

Abstract
The role of metabotropic l-glutamate (mGlu) receptors in supralinear Ca2+ signaling was investigated in cultured hippocampal cells using Ca2+ imaging techniques and whole-cell voltage-clamp recording. In neurons, but not glia, global supralinear Ca2+ release from intracellular stores was observed when the mGlu receptor agonist (RS)-3,5-dihydroxyphenylglycine (DHPG) was combined with elevated extracellular K+ levels (10.8 mm), moderate depolarization (15–30 mV), or NMDA (3 μm). There was a delay (2–8 min) before the stores were fully charged, and the enhancement persisted for a short period (up to 10 min) after removal of the store-loading stimulus. Studies with the mGlu receptor antagonist 2-methyl-6-(phenylethynyl)-pyridine demonstrated that these effects were mediated by activation of the mGlu5 receptor subtype. The L-type voltage-gated Ca2+ channel antagonist nifedipine (10 μm) substantially reduced responses to DHPG obtained in the presence of elevated extracellular K+ but not NMDA. This suggests that the Ca2+ that is required to load the stores can enter either through L-type voltage-gated Ca2+ channels or directly through NMDA receptors. The findings that both depolarization and NMDA receptor activation can facilitate mGlu receptor Ca2+ signaling adds considerable flexibility to the processes that underlie activity-dependent changes in synaptic strength. In particular, a temporal separation between the store-loading stimulus and the activation of mGlu receptors could be used as a recency detector in neurons.
Keywords: mGlu, NMDA, Ca2+ stores, Ca2+ release, recency detector, supralinear
Local and global elevations in neuronal cytosolic Ca2+ are important for a variety of physiological and pathological processes, including synaptic plasticity and gene expression (Berridge, 1998). The role of NMDA receptors in hippocampal Ca2+signaling has been the subject of intense investigation (Mayer et al., 1987; Regehr and Tank, 1992; Segal and Manor, 1992; Alford et al., 1993; Perkel et al., 1993; Malinow et al., 1994; Petrozzino et al., 1995; Schiller et al., 1998; Emptage et al., 1999; Yuste et al., 1999;Kovalchuk et al., 2000), fueled by the role of these receptors in processes such as long-term potentiation (LTP) and long-term depression (LTD) (Bliss and Collingridge, 1993; Bear and Abraham, 1996). Of particular significance for hebbian plasticity, the biophysical properties of NMDA receptors enable them to act as coincidence detectors, whereby they only generate an effective response in the presence of a postsynaptic depolarization (Nowak et al., 1984; Mayer et al., 1984). These characteristics, coupled with their permeability to Ca2+ (Jahr and Stevens, 1987; Mayer et al., 1987; Ascher and Nowak, 1988), make them ideally suited to converting changes in synaptic activity into intracellular Ca2+ signals (Bliss and Collingridge, 1993).
Recently, interest has grown in the possibility that metabotropicl-glutamate (mGlu) receptors could also act as coincidence detectors by generating supralinear Ca2+signals during membrane depolarization (Emptage, 1999; Nakamura et al., 1999). The physiological relevance of such interactions is indicated by findings that synaptic activity can lead to mGlu receptor-dependent Ca2+ signals (Frenguelli et al., 1993;Miller et al., 1996; Takechi et al., 1998; Nakamura et al., 1999;Yeckel et al., 1999) and that inhibition of mGlu receptors can, under some experimental conditions, result in blockade of the induction of LTP and LTD (Bortolotto et al., 1999). It is therefore important to understand the mechanisms that enable mGlu receptor-mediated supralinear Ca2+ signaling.
In the present study, we have used Ca2+imaging and whole-cell recording techniques to investigate the interactions between depolarization and/or NMDA receptor activation with mGlu receptors in the mediation of supralinear Ca2+ signals in cultured hippocampal neurons. We find that activation of mGlu5receptors in these cells releases substantial amounts of Ca2+ from intracellular stores, provided that they have been charged with Ca2+. There is also an initial delay while the Ca2+ stores charge, and the enhancement persists for a period after removal of the store-loading stimulus. We find that the Ca2+ that is required to load the stores can enter either through L-type voltage-gated Ca2+ channels (VGCCs) or directly through NMDA receptors. This ménage à trois, in which depolarization can facilitate both NMDA and mGlu receptor Ca2+ signaling whereas NMDA receptor activation can facilitate mGlu receptor Ca2+ signaling, adds considerable flexibility to the processes involved in synaptic plasticity. In addition, the transient “memory” of previous neuronal activity contained within the Ca2+ stores could act as a mechanism for recency detection.
MATERIALS AND METHODS
Materials. The following materials were used:d(−)-2-amino-5-phosphonopentanoic acid (d-AP-5), (RS)-3,5-dihydroxyphenylglycine (DHPG), 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), and 2-methyl-6-(phenylethynyl)-pyridine (MPEP) were obtained from Tocris Cookson (Bristol, UK); cytosine arabinofuranoside, dialyzed fetal bovine serum, EGTA, fura-2, fura-2 AM, fluo-3 AM, thapsigargin, nifedipine, ionomycin, pronase E, protease type X, and tetrodotoxin were purchased from Sigma (Dorset, UK); minimal essential medium was purchased from Life Technologies (Paisley, UK).
Cell culture. Cultures of rat hippocampal neurons were prepared as described previously (Richmond et al., 1996). Briefly, rat pups (2 d old) were killed by cervical dislocation. After dissection, hippocampal tissue was treated with a mixture of pronase E and protease type X (both at 0.5 mg/ml) for 30 min in a HEPES-buffered saline (HBS) of the following composition (in mm): NaCl 130, HEPES 10, KCl 5.4, CaCl2 1.8, MgCl2 1.0, andd-glucose 25, pH 7.4. The tissue was dissociated by trituration, centrifuged, and then plated onto culture dishes (35 mm) that had been pretreated with poly-l-lysine (20 mg/ml for 3 hr). Cultures were then incubated at 37°C in medium consisting of 90% minimal essential medium, 10% dialyzed fetal bovine serum, and 2 mml-glutamine. Cells were maintained in a humidified atmosphere of 5% CO2 in air at 37°C for up to 3 weeks. After 3–5 d in culture, cytosine arabinofuranoside (final concentration of 5 μm) was added to inhibit glial cell proliferation.
Dye loading and subsequent experiments were performed in HBS at room temperature. Experiments involving NMDA were performed in Mg2+-free medium with 10 μmglycine. Culture dishes with attached cells were incubated with the Ca2+-sensitive dyes fura-2 AM (6 μm; 40–60 min) or fluo-3 AM (10 μm; 40–60 min). To block indirect actions of mGlu receptor activation via synaptically driven Ca2+ transients, all experiments were performed in the presence of tetrodotoxin (0.5 μm). Compounds were applied directly to the perfusate (1.5–2 ml/min). Cells were studied between 6 and 21 d in culture. Data were obtained from neurons and glia, which were identified by their morphological and functional characteristics, including sensitivity to NMDA and depolarization with high extracellular K+.
Microscopy and data analysis. In most experiments, a standard, conventional imaging system [PerkinElmer Life Sciences (Santa Clara, CA) or Improvision, Lexington, MA] and the Ca2+-sensitive dye fura-2 were used to measure changes in intracellular Ca2+levels. Ratiometric images (350/380 or 360/380 nm excitation) were collected at 3–10 sec intervals. This was increased to 0.5–1 sec intervals during mGlu receptor-mediated responses. Ratio values were calculated for each pixel in the frame, after subtraction of background fluorescence intensities. Numerical data were derived from somatic measurements unless otherwise indicated. In some experiments, the ratio values have been converted to estimated measurements of intracellular Ca2+ concentration using a method similar to that of Irving and Collingridge (1998) and the equations ofGrynkiewicz et al. (1985). The calibration was performed in situ, in which Rmax,Rmin, and β values were determined using solutions containing a Ca2+ionophore (10 μm ionomycin) with either nominally Ca2+-free saline plus 1 mm EGTA or saline containing 1.8 mm Ca2+. Because of the uncertainties associated with the accurate calibration of Ca2+ levels in cells, some of the data are presented as changes in fluorescence ratio rather than intracellular Ca2+ concentrations. High-resolution confocal imaging was performed using a Bio-Rad (Hercules, CA) Microradiance imaging system connected to an Olympus Optical (Tokyo, Japan) BX50 WI microscope (60× objective). Cells were loaded with the single wavelength, intensity modulating Ca2+ indicator fluo-3AM and were excited at 488 nm with emission detected at 550 nm. Data were analyzed both on-line and off-line using Time-Course software (Bio-Rad). Experiments were performed on at least two sets of cultures obtained from different rats, with up to 30 neurons being analyzed per experiment. Statistical analysis was performed using a paired Student's t test.p < 0.05 was considered significant. In protocols investigating the effects of blocking agents on responses to DHPG, only cells that exhibited <25% variability between two successive control responses were selected for analysis.
Electrophysiology. Whole-cell recordings were made at room temperature (22–25°C) from neurons using an Axopatch-200B amplifier (Axon Instruments, Foster City, CA) with patch pipettes of 3–10 MΩ resistance. Pipette solutions contained (in mm): KCl 20, K-gluconate 115, MgATP 4, GTP 0.3, phosphocreatine 10, HEPES 10, and fura-2 0.15, pH adjusted to 7.2 with KOH. The bathing solution contained (in mm): NaCl 130, HEPES 10, KCl 5.4, CaCl2 1.8, MgCl2 1.0, and d-glucose 25, pH 7.4. Experiments involving NMDA were performed in Mg2+-free medium with 10 μm glycine. Cells were initially held at −60 mV and were low-pass filtered at 1–5 kHz. Series resistances ranged from 15 to 30 MΩ and were not compensated. Solution changes were achieved using a gravity feed system at a rate of 1.5–2 ml/min.
RESULTS
Activation of mGlu receptors combined with depolarization generates supralinear Ca2+ signals
The group I mGlu receptor agonist DHPG (100 μm; applied for 90 sec) (Conn and Pin, 1997) elicited little (<100 nm; 31 of 68 cells) or no (22 of 68 cells) increase in intracellular Ca2+ levels under control conditions (5.4 mm K+; 1.8 mm Ca2+) in the majority of hippocampal neurons tested. When present, neuronal responses to DHPG comprised a single Ca2+ transient, with a rapid rising phase and slower decay. Reproducible responses to DHPG could be obtained providing sufficient time separated successive agonist applications (10–15 min).
To determine whether DHPG-induced responses were facilitated by membrane depolarization, we doubled the extracellular K+ concentration to 10.8 mm. This proved remarkably effective, such that DHPG induced responses in almost all neurons investigated (60 of 68 cells). Indeed, in a proportion of these neurons, evoked responses were only observed in the presence of elevated extracellular K+ (19 of 68 cells). Overall, DHPG-induced responses in the presence of 10.8 mm K+ were increased from a mean ± SEM peak amplitude of 87 ± 21 to 322 ± 45 nm (p < 0.01) (Fig.1). Doubling the extracellular K+ concentration usually resulted in a small increase in intracellular Ca2+levels (90 ± 8 nm above basal;p < 0.01; n = 60). In neurons in which there was no detectable elevation in intracellular Ca2+ levels during exposure to 10.8 mm K+, a marked enhancement of responses to DHPG was still observed (Fig.1A). Thus, the combination of DHPG with a modest K+ elevation generated a marked, supralinear increase in intracellular Ca2+levels, in which the peak Ca2+ rise was much greater than the summation of the individual responses.
Fig. 1.
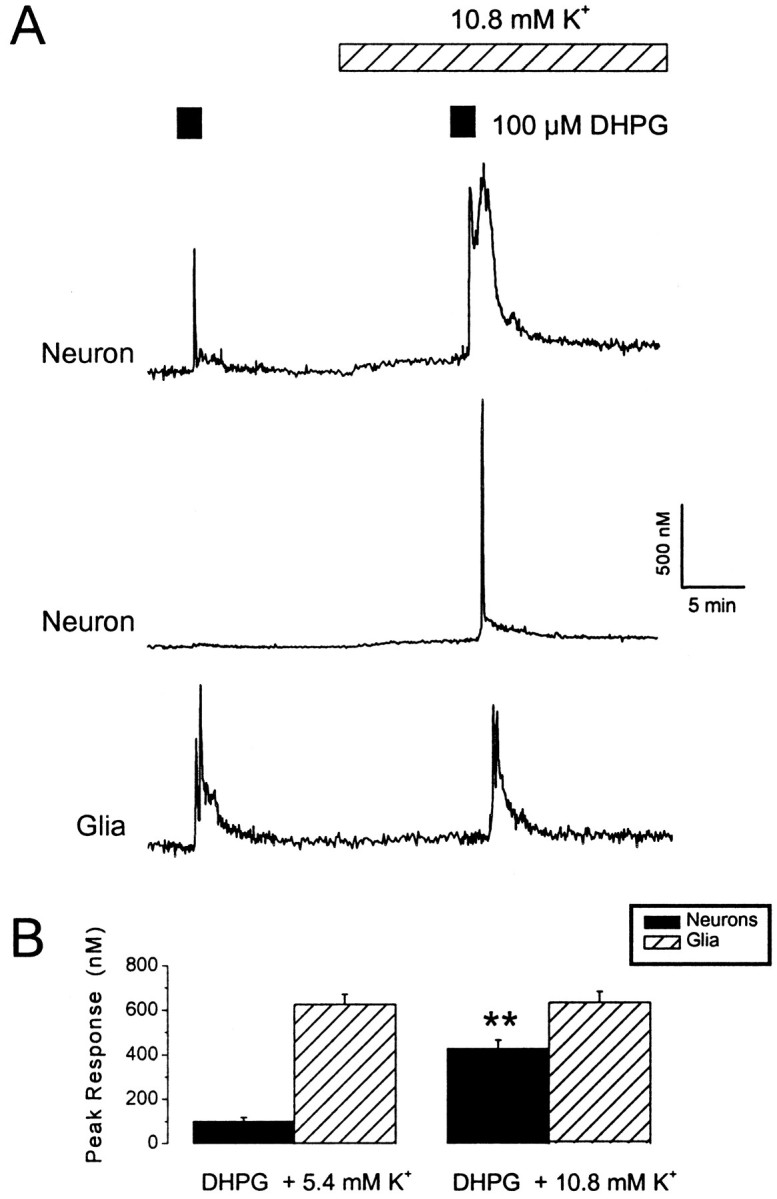
Neuronal responses to DHPG are enhanced in the presence of elevated extracellular K+.A, Traces showing changes in intracellular Ca2+ levels within the somas of two neurons and a glial cell. Cells were exposed to DHPG under control conditions (100 μm; 1.5 min exposure; 5.4 mm extracellular K+) or in the presence of a moderate elevation in the extracellular K+ concentration (total of 10.8 mm). Note the marked enhancement of the neuronal response to DHPG with minimal effects of elevated K+ on resting Ca2+ levels. B, A histogram illustrating pooled data for the effects of elevated extracellular K+ levels on DHPG responses in neurons (black bars) and glia (hatched bars). **p < 0.01.
The temporal profile of mGlu receptor-mediated responses was also modified in the presence of elevated K+. The duration of the response to DHPG in neurons was usually prolonged, and, in some cells, pronounced oscillatory responses were observed. In addition, the latency between agonist exposure and response was reduced; the time from the start of perfusion with DHPG to 50% of the peak response was 40 ± 3 sec in 5.4 mmK+ and 27 ± 3 sec in 10.8 mm K+ (n = 15; these values include a dead space time of ∼20 sec; p< 0.01). A separate series of experiments, using confocal imaging of fluo-3 AM-loaded cells, investigated the cellular localization of mGlu receptor-mediated responses under optimal conditions for the release of Ca2+ from intracellular stores. In the presence of 10.8 mmK+, DHPG-induced responses were observed throughout the neuron, including fine processes (n = 6; data not shown).
In contrast to the data obtained with the neurons under control conditions, DHPG elicited much larger responses in glial cells present in the same cultures (624 ± 47 nm; n= 21) (Fig. 1). Glial responses consisted of an initial spike, which was usually followed by a plateau phase and an oscillatory Ca2+ signal. Intracellular Ca2+ levels in glial cells were not significantly affected by doubling extracellular K+ levels (p > 0.05; n = 31), and glial responses to DHPG were not facilitated by this treatment (p > 0.05;n = 31) (Fig. 1). Thus, in this preparation, the combination of mGlu receptor activation and elevated extracellular K+ levels generated supralinear Ca2+ signals in neurons but not glia.
Supralinear interactions between mGlu and NMDA receptors
We also studied interactions between NMDA receptor-mediated Ca2+ influx and DHPG-induced Ca2+ release. Exposure of cells to NMDA (3 μm) elevated intracellular Ca2+ levels in neurons by 228 ± 18 nm (p < 0.01; n = 47) but was without effect in glia (p > 0.5;n = 13). In neurons, the combined presence of NMDA and DHPG greatly enhanced the intracellular Ca2+ response (Fig.2). Moreover, in seven neurons that exhibited no detectable response to a DHPG exposure under control conditions, a response to DHPG was observed in the presence of NMDA (3 μm). Overall, the mean, peak neuronal response to DHPG in control medium was 107 ± 8 nm, whereas in the presence of NMDA, it was 478 ± 70 nm (p < 0.05; 45 cells). As with exposure to high extracellular K+, the temporal profile of DHPG responses was modified in the presence of NMDA. The duration of responses to DHPG in neurons was prolonged, and, in some cells, pronounced oscillatory responses were observed. In glial cells, NMDA had no significant effect on the magnitude of responses to DHPG (p > 0.05; n = 13). Thus, the effects of elevated extracellular K+and NMDA on DHPG-induced responses were very similar.
Fig. 2.
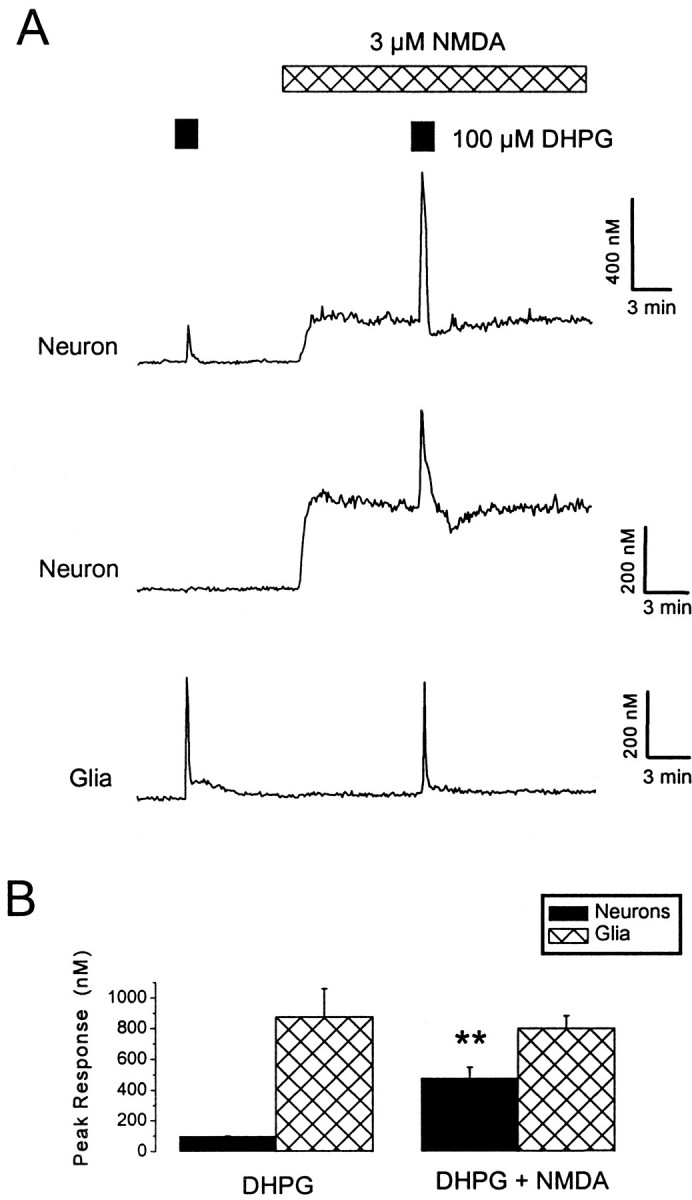
Supralinear interactions between NMDA and DHPG receptor-mediated Ca2+ signals. A, DHPG (100 μm) was applied under control conditions and in the presence of NMDA (3 μm). Neuronal, but not glial, responses to DHPG were greatly enhanced during exposure to NMDA. Themiddle trace illustrates a neuron that only exhibited a response to DHPG in the presence of NMDA. B, A histogram illustrating pooled data for the effects of NMDA on DHPG responses in neurons (black bars) and glia (hatched bars). **p < 0.01.
DHPG responses are attributable to mGlu5receptor-mediated Ca2+ mobilization
Group I mGlu receptors comprise two subtypes, mGlu1 and mGlu5, both of which are expressed in hippocampal neurons (Conn and Pin, 1997). We found that DHPG responses, evoked in the presence of elevated K+, were greatly inhibited in neurons (by 86 ± 6%; p < 0.01; n = 18) and abolished in glia (p < 0.01; n= 16) by the specific mGlu5 receptor antagonist MPEP (1 μm) (Gasparini et al., 1999) (Fig.3). A combination of the ionotropic glutamate receptor antagonists NBQX (2 μm) andd-AP-5 (50 μm) had no significant effect on the magnitude of responses to DHPG in the presence of 10.8 mmK+ (n = 11;p > 0.05). Thus, mGlu5 receptors are the predominant subtype involved in the present investigation.
Fig. 3.

Antagonism of DHPG responses by the mGlu receptor antagonist MPEP. A, Traces from a neuron and a glial cell that were exposed to successive applications of DHPG (100 μm) in the presence of 10.8 mmK+. Responses to DHPG were blocked in the presence of MPEP (1 μm) and recovered on washout.B, A histogram illustrating pooled data for the effects on DHPG responses in neurons (black bars) and glia (hatched bars). **p < 0.01.
DHPG responses evoked in the presence of either elevated extracellular K+ or NMDA are reminiscent of Ca2+ release from intracellular stores (Murphy and Miller, 1989; Irving and Collingridge, 1998). This was confirmed using thapsigargin (1 μm) (Fig.4), an agent that irreversibly depletes intracellular Ca2+ stores by inhibition of the sarcoplasmic/endoplasmic Ca2+ ATPase (SERCA) (Law et al., 1990) and prevents Ca2+ mobilization after their spontaneous emptying (Irving et al., 1992). Responses to DHPG in the presence of elevated K+ were inhibited by 72 ± 7% (p < 0.01; n = 23) in neurons and by 97 ± 3% (p < 0.01;n = 9) in glia after exposure to thapsigargin (2 μm; applied for 8 min). Likewise, neuronal responses to DHPG in the presence of NMDA were inhibited by 83 ± 6% (p < 0.01; n = 20) after exposure to thapsigargin.
Fig. 4.
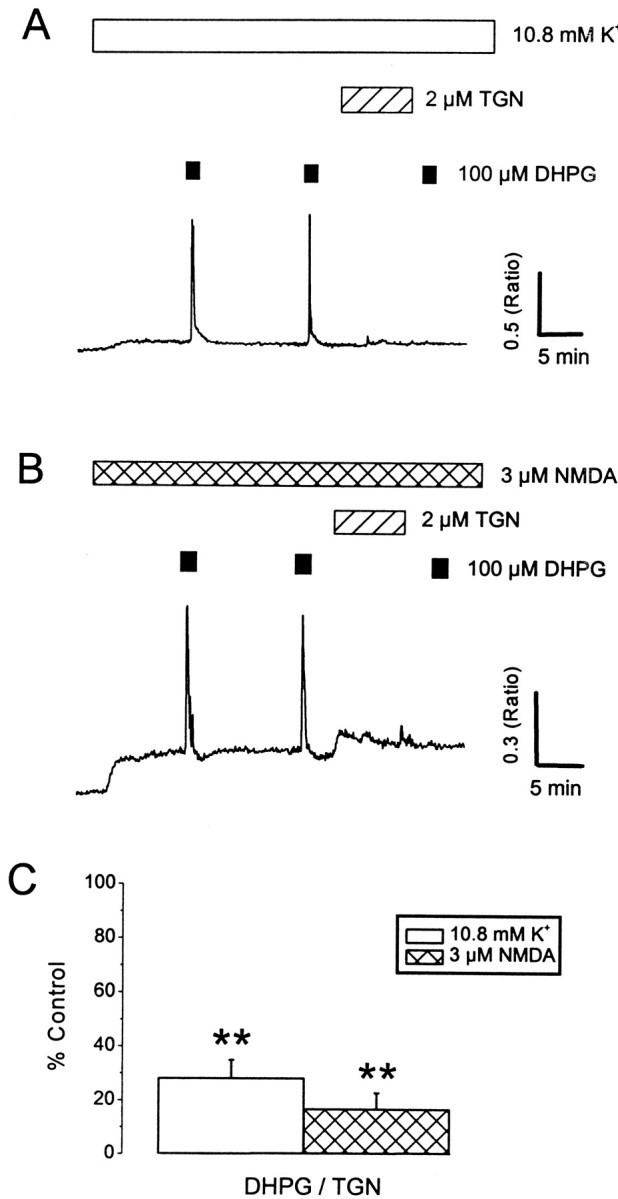
Involvement of Ca2+ stores in the supralinear responses. A, Neurons were exposed to successive applications of 100 μm DHPG in the presence of 10.8 mm K+ (A) or NMDA (3 μm; B). Treatment with thapsigargin (TGN; 2 μm; 8 min) blocked subsequent responses to DHPG. C, A histogram illustrating pooled data for the effects of thapsigargin on DHPG responses in the presence of 10.8 mm K+(white bars) and NMDA (hatched bars). **p < 0.01.
Treatment of neurons with thapsigargin during exposure to 10.8 mm K+ or NMDA was only occasionally linked with a small elevation in intracellular Ca2+ levels, which slowly returned to baseline levels. This suggests three things: first, that Ca2+ release from intracellular stores is not contributing to the modest increases in Ca2+ levels that are associated with exposure to high extracellular K+ or NMDA; second, that the stores discharge spontaneously; and third, that this occurs sufficiently slowly for the Ca2+ to be rapidly excluded from the cytosol (Garaschuk et al., 1997). In glial cells, thapsigargin induced a marked elevation in intracellular Ca2+ levels that decayed slowly but remained at a level above the initial baseline (data not shown). This sustained increase in Ca2+ levels presumably reflects the presence of a powerful store-operated Ca2+ influx pathway in glia (Simpson and Russell, 1997). However, the absence of a similar Ca2+ elevation in neurons does not rule out the presence of a store-operated Ca2+influx pathway in these cells.
Neuronal Ca2+ stores have a high capacity to sequester Ca2+
The previous experiments suggest that Ca2+ stores are the primary source of the supralinear response when DHPG was combined with depolarization or NMDA exposure. Under these conditions, the Ca2+content of neuronal Ca2+ stores was tested using a high concentration of caffeine (50 mm) to directly activate Ca2+-induced, Ca2+ release channels through a mechanism that is independent of cytosolic Ca2+levels (Sitsapesan and Williams, 1990; Irving and Collingridge, 1998). A brief exposure of neurons to caffeine (90 sec) under control conditions elicited little (<100 nm; 30 of 59 cells) or no (17 of 59 cells) increase in intracellular Ca2+levels. However, in the presence of 10.8 mmK+ or NMDA (3 μm; Mg2+-free medium), responses to caffeine were markedly enhanced (Fig.5A–C) and were observed in almost all neurons investigated (n = 53 of 59). Overall, caffeine responses were increased from a mean peak amplitude of 52 ± 10 to 199 ± 31 nm(p < 0.01; n = 31) in the presence of 10.8 mmK+ and from 69 ± 12 to 281 ± 38 nm in the presence of NMDA (3 μm; p < 0.01;n = 28). In control experiments, ryanodine (10 μm) abolished responses to caffeine (50 mm; n = 6). Caffeine elicited no detectable Ca2+ rise in glial cells under any condition (n = 40).
Fig. 5.
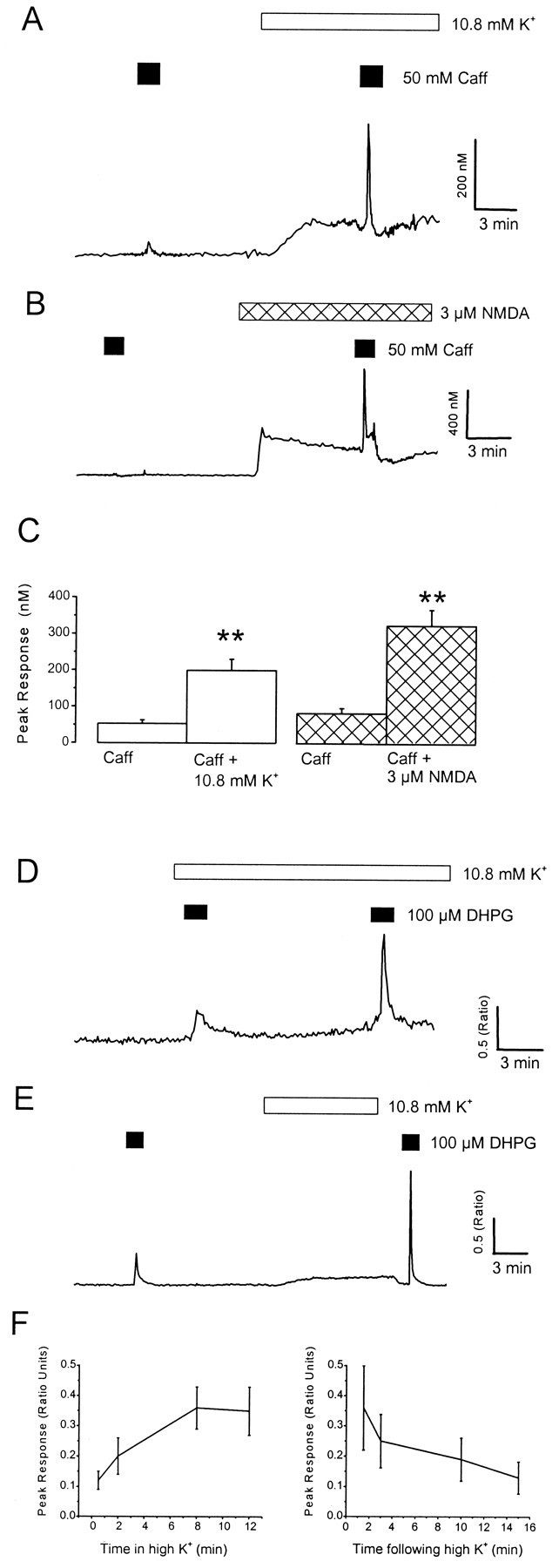
Loading and unloading neuronal Ca2+ stores. A–C, The Ca2+ content of neuronal Ca2+stores was tested using a high concentration of caffeine.A, The neuron was exposed to caffeine (Caff; 50 mm) in control medium and then in the presence of high extracellular K+ (10.8 mm). B, The neuron was exposed to caffeine in Mg2+-free medium and then in the presence of NMDA (3 μm). Note the similar enhancement of neuronal responses to caffeine during exposure to elevated extracellular K+ or NMDA. C, A histogram illustrating pooled data for the effects of 10.8 mmK+ (white bars) and NMDA (hatched bars) on caffeine responses. **p < 0.01. These data indicate that neuronal Ca2+ stores have a high capacity to sequester Ca2+. D–F, Time course of loading and discharging of Ca2+ stores. D, The cell was exposed to 100 μm DHPG close to the onset (30 sec) of perfusion with elevated K+-containing medium (10.8 mm) and then applied 12 min later.E, The cell was exposed to DHPG under control conditions and then a short period (90 sec) after removal of elevated K+ (applied for 10 min). F, Pooled data illustrating the effects of duration of exposure to 10.8 mm K+ (n = 19) and time after removal of 10.8 mm K+ on the magnitude of responses to DHPG (n = 11).
These data indicate that Ca2+ stores are functionally depleted under control conditions. If the supralinear responses reflect the loading of Ca2+stores, then the potentiation would also be expected to persist for a period after a store-loading paradigm and there would be an initial delay in the enhancement of responses to DHPG as the stores charge with Ca2+. Thus, the magnitude of responses to DHPG was compared after various times in the presence of, or after, depolarization with 10.8 mmK+. A delay of 2–8 min was observed before maximal levels of potentiation were achieved after exposure to elevated K+, and a degree of facilitation persisted for up to 10 min after removal of the store-loading stimulus (Fig. 5D–F). These results are consistent with supralinear Ca2+ signaling reflecting the loading state of Ca2+ stores.
Role of L-type voltage-gated Ca2+ channels
Because in neurons the state of filling of Ca2+ stores has such an influence on the DHPG-induced response, it is important to determine the pathways by which the Ca2+ stores can be filled. The influx of Ca2+ through L-type VGCCs has been reported previously to charge Ca2+stores during depolarization (Irving and Collingridge, 1998). Therefore, the contribution of L-type VGCCs to the loading of DHPG-sensitive intracellular Ca2+ stores under control conditions, in the presence of elevated extracellular K+ or NMDA (3 μm), was investigated. The L-type Ca2+ channel antagonist nifedipine (10 μm) did not significantly affect either neuronal (n = 12) or glial (n = 6) responses to DHPG obtained under control conditions (p > 0.05). However, responses in the presence of elevated K+ were strongly inhibited in its presence (83 ± 9% inhibition; n= 9; p < 0.01) (Fig. 6). In contrast, responses to DHPG in the presence of NMDA were only slightly reduced by nifedipine (13 ± 9% inhibition;n = 9; p < 0.05) (Fig. 6). Nifedipine had little effect on resting Ca2+ levels in control medium but lowered intracellular Ca2+ levels to near control values in which an increase in Ca2+ was observed during exposure to 10.8 mmK+. In the presence of NMDA, nifedipine only reduced Ca2+ levels slightly (Fig.6). These results suggest that the same Ca2+ stores can be filled by at least three separate pathways; one that is active at rest, and one involving L-type Ca2+, which accounts for most of the effect of a small depolarization and a pathway that is mainly used after activation of NMDA receptors.
Fig. 6.
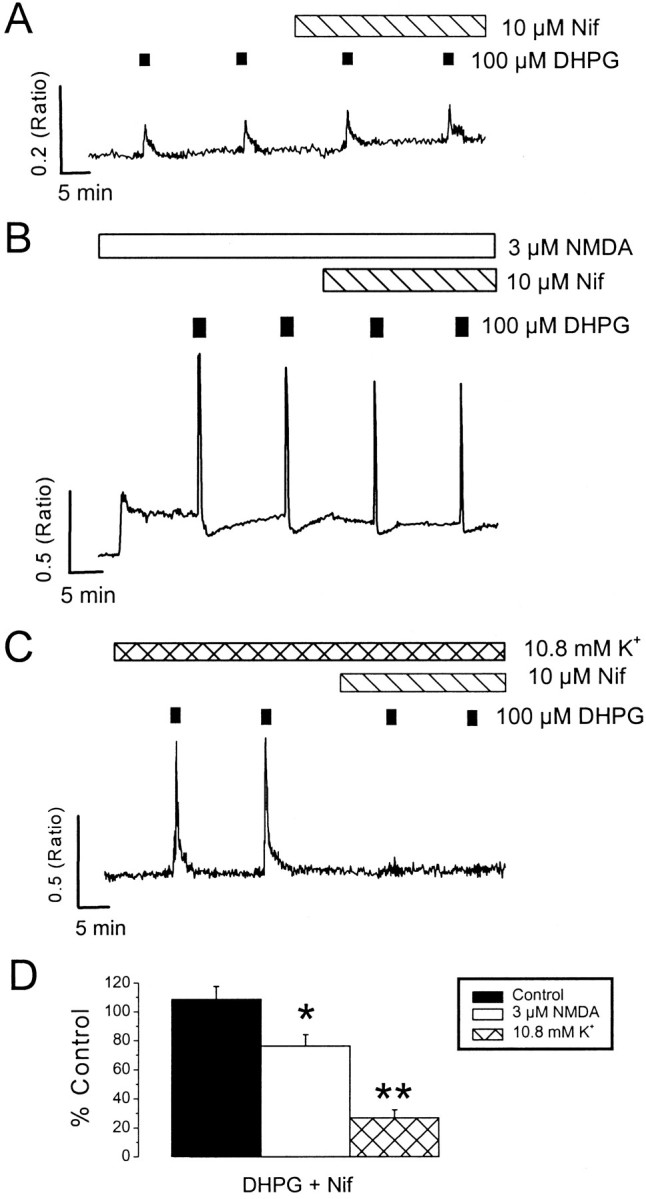
Role of L-type voltage-gated Ca2+ channels in charging Ca2+stores. Neurons were exposed to four successive applications of DHPG (100 μm) in control media (A), in the presence of NMDA (3 μm; B), or in the presence of 10.8 mm K+(C). The final two DHPG applications were made in the presence of nifedipine (Nif; 10 μm).D, A histogram illustrating pooled data for the effects of nifedipine on responses to DHPG in control media (black bars), in the presence of NMDA (3 μm;white bars), or in the presence of 10.8 mmK+ (hatched bars). *p < 0.05; **p < 0.01. Note the strong inhibition of responses to DHPG obtained in the presence of 10.8 mm K+ by nifedipine. Only neurons that responded to DHPG were selected for analysis.
Two activity-dependent mechanisms for Ca2+store loading
To confirm that postsynaptic depolarization alone is sufficient to load Ca2+ stores, experiments were performed using combined whole-cell recording and Ca2+ imaging. In current-clamp experiments, exposure of neurons to 10.8 mmK+ resulted in a small depolarization of 9.6 ± 0.6 mV from a mean resting potential of −52 ± 1.7 mV (n = 12). Thus, the ability of modest depolarization to enhance DHPG responses was tested directly under voltage-clamp conditions. In seven neurons, responses to DHPG were enhanced from 0.33 ± 0.12 to 0.64 ± 0.14 ratio units (p < 0.05; n = 10) by a 15–30 mV depolarization, relative to the initial holding potential of −60 mV (Fig. 7A).
Fig. 7.
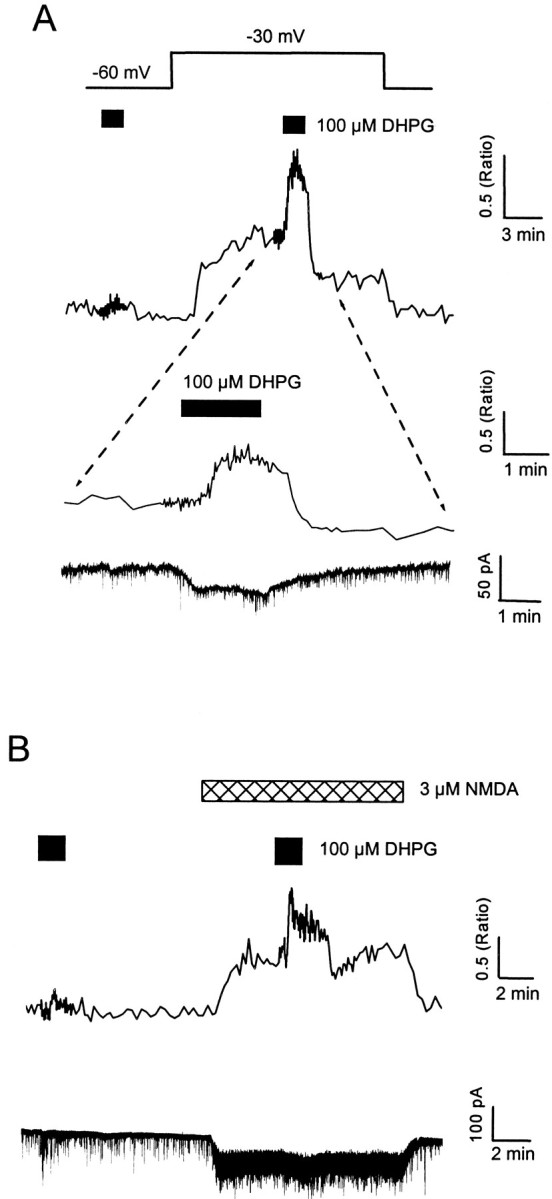
Multiple pathways for loading neuronal Ca2+ stores. A, B, Combined whole-cell voltage-clamp recordings and Ca2+ imaging, with fura-2 (150 μm) in the patch pipette. A, The neuron was exposed to 100 μm DHPG at a holding potential of −60 mV, the holding potential was then changed to −30 mV, as indicated by the voltage step, and DHPG was reapplied. The expanded region shows the Ca2+ response to DHPG during depolarization (top trace) associated with a small inward current (bottom trace). Similar currents were present in other cells and ranged from 3 to 45 pA. Note the absence of a detectable Ca2+ response to DHPG at −60 mV. B, The neuron was exposed to 100 μm DHPG at a holding potential of −60 mV in control Mg2+-free media and then in the presence of NMDA (3 μm). Exposure to NMDA itself was associated with an inward current (40.1 ± 10.8 pA;n = 10) and an increase in membrane noise. The spontaneous, spike-like activity on the electrical traces represent miniature synaptic currents.
The nifedipine-insensitive enhancement of DHPG responses in the presence of NMDA could be attributable to activation of a different class of voltage-gated Ca2+ channel(s) or the result of Ca2+ permeation through NMDA channels. This was tested directly by holding cells at −60 mV throughout the experiment. Responses to DHPG were enhanced from 0.36 ± 0.17 to 0.69 ± 0.14 ratio units (p < 0.05; n = 10) in the presence of NMDA (3 μm) (Fig.7B).
Collectively, these data show that activation of mGlu5 receptors can release Ca2+ from intracellular stores. These stores are functionally depleted at rest but can be charged with Ca2+ either via modest depolarization, which activates L-type Ca2+ channels, or by Ca2+ entry through NMDA receptors (Fig.8).
Fig. 8.

Scheme illustrating the different pathways for loading IP3-sensitive Ca2+ stores in hippocampal neurons. A, At rest, background loading of Ca2+ stores is via a dihydropyridine-insensitive basal influx pathway. Activity-dependent Ca2+ influx via L-type VGCCs during depolarization (B) or by stimulation of NMDA receptor-operated channels (C) substantially increases the Ca2+ loading of the stores and allows a strong Ca2+ signal on activation of IP3receptors. The ability of IP3-linked agonists to mobilize Ca2+ is primarily related to the level of Ca2+ contained within the stores, which is governed by the net flux of Ca2+ into the cell, the activity of the SERCA pump, and by the activity of the release channels.
DISCUSSION
Supralinear Ca2+ responses are observed in neurons when mGlu receptor activation is combined with depolarization or exposure to NMDA, and this interaction reflects the loading state of intracellular Ca2+stores. Group I mGlu receptor-mediated responses are also observed in glial cells (Bernstein et al., 1998); however, they are not modulated by depolarization or exposure to NMDA.
Multiple pathways for loading Ca2+ stores
The reduced magnitude of neuronal Ca2+ mobilizing responses under control conditions suggests that the Ca2+ stores are only partially loaded (Garaschuk et al., 1997) or functionally empty (Shmigol et al., 1996; Irving and Collingridge, 1998; Koizumi et al., 1999) at rest. Reproducible responses to DHPG were observed in glia under control conditions, suggesting that their Ca2+ stores are able to replenish spontaneously (Simpson and Russell, 1997). When present, reproducible responses were also observed in neurons at rest. In many types of nonexcitable cells, store depletion triggers a Ca2+ release-activated Ca2+ current (ICRAC) (Parekh and Penner, 1997), which is thought to underlie “capacative” Ca2+ influx and store refilling (Putney, 1986). In addition, a novel noncapacitative Ca2+ entry channel (IARC) has been described recently in human embryonic kidney cells (Mignen and Shuttleworth, 2000), which could also allow store refilling during pulsatile Ca2+ release. Similar pathways may be involved in store refilling after Ca2+release under resting conditions in mammalian neurons (Garaschuk et al., 1997; Usachev and Thayer, 1999).
Neurons express many different types of Ca2+-permeable channels in their plasma membranes, which could, in principle, link with their Ca2+ stores. The present data show that activation of VGCCs or NMDA receptors can add substantial additional loading to neuronal Ca2+ stores. The involvement of L-type Ca2+ channels in the depolarization-induced loading of IP3-sensitive Ca2+ stores has been implicated in our previous work characterizing muscarinic responses in cultured hippocampal neurons (Irving and Collingridge, 1998). The lack of dihydropyridine sensitivity of neuronal DHPG responses under control conditions is similar to that observed with responses to caffeine in hippocampal slices (Garaschuk et al., 1997). The enhancement of caffeine responses by depolarization or NMDA receptor activation extends on previous studies investigating the properties of caffeine-sensitive Ca2+ stores (Friel and Tsien 1992; Shmigol et al., 1994; Garaschuk et al., 1997; Koizumi et al., 1999). The present results can be explained by the development of a simple scheme (Irving and Collingridge, 1998), whereby the loading state of the intracellular Ca2+ stores in neurons is regulated dynamically by Ca2+influx across the plasma membrane (Fig. 8) (see also Verkhratsky and Petersen, 1998).
Supralinear Ca2+ signaling
Classically, the biophysical properties of NMDA receptors enable them to act as coincidence detectors (Mayer et al., 1984; Nowak et al., 1984). The present data suggests that the group I mGlu receptor-mediated Ca2+ signaling could also detect changes in neuronal activity by sensing membrane depolarization or NMDA receptor activation. Although this functionality can arise at a number of different levels, the loading state of intracellular Ca2+ stores represents the primary mechanism underlying this action. There are two inter-related processes whereby Ca2+ stores could influence the response to phosphoinositidase C (PIC)-linked agonists. The magnitude of the response will be dependent on the quantity of Ca2+ available for release, and the sensitivity of Ca2+ release channels may themselves be regulated by luminal Ca2+levels (Missiaen et al., 1992; Koizumi et al., 1999). Indeed, increased store loading in PC12 cells leads to an enhancement in the frequency and coupling between elementary release sites leading to the generation of global Ca2+ signals (Koizumi et al., 1999). An action of luminal Ca2+ on the sensitivity of neuronal Ca2+ release channels is also indicated by the finding that relatively short periods of thapsigargin exposure, under conditions in which Ca2+ stores are fully charged, can block subsequent responses to DHPG without necessarily increasing Ca2+ levels. Because the period of exposure to thapsigargin is relatively short, the stores themselves are likely to be only partially depleted. However, this effect is presumably sufficient to inhibit IP3-mediated Ca2+ release.
The contribution of other processes, such as the effects of cytosolic Ca2+ on the sensitivity of the IP3 receptor (Bezprozvanny et al., 1991, Nakamura et al., 1999) and PIC activation (Irving et al., 1992; Challiss et al., 1994), could also have a role. The observation that there is a reduction in the response latency to DHPG in elevated K+ relative to that in control medium may indicate higher levels of IP3 formation (Carter and Ogden, 1997). Importantly, however, the marked facilitation of responses to DHPG by modest depolarization does not necessarily require increased cytosolic Ca2+ levels. This suggests further that the loading of intracellular stores is the key factor in the observed supralinearity. For example, the enhancement of DHPG responses for a period after depolarization is indicative of store loading (Garaschuk et al., 1997; Irving and Collingridge, 1998) and is present when Ca2+ levels have returned to baseline. In addition, the store-loading stimulus itself need not increase cytosolic Ca2+ levels. If during depolarization the buffering capacity of the cell (including Ca2+ pumps on the endoplasmic reticulum and plasma membrane) can effectively handle the increased Ca2+ influx, luminal Ca2+ levels will rise without an associated elevation in the cytoplasmic Ca2+ concentration (Irving and Collingridge, 1998). Moreover, the idea that loading (or refilling) of Ca2+ stores can occur without any measurable rise in cytosolic Ca2+ levels is supported by studies on pancreatic acinar cells (Mogami et al., 1997).
In pyramidal cells (Bianchi et al., 1999) and interneurons (Woodhall et al., 1999) in hippocampal slices, group I mGlu receptor activation can elevate Ca2+ levels by membrane depolarization and activation of VGCCs. It is possible that a component of the DHPG-induced Ca2+ rise observed in the present investigation could also be mediated by recruitment of VGCCs; however, a number of observations indicate that Ca2+ release from intracellular stores is the principle source of the Ca2+elevation. Thus, DHPG responses were sensitive to thapsigargin, were observed under voltage-clamp conditions, and were only associated with small inward currents, which often exhibited a different time course from the Ca2+ transients. Brief exposure of cells to DHPG was not associated with the prolonged Ca2+ rises observed in pyramidal cells byBianchi et al. (1999).
Activation of group I mGlu receptors can also directly enhance responses to NMDA through a mechanism that is independent of Ca2+ release from stores in rat hippocampal slices (Harvey and Collingridge, 1993; Fitzjohn et al., 1996) and augment mGlu receptor-mediated currents in CA3 pyramidal neurons in cultured slices (Lüthi et al., 1994). It is unlikely that this effect contributes much to the supralinear responses observed when DHPG was applied in the presence of NMDA because the responses were markedly inhibited after thapsigargin exposure. In addition, a similar interaction was observed when NMDA was combined with a high concentration of caffeine, which directly stimulates Ca2+ release from intracellular stores (Sitsapesan and Williams, 1990; Irving and Collingridge, 1998).
Physiological implications
In neurons, group I mGlu receptors may have a key role in detecting changes in neuronal activity through the generation of supralinear signals. This action reflects the characteristics of mGlu receptor-mediated Ca2+ signaling and the loading state of intracellular stores. Consequently, it need not be specific to mGlu receptors and could be exhibited by other PIC-coupled receptors in these cells (Irving and Collingridge, 1998). The global nature of the supralinear responses is ideally suited to conveying Ca2+ signals from neurites and spines to the nucleus (Berridge, 1998; Nakamura et al., 1999), which may be involved in changes in gene expression associated with synaptic plasticity (Stanton and Sarvey, 1984; Frey and Morris, 1997). Although depolarization itself can enhance Ca2+influx through NMDA receptor-operated channels, the present data show that additional, powerful mechanisms exist for the generation of supralinear Ca2+ rises. A combination of NMDA and mGlu receptor activation leading to supralinear Ca2+ signaling may be involved during the induction of LTP in area CA1 of the hippocampus during weak stimulation paradigms, whereas NMDA receptor activation alone may be sufficient for synaptic plasticity during robust stimulation (Wilsch et al., 1998). A linkage between depolarization and mGlu receptor-mediated Ca2+ signaling could also act as a distinct mechanism for detecting changes in synaptic activity. Moreover, a synergistic interaction between depolarization and mGlu receptor-mediated Ca2+ release from stores may have a role in coincidence detection associated with backpropagating action potentials (Nakamura et al., 1999). The present data show that the mechanism of detection involving mGlu receptor-mediated Ca2+ signaling has different temporal properties compared with the NMDA receptor. There is an initial delay while the Ca2+ stores charge, and the effect persists for a period after removal of the store-loading stimulus. Thus, this process could act as a mechanism for recency detection by integrating activity that is close in time but not necessarily temporally coincident. In summary, the expression of multiple routes for the generation of supralinear Ca2+ signals adds considerable flexibility to the processes that underlie activity-dependent changes in synaptic strength.
Footnotes
This work was supported by Wellcome Trust Grant 47368.
Correspondence should be addressed to Andrew J. Irving at the above address. E-mail: a.j.irving@abdn.ac.uk.
REFERENCES
- 1.Alford S, Frenguelli BG, Schofield JG, Collingridge GL. Characterization of Ca2+ signals induced in hippocampal CA1 neurones by the synaptic activation of NMDA receptors. J Physiol (Lond) 1993;469:693–716. doi: 10.1113/jphysiol.1993.sp019838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ascher P, Nowak L. The role of divalent cations in the N-methyl-d-aspartate responses of mouse central neurones in culture. J Physiol (Lond) 1988;399:247–266. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bear MF, Abraham WC. Long-term depression in the hippocampus. Annu Rev Neurosci. 1996;19:437–462. doi: 10.1146/annurev.ne.19.030196.002253. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein M, Behnisch T, Balschun D, Reymann KG, Reiser G. Pharmacological characterisation of metabotropic glutamatergic and purinergic receptors linked to Ca2+ signalling in hippocampal astrocytes. Neuropharmacology. 1998;37:169–178. doi: 10.1016/s0028-3908(98)00012-4. [DOI] [PubMed] [Google Scholar]
- 5.Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- 6.Bezprozvanny I, Watras J, Ehrlich BE. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature. 1991;351:751–754. doi: 10.1038/351751a0. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi R, Young SR, Wong RKS. Group I mGluR activation causes voltage-dependent and-independent Ca2+ rises in hippocampal pyramidal cells. J Neurophysiol. 1999;81:2903–2913. doi: 10.1152/jn.1999.81.6.2903. [DOI] [PubMed] [Google Scholar]
- 8.Bliss T, Collingridge G. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 9.Bortolotto ZA, Fitzjohn SM, Collingridge GL. Roles of metabotropic glutamate receptors in LTP and LTD in the hippocampus. Curr Opin Neurobiol. 1999;9:299–304. doi: 10.1016/s0959-4388(99)80044-0. [DOI] [PubMed] [Google Scholar]
- 10.Carter TD, Ogden D. Kinetics of Ca2+ release by InsP3 in pig single aortic endothelial cells: evidence for an inhibitory role of cytosolic Ca2+ in regulating hormonally evoked Ca2+ spikes. J Physiol (Lond) 1997;504:17–33. doi: 10.1111/j.1469-7793.1997.00017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Challiss RAJ, Mistry R, Gray DW, Nahorski SR. Modulatory effects of NMDA on phosphoinositide responses evoked by the metabotropic glutamate receptor agonist 1S,3R-ACPD in neonatal rat cerebral cortex. Br J Pharmacol. 1994;112:231–239. doi: 10.1111/j.1476-5381.1994.tb13057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 13.Emptage N, Bliss TV, Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999;22:115–124. doi: 10.1016/s0896-6273(00)80683-2. [DOI] [PubMed] [Google Scholar]
- 14.Emptage NJ. Calcium on the up: supralinear calcium signaling in central neurons. Neuron. 1999;24:495–497. doi: 10.1016/s0896-6273(00)81103-4. [DOI] [PubMed] [Google Scholar]
- 15.Fitzjohn SM, Irving AJ, Palmer MJ, Harvey J, Lodge D, Collingridge GL. Activation of group I mGluRs potentiate NMDA responses in rat hippocampal slices. Neurosci Lett. 1996;203:1–3. doi: 10.1016/0304-3940(96)12301-6. [DOI] [PubMed] [Google Scholar]
- 16.Frenguelli BG, Potier B, Slater NT, Alford S, Collingridge GL. Metabotropic glutamate receptors and calcium signalling in dendrites of hippocampal CA1 neurones. Neuropharmacology. 1993;32:1229–1237. doi: 10.1016/0028-3908(93)90017-w. [DOI] [PubMed] [Google Scholar]
- 17.Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- 18.Friel DD, Tsien RW. A caffeine- and ryanodine-sensitive Ca2+ store in bullfrog sympathetic neurones modulates effects of Ca2+ entry on [Ca2+]i. J Physiol (Lond) 1992;450:217–246. doi: 10.1113/jphysiol.1992.sp019125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garaschuk O, Yaari Y, Konnerth A. Release and sequestration of calcium by ryanodine-sensitive stores in rat hippocampal neurons. J Physiol (Lond) 1997;502:13–30. doi: 10.1111/j.1469-7793.1997.013bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, Kuhn R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 21.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 22.Harvey J, Collingridge GL. Signal transduction pathways involved in the acute potentiation of NMDA responses by 1S,3R-ACPD in rat hippocampal slices. Br J Pharmacol. 1993;109:1085–1090. doi: 10.1111/j.1476-5381.1993.tb13733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Irving AJ, Collingridge GL. A characterisation of muscarinic receptor-mediated Ca2+ mobilisation in cultured rat hippocampal neurons. J Physiol (Lond) 1998;511:747–759. doi: 10.1111/j.1469-7793.1998.747bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irving AJ, Collingridge GL, Schofield JG. l-Glutamate and acetylcholine mobilise Ca2+ from the same intracellular pool in cerebellar granule cells using transduction mechanisms with different Ca2+ sensitivities. Cell Calcium. 1992;13:293–301. doi: 10.1016/0143-4160(92)90064-y. [DOI] [PubMed] [Google Scholar]
- 25.Jahr CE, Stevens CF. Glutamate activates multiple single channel conductances in hippocampal neurons. Nature. 1987;325:522–525. doi: 10.1038/325522a0. [DOI] [PubMed] [Google Scholar]
- 26.Koizumi S, Bootman MD, Bobanovic LK, Schell MJ, Berridge MJ, Lipp P. Characterization of elementary Ca2+ release signals in NGF-differentiated PC12 cells and hippocampal neurons. Neuron. 1999;22:125–137. doi: 10.1016/s0896-6273(00)80684-4. [DOI] [PubMed] [Google Scholar]
- 27.Kovalchuk Y, Eilers J, Lisman J, Konnerth A. NMDA receptor-mediated subthreshold Ca2+ signals in spines of hippocampal neurons. J Neurosci. 2000;20:1791–1799. doi: 10.1523/JNEUROSCI.20-05-01791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Law GL, Pachter JA, Thastrup OM, Hanley R, Dannies PS. Thapsigargin, but not caffeine, blocks the ability of thyrotropin-releasing hormone to release Ca2+ from an intracellular store in GH4C1 pituitary cells. Biochem J. 1990;267:359–364. doi: 10.1042/bj2670359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lüthi A, Gähwiler BH, Gerber U. Potentiation of a metabotropic glutamatergic response following NMDA receptor activation in rat hippocampus. Pflügers Arch. 1994;427:197–202. doi: 10.1007/BF00585965. [DOI] [PubMed] [Google Scholar]
- 30.Malinow R, Otmakhov N, Blum KI, Lisman J. Visualizing hippocampal synaptic function by optical detection of Ca2+ entry through the N-methyl-d-aspartate channel. Proc Natl Acad Sci USA. 1994;91:8170–8174. doi: 10.1073/pnas.91.17.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 32.Mayer ML, MacDermott AB, Westbrook GL, Smith SJ, Barker JL. Agonist- and voltage-gated calcium entry in cultured mouse spinal cord neurons under voltage clamp measured using arsenazo III. J Neurosci. 1987;7:3230–3244. doi: 10.1523/JNEUROSCI.07-10-03230.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mignen O, Shuttleworth TJ. I(ARC), a novel arachidonate-regulated, noncapacitative Ca2+ entry channel. J Biol Chem. 2000;275:9114–9119. doi: 10.1074/jbc.275.13.9114. [DOI] [PubMed] [Google Scholar]
- 34.Miller LD, Petrozzino JJ, Golarai G, Connor JA. Ca2+ release from intracellular stores induced by afferent stimulation of CA3 pyramidal neurons in hippocampal slices. J Neurophysiol. 1996;76:554–562. doi: 10.1152/jn.1996.76.1.554. [DOI] [PubMed] [Google Scholar]
- 35.Missiaen L, De Smedt H, Droogmans G, Casteels R. Ca2+ release induced by inositol 1,4,5-trisphosphate is a steady-state phenomenon controlled by luminal Ca2+ in permeabilized cells. Nature. 1992;357:599–602. doi: 10.1038/357599a0. [DOI] [PubMed] [Google Scholar]
- 36.Mogami H, Nakano K, Tepikin AV, Petersen OH. Ca2+ flow via tunnels in polarized cells: recharging of apical Ca2+ stores by focal Ca2+ entry through basal membrane. Cell. 1997;88:49–55. doi: 10.1016/s0092-8674(00)81857-7. [DOI] [PubMed] [Google Scholar]
- 37.Murphy SN, Miller RJ. Two distinct quisqualate receptors regulate Ca2+ homeostasis in hippocampal neurons in vitro. Mol Pharmacol. 1989;35:671–680. [PubMed] [Google Scholar]
- 38.Nakamura T, Barbara J-G, Nakamura K, Ross WN. Synergistic release of Ca2+ from IP3-sensitive stores evoked by synaptic activation of mGluRs paired with backpropagating action potentials. Neuron. 1999;24:727–737. doi: 10.1016/s0896-6273(00)81125-3. [DOI] [PubMed] [Google Scholar]
- 39.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 40.Parekh AB, Penner R. Store depletion and calcium influx. Physiol Rev. 1997;77:901–390. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- 41.Perkel DJ, Petrozzino JJ, Nicoll RA, Connor JA. The role of Ca2+ entry via synaptically activated NMDA receptors in the induction of long-term potentiation. Neuron. 1993;11:817–823. doi: 10.1016/0896-6273(93)90111-4. [DOI] [PubMed] [Google Scholar]
- 42.Petrozzino JJ, Pozzo Miller LD, Connor JA. Micromolar Ca2+ transients in dendritic spines of hippocampal pyramidal neurons in brain slice. Neuron. 1995;14:1223–1231. doi: 10.1016/0896-6273(95)90269-4. [DOI] [PubMed] [Google Scholar]
- 43.Putney JW. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- 44.Regehr WG, Tank DW. Calcium concentration dynamics produced by synaptic activation of CA1 hippocampal pyramidal cells. J Neurosci. 1992;12:4202–4223. doi: 10.1523/JNEUROSCI.12-11-04202.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richmond SA, Irving AJ, Molnár E, McIlhinney RAJ, Michelangeli F, Henly JM, Collingridge GL. Localisation of the glutamate receptor subunit GluR1 on the surface of living and within cultured hippocampal neurones. Neuroscience. 1996;75:69–82. doi: 10.1016/0306-4522(96)00217-5. [DOI] [PubMed] [Google Scholar]
- 46.Schiller J, Schiller Y, Clapham DE. NMDA receptors amplify calcium influx into dendritic spines during associative pre- and postsynaptic activation. Nat Neurosci. 1998;1:114–118. doi: 10.1038/363. [DOI] [PubMed] [Google Scholar]
- 47.Segal M, Manor D. Confocal microscopic imaging of [Ca2+]i in cultured rat hippocampal neurons following exposure to N-methyl-d-aspartate. J Physiol (Lond) 1992;448:655–676. doi: 10.1113/jphysiol.1992.sp019063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shmigol A, Kirischuk S, Kostyuk P, Verkhratsky A. Different properties of caffeine-sensitive Ca2+ stores in peripheral and central mammalian neurons. Pflügers Arch. 1994;426:174–176. doi: 10.1007/BF00374686. [DOI] [PubMed] [Google Scholar]
- 49.Shmigol A, Svichar N, Kostyuk P, Verkhratsky A. Gradual caffeine-induced Ca2+ release in mouse dorsal root ganglia neurons is controlled by cytoplasmic and luminal Ca2+. Neuroscience. 1996;73:1061–1067. doi: 10.1016/0306-4522(96)00108-x. [DOI] [PubMed] [Google Scholar]
- 50.Simpson PB, Russell JT. Role of sarcoplasmic/endoplasmic-reticulum Ca2+-ATPases in mediating Ca2+ waves and local Ca2+-release microdomains in cultured glia. Biochem J. 1997;325:239–247. doi: 10.1042/bj3250239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sitsapesan R, Williams AJ. Mechanisms of caffeine activation of single Ca2+ release channels of sheep cardiac sarcoplasmic reticulum. J Physiol (Lond) 1990;423:425–439. doi: 10.1113/jphysiol.1990.sp018031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stanton PK, Sarvey JM. Blockade of long-term potentiation in rat hippocampal CA1 region by inhibitors of protein synthesis. J Neurosci. 1984;4:3080–3088. doi: 10.1523/JNEUROSCI.04-12-03080.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takechi H, Eilers J, Konnerth A. A new class of synaptic response involving calcium release in dendritic spines. Nature. 1998;396:757–760. doi: 10.1038/25547. [DOI] [PubMed] [Google Scholar]
- 54.Usachev YM, Thayer SA. Ca2+ influx in resting rat sensory neurons that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J Physiol (Lond) 1999;519:115–130. doi: 10.1111/j.1469-7793.1999.0115o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Verkhratsky A, Petersen OH. Neuronal Ca2+ stores. Cell Calcium. 1998;24:333–343. doi: 10.1016/s0143-4160(98)90057-4. [DOI] [PubMed] [Google Scholar]
- 56.Wilsch VW, Behnisch T, Jäger T, Reymann KG, Balschun D. When are class I metabotropic glutamate receptors necessary for long-term potentiation? J Neurosci. 1998;18:6071–6080. doi: 10.1523/JNEUROSCI.18-16-06071.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woodhall G, Gee CE, Robitaille R, Lacaille JC. Membrane potential and intracellular Ca2+ oscillations activated by mGluRs in hippocampal stratum oriens/alveus interneurons. J Neurophysiol. 1999;81:371–382. doi: 10.1152/jn.1999.81.1.371. [DOI] [PubMed] [Google Scholar]
- 58.Yeckel MF, Kapur A, Johnston D. Multiple forms of LTP in hippocampal CA3 neurons use a common postsynaptic mechanism. Nat Neurosci. 1999;2:625–633. doi: 10.1038/10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yuste R, Majewska A, Cash SS, Denk W. Mechanisms of calcium influx into hippocampal spines: heterogeneity among spines, coincidence detection by NMDA receptors, and optical quantal analysis. J Neurosci. 1999;19:1976–1987. doi: 10.1523/JNEUROSCI.19-06-01976.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]