

Abstract
A photoinduced carboxylation of alkyl halides with CO2 at remote sp3 C−H sites enabled by the merger of photoredox and Ni catalysis is described. This protocol features a predictable reactivity and site selectivity that can be modulated by the ligand backbone. Preliminary studies reinforce a rationale based on a dynamic displacement of the catalyst throughout the alkyl side chain.
Keywords: carbon dioxide, carboxylation, chain-walking, nickel, photocatalysis
Metal‐catalyzed reductive carboxylation reactions of organic (pseudo)halides with abundant and inexpensive carbon dioxide (CO2)1 have provided new vistas for preparing industrially‐relevant carboxylic acids in the absence of stoichiometric organometallic reagents.2 Although this area of expertise has reached remarkable levels of sophistication, the vast majority of sp3 carboxylation reactions primarily rely on ipso‐CO2 insertions at prefunctionalized sites (Scheme 1, path a).3, 4
Scheme 1.

Merging Ni and photoredox catalysts for sp3 C−H carboxylation.
From both a conceptual and practical standpoint, the ability to expand the boundaries of CO2 fixation into unactivated sp3 C−H sites would be a particularly attractive scenario.5 Unfortunately, the available sp3 C−H carboxylation portfolio indicates that a vast number of daunting challenges remain.6 At present, photochemical techniques7 remain confined to the activation of sp3 C−H bonds adjacent to heteroatoms or aromatic rings, invariably requiring high‐intensity UV‐light irradiation (Scheme 1, path b).8 The latter is particularly problematic given the wide number of functional groups that absorb in the UV region, leading to deleterious side‐reactions arising from photoexcitation of the substrate itself, thus reinforcing the need for a sp3 C−H carboxylation technique with improved flexibility, generality and practicality. A significant step‐forward in sp3 C−H carboxylation has been recently reported by our group within the context of chain‐walking reactions; however, stoichiometric amounts of metal salts are inevitably required,4d, 9 thus hampering the implementation of these processes in industrial endeavors.10 Prompted by our interest in nickel catalysis and visible‐light‐induced processes,3b–3g, 4b–4d, 9 we wondered whether the merger of these two techniques might enable a CO2 insertion at remote sp3 C−H sites in the absence of stoichiometric metal salts, thus offering an unrecognized opportunity in metallaphotoredox11, 12 and carboxylation reactions.1 Herein, we report the successful realization of this goal by using alkyl halides as precursors (Scheme 1, bottom). The protocol is characterized by a site‐selectivity pattern that can be modulated by a subtle modification of the ligand backbone, thus resulting in the functionalization at benzylic or even at primary sp3 C−H sites, arguably the strongest C−H bonds in the alkyl series. Preliminary mechanistic studies suggest that a dynamic displacement of the nickel catalyst throughout the hydrocarbon side chain through NiII intermediates comes into play.13
Our investigations began by studying the Ni‐catalyzed carboxylation of homobenzylic bromide (1 a) with CO2 (1 bar; Scheme 2). After systematic evaluation of all reaction parameters,14 we found that a protocol based on (L1)NiBr2, organic photocatalyst 4‐CzIPN, K2CO3 and Hantzsch ester (HEH) as the terminal reductant provided the best results, giving rise to the targeted carboxylic acid in 56 % isolated yield with a 90:10 branched/linear selectivity (b/l; entry 1). As initially anticipated, subtle modifications on the ligand backbone resulted in a markedly decrease in reactivity; although the inclusion of substituents adjacent to the nitrogen atom was shown to be critical for success,15 it became apparent that aryl or alkyl groups at C4 and/or C7 had a deleterious effect in both selectivity and reactivity (entries 2–6). Intriguingly, the utilization of desiccants led to lower yields of 2 a (entry 9), thus revealing a non‐innocent role exerted by water.4d Unlike related Ni‐catalyzed carboxylations of organic (pseudo)halides,3 the presence of additives, such as LiBr, was not necessary for the reaction to occur (entry 10); notably, K2CO3 provided better results than Cs2CO3 (entry 11), showing the influence that the escorting cation might have on reactivity. As anticipated, control experiments in the absence of either (L1)NiBr2, 4‐CzIPN or light resulted in no conversion to 2 a (entry 12).
Scheme 2.
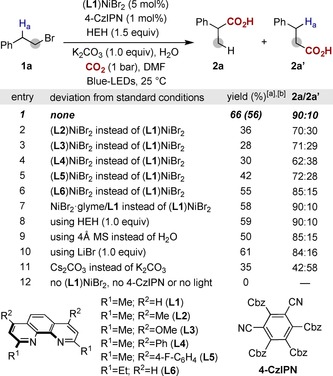
Optimization of the reaction conditions. 1 a (0.20 mmol), (L1)NiBr2 (5 mol %), 4‐CzIPN (1 mol %), HEH (1.5 equiv), K2CO3 (1.0 equiv), H2O (5.0 equiv), CO2 (1 bar), Blue‐LEDs in DMF (0.1 m) at 25 °C for 5 h. [a] Yields determined by NMR using 1,3,5‐trimethoxybenzene as standard. [b] Isolated yield, average of two independent runs. 4‐CzIPN: 2,4,5,6‐tetra(carbazol‐9‐yl)‐isophthalonitrile; DMF=N,N‐dimethylformamide; Cbz=carbazole; HEH=diethyl 1,4‐dihydro‐2,6‐dimethyl‐3,5‐pyridinedicarboxylate.
With optimized conditions in hand, we next set out to explore the generality of our light‐induced Ni‐catalyzed carboxylation at benzylic sp3 C−H sites. As shown in Scheme 3, similar reactivity and site selectivity were obtained for a plethora of homobenzyl bromides independent of whether they possessed electron‐rich or electron‐poor substituents on the aryl ring. However, it is worth noting that electron‐donating groups provided the best yields of the series (2 e, 2 g, 2 h).16 As shown for 2 a, the reaction can be scaled‐up without significant erosion in yield or site selectivity. Importantly, phenols (2 g), amides (2 h), ketones (2 j) or esters (2 k) do not interfere with productive carboxylation at the benzylic sp3 C−H site. Phenol (2 g) gave higher yields, most likely through Lewis‐acidic assistance of K+ during the CO2 insertion (Scheme 3, middle).17 We found that our protocol can be extended to electron‐rich unprotected indoles (2 l) or secondary homobenzylic bromides (2 m), albeit in lower yields. The latter result is particularly interesting, particularly if one takes into consideration the inherent structural limitations observed in otherwise related carboxylation of benzyl halides possessing α‐substituents other than methyl groups.3f–3h
Scheme 3.
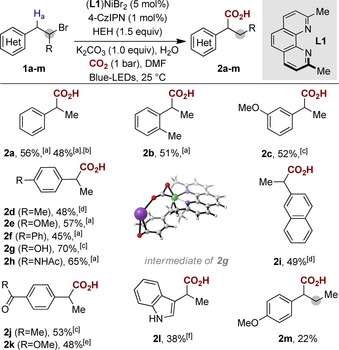
Benzylic sp3 C−H carboxylation by merging Ni and photoredox catalysis. Conditions: see Scheme 2 (entry 1); yields of isolated products, average of two independent runs. [a] b/l=90:10. [b] 1.0 mmol scale. [c] b/l=85:15. [d] b/l=93:7. [e] Isolated as methyl ester. [f] b/l=80:20. For additional substrates, see Supporting Information.14
Although the results summarized in Scheme 3 clearly illustrated the feasibility of a benzylic sp3 C−H carboxylation in the absence of metal reductants, there was a reasonable doubt on whether our protocol could be extended to carboxylation events at distal, primary sp3 C−H sites. Undoubtedly, the successful realization of such a void terrain would unlock new fundamental reactivity within the metallaphotoredox arena11, 12 while expanding our repertoire when activating strong primary sp3 C−H bonds in the presence of a priori more reactive sites.18 As expected, the ligand had a non‐negligible impact on both efficiency and site selectivity.15 Indeed, the reaction of 2‐bromoheptane (3 a) under the optimized conditions of Scheme 3 based on a Ni/L1 regime resulted in traces, if any, of 1‐octanoic acid (4 a). However, a protocol based on Ni/L7 furnished 4 a in 42 % yield and with exquisite site selectivity (99:1).19 Importantly, although an excellent site selectivity was found for a Ni‐catalyzed protocol based on 2,9‐dihexyl‐4,7‐diphenyl‐1,10‐phenanthroline as ligand and Mn as terminal reductant,9 a photochemical event based on such ligand backbone resulted in a significant erosion in yield and site‐selectivity, thus showing the subtleties of our photocatalytic chain‐walking carboxylation.14 As shown in Scheme 4, a variety of linear carboxylic acids could be prepared from a range of alkyl bromides in excellent site selectivities through formal [1,n]‐migration of the Ni atom throughout the side chain. Although modest yields, the outcome of our remote Ni/photoredox carboxylation at primary sp3 C−H sites should be assessed against the challenge that it addresses. Particularly noteworthy was the site‐selectivity pattern observed for 4 g–4 j, with CO2 insertion occurring at the strongest, primary sp3 C−H sites. Given that primary alkyl radicals are beyond reach through hydrogen‐atom transfer (HAT),20 these results constitute an orthogonal gateway with existing UV‐mediated carboxylations occurring at weaker benzylic sites or adjacent to heteroatoms through open‐shell species (Scheme 1, path b).8 Equally interesting was the functional group compatibility in the presence of esters (4 d), nitriles (4 f), ketones (4 j), alkyl chlorides (4 e) or amides (4 i). Notably, the alkyl bromide derived from a nonsteroidal anti‐inflammatory drug such as Nabumetone delivered 4 h in a 99:1 ratio.21 Similarly, 4 b was obtained as a single regioisomer, indicating that the reaction took place at the most accessible sp3 C−H site. Although competitive C3‐carboxylation events might occur with electron‐rich indoles,22 this was not the case (4 g). Particularly rewarding was the ability to convert n‐heptane into 4 a (l/b=99:1) through bromination/chain‐walking photocarboxylation (Scheme 4, bottom), standing as a testament to the prospective impact of our protocol to repurpose chemical feedstocks (alkanes and CO2) in a controllable fashion.
Scheme 4.
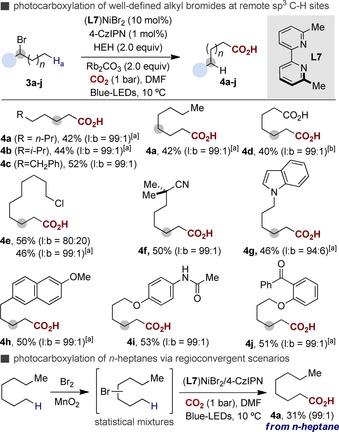
Catalytic carboxylation at remote sp3 C−H sites by merging Ni & photoredox catalysis. Conditions: 3 (0.25 mmol), (L7)NiBr2 (10 mol %), 4‐CzIPN (1 mol %), HEH (2.0 equiv), Rb2CO3 (2.0 equiv), CO2 (1 bar), DMF (0.08 m), blue LEDs in DMF at 10 °C for 20 h; yields of isolated products, average of two independent runs. [a] TBAI (1.0 equiv) as additive. [b] Obtained upon hydrolytic workup using methyl 5‐bromohexanoate. Regioconvergent photocarboxylation of n‐heptane: Br2 (0.25 mmol), MnO2 (0.50 mmol) in n‐heptane (1.25 mL) followed by the conditions highlighted above for 4 a.
Although unraveling the underpinnings of our Ni/photoredox chain‐walking carboxylation at sp3 C−H sites should await further investigations, we decided to shed light into the mechanism through combined experimental and theoretical studies (Scheme 5).14 Synergistic spectroelectrochemical measurements and in situ UV/Vis spectroscopy on (L1)NiBr2 and (L7)NiBr2 revealed that low‐valent NiI and Ni0 species are formed during light irradiation in the presence of 4‐CzIPN and Hantzsch ester.14, 23 The assumption that the benzylic carboxylation featured a rather facile β‐H elimination from cationic NiII species was experimentally corroborated by detecting styrenes in small amounts, the concentration of which decays to zero after consumption of the alkyl bromide.14 In addition, olefins were not detected in the absence of Ni/L1 or Ni/L7, arguing against base‐promoted E2‐elimination/olefin carboxylation event. DFT calculations confirmed that species B–D had similar energy and that these species coexist in rapid equilibrium via rather facile β‐H elimination from cationic NiII intermediates.24, 25 Importantly, DFT calculations revealed a rather unfavorable CO2 insertion for NiII species D, either through outer‐ or inner‐sphere mechanisms, reinforcing the notion that CO2 takes place at a NiI center (E) generated upon single‐electron transfer (SET), thus giving rise to a NiI‐carboxylate F.26 A final SET can provide 4 a‐I while recovering back the propagating Ni0/L7 (A) species. Although the activation energy for the SET reduction could not be obtained by DFT calculations, the absence of a kinetic isotope effect and the preferential regioselectivity observed at remote primary sp3 C−H sites (4 a‐I vs. 4 a‐I′) suggests that the formation of NiI species might be the rate‐determining step of the reaction. A similar rationale can be drawn for homobenzylic bromides, with an energetic profile that favors CO2 incorporation at benzylic sp3 C−H sites.14 In this case, the selectivity at benzylic sites can be explained by both kinetic and thermodynamic grounds.14
Scheme 5.

Mechanistic rationale for the photocarboxylation of 3 a.
In summary, we have described the merger of photoredox and Ni catalysis as a platform for enabling carboxylation at remote sp3 C−H sites under atmospheric pressure of CO2, thereby obviating the need for stoichiometric metal reductants. The salient features of this method are the mild conditions and a site‐selectivity pattern that can be modulated by the nature of the ligand employed, offering an unrecognized opportunity in the metallaphotoredox arena for the activation of sp3 C−H bonds. Further work along these lines is currently in progress.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank ICIQ and MINECO (CTQ2015‐65496‐R) for support. B.S. and P.B. thank European Union Horizon 2020 under the Marie Sklodowska‐Curie grant agreement (795961) and Elitenetzwerk Bayern (SynCat) for financial support. We thank C. Riesinger for the initial optimization of the reaction conditions, F. Fricke for the help with substrate scope, R. Hoheisel for spectroelectrochemical measurements, and B. F. Gu for helpful discussions.
B. Sahoo, P. Bellotti, F. Juliá-Hernández, Q.-Y. Meng, S. Crespi, B. König, R. Martin, Chem. Eur. J. 2019, 25, 9001.
Contributor Information
Dr. Stefano Crespi, Email: Stefano.Crespi@chemie.uni-regensburg.de.
Prof. Dr. Burkhard König, Email: Burkhard.Koenig@chemie.uni-regensburg.de.
Prof. Ruben Martin, Email: rmartinromo@iciq.es.
References
- 1.For selected reviews, see:
- 1a. Tortajada A., Juliá-Hernández F., Börjesson M., Moragas T., Martin R., Angew. Chem. Int. Ed. 2018, 57, 15948–15982; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16178–16214; [Google Scholar]
- 1b. Juliá-Hernández F., Gaydou M., Serano E., van Gemmeren M., Martin R., Top. Curr. Chem. 2016, 374, 45; [DOI] [PubMed] [Google Scholar]
- 1c. Börjesson M., Moragas T., Gallego D., Martin R., ACS Catal. 2016, 6, 6739–6749; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1d. Liu Q., Wu L., Jackstell R., Beller M., Nat. Commun. 2015, 6, 5933; [DOI] [PubMed] [Google Scholar]
- 1e. Tsuji Y., Fujihara T., Chem. Commun. 2012, 48, 9956–9964. [DOI] [PubMed] [Google Scholar]
- 2. Maag H., Prodrugs of Carboxylic Acids, Springer, New York, 2007. [Google Scholar]
- 3.For selected catalytic reductive ipso-carboxylations of organic (pseudo)halides at sp3 sites:
- 3a. Liao L.-L., Cao G.-M., Ye J.-H., Sun G.-Q., Zhou W.-J., Gui Y.-Y., Yan S.-S., Shen G., Yu D.-G., J. Am. Chem. Soc. 2018, 140, 17338–17342; [DOI] [PubMed] [Google Scholar]
- 3b. Meng Q.-Y., Wang S., König B., Angew. Chem. Int. Ed. 2017, 56, 13426–13430; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 13611–13615; [Google Scholar]
- 3c. Börjesson M., Moragas T., Martin R., J. Am. Chem. Soc. 2016, 138, 7504–7507; [DOI] [PubMed] [Google Scholar]
- 3d. Moragas T., Gaydou M., Martin R., Angew. Chem. Int. Ed. 2016, 55, 5053–5057; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 5137–5141; [Google Scholar]
- 3e. Liu Y., Cornella J., Martin R., J. Am. Chem. Soc. 2014, 136, 11215–11215; [DOI] [PubMed] [Google Scholar]
- 3f. Correa A., León T., Martin R., J. Am. Chem. Soc. 2014, 136, 1062–1069; [DOI] [PubMed] [Google Scholar]
- 3g. León T., Correa A., Martin R., J. Am. Chem. Soc. 2013, 135, 1221–1224; [DOI] [PubMed] [Google Scholar]
- 3h. Zhang S., Chen W.-Q., Yu A., He L.-N., ChemCatChem 2015, 7, 3972–3977; [Google Scholar]
- 3i. Nogi K., Fujihara T., Terao J., Tsuji Y., Chem. Commun. 2014, 50, 13052–13055. [DOI] [PubMed] [Google Scholar]
- 4.For selected metal-catalyzed carboxylation events at sp3 sites with prefunctionalized olefins as starting precursors:
- 4a. Hou J., Ee A., Cao H., Ong H.-W., Xu J.-H., Wu J., Angew. Chem. Int. Ed. 2018, 57, 17220–17224; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 17466–17470; [Google Scholar]
- 4b. Meng Q.-Y., Wang S., Huff G. S., König B., J. Am. Chem. Soc. 2018, 140, 3198–3201; [DOI] [PubMed] [Google Scholar]
- 4c. Yatham V. R., Shen Y., Martin R., Angew. Chem. Int. Ed. 2017, 56, 10915–10919; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11055–11059; [Google Scholar]
- 4d. Gaydou M., Moragas T., Juliá-Hernández F., Martin R., J. Am. Chem. Soc. 2017, 139, 12161–12164; [DOI] [PubMed] [Google Scholar]
- 4e. Seo H., Liu A., Jamison T. F., J. Am. Chem. Soc. 2017, 139, 13969–13972; [DOI] [PubMed] [Google Scholar]
- 4f. Ye J.-H., Miao M., Huang H., Yan S.-S., Yin Z.-B., Zhou W.-J., Yu D.-G., Angew. Chem. Int. Ed. 2017, 56, 15416–15420; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 15618–15622; [Google Scholar]
- 4g. Murata K., Numasawa N., Shimomaki K., Takaya J., Iwasawa N., Chem. Commun. 2017, 53, 3098–3101; [DOI] [PubMed] [Google Scholar]
- 4h. Wu L., Liu Q., Fleischer I., Jackstell R., Beller M., Nat. Commun. 2014, 5, 3091; [DOI] [PubMed] [Google Scholar]
- 4i. Ostapowicz T. G., Schmitz M., Krystof M., Klankermayer J., Leitner W., Angew. Chem. Int. Ed. 2013, 52, 12119–12123; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 12341–12345; [Google Scholar]
- 4j. Greenhalgh M. D., Thomas S. P., J. Am. Chem. Soc. 2012, 134, 11900–11903; [DOI] [PubMed] [Google Scholar]
- 4k. Williams C. M., Johnson J. B., Rovis T., J. Am. Chem. Soc. 2008, 130, 14936–14937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For selected reviews on C−H carboxylation techniques:
- 5a. Hong J., Li M., Zhang J., Sun B., Mo F., ChemSusChem 2019, 12, 6–39; [DOI] [PubMed] [Google Scholar]
- 5b. Luo J., Larrosa I., ChemSusChem 2017, 10, 3317–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected sp2 C−H carboxylation scenarios, see:
- 6a. Luo J., Preciado S., Larrosa I., Chem. Eur. J. 2016, 22, 6798–6802; [DOI] [PubMed] [Google Scholar]
- 6b. Mizuno H., Takaya J., Iwasawa N., J. Am. Chem. Soc. 2011, 133, 1251–1253; [DOI] [PubMed] [Google Scholar]
- 6c. Boogaerts I. I. F., Nolan S. P., J. Am. Chem. Soc. 2010, 132, 8858–8859; [DOI] [PubMed] [Google Scholar]
- 6d. Zhang L., Cheng J., Ohishi T., Hou Z., Angew. Chem. Int. Ed. 2010, 49, 8670–8673; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8852–8855. [Google Scholar]
- 7.For recent reviews on photocarboxylation reactions:
- 7a. Yeung C. S., Angew. Chem. Int. Ed. 2019, 58, 5492–5502; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5546–5556; [Google Scholar]
- 7b. Hou J., Li J.-S., Wu J., Asian J. Org. Chem. 2019, 7, 1439–1447. [Google Scholar]
- 8.
- 8a. Seo H., Katcher M. H., Jamison T. F., Nat. Chem. 2017, 9, 453–456; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Masuda Y., Ishida N., Murakami M., J. Am. Chem. Soc. 2015, 137, 14063–14066. [DOI] [PubMed] [Google Scholar]
- 9. Juliá-Hernández F., Moragas T., Cornella J., Martin R., Nature 2017, 545, 84–88. [DOI] [PubMed] [Google Scholar]
- 10.Stoichiometric metallic waste is toxic and has to be treated and recycled in industrial processes. See for example: Orrù R., Sannia M., Cincotti A., Cao G., Chem. Eng. Sci. 1999, 54, 3053. [Google Scholar]
- 11.For selected reviews in visible light photoredox catalysis:
- 11a. Milligan J. A., Phelan J. P., Badir S. O., Molander G. A., Angew. Chem. Int. Ed. 2019, 58, 6152–6163; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 6212–6224; [Google Scholar]
- 11b. Fabry D. C., Rueping M., Acc. Chem. Res. 2016, 49, 1969; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11c. Hopkinson M. N., Tlahuext-Aca A., Glorius F., Acc. Chem. Res. 2016, 49, 2261; [DOI] [PubMed] [Google Scholar]
- 11d. Kärkäs M. D., Porco J. A., Stephenson C. R. J., Chem. Rev. 2016, 116, 9683; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11e. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075; [DOI] [PubMed] [Google Scholar]
- 11f. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For selected reviews on dual catalysis in photochemical endeavors:
- 12a. Marzo L., Pagore S. K., Reiser O., König B., Angew. Chem. Int. Ed. 2018, 57, 10034–10072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10188–10228; [Google Scholar]
- 12b. Twilton J., Le C., Zhang P., Shaw M. H., Evans R. W., MacMillan D. W. C., Nat. Chem. Rev. 2017, 1, 0052; [Google Scholar]
- 12c. Skubi K. L., Blum T. R., Yoon T. P., Chem. Rev. 2016, 116, 10035; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12d. Hopkinson M. N., Sahoo B., Li J.-L., Glorius F., Chem. Eur. J. 2014, 20, 3874–3886. [DOI] [PubMed] [Google Scholar]
- 13.For recent reviews on chain-walking strategies:
- 13a. Sommer H., Juliá-Hernández F., Martin R., Marek I., ACS Cent. Sci. 2018, 4, 153–165; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Vasseur A., Bruffaerts J., Marek I., Nat. Chem. 2016, 8, 209–219; [DOI] [PubMed] [Google Scholar]
- 13c. Larionov E., Li H., Mazet C., Chem. Commun. 2014, 50, 9816–9826. For selected recent reports on chain-walking strategies: [DOI] [PubMed] [Google Scholar]
- 13d. Zhou F., Zhang Y., Xu X., Zhu S., Angew. Chem. Int. Ed. 2019, 58, 1754–1758; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 1768–1772; [Google Scholar]
- 13e. Sun S.-Z., Börjesson M., Martin-Montero R., Martin R., J. Am. Chem. Soc. 2018, 140, 12765–12769; [DOI] [PubMed] [Google Scholar]
- 13f. Peng L., Li Z., Yin G., Org. Lett. 2018, 20, 1880–1883; [DOI] [PubMed] [Google Scholar]
- 13g. Peng L., Li Y., Li Y., Wang W., Pang H., Yin G., ACS Catal. 2018, 8, 310–313; [Google Scholar]
- 13h. Chen F., Chen K., Zhang Y., He Y., Wang Y.-M., Zhu S., J. Am. Chem. Soc. 2017, 139, 13929–13935. [DOI] [PubMed] [Google Scholar]
- 14.For details, see Supporting Information.
- 15.For selected references in which ortho-substituents on the nitrogen ligand exerted a critical influence on catalytic carboxylation reactions:
- 15a.Ref. [3b]–[3f], [4b]–[4e], [7];
- 15b. Nogi K., Fujihara T., Terao J., Tsuji Y., J. Org. Chem. 2015, 80, 11618. [DOI] [PubMed] [Google Scholar]
- 16.Such a high reactivity can be interpreted on the basis of an increased nucleophilicity of the benzyl C−Ni bond and a more favorable chain-walking event.
- 17.For Kolbe-like reaction mechanism involving potassium-phenoxide-CO2 complex: Marković Z., Marković S., Manojlović N., Predović-Simović J., J. Chem. Inf. Model. 2007, 47, 1520–1525. [DOI] [PubMed] [Google Scholar]
- 18.For selected reviews on sp3 C−H functionalization:
- 18a. He J., Wasa M., Chan K. S. L., Shao Q., Yu J.-Q., Chem. Rev. 2017, 117, 8754–8786; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Baudoin O., Acc. Chem. Res. 2017, 50, 1114–1123; [DOI] [PubMed] [Google Scholar]
- 18c. He G., Wang B., Nack W. A., Chen G., Acc. Chem. Res. 2016, 49, 635–645; [DOI] [PubMed] [Google Scholar]
- 18d. Dastbaravardeh N., Christakakou M., Haider M., Schnürch M., Synthesis 2014, 46, 1421–1439; [Google Scholar]
- 18e. Rouquet G., Chatani N., Angew. Chem. Int. Ed. 2013, 52, 11726–11743; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11942–11959. [Google Scholar]
- 19.Although tentative, tetrabutylammonium iodide (TBAI) might facilitate electron-transfer processes to the metal center. See for example: Iyoda M., Otsuka H., Sato K., Nisato N., Oda M., Bull. Chem. Soc. Jpn. 1990, 63, 80 Particularly noteworthy is the observation that related chain-walking scenarios based on Ni/L7 results in benzylic sp3 C−H arylation with organic halides (Ref. [13h]). Although tentative, we believe that the site-selectivity pattern observed in our chain-walking photocarboxylation based on Ni/L7 is due to a favorable insertion of CO2 into the harder primary sp3 C−Ni bond when compared to the softer benzylic sp3 site. [Google Scholar]
- 20.For an excellent review on HAT processes, see: Capaldo L., Ravelli D., Eur. J. Org. Chem. 2017, 2017, 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The photochemical Ni/L1 carboxylation of 3 c and 3 h resulted in <6 % yield of 4 c and 4 h, respectively. Under the limits of detection, no benzylic sp3 C−H carboxylation was found in the crude mixtures. Interestingly, the photocarboxylation of 1 a under a Ni/L7 regime resulted in the preferential carboxylation at primary sp3 site. See Ref. [14].
- 22.
- 22a. Nemoto K., Tanaka S., Konno M., Onozawa S., Chiba M., Tanak Y., Sasai Y., Okubo R., Hattori T., Tetrahedron 2016, 72, 734–745; [Google Scholar]
- 22b. Xin Z., Lescot C., Friis S. D., Daasbjerg K., Skrydstrup T., Angew. Chem. Int. Ed. 2015, 54, 6862–6866; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6966–6970; [Google Scholar]
- 22c. Yoo W.-J., Capdevila M. G., Du X., Kobayashi S., Org. Lett. 2012, 14, 5326–5329. [DOI] [PubMed] [Google Scholar]
- 23.For related spectroelectrochemical studies with (bipy)NiIIX2: Sun R., Qin Y., Ruccolo S., Schnedermann C., Costentin C., Nocera D. G., J. Am. Chem. Soc. 2019, 141, 89–93. [DOI] [PubMed] [Google Scholar]
- 24.Deuterium labeling studies confirmed this observation. See Ref. [14].
- 25.The transition state for the β-hydride elimination from putative alkyl-NiI species could not be optimized, suggesting that a scenario consisting of an iterative sequence of β-hydride elimination/migratory insertion from NiI intermediates is highly unlikely.
- 26.For the role of NiI in carboxylation reactions:
- 26a. Charboneau D. J., Brudvig G. W., Hazari N., Lant H. M. C., Sadjari A. K., ACS Catal. 2019, 9, 3228–3241; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Menges F. S., Craig S. M., Tötsch N., Bloomfield A., Ghosh S., Krüger H.-J., Johnson M. A., Angew. Chem. Int. Ed. 2016, 55, 1282–1285; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 1304–1307. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary