, suggesting a local activation of the protein (Fig.1C). Then, a strong nuclear staining was observed 10 min after cocaine, supporting a nuclear translocation of activated ERK proteins at this time point (Fig. 1C).
Fig. 1.
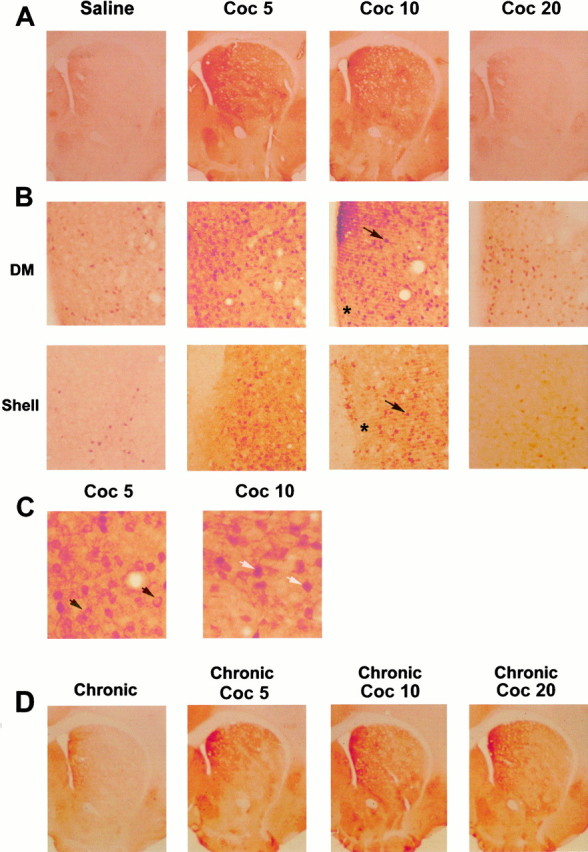
Acute cocaine injection activates ERK in the striatum. Immunocytochemical detection of activated ERK proteins was detected using a specific anti-active antibody, 5 (Coc 5), 10 (Coc 10), and 20 (Coc 20) min after cocaine injection (20 mg/kg, i.p.). A, Low magnification (50×) showing P-ERK immunoreactivity in the whole striatum. Note the strong immunoreactivity at Coc 5 and Coc 10 in the whole striatum and its decrease at Coc 20 (data are representative of 5 independent animals in each group). B, Higher magnification (200×) of P-ERK immunoreactivity in the dorsomedial striatum (DM) and the shell of NA. In both striatal regions, this immunoreactivity corresponds to both neuropil (asterisk) and cell bodies (arrows).C, Whereas at Coc 5 most of cell bodies showed a cytoplasmic (black arrows) P-ERK immunolabeling, at Coc 10, the majority of P-ERK was nuclear (white arrows).D, P-ERK immunoreactivity at low magnification showing ERK activation in chronically treated mice (6 d, once daily) with a challenge of cocaine (Coc 5, Coc 10, Coc 20) performed on day 7 (data are representative of 4 independent animals in each group).
ERK activation was rapid, but transient, because a strong decrease of cocaine-induced ERK activation was found in the dorsal striatum and NA 20 min after cocaine injection (Fig. 1A,B). One hour after cocaine injection, P-ERK immunostaining returned to basal levels (data not shown). The kinetics of ERK activation was slightly modified by previous chronic administration of cocaine (20 mg/kg, 6 d, once daily), with a strong increase of P-ERK immunostaining occurring from 5 to 20 min (Fig. 1D). Noteworthy was the lack of P-ERK induction when chronic cocaine treatment was followed by saline. In summary, acute and challenge of cocaine injection after chronic treatment are both able to induce a rapid and strong activation of ERK in the different subregions of the striatum.
ERK activation induced by acute cocaine depends on dopamine receptors
We then evaluated the involvement of dopaminergic receptors in cocaine-induced ERK activation at its peak time point of activation (i.e., 10 min). One-way ANOVA (between subjects) revealed significant differences in number of P-ERK positive cells in the following striatal compartments: dorsolateral (F(5,12) = 62.8; p < 0.01), dorsomedial (F(5,12) = 66.4; p < 0.01), core (F(5,12) = 44.1;p < 0.01), and shell (F(5,12) = 43.9; p < 0.01). Post hoc comparisons (Newman–Keuls) showed a significant increase in P-ERK-immunoreactive cells after cocaine administration in dorsal striatum and NA (p < 0.01) (Fig. 2A). The blockade of D1 receptors by administration of a selective antagonist, SCH 23390 (0.1 mg/kg), resulted in a total inhibition of cocaine-induced ERK activation throughout the dorsal striatum (dorsolateral and dorsomedial, p < 0.01) and NA (core and shell, p < 0.01) (Fig. 2A,B). This result shows that ERK activation was dependent on DA via D1 receptor activation. Moreover, basal P-ERK immunoreactivity found in control mice was significantly decreased by SCH 23390 in dorsomedial striatum (p < 0.05), implicating D1 receptors in spontaneous tonic control of ERK activity (Fig.2B).
Fig. 2.

Cocaine-induced ERK activation depends on DA-D1 receptors. SCH 23390 (0.1 mg/kg, i.p.) or raclopride (0.3 mg/kg, i.p.) were injected 15 min before cocaine. A, Low magnification of P-ERK immunolabeling in the striatum in cocaine-injected mice, in the presence or not of DA antagonists. Note the total inhibition of cocaine-induced P-ERK immunolabeling by DA-D1 antagonist. B, P-ERK-immunoreactive cells were counted in the dorsomedial (DM) and dorsolateral (DL) striatum, core, and shell of the NA from three independent mice in each group. Data represents the total number of P-ERK-positive cells in each region, delimited as depicted ininsert.C, The same doses of DA antagonists were used to analyze locomotor response induced by cocaine (n = 8 mice for each group). Statistical comparisons for B and C: ★p < 0.05; ★★ p < 0.001 when comparing with saline group. ⋆ p < 0.05; ⋆⋆ p < 0.001 when comparing with cocaine group (Newman–Keuls test).
The possible involvement of D2 receptor subtype in cocaine-induced ERK activation was also analyzed. Raclopride injection (0.3 mg/kg) alone induced a slight but not significant activation in number of P-ERK-positive neurons in the shell of NA. Raclopride failed to block cocaine-induced P-ERK immunoreactivity in the NA (core and shell) but partially decreased it in the dorsal striatum (dorsomedial,p < 0.05; dorsolateral, p < 0.01), suggesting that D2 receptors could also play some role in these cocaine effects (Fig. 2A,B). Taken together, these results show that cocaine-induced ERK activation critically involves D1 receptors, whereas contribution of D2 receptors seems to be restricted to dorsal striatum.
Doses of DA antagonists used above to analyze ERK activity were also tested in hyperlocomotion induced by cocaine. As revealed by one-way ANOVA, significant effects of treatment on locomotor responses are observed under these experimental conditions (F(5,43) = 87.4; p < 0.001). Post hoc comparison (Newman–Keuls) showed that SCH 23390 (0.1 mg/kg) completely abolished hyperlocomotion induced by cocaine (20 mg/kg) (p < 0.01), whereas raclopride (0.3 mg/kg) poorly affected this response (p < 0.01) (Fig. 2C). These data are consistent with literature data (Cabib et al., 1991; Xu et al., 1994a) showing that D1 receptors are preferentially involved in cocaine-induced hyperlocomotion.
Contribution of NMDA receptor in cocaine-induced ERK activation
Because NMDA receptor activation plays a role in amphetamine-induced gene regulation (Konradi et al., 1996) and is known to activate ERK pathway (English and Sweatt, 1996), the involvement of NMDA receptors in cocaine-induced ERK activation was studied. The noncompetitive NMDA receptor antagonist MK 801 (0.15 mg/kg) did not modify basal P-ERK immunoreactivity but decreased cocaine-induced P-ERK immunoreactivity in the striatum (Fig.3A). Statistical comparisons (Fig. 3B) showed a significant decrease in the dorsomedial (F(3,8) = 48.5; p < 0.01) and dorsolateral striatum (F(3,8) = 54.8; p < 0.01), as well as in the core (F(3,8)= 26; p < 0.01) and shell (F(3,8) = 19.7; p < 0.01) of NA. Thus, ERK activation by cocaine involves a participation of NMDA glutamate receptor. Interestingly, the neuropil still remained highly labeled after MK801 treatment, suggesting the involvement of NMDA receptors in the control of P-ERK activation at the postsynaptic level rather than presynaptically (Fig. 3A). The effects of MK 801 (0.15 mg/kg) were also evaluated in cocaine-induced hyperlocomotion (F(3,27) = 39.9;p < 0.01). Despite its effect on ERK activation, MK 801, at the dose used here, did not modify hyperlocomotion induced by cocaine (Fig. 3C).
Fig. 3.
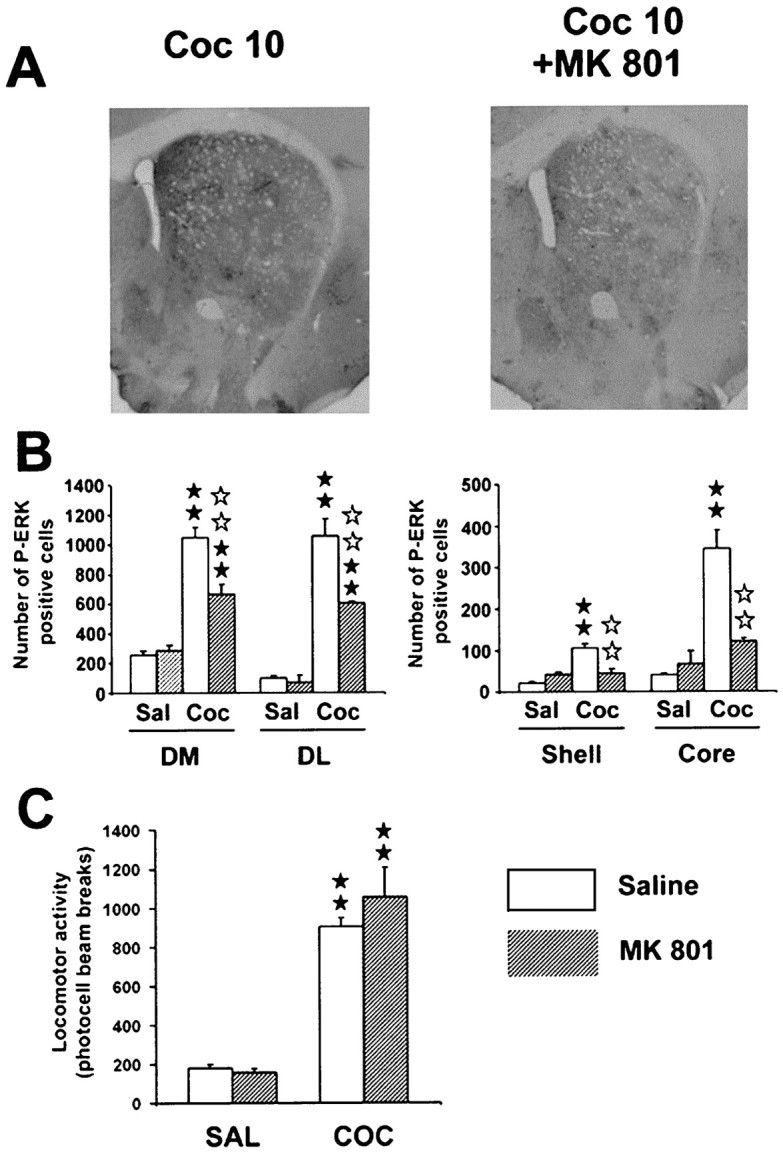
Partial contribution of glutamatergic NMDA receptors on cocaine-induced ERK activation. MK 801 (0.15 mg/kg, i.p.) was injected 30 min before cocaine.A, Low magnification of P-ERK immunolabeling in the striatum in cocaine-injected mice, in the presence or not of MK 801. Note the slight decrease of cocaine-induced P-ERK immunolabeling.B, P-ERK-immunoreactive cells were counted in the four striatal subregions as described Figure 3 (n = 3 independent mice for each group). C, The same dose of MK 801 was used to analyze locomotor response induced by cocaine (n = 8 mice for each group). Statistical comparisons for B and C were performed as described Figure 2.
Cocaine-induced locomotor activity is partially linked to ERK activity
Because ERK activation was completely prevented by D1 antagonist (Fig. 2A), we then tested whether ERK activation was involved in cocaine-induced locomotor activity. For this purpose, we assessed the effects of SL327, a selective MEK inhibitor (Atkins et al., 1998). This compound is a structural analog of the highly selective and efficient MEK inhibitor U0126 (Favata et al., 1998). Although recent studies report that this compound has no effect on a variety of other kinases, including PKA, PKC, or CamKII (Atkins et al., 1998; Selcher et al., 1999) (J. M. Trzaskos unpublished results), we analyzed whether it could have any D1 receptor blocking activity. Adenylyl cyclase activity induced by dopamine (10−4m) was tested from mouse brain slices in the presence of various concentrations of SL 327 (0.3, 3, 10, 30, 100 and 300 μm). Table 1shows that, whatever the concentration used, this compound has no effect on DA-induced cAMP production. When administered systemically, SL327 (50 mg/kg) totally abolished cocaine-induced ERK activation in the whole striatum (Fig.4A). However, cocaine-induced hyperlocomotion (F(3,28) = 59.2; p < 0.01) was only partially decreased, by SL327 (50 mg/kg), as shown in Figure 4B. We noted a complete blockade of rearing behavior induced by cocaine after SL327 administration (data not shown). These results suggest that ERK activation plays a role in the control of locomotor response induced by cocaine.
Table 1.
Effects of SL 327 on dopamine-induced cAMP production on striatal mouse slices
| Dopamine (m) | SL 327 (μm) | % increase |
|---|---|---|
| 10−4 | 0 | 124 ± 9 |
| 0 | 10 | 12 ± 8 |
| 0 | 100 | 7 ± 6 |
| 10−4 | 0.3 | 129 ± 21 |
| 10−4 | 3 | 126 ± 3 |
| 10−4 | 10 | 167 ± 42 |
| 10−4 | 30 | 163 ± 42 |
| 10−4 | 100 | 143 ± 22 |
| 10−4 | 300 | 140 ± 30 |
Adenylyl cyclase activity stimulated by dopamine was measured in presence of increasing doses of SL327 (0.3–300 μm). Results were calculated in picomoles of cAMP formed per minute per milligram of protein and expressed in percentage of increase when compared to the basal adenylyl cyclase activity (n = 3 individual experiments run in triplicate). Note the lack of effects of SL327 on both the basal and dopamine-induced cAMP levels.
Fig. 4.

SL 327, an inhibitor of ERK activation, partially inhibits cocaine-induced hyperlocomotion. SL 327 (50 mg/kg, i.p.) was injected 1 hr before cocaine. A, Note the total inhibition of cocaine-induced P-ERK immunoreactivity by SL 327. B, Effect of SL 327 on cocaine-induced hyperlocomotion (n = 8 mice for each group). Statistical comparisons were performed as described Figure 2.
Induction of c-fos and Elk-1 hyperphosphorylation by acute cocaine is dependent on ERK activation
Hyperphosphorylation of ERK was clearly nuclear 10 min after cocaine administration (Fig. 1), suggesting a possible involvement of ERK activation in IEG upregulation (for review, seeGrewal et al., 1999). Because c-fos is a prime marker of gene expression after cocaine treatment, and its transcriptional regulation is critically controlled by ERK in the striatum (Sgambato et al., 1998;Vanhoutte et al., 1999), we analyzed cocaine-induced c-fos expression after inhibition of ERK by SL327 (50 mg/kg). A strong induction of c-fos expression in the dorsal striatum and the NA was found 1 hr after cocaine administration, as previously described (Graybiel et al., 1990;Moratalla et al.; 1993) (Fig. 5). When SL327 was injected before cocaine injection, c-fos immunolabeling returned to baseline levels in the dorsal striatum and strongly decreased in the shell of the NA (Fig. 5).
Fig. 5.
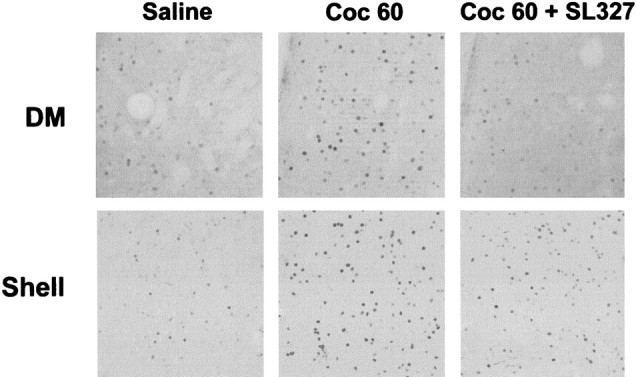
SL 327 inhibits c-fos induction by cocaine. c-fos expression was analyzed 1 hr after cocaine injection in the dorsomedial striatum (DM) and in the shell of the NA. SL 327 was injected as described Figure 4. Note the total inhibition of c-fos immunoreactivity in the DM and the strong but not total inhibition in the shell (data are representative of 4 independent mice for each group).
Although CREB activation can be linked to ERK activation in various model systems (Xing et al., 1996; Impey et al., 1998; Davis et al., 2000) and its involvement in cocaine responses has been reported (Carlezon et al., 1998), no data yet describes whether Elk-1, a direct nuclear target of ERKs is also activated after cocaine treatment. A hyperphosphorylation of Elk-1 was found in a strict spatiotemporal correlation with ERK activation, i.e., in both the dorsal striatum and NA 10 min after cocaine injection (Fig.6). Furthermore, P-Elk1 immunoreactivity was prevented by SL327, demonstrating that Elk1 activation is completely dependent on ERK activation. Altogether, these data suggest that ERK activation, targeting the transcription factor Elk-1, is involved in cocaine-induced c-fos expression in the striatum.
Fig. 6.
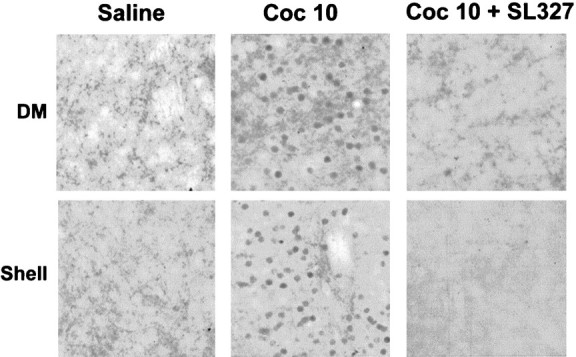
Cocaine-induced Elk-1 hyperphosphorylation depends on ERK activation. Elk-1 hyperphosphorylation was analyzed using an anti-active antibody (anti phospho-Ser383-Elk-1) 10 min after cocaine injection in the absence or presence of SL 327. Note the strong induction of Elk-1 activation by cocaine in the dorsomedial striatum (DM) and in the shell of the NA. This activation is totally inhibited by SL 327 (data are representative of 4 independent mice for each group).
Inhibition of ERK activity impairs rewarding properties induced by cocaine
Expression of transcription factors encoded by IEGs has been hypothesized to initiate downstream molecular events, which in turn are involved in the instatement of cocaine-rewarding properties (for review, see Berke and Hyman, 2000). The place preference conditioning paradigm was used to assess the role of ERK activation on the rewarding effects of cocaine. Two-way ANOVA revealed a significant effect of treatment (F(3,28) = 8.079;p < 0.01) and phase–treatment interaction (F(3,28) = 8.21; p < 0.01) with no main effect of phase alone (F(1,28) = 3.649; NS). Comparison of preconditioning times spent in the drug-paired compartment did not reveal any significant difference between groups, indicating the unbiased characteristics of the experimental design. After conditioning, mice treated with cocaine (20 mg/kg) spent more time in the cocaine-paired compartment, revealing a clear place preference as revealed by one-way ANOVA (F(3,28) = 8.22; p < 0.01) and post hoc comparisons (p < 0.01) of score values (Fig.7A,B). Interestingly, pretreatment with SL 327 during the conditioning phase completely abolished the place preference induced by cocaine (p < 0.01) (Fig. 7A,B). No conditioned response or locomotor modification was observed in mice treated with SL327 alone (Fig. 7A,B). These results suggest a critical involvement of ERK signaling cascade on the rewarding effects of cocaine.
Fig. 7.

Lack of cocaine-induced place preference in mice injected with SL 327. A, Time spent in drug-associated compartment during the preconditioning (white bars) and the testing phase (hatched bars). Values represent mean ± SEM from n = 8 independent mice per group. B, Scores calculated as the difference between postconditioning and preconditioning time spent in the compartment associated with cocaine. ★★ p < 0.001 when comparing with saline group. ⋆⋆ p < 0.001 when comparing with cocaine group (Newman–Keuls test).
DISCUSSION
The ERK pathway that involves a complex intracellular signaling cascade, including the small GTP-binding protein Ras and the kinases Raf and MEK, controls various neurobiological effects, including neuronal differentiation (for review, see Seger and Krebs, 1995) and synaptic plasticity in the adult CNS (for review, see Orban et al., 1999). Initially described in response to neurotrophin receptors, the Ras–ERK pathway can also integrate second messenger systems, such as calcium, PKA, and DAG, which may explain its crucial role in activity-dependent regulation of neuronal function (for review, seeGrewal et al., 1999).
This work reports that acute cocaine administration strongly activates ERK, which is rapid and transient. This activation occurred in the striatum but not in the ventral tegmental area (VTA) (data not shown). We did not find any ERK activation in the striatum after chronic cocaine administration (analyzed 12 hr after the last injection), a result in agreement with previous studies (Berhow et al., 1996). In this case, authors described a specific ERK activation in the VTA, which was thought to result from stimulation of tyrosine kinase receptors by neurotrophins such as BDNF (Berhow et al., 1996) or neurotrophin-3 (Pierce et al., 1999). However, it must be noticed that ERK activation was found in the striatum when analyzed rapidly (5 min) after a challenge cocaine injection on day 7, and this activation was more sustained than after an acute injection, suggesting a sensitization of this pathway by previous chronic cocaine injection. Altogether these data suggest that ERK signaling in the striatum could be involved in the control of cocaine-induced effects.
ERK activation was critically linked to D1 receptors. Several second messengers could be responsible for the link between D1 receptors and ERK, such as the small Ras-related G protein Rap1, activated by PKA, and the subsequent activation of B Raf isoform (Vossler et al., 1997;Yao et al., 1998; York et al., 1998). Another possible intermediate between D1 receptor and ERK activation could be Calcyon, a D1 receptor-interacting protein, expressed in the striatum and stimulating intracellular calcium release (Lezcano et al., 2000), which is known to activate the Ras–ERK pathway (Lev et al., 1995). An interesting observation was the different subcellular localization of activated ERK proteins during the time course. Indeed, ERK activation was first observed in cytoplasmic compartments, suggesting an early local role of the protein at the membrane–cytoplasmic level. As a matter of fact, activated ERKs can target a number of membrane-associated proteins such as EGR receptors, phospholipase A2, or cytoskeletal proteins, including microtubule-associated proteins (MAPs) and neurofilaments (for review, see Grewal et al., 1999). The later nuclear activation suggests a second effect in gene expression (see below). Another localization of activated ERK likely corresponded to presynaptic terminals, as attested by the strong immunolabeling observed in the neuropil. This presynaptic activation could be possibly related to the facilitating role of ERK in neurotransmitter release via phosphorylation of synapsins (Jovanovic et al., 2000).
The D1 antagonist SCH 23390 completely reversed ERK activation and hyperlocomotion induced by acute cocaine. However, the MEK inhibitor only partially blocked cocaine locomotor response, although the increase observed in rearing behavior was completely abolished (data not shown). These data suggest that D1 receptors control cocaine-induced striatal activity and hyperlocomotion via both ERK-dependent and ERK-independent pathways. The ERK-dependent control of striatal synaptic activity remains to be established, however various presynaptic or postsynaptic substrates could participate in this control (Grewal et al., 1999; Jovanovic et al., 2000). Concerning the ERK-independent pathway, it is now well known that D1 receptors can regulate striatal neuronal excitability by activation of PKA and subsequent modification of the phosphorylation state of various substrates (such as L type calcium channels, DARPP 32, NMDA receptors, or electronic ion pumps) (for review, see Greengard et al., 1999).
Glutamate receptors of NMDA subtype strongly contributed to ERK activation by cocaine, in particular in the NA where MK 801 totally abolished cocaine-induced ERK activation in neurons but not in the neuropil. This result further illustrates the participation of NMDA receptors in D1-mediated intracellular events previously showed byKonradi et al. (1996). Whether this cooperation occurs at the level of neuronal circuitry or intrinsically in striatal neurons remains to be established. However, the lack of P-ERK inhibition in the neuropil after MK 801 argues for a preferential postsynaptic effect, probably through glutamatergic cortical inputs (Spencer, 1976). No modification of cocaine-induced hyperlocomotion was found after MK 801 blockade. In this regard, it must be noted that controversial results have been previously reported on the contribution of NMDA receptors in acute behavioral responses induced by cocaine, depending on doses, animal species, and experimental conditions (Pulvirenti et al., 1991; Witkin, 1993; Wolf et al., 1994).
Cocaine-induced locomotor activity was partially inhibited by the D2 antagonist raclopride. Interestingly, raclopride also decreased ERK activation, but only in the dorsolateral striatum, a striatal region involved in the motor responses of cocaine (Canales and Graybiel, 2000). Recent evidences indicate that D2 agonists can activate ERK in striatal slices independently of βγ subunits of G-proteins but via coupling of a Gq-protein to PLCβ pathway and mobilization of intracellular calcium stores (Yan et al., 1999). Although D1 and D2 receptors are classically considered to be localized in distinct striatal subpopulations (Gerfen et al., 1990), our observations suggest a synergism of both D1 and D2 receptors in the same striatal subpopulation. Interestingly, recent anatomical and physiological evidences show their colocalization in striatal neurons (Aizman et al., 2000).
Activated ERKs were translocated to the nucleus 10 min after cocaine injection. A prime nuclear target of activated ERK is the ternary complex factor Elk-1, which acts as a transcriptional activator, via the SRE of various IEG, such as c-fos. Elk-1 was strongly phosphorylated after cocaine treatment, in a strict spatiotemporal correlation with ERK activation. The MEK inhibitor SL 327 totally abolished Elk-1 activation, supporting its complete dependence on ERK pathway. Although Elk-1 is a common substrate of the different MAP kinase pathways in cell model systems (Davis, 1993), neither N-terminal Jun kinase (JNK) nor p38 were activated by cocaine in this study (data not shown). The lack of JNK activation is in agreement with previous data indicating that neither dopamine nor forskolin activate JNK on striatal primary neuronal cells (Schwarzschild et al., 1997).
After nuclear translocation, activated ERK controls IEG transcription in neuronal cells (Davis et al., 2000). In agreement with this, a strong inhibition of cocaine-induced c-fos expression was found after treatment with the MEK inhibitor SL327. Because this compound has no effect on a variety of other kinases, including PKA, PKC, or CamKII (Atkins et al., 1998; Selcher et al., 1999) (Trzaskos, unpublished results), or adenylyl cyclase production, this effect on c-fos expression cannot be readily attributed to nonspecific actions of the drug. It must be noticed, however, that although total in the dorsomedial striatum, c-fos inhibition was only partial in the shell of NA. Therefore, the transcriptional regulation of c-fos seems to be under a combined control of an ERK-dependent and -independent pathway, at least in this striatal region. In this way, it is now well established that cAMP–PKA pathway directly controls CREB phosphorylation, which targets the CRE site of the c-fos promoter (Dash et al., 1991). Interestingly, consistent with our data, results from mutant mice in which the CRE response is affected indicate that this site is not sufficient to induce full expression of c-fos proteins (Robertson et al., 1995; Maldonado et al., 1996).
An important observation was the abolishment of cocaine-induced conditioned place preference after blockade of ERK by SL327. The locomotor sensitization observed in mice receiving a repeated cocaine administration during this conditioned treatment was prevented (data not show). In agreement with this, inhibition of ERK in the VTA, a brain structure closely related to drugs of abuse-induced rewarding effect (Koob, 1992), blocked cocaine-induced behavioral sensitization (Pierce et al., 1999). By controlling the expression of tyrosine hydroxylase, ERK activation in the VTA was thought to be involved in increased DA neuronal activity (Berhow et al., 1996). Thus, the ERK pathway could be involved in cocaine-induced behavioral rewarding effects via different mechanisms: (1) control of genes encoding transcription factors (such as c-fos) in the striatum, and (2) control of genes associated with DA neuronal activity after neurotrophin receptor activation.
In summary, the present results reveal that cocaine-induced ERK activation plays an important role in striatal c-fos expression and some of its behavioral responses related to the development of addiction. These findings provide a new mechanism to explain the neurobiological substrate of cocaine rewarding properties that could be important to better understand the different neural pathways involved in its addictive properties.
Footnotes
This work was supported by the University Pierre et Marie Curie and the Centre National de la Recherche Scientifique for J.C. and European Commission (BIOMED-2 # 98–227) for R.M. E.V. was supported by a grant from “La fondation des Treilles”. We are grateful to J. M. Trzaskos, J. L. Hytrek, A. C. Tabaka, J. S. Piecara, and C. Teleha for the generous gift of SL327. We are grateful to Jean Antoine Girault for helpful discussions and support for adenyl cyclase measurements.
Correspondence should be addressed to Jocelyne Caboche, Laboratoire de Neurochimie-Anatomie, Institut des Neurosciences, Unité Mixte de Recherche 7624, Centre National de la Recherche Scientifique, Université Pierre et Marie Curie, 75005 Paris, France. E-mail:Jocelyne.Caboche@snv.jussieu.fr.
REFERENCES
- 1.Aizman O, Brismar H, Uhlen P, Zettergren E, Levey AI, Forssberg H, Greengard P, Aperia A. Anatomical and physiological evidence for D1 and D2 dopamine receptor colocalization in neostriatal neurons. Nat Neurosci. 2000;3:226–230. doi: 10.1038/72929. [DOI] [PubMed] [Google Scholar]
- 2.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 3.Berhow MT, Hiroi N, Nestler EJ. Regulation of ERK (extracellular signal regulated kinase), part of the neurotrophin signal transduction cascade, in the rat mesolimbic dopamine system by chronic exposure to morphine or cocaine. J Neurosci. 1996;16:4707–4715. doi: 10.1523/JNEUROSCI.16-15-04707.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berke JD, Hyman SE. Addiction, dopamine, the molecular mechanisms of memory. Neuron. 2000;25:515–532. doi: 10.1016/s0896-6273(00)81056-9. [DOI] [PubMed] [Google Scholar]
- 5.Berke JD, Paletzki RF, Aronson GJ, Hyman SE, Gerfen CR. A complex program of striatal gene expression induced by dopaminergic stimulation. J Neurosci. 1998;18:5301–5310. doi: 10.1523/JNEUROSCI.18-14-05301.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bockaert J, Tassin JP, Thierry AM, Glowinski J, Premont J. Characteristics of dopamine and beta-adrenergic sensitive adenylate cyclases in the frontal cerebral cortex of the rat. Comparative effects of neuroleptics on frontal cortex and striatal dopamine sensitive adenylate cyclase. Brain Res. 1977;122:71–86. doi: 10.1016/0006-8993(77)90663-1. [DOI] [PubMed] [Google Scholar]
- 7.Cabib S, Castellano C, Cestari V, Filibeck U, Puglisi-Allegra S. D1 and D2 receptor antagonists differently affect cocaine-induced locomotor hyperactivity in the mouse. Psychopharmacology. 1991;105:335–339. doi: 10.1007/BF02244427. [DOI] [PubMed] [Google Scholar]
- 8.Canales JJ, Graybiel AM. A measure of striatal function predicts motor stereotypy. Nat Neurosci. 2000;3:377–383. doi: 10.1038/73949. [DOI] [PubMed] [Google Scholar]
- 9.Carlezon WA, Jr, Thome J, Olson VG, Lane-Ladd SB, Brodkin ES, Hiroi N, Duman RS, Neve RL, Nestler EJ. Regulation of cocaine reward by CREB. Science. 1998;282:2272–2275. doi: 10.1126/science.282.5397.2272. [DOI] [PubMed] [Google Scholar]
- 10.Chen RH, Sarneki C, Blenis J. Nuclear localization and regulation of erk- and rsk- encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dash PK, Karl KA, Prywes R, Kandel ER. cAMP response element-binding protein is activated by Ca2+/calmodulin as well as cAMP dependent protein kinase. Proc Natl Acad Sci USA. 1991;88:5061–5065. doi: 10.1073/pnas.88.11.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 13.Davis S, Vanhoutte P, Pagès C, Caboche J, Laroche S. The MAPK/ERK cascade targets both Elk-1 cAMP response element-binding protein to control long-term potentiation-dependent gene expression in the dentate gyrus in vivo. J Neurosci. 2000;20:4563–4572. doi: 10.1523/JNEUROSCI.20-12-04563.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 16.English JD, Sweatt JD. Requirement for the mitogen-activated protein kinase cascade in hippocampal long-term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- 17.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. Identification of a novel inhibitor of Mitogen-activated Protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 18.Fitzgerald LW, Ortiz J, Hamedani AG, Nestler EJ. Regulation of glutamate receptor subunit expression by drugs of abuse and stress: common adaptation among cross-sensitizing agents. J Neurosci. 1996;16:274–282. doi: 10.1523/JNEUROSCI.16-01-00274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gerfen CR, Engber TM, Mahan LC, Susel Z, Chase TN, Monsma FJ, Jr, Sibley DR. D1 and D2 dopamine receptor-regulated gene expression of striatonigral and striatopallidal neurons [see comments]. Science. 1990;250:1429–1432. doi: 10.1126/science.2147780. [DOI] [PubMed] [Google Scholar]
- 20.Gille H, Sharrocks AD, Shaw PE. Phosphorylation of transcription factor p62TCF by MAP kinase stimulates ternary complex formation at c-fos promoter. Nature. 1992;358:414–417. doi: 10.1038/358414a0. [DOI] [PubMed] [Google Scholar]
- 21.Gille H, Kortenjann M, Thoma O, Moomaw C, Slaughter C, Cobb M, Shaw PE. ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 1995;14:951–962. doi: 10.1002/j.1460-2075.1995.tb07076.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graybiel A, Moratalla R, Robertson HA. Amphetamine and cocaine induce drug-specific activation of the c-fos gene in striosome-matrix compartments and limbic subdivisions of the striatum. Proc Natl Acad Sci USA. 1990;87:6912–6916. doi: 10.1073/pnas.87.17.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- 24.Grewal SS, York RD, Stork PJ. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- 25.Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev. 1998;28:370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- 26.Hill CS, Marais R, John S, Wynne J, Dalton S, Treisman R. Functional analysis of a growth factor-responsive transcription factor complex. Cell. 1993;73:395–406. doi: 10.1016/0092-8674(93)90238-l. [DOI] [PubMed] [Google Scholar]
- 27.Hope B, Kosofsky B, Hyman SE, Nestler EJ. Regulation of immediate early gene expression and AP-1 binding in the rat nucleus accumbens by chronic cocaine. Proc Natl Acad Sci USA. 1992;89:5764–5768. doi: 10.1073/pnas.89.13.5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hope BT, Nye HE, Kelz MB, Self DW, Iadarola MJ, Nakabeppu Y, Duman RS, Nestler EJ. Induction of a long-lasting AP-1 complex composed of altered Fos-like proteins in brain by chronic cocaine and other chronic treatments. Neuron. 1994;13:1235–1244. doi: 10.1016/0896-6273(94)90061-2. [DOI] [PubMed] [Google Scholar]
- 29.Hipskind RA, Baccarini M, Nordheim A. Transient activation of RAF-1, MEK, and ERK2 coincides kinetically with ternary complex factor phosphorylation and immediate-early gene promoter activity in vivo. Mol Cell Biol. 1994a;14:6219–6231. doi: 10.1128/mcb.14.9.6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hipskind RA, Büscher D, Nordheim A, Baccarini M. Ras/MAP kinase-dependent and-independent signaling pathways target distinct ternary complex factors. Genes Dev. 1994b;8:1803–1816. doi: 10.1101/gad.8.15.1803. [DOI] [PubMed] [Google Scholar]
- 31.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 32.Janknecht R, Zinck R, Ernst WH, Nordheim A. Functional dissection of the transcription factor Elk-1. Oncogene. 1994;9:1273–1278. [PubMed] [Google Scholar]
- 33.Jovanovic JN, Czernik AJ, Fienberg AA, Greengard P, Sihra TS. Synapsins as mediators of BDNF-enhanced neurotransmitter release. Nat Neurosci. 2000;3:323–329. doi: 10.1038/73888. [DOI] [PubMed] [Google Scholar]
- 34.Kelz MB, Chen J, Carlezon WA, Jr, Whisler K, Gilden L, Bechmann AM, Steppen C, Zhang YJ, Marotti L, Self DW, Tkatch T, Baranauskas G, Surmeier DJ, Neve RL, Duman RS, Picciotto MR, Nestler EJ. Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature. 1999;401:272–276. doi: 10.1038/45790. [DOI] [PubMed] [Google Scholar]
- 35.Konradi C, Cole RL, Heckers S, Hyman SE. Amphetamine regulates gene expression in rat striatum via transcription factor CREB. J Neurosci. 1994;14:5623–5634. doi: 10.1523/JNEUROSCI.14-09-05623.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Konradi C, Leveque JC, Hyman SE. Amphetamine and dopamine-induced immediate early gene expression in striatal neurons depends on postsynaptic NMDA receptors and calcium. J Neurosci. 1996;16:4231–4239. doi: 10.1523/JNEUROSCI.16-13-04231.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koob GF. Drugs of abuse: anatomy, pharmacology, and function of reward pathways. Trends Pharmacol Sci. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- 38.Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase PYK2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 39.Lezcano N, Mrzljak L, Eubanks S, Levenson R, Goldman-Rakic P, Bergson C. Dual signaling regulated by calcyon, a D1 dopamine receptor interacting protein. Science. 2000;287:1660–1664. doi: 10.1126/science.287.5458.1660. [DOI] [PubMed] [Google Scholar]
- 40.Maldonado R, Blendy JA, Tzavara E, Gass P, Roques BP, Hanoune J, Schutz G. Reduction of morphine abstinence in mice with a mutation in the gene encoding CREB [see comments]. Science. 1996;273:657–659. doi: 10.1126/science.273.5275.657. [DOI] [PubMed] [Google Scholar]
- 41.Maldonado R, Saiardi A, Valverde O, Samad TA, Roques BP, Borrelli E. Absence of opiate rewarding effects in mice lacking dopamine D2 receptors. Nature. 1997;388:586–589. doi: 10.1038/41567. [DOI] [PubMed] [Google Scholar]
- 42.Marais R, Wynne J, Treisman R. The SRF accessory protein Elk-1 contains a growth factor-regulated transcriptional activation domain. Cell. 1993;73:381–393. doi: 10.1016/0092-8674(93)90237-k. [DOI] [PubMed] [Google Scholar]
- 43.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP Kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 44.Moratalla R, Vickers EA, Robertson HA, Cochran BH, Graybiel AM. Coordinate expression of c-fos and jun B is induced in the rat striatum by cocaine. J Neurosci. 1993;13:423–433. doi: 10.1523/JNEUROSCI.13-02-00423.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moratalla R, Xu M, Tonegawa S, Graybiel AM. Cellular responses to psychomotor stimulant and neuroleptic drugs are abnormal in mice lacking the D1 dopamine receptor. Proc Natl Acad Sci USA. 1996;93:14928–4933. doi: 10.1073/pnas.93.25.14928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nestler EJ, Aghajanian GK. Molecular and cellular basis of addiction. Science. 1997;278:58–63. doi: 10.1126/science.278.5335.58. [DOI] [PubMed] [Google Scholar]
- 47.Orban PC, Chapman PF, Brambilla R. Is the Ras-MAPK signalling pathway necessary for long-term memory formation? Trends Neurosci. 1999;22:38–44. doi: 10.1016/s0166-2236(98)01306-x. [DOI] [PubMed] [Google Scholar]
- 48.Pierce RC, Pierce-Bancroft AF, Prasad BM. Neurotrophin-3 contributes to the initiation of behavioral sensitization to cocaine by activating the Ras/Mitogen-activated protein kinase signal transduction cascade. J Neurosci. 1999;19:8685–8695. doi: 10.1523/JNEUROSCI.19-19-08685.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pulvirenti L, Swerdlow NR, Koob GF. Nucleus accumbens NMDA antagonist decreases locomotor activity produced by cocaine, heroin or accumbens dopamine, but not caffeine. Pharmacol Biochem Behav. 1991;40:815–845. doi: 10.1016/0091-3057(91)90095-j. [DOI] [PubMed] [Google Scholar]
- 50.Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD. The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element binding protein phosphorylation in area CA1 of hippocampus. J Neurosci. 1999;19:4337–4348. doi: 10.1523/JNEUROSCI.19-11-04337.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robertson LM, Kerpolla TK, Vendrell M, Luk D, Smeyne RJ, Bocchiara C, Morgan JI, Curran T. Regulation of c-fos expression in transgenic mice requires multiple interdependent transcription control elements. Neuron. 1995;14:214–252. doi: 10.1016/0896-6273(95)90282-1. [DOI] [PubMed] [Google Scholar]
- 52.Salomon Y, Londos C, Rodbell M. A highly sensitive adenylate cyclase assay. Anal Biochem. 1974;58:541–548. doi: 10.1016/0003-2697(74)90222-x. [DOI] [PubMed] [Google Scholar]
- 53.Schwarzschild MA, Col RL, Hyman SE. Glutamate, but not dopamine, stimulates stress-activated protein kinase and AP-1-mediated transcription in striatal neurons. J Neurosci. 1997;17:3455–3466. doi: 10.1523/JNEUROSCI.17-10-03455.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seger R, Krebs EG. The MAPK signaling pathway. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 55.Selcher JC, Atkins CM, Trzaskos JM, Paylor R, Sweatt JD. A necessity for MAP kinase activation in mammalian spatial learning. Learn Mem. 1999;6:478–490. doi: 10.1101/lm.6.5.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spencer HJ. Antagonism of cortical excitation of striatal neurons by glutamic acid diethylester: evidence for glutamic acid as an excitatory transmitter in the rat striatum. Brain Res. 1976;102:91–101. doi: 10.1016/0006-8993(76)90577-1. [DOI] [PubMed] [Google Scholar]
- 57.Sgambato V, Pages C, Rogard M, Besson MJ, Caboche J. Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. 1998;18:8814–8825. doi: 10.1523/JNEUROSCI.18-21-08814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Treisman R. Journey to the surface of the cell: Fos regulation and the SRE. EMBO J. 1995;14:4905–4913. doi: 10.1002/j.1460-2075.1995.tb00173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vanhoutte P, Barnier JV, Guibert B, Pagès C, Besson MJ, Hipskind RA, Caboche J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. 1999;19:136–146. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vincent SR, Sebben M, Dubuis A, Bockaert J. Neurotransmitter regulation of MAP kinase signaling in striatal neurons in primary culture. Synapse. 1998;29:29–36. doi: 10.1002/(SICI)1098-2396(199805)29:1<29::AID-SYN3>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 61.Vossler MR, Yao H, York RD, Pan M-G, Rim CS, Stork PJS. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–62. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 62.Xing J, Ginty DD, Greenberg ME. Coupling of the ras-MAPK pathway to gene activation by RSK2, a growth-factor-regulated CREB kinase. Science. 1996;273:959–963. doi: 10.1126/science.273.5277.959. [DOI] [PubMed] [Google Scholar]
- 63.Xu M, Hu XT, Cooper DC, Moratalla R, Graybiel AM, White FJ, Tonegawa S. Elimination of cocaine-induced hyperactivity and dopamine-mediated neurophysiological effects in dopamine D1 receptor mutant mice. Cell. 1994a;79:945–955. doi: 10.1016/0092-8674(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 64.Xu M, Moratalla R, Gold LH, Hiroi N, Koob GF, Graybiel AM, Tonegawa S. Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell. 1994b;79:729–742. doi: 10.1016/0092-8674(94)90557-6. [DOI] [PubMed] [Google Scholar]
- 65.Yan Z, Feng J, Fienberg AA, Greengard P. D(2) dopamine receptors induce mitogen-activated protein kinase and cAMP response element-binding protein phosphorylation in neurons. Proc Natl Acad Sci USA. 1999;96:11607–11612. doi: 10.1073/pnas.96.20.11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yao H, York RD, Misra-Press A, Carr DW, Stork PJ. The cyclic adenosine monophosphate-dependent protein kinase (PKA) is required for the sustained activation of mitogen-activated kinases and gene expression by nerve growth factor. J Biol Chem. 1998;273:8240–8247. doi: 10.1074/jbc.273.14.8240. [DOI] [PubMed] [Google Scholar]
- 67.York RD, Yao H, Dillon T, Ellig CL, Eckert SP, McCleskey EW, Stork PJ. Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature. 1998;392:622–626. doi: 10.1038/33451. [DOI] [PubMed] [Google Scholar]
- 68.Young ST, Porrino LJ, Iadarola MJ. Cocaine induces striatal c-fos-immunoreactive proteins via dopaminergic D1 receptors. Proc Natl Acad Sci USA. 1991;88:1291–1295. doi: 10.1073/pnas.88.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Witkin JM. Blockade of the locomotor stimulant effects of cocaine and methamphetamine by glutamate antagonists. Life Sci. 1993;53:405–410. doi: 10.1016/0024-3205(93)90496-p. [DOI] [PubMed] [Google Scholar]
- 70.Wolf ME, Xue CJ, White FJ, Dahlin SL. MK-801 does not prevent acute stimulatory effects of amphetamine or cocaine on locomotor activity or extracellular dopamine levels in rat nucleus accumbens. Brain Res. 1994;15:223–231. doi: 10.1016/0006-8993(94)90776-5. [DOI] [PubMed] [Google Scholar]
- 71.Zinck R, Cahill MA, Kracht M, Sachsenmaier C, Hipskind RA, Nordheim A. Protein synthesis inhibitors reveal differential regulation of mitogen-activated protein kinase and stress-activated protein kinase pathways that converge on Elk-1. Mol Cell Biol. 1995;15:4930–4938. doi: 10.1128/mcb.15.9.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]