

Abstract
Neuronal death evoked by DNA damage requires cyclin-dependent kinase 4 (Cdk4) and 6 activity and is accompanied by elevation of cyclin D1-associated kinase activity. Because Cdk4/6 phosphorylates retinoblastoma protein (pRb) family members that then modulate the transcriptional activity of E2F/DP1 complexes, we examined the involvement of these components in DNA damage-evoked neuronal death. Camptothecin induced rapid pRb and p107 phosphorylation at a Cdk4/6 phosphorylation site followed by selective loss of Rb and p107. The CDK inhibitor flavopiridol suppressed pRb and p107 phosphorylation and loss, implicating CDK activity in these events. Moreover, the loss of pRb and p107 appeared to be mediated by caspases because it was blocked by general caspase inhibitors. The role of phosphorylation and pRb and p107 loss in the death pathway was indicated by observations that virally mediated expression of pRb mutated at sites of phosphorylation, including the Cdk4/6 site, inhibited death. Finally, expression of dominant-negative versions of DP1, known to compromise E2F transcriptional activity, protects cortical neurons from death induced by camptothecin and sympathetic neurons from death evoked by UV treatment. Taken together, these results implicate the CDK–pRb/E2F/DP pathway as a required element in the neuronal death evoked by DNA damage.
Keywords: apoptosis, pRb, E2F, DP1, neuronal, cyclin
Although neuronal apoptosis is an important component of brain development (Oppenheim, 1991), a growing body of evidence suggests that it also participates in the progression of neuropathological conditions such as stroke and neurodegenerative disease (Anderson et al., 1996; Stefanis et al., 1997; Chen et al., 1998; Cotman, 1998). Although numerous etiologies and putative death initiators have been described, the signaling pathways that control neuronal death are not fully understood.
Multiple lines of evidence indicate that cell cycle elements play an important role in some but not all paradigms of neuronal death. For example, NGF deprivation leads to increased cdc2 activity and cyclin B expression in neuronal pheochromocytoma 12 (PC12) cells (Gao and Zelenka, 1995), as well as elevated cyclin D1 transcript levels in sympathetic neurons (Freeman et al., 1994). Others observe increases in cell cycle molecules in affected regions of brains from Alzheimer's disease patients and in brains exposed to ischemic conditions (McShea et al., 1997; Nagy et al., 1997; Vincent et al., 1997; Busser et al., 1998). In addition, cyclin D1-associated kinase activity is elevated in cortical neurons treated with the DNA-damaging agent camptothecin (Park et al., 1998b). With respect to functional inhibition studies, we have shown that G1/S blockers (Farinelli and Greene, 1996) and the CDK inhibitors suppress the death of sympathetic and cortical neurons evoked by trophic factor deprivation (Park et al., 1996a) and/or DNA-damaging conditions (Park et al., 1997a, 1998a). Finally, in the latter systems, virally mediated expression of cyclin-dependent kinase (CDK) inhibitors such as p16 and p27 or of dominant-negative forms of Cdk4 and 6 but not Cdk2 or 3 inhibit death (Park et al., 1997b,1998b).
Our observations on the importance of Cdk4/6 activity in certain paradigms of neuronal death raise the questions of the target(s) of this activity and whether such a target(s) plays required roles in the death mechanism. The only presently known in vivo substrates of activated Cdk4/6 are the tumor suppresser pRb and its related family member p107 (Chellappan et al., 1991; Beijersbergen et al., 1995; Weinberg, 1995). Although pRb appears to interact with numerous proteins, its regulation of the cell cycle-related transcription factor E2F is best understood. pRb is normally hypophosphorylated in quiescent cells such as neurons and binds to and inhibits the transcriptional activity of E2F. In cells undergoing G1-to-S cell cycle transition, pRb becomes hyperphosphorylated. Numerous studies indicate that a consequence of Rb hyperphosphorylation is the release and activation of E2F (Chellappan et al., 1991; Suzuki-Takahashi et al., 1995). The E2F family of transcription factors has a diverse set of cellular functions (Nevins, 1992; La Thangue, 1994) that include the control of cell proliferation. In this case, transcriptional activation requires association of E2F with its obligate binding partner DP1-2 (Bandara et al., 1993; Girling et al., 1993; Wu et al., 1996).
In addition to cell cycle progression, there are also reports implicating Rb and E2F in apoptosis. For example, Rb null mice show neurological deficits and neuronal apoptosis (Jacks et al., 1992;Macleod et al., 1996). Furthermore, overexpression of E2F in proliferating cells results in apoptosis (Qin et al., 1994; Hiebert et al., 1995). Finally, there is evidence that E2F, DP1, and their complexes interact with and regulate other proteins such as MDM2 and p53 (Martin et al., 1995; O'Connor et al., 1995; Sorensen et al., 1996).
These observations lead us to the hypothesis that the neuronal death evoked by DNA damage is mediated via Cdk4/6 activation, consequent pRb phosphorylation, loss of pRb function, and activation of E2F/DP complexes. To test this model, we determined whether changes in pRb phosphorylation occur during the death of camptothecin-treated neurons. In addition, we examined the potential changes in the expression levels of cell cycle control elements including CDKs, cyclins, and Rb family members. Finally, we examined the potential role of E2F/DP in the death of neurons evoked by DNA damage.
MATERIALS AND METHODS
Materials.(−)cis-5,7-Dihydroxy-2-(2-chlorophenyl)-8[4-(3-hydroxy-1-methyl)piperidinyl]-4H-benzopyran-4-one (L86-8275; flavopiridol) was a generous gift from Dr. Peter J. Worland (National Cancer Institute). Camptothecin was obtained from Sigma (St. Louis, MO). Boc-aspartyl- (ome)-fluoromethyl ketone (BAF) and zVAD-fluoromethylketone (zVAD-fmk) were purchased from Enzyme Systems Products (Dublin, CA). Adenovirus expressing β-galactosidase (LacZ) was a kind gift from Dr. Ruth Slack (University of Ottawa).
Generation of recombinant Sindbis viruses. The cDNAs for the coding regions of wild-type (WT) human DP1, dominant-negative (DN) (Δ103–126) DP1 (containing a deletion of the DNA-binding domain), the DN (Δ1–126) DP1 truncation mutant containing a deletion of amino acids 1–126, and (Δ233–272) DP1 containing a deletion of the E2F-binding domain (233–272) were inserted into the BSTE11 site of the DSTEQ12 vector (Joe et al., 1996;Park et al., 1997c) The dominant-negative forms of DP1 have been reported previously to inactivate the transcriptional activity of E2F (Wu et al., 1996). These would be expected to interfere with complexes of all E2F/DP family members. FLAG tags (ATGGACTACAAGGACGATG-ATGACAAA) were introduced at the 3′ end of the coding region of WT DP1, DN (Δ103–126) DP1, and (Δ233–272) DP1. Control nonexpressing vectors of each DP1 construct were generated by eliminating the initiating codon. All mutations, deletions, and FLAG tags were introduced by PCR and confirmed by sequencing. Viral particles were generated by in vitro transcription and transfection into BHK cells and titered by plaque assay as described previously (Joe et al., 1996).
Generation of recombinant adenovirus. The cDNA for the coding regions of the ΔK11 Rb mutant (full-length mouse Rb containing T246 to A, T350 to R, S601 to A, S605 to A, S773 to A, S781 to A, S788 to A, S800 to A, S804 to E, T814 to A, and T819 to A mutations; a kind gift from Dr. Eldad Zacksenhaus, University of Toronto) (Brown et al., 1999) or green fluorescent protein (GFP) was subcloned into pAdlox vector (Hardy et al., 1997) and recombined with adenoviral backbone DNA in CRE8 cells as described previously (Hardy et al., 1997). The ΔK11 Rb construct was derived from ΔP34Rb (Hamel et al., 1992) by the addition of three additional phosphorylation site mutations (S773, T814, and T819 to A mutations).
Culture and survival of cortical neurons. Mouse (embryonic day 16 for Rb expression experiments) or rat (embryonic day 18) cortical neurons were cultured as described previously (Park et al., 1998b) and grown in defined serum-free medium. These cultures typically contain >99% neurons as assessed by staining with antibody directed against phosphorylated neurofilament (RT97; Developmental Studies Hybridoma Bank, University of Iowa). The neurons were plated into 6-well dishes (∼3,000,000 cells/well) or 24-well plates (200,000 cells/well) coated with poly-l-lysine (100 μg/ml) in serum-free medium [N2:EMEM (1:1) supplemented with 6 mg/mld-glucose, 100 μg/ml transferrin, 25 μg/ml insulin, 20 nm progesterone, 60 μm putrescine, and 30 nm selenium]. For Westerns (6-well dishes), the neurons were treated with camptothecin (10 μm) alone or with flavopiridol (1 μm), BAF (100 μm), or zVAD-fmk (100 μm) in serum-free medium 2–3 d after plating. At appropriate times of culture under the conditions described in the text, cells were washed twice in PBS and collected by trituration in PBS. The neurons were centrifuged in an Eppendorf tabletop microfuge for 3 min at 10,000 rpm. The neurons were then lysed in sample buffer as described previously (Park et al., 1998b). Alternatively, neurons were infected 1 d after plating (in 24-well plates) with Sindbis virus at a multiplicity of infection (MOI) of ∼2 or with adenovirus at an MOI of 150 and incubated overnight. The medium was then exchanged with serum-free medium supplemented with 10 μm camptothecin where appropriate. At the indicated times of culture under the conditions described in the text, cells were lysed, and the numbers of viable cells were evaluated as described previously (Park et al., 1998b). Briefly, cells were lysed in 200 μl of cell lysis buffer [0.1× PBS, pH 7.4 (0.4 mmNa2HP04, 0.15 mm KH2P04, 13.5 mm NaCl, and 0.25 mm KCl)] containing 0.5% Triton X-100, 2 mm MgCl2, and cetyldimethylethylammonium bromide (0.5 gm/100 ml) that disrupts cells but leaves the nuclei intact. Ten microliters of sample from each culture were loaded onto a hemacytometer, and the number of healthy intact nuclei was evaluated by phase microscopy. Nuclei that displayed characteristics of blebbing, disruption of nuclear membrane, phase-bright apoptotic bodies, and chromatin margination were excluded. All experimental points are expressed as a percentage of the cells plated on day 0 and are reported as the mean ± SEM (n = 3).
Rat sympathetic neuron culture and survival assay. Primary cultures of sympathetic neurons were generated from dissociated superior cervical ganglia of postnatal day 1 Sprague Dawley rats as described previously (Park et al., 1996b). The cells were plated in 0.5 ml of medium per well in collagen-coated 24-well dishes at a density of ∼0.5 ganglia per well (∼10,000 neurons/well). The growth medium was RPMI 1640 medium supplemented with 10% heat-inactivated horse serum and 60 ng/ml mouse NGF (Sigma). To eliminate non-neuronal cells, we added a mixture of uridine and 5-fluorodeoxyuridine (10 μm each) to the cultures on the following day. On the third day after plating, the neurons were infected with Sindbis virus (1–2 plaque-forming units per cell) in 0.2 ml of RPMI 1640 medium containing 2% heat-inactivated horse serum. After 1 hr of infection, 0.3 ml of RPMI 1640 medium containing 16% heat-inactivated horse serum was added. The cultures were then left to incubate overnight before UV irradiation (300 J/m2). Neurons were UV irradiated in 200 μl of medium containing NGF using a Stratolinker (Stratagene, La Jolla, CA). After irradiation, an additional 300 μl of medium containing NGF was added to each well. At the indicated times, the numbers of viable, phase-bright neurons were determined by strip counting as described previously (Park et al., 1996b). Briefly, a 24-well plate was placed onto an adjustable stage. From a defined point (consistent throughout the course of the experiment) the same field of neurons (consisting of the diameter of each well) was assessed by phase microscopy. Phase-bright, intact neurons were scored as viable, whereas those with apoptotic bodies or phase-dark shrunken cell bodies were counted as dying or dead. All experimental points were performed in triplicate and are reported as the mean ± SEM. All points are expressed relative to the number of neurons originally plated.
Western blot analyses. The neurons or rat fibroblasts were harvested in sample buffer as described above. Twenty-five micrograms of protein were loaded onto 10% SDS-polyacrylamide gels (except for the p16 analysis in which a 15% gel was used) and transferred onto nitrocellulose membrane as described previously (Park et al., 1998b). Blots were probed with primary antibody [cyclin D1, cyclin A, cyclin E, p107, Cdk2, Cdk3, Ckd4, Cdk6, p16, p21, and p27 from Santa Cruz Biotechnology, Santa Cruz, CA (1:300); phospho-Rb (ser795) from New England Biolabs, Beverly, MA (1:1000); pRb from PharMingen, San Diego, CA (1:1000); anti-β-actin (1:5000) from Sigma, and anti-FLAG (1:40) from VWR Scientific)] in PBS supplemented with 3% BSA overnight. Blots were washed four times for 25 min each in PBS containing 0.2% Tween 20. The membranes were then probed with the appropriate secondary antibody (Amersham, Arlington Heights, IL, or Bio-Rad, Hercules, CA) for 1 hr in PBS containing 5% milk. The membranes were visualized by ECL detection (Amersham).
Immunofluorescence. Sympathetic neurons or cortical neurons were dissociated and cultured, as described above, in six-well plates at a density of 2 ganglia/well (sympathetic neurons) or 1,000,000 cells/well (cortical neurons). After various times of infection with the indicated viruses, neurons were fixed with 100% ethanol for 20 min at −20°C or 4% paraformaldehyde for 30 min at 4°C and incubated with anti-FLAG primary antibody (1:500 dilution) or anti-pRb antibody (1:500 dilution). FITC-conjugated horse anti-mouse (1:50 dilution) or indocarbocyanine-conjugated goat anti-mouse (1:200 dilution) was used as the secondary antibody.
RESULTS
pRb/p107 is phosphorylated and rapidly lost during the death of cortical neurons evoked by camptothecin
We demonstrated previously that camptothecin (10 μm) induces apoptotic death of cortical neurons maintained in serum-free defined medium (Morris and Geller, 1996; Park et al., 1998a,b). In this death paradigm, cyclin D1-associated kinase activity rises greatly within 1 hr after camptothecin treatment (Park et al., 1998b). This increase is before caspase 3-like activation that is detectable ∼4 hr after camptothecin treatment and morphological death that is observed 10–12 hr after drug treatment (Morris and Geller, 1996; Stefanis et al., 1999). Because of the increase in cyclin D1-associated kinase activity and the inhibition of camptothecin-evoked neuronal death by pharmacological CDK inhibitors or by expression of CDK inhibitors (CKIs) or DN Cdk4/6 (Park et al., 1997a, 1998b), we have suggested that Cdk4/6 activity may be required for death signaling in this paradigm. Accordingly, we examined whether Rb phosphorylation increases during a time course of camptothecin treatment by Western blot analyses using an antibody directed against the phosphoepitope of pRb and p107 equivalent to S795 of human pRb, a site known to be phosphorylated by Cdk4/6. The antibody does not, however, distinguish between the phosphorylation of pRb or p107, and so we refer to the detected species as pRb/p107. As a control, the antibody detects the increase in pRb/107 phosphorylation in fibroblasts after they are switched from low- to high-serum conditions (data not shown). As shown in Figure 1, camptothecin treatment elicits a rapid phosphorylation of pRb/p107. This phosphorylation peaks in intensity at ∼1–2 hr after the start of camptothecin treatment. The time course of pRb/p107 phosphorylation closely follows the time course of cyclin D1-associated kinase activity that also is greatly elevated at 1 hr after the start of camptothecin addition (Park et al., 1998b).
Fig. 1.
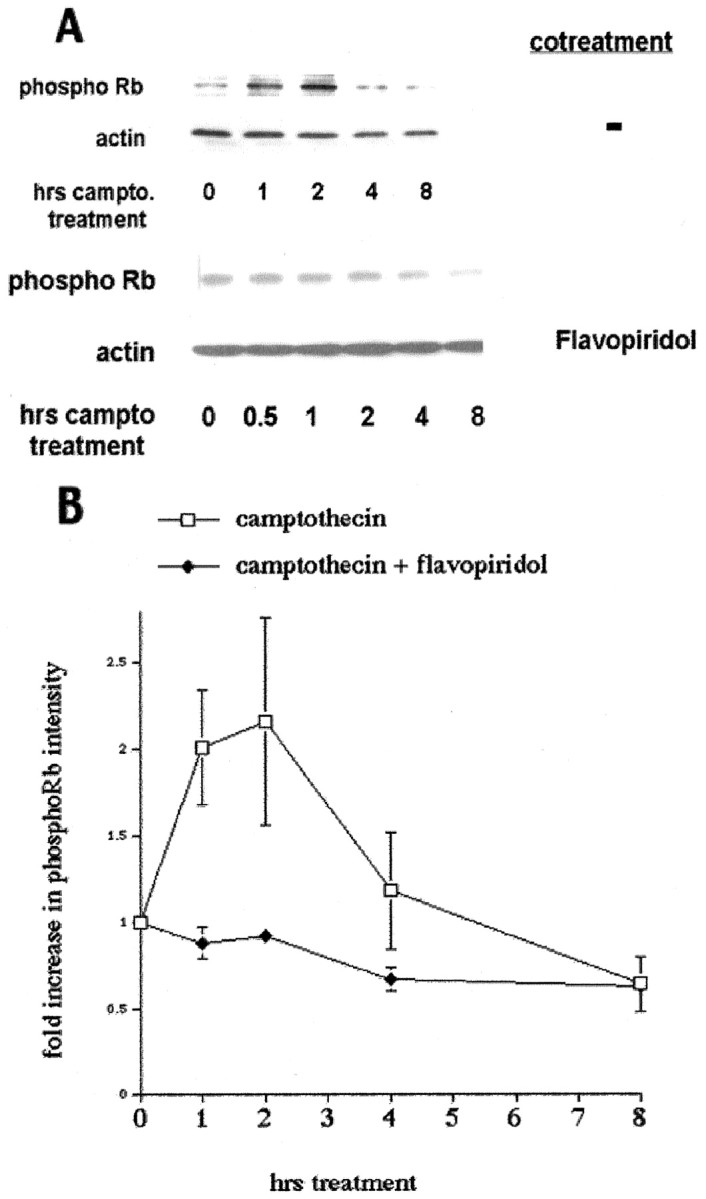
pRb/p107 is phosphorylated during the death of cortical neurons evoked by camptothecin. A, Top, Western immunoblots (probed with anti-phospho-Rb antibody) of whole-cell lysates of cortical neurons after various periods of treatment with camptothecin (10 μm). Bottom, Cotreatment with flavopiridol (1 μm) is indicated.campto, Camptothecin. B, Densitometric analyses of Western immunoblots of cortical neurons treated with camptothecin alone or cotreated with flavopiridol and probed with anti-phospho-Rb antibody. All data points are the mean ± SEM of data from three separate experiments and are expressed relative to the initial amount of phosphorylated pRb/p107 at time 0.
The CDK inhibitor flavopiridol inhibits Cdk4 activity and protects cortical neurons from camptothecin-induced death (Park et al., 1997a). As such, we predicted that cotreatment of neurons with flavopiridol and camptothecin should block the phosphorylation observed with camptothecin treatment alone. As shown in Figure 1, flavopiridol cotreatment inhibited the camptothecin-induced induction of pRb/p107 phosphorylation.
We next examined the levels of pRb and p107 during camptothecin treatment of cortical neurons. As shown in Figure2, the levels of pRb and p107 are significantly reduced after 3–4 hr of camptothecin treatment. The observed losses of pRb and p107 are not caused by generalized loss of proteins because little or no reduction of other cell cycle proteins (see below) or β-actin (Figs. 1, 2) is observed under the same conditions. Moreover, this loss is inhibited by cotreatment with flavopiridol, thus indicating that inhibition of CDK activity blocks the degradation of pRb and p107. Such observations raise the possibility that pRb/p107 phosphorylation in this system might target these proteins for degradation.
Fig. 2.

pRb and p107 levels decrease during the death of cortical neurons evoked by camptothecin. A, Top, C, Top, Representative Western immunoblots of whole-cell extracts of cortical neurons treated with camptothecin for the indicated times and probed with antibodies for pRb (A) and p107 (C). Middle,Bottom, Cotreatment with flavopiridol (1 μm; middle) and BAF (100 μm;bottom) is indicated. Blots were stripped and reprobed for β-actin as a loading control. B, D, Densitometric analyses of Western immunoblots of cortical neurons treated with camptothecin alone or cotreated with flavopiridol or BAF and probed for pRb (B) or p107 (D). All data points are the mean ± SEM of data from three separate experiments and are expressed relative to the initial amount of protein at time 0.
We observed recently that caspase activity is detectable in cortical cultures ∼4 hr after the start of camptothecin treatment (Stefanis et al., 1999). This is similar to the time at which losses of pRb and p107 are observed. In addition, general caspase inhibitors significantly delay death in this experimental paradigm (Stefanis et al., 1999). To evaluate the possible involvement of caspase activity in the observed loss of pRb and p107, we cotreated cultures with camptothecin and caspase inhibitors. BAF (Fig. 2) and zVAD-fmk (data not shown) significantly inhibited the loss of pRb and p107. Exposure to flavopiridol, BAF, or zVAD-fmk alone had no effect on the levels of pRb or p107. These results indicate that caspase activation contributes either indirectly or directly to the degradation of pRb and p107.
Early changes in the cyclins, CDKs, and CKIs are not observed in camptothecin-treated cortical neurons
The inhibition of camptothecin-induced neuronal death by CDK inhibitors, along with the observed increase in cyclin D1-associated kinase activity and pRb/p107 phosphorylation, suggests the importance of Cdk4/6 signaling in the control of neuronal apoptosis. We therefore examined the expression of additional cell cycle regulators during camptothecin-evoked cortical neuronal death. As shown in Figure3, the levels of cyclin D1 and cyclin E did not change during camptothecin treatment. Cyclin A was undetectable (data not shown). No difference in expression levels was detected with flavopiridol, zVAD-fmk, or BAF cotreatment or with flavopiridol, zVAD, or BAF alone. These results indicate that elevation of cyclin D1 levels could not account for the increase in observed cyclin D1-associated kinase activity.
Fig. 3.
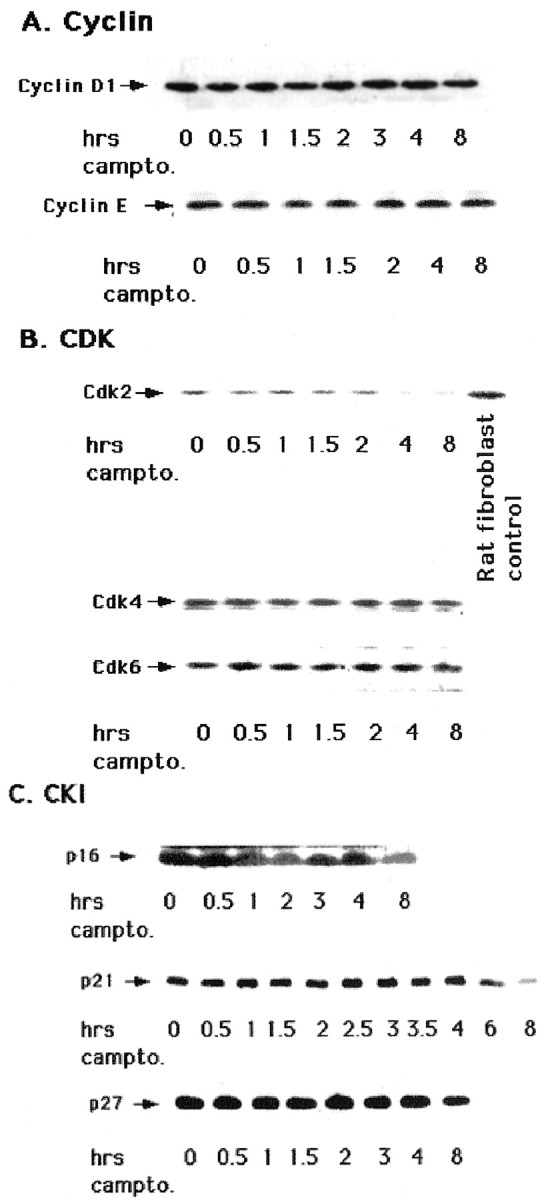
Levels of cyclins, CDKs, and CKIs do not increase during the death of cortical neurons evoked by camptothecin.A—C, Western immunoblot analyses of whole-cell extracts of cortical neurons treated with camptothecin for the indicated times and probed with the following antibodies: cyclin D1 and cyclin E (A); Cdk2, Cdk4, and Cdk6 (B); and p16, p21, and p27 (C). For Cdk2 blots, an equal amount (25 μg) of rat fibroblast cell extract was used as a positive control.
We next explored the expression levels of CDKs in camptothecin-treated cortical neuron cultures. Detection of Cdk2 was inconsistent, but when it was observed by Western blot analyses as shown in Figure 3, the levels were significantly less than that in proliferating rat fibroblast cultures. The levels of Cdk2 protein did not rise in cultures treated with camptothecin and, when detectable, decreased ∼4 hr after treatment. Similarly, levels of Cdk4 and 6, which were readily detectable, did not change significantly during camptothecin-induced cortical neuronal death. As with cyclins, no significant change in the protein levels of Cdk4 and 6 was observed in cultures cotreated with flavopiridol, zVAD-fmk, or BAF or treated with flavopiridol, zVAD, or BAF alone (data not shown).
Finally, changes in the levels of the CKIs p16, p21, and p27 were not observed during the first 4 hr of camptothecin treatment and, if present, only occurred considerably later (Fig. 3). Taken together, our results indicate that loss of pRb and p107 is not generalized to other cell cycle regulators and that activation of cyclin D1-associated kinase activity and subsequent pRb/p107 phosphorylation cannot be accounted for by changes in the levels of the cell cycle regulators examined.
Expression of pRb suppresses the death of cortical neurons evoked by camptothecin
Our observations raise the possibility that phosphorylation and loss of pRb/p107 play important roles in the apoptotic death of cortical neurons evoked by camptothecin. If this is the case, we would predict that exogenous expression of pRb would inhibit such death. To test this, we used recombinant adenovirus to express a mutant of pRb (ΔK11 Rb) in which 11 of the 16 putative phosphorylation sites are mutated (Brown et al., 1999). Among the sites mutated is the putative Cdk4/6 phosphorylation site (Kitagawa et al., 1996). Expression in the neurons was confirmed by Western blot analyses and immunofluorescence (Fig. 4). Approximately 61% of the neurons in cultures infected with ΔK11 Rb-expressing adenovirus showed increased Rb immunoreactivity (n = 243). By Western blot analyses, we observed expression at four to five times the levels of endogenous pRb, and this expression was maintained for at least 10 hr after camptothecin treatment (Fig. 4). As shown in Figure5, expression of ΔK11 Rb significantly blocked death at 16 hr of camptothecin exposure [∼52 vs 18% survival in the noninfected or GFP-infected or LacZ-infected (data not shown) controls]. Protection was transient. Minimal protection (18 ± 2 vs 3 ± 1% in the controls) was observed at 30 hr, and no protection was observed 48 hr after camptothecin treatment (data not shown). Thus, these results along with our observations of increased pRb/p107 phosphorylation and loss of pRb/p107 suggest that pRb members may play an important regulatory role in the death of cortical neurons evoked by DNA damage.
Fig. 4.
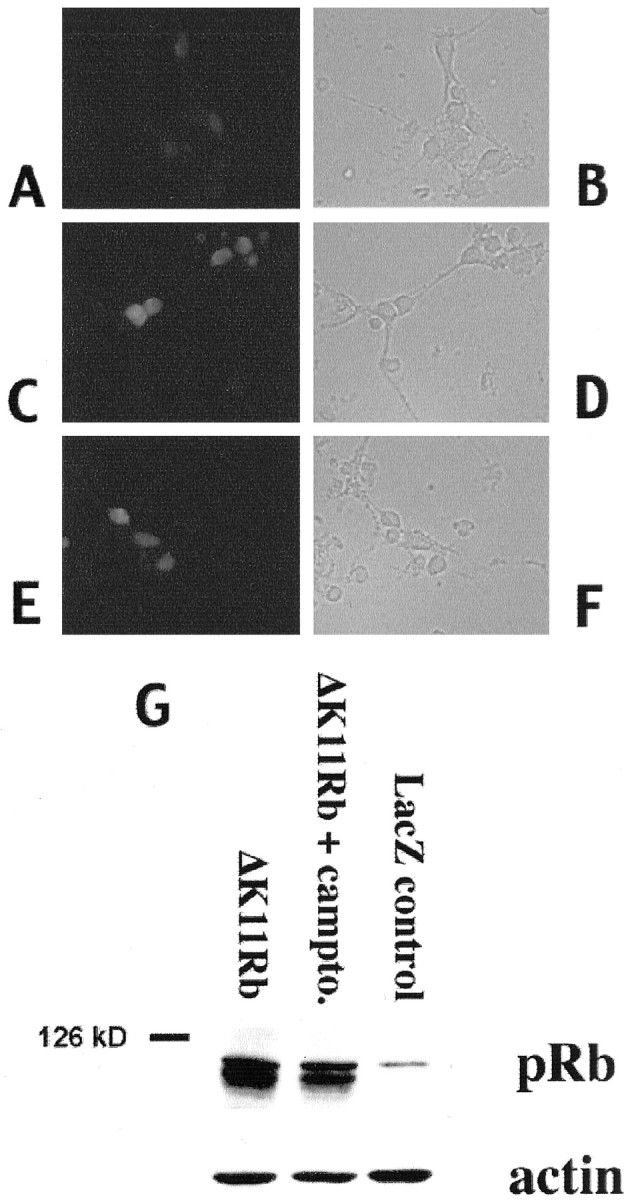
Immunofluorescence (A–F) and Western immunoblot (G) analyses of ΔK11 Rb expression. A—F, Immunofluorescence (A, C, E) staining with an antibody directed against pRb or corresponding light micrographs (B, D, F) of cortical neurons in culture infected with ΔK11 Rb-expressing virus (C–F) or control (A, B). Neurons were fixed and stained with anti-pRb antibody 24 hr after infection (A–D) and after 12 hr of camptothecin treatment (E, F). G, Western immunoblot analysis of whole-cell extracts of cortical neurons infected with adenovirus expressing ΔK11 Rb (no treatment or 10 hr camptothecin treatment) or LacZ as indicated. The blots were analyzed using an anti-pRb antibody, stripped, and reprobed with anti-β-actin.
Fig. 5.

Expression of ΔK11 Rb suppresses the death of cortical neurons evoked by camptothecin. Neuronal cultures were uninfected or infected for 24 hr with adenovirus expressing ΔK11 Rb or GFP before camptothecin treatment for 16 hr. Each point is the mean ± SEM of combined data from three independent experiments (each independent experiment performed in triplicate) and is expressed relative to the number of neurons present at the initial time of camptothecin treatment. * indicates significance (p < 0.02, Student's ttest) compared with GFP-infected cultures treated with camptothecin.
Expression of DN DP1 inhibits the death of neurons evoked by DNA damage
Because pRb family members bind to and affect the transcriptional activity of E2F/DP complexes, we next explored whether such complexes play a role in the death of cortical neurons evoked by DNA damage. This was accomplished by expression of dominant-negative versions of the E2F-binding protein DP1 using the Sindbis virus. We have shown previously that recombinant Sindbis virus can be used for heterologous gene expression and the study of neuronal death mediated by NGF deprivation and DNA damage (Park et al., 1997b, 1998b). DP1 is required for E2F activation (Bandara et al., 1993; Girling et al., 1993; Wu et al., 1996), and Wu et al. (1996) have shown that expression of mutant forms of DP1 that lack the DNA-binding domain either by truncation of 126 amino acids at the N terminal (Δ1–126 DN DP1) or by internal deletion of the DNA-binding domain (Δ103–126 DN DP1) mediates inhibition of E2F-mediated transactivation and cell cycle progression. It was also reported that a DP1 mutant with a deletion of the E2F-binding domain (Δ233–272 DP1) has no such dominant-negative effect (Wu et al., 1996), and this construct was therefore used as one negative control in our experiments.
We first tested whether expression of either (Δ1–126) DN DP1 or (Δ103–126) DN DP1, both of which contain a DNA-binding domain deletion, inhibits the death of cortical neurons evoked by DNA damage. Parallel cultures were infected with control viruses. All constructs except the truncation DN mutant (Δ1–126) DN DP1 were FLAG epitope tagged (F) to permit detection of the expressed protein. Expression was confirmed by immunofluorescence and Western blot analyses (Fig.6) in which comparable expression levels were observed for DN and control DP1 constructs. Approximately 61% of neurons were FLAG immunopositive for (Δ103–126) DN DP1 (n = 178), 63.4% of neurons were positive for Δ233–272 DP1 expression, and 80% were positive for WT DP1 (n = 147). Expression of all three DP1 constructs was both cytoplasmic and nuclear. As shown in Figure7, camptothecin alone produced 90% death at 24 hr of treatment. Expression of each of the DN versions of DP1 significantly inhibited camptothecin-induced death (50–70% survival with DN DP1 expression). Infection with control viruses expressing (Δ233–272) DP1F, WT DP1, or “stop” sequences that contain deletions of the initiation codon to prevent protein expression had small, but consistent, protective effects in comparison with cultures treated with camptothecin alone, but the level of protection was significantly lower than that achieved with the DN constructs. Cortical neurons protected by DN DP1 expression show the flat phase-dark morphology typical of healthy cells, whereas cultures treated with camptothecin alone or with camptothecin and a control virus show membrane blebbing and numerous ghost-like bodies (Fig.8). Neuritic processes, however, did not appear to be spared in camptothecin-treated cultures protected by DN DP1 expression (Fig. 8).
Fig. 6.
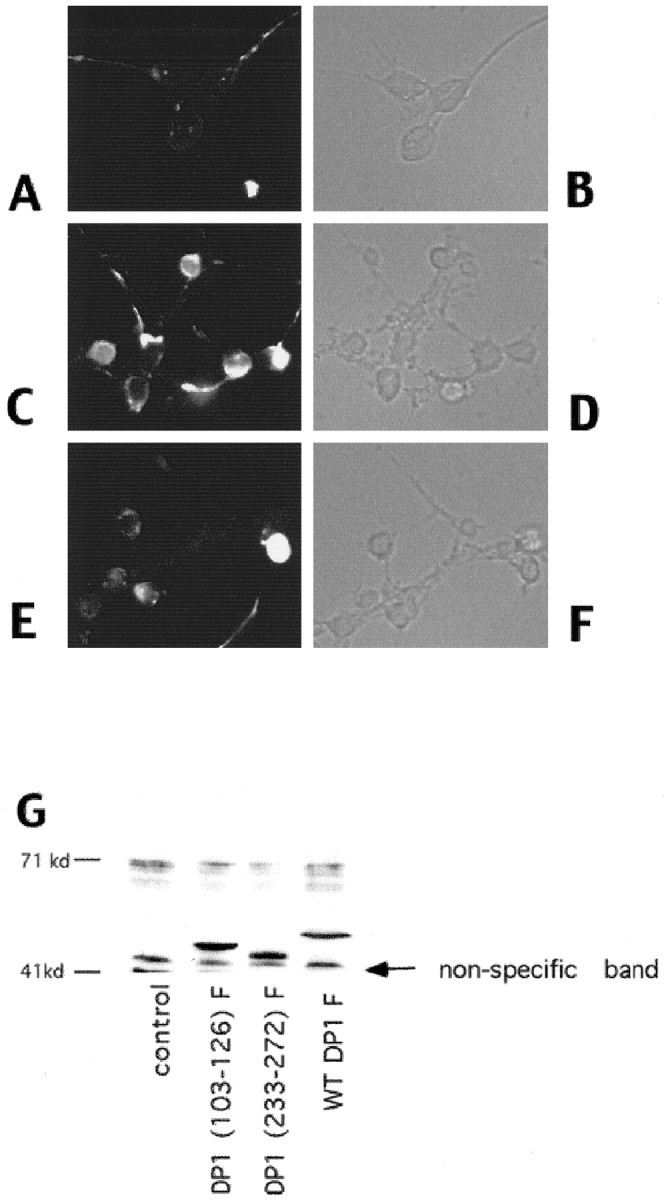
A—F, Immunofluorescence (A, C, E) or corresponding phase (B, D, F) images of neurons infected with control virus (A, B) or with virus encoding (Δ103–126) DN DP1F (C–F). Neurons were fixed and stained with anti-FLAG antibody 24 hr after infection (A–D) and after 12 hr of camptothecin treatment (E, F). G, Western immunoblot analyses of whole-cell extracts of cortical neurons infected with Sindbis viruses expressing WT or the indicated mutant FLAG-tagged DP1 constructs. control indicates samples in which the neurons were infected with a control virus. Theblots were analyzed using an anti-FLAG antibody. A nonspecific immunoreactive band is marked as indicated. Neurons were lysed 24 hr after infection with the indicated viruses.
Fig. 7.

Expression of DN DP1 suppresses the death of cultured cortical neurons treated with camptothecin. Control viruses for each vector were generated by removal of the start codon from the insert. F denotes that the protein has a FLAG epitope attached to the C terminal. Effects of expression of (Δ1–126) DN DP1, a dominant-negative mutant containing an N-terminal truncation from amino acids 1–126 (A), (Δ103–126) DN DP1F, a FLAG-tagged dominant-negative mutant containing a DNA-binding domain deletion of amino acids 103–126 (B), (Δ233–272) DP1F, a FLAG-tagged mutant containing the E2F-binding domain deletion of amino acids 233–272 (C), and WT DP1 and respective controls (D) on the survival of cortical neurons after camptothecin treatment for 1 d. A, Graph in which each point is the mean ± SEM of combined data from three independent experiments (each independent experiment performed in triplicate) and is expressed relative to the number of neurons present in each culture at the initial time of camptothecin (10 μm) treatment. B—D, Representative experiments in which each point is the mean ± SEM of three cultures from the same experimental trial. Similar results (B–D) were obtained in two independent experiments.
Fig. 8.
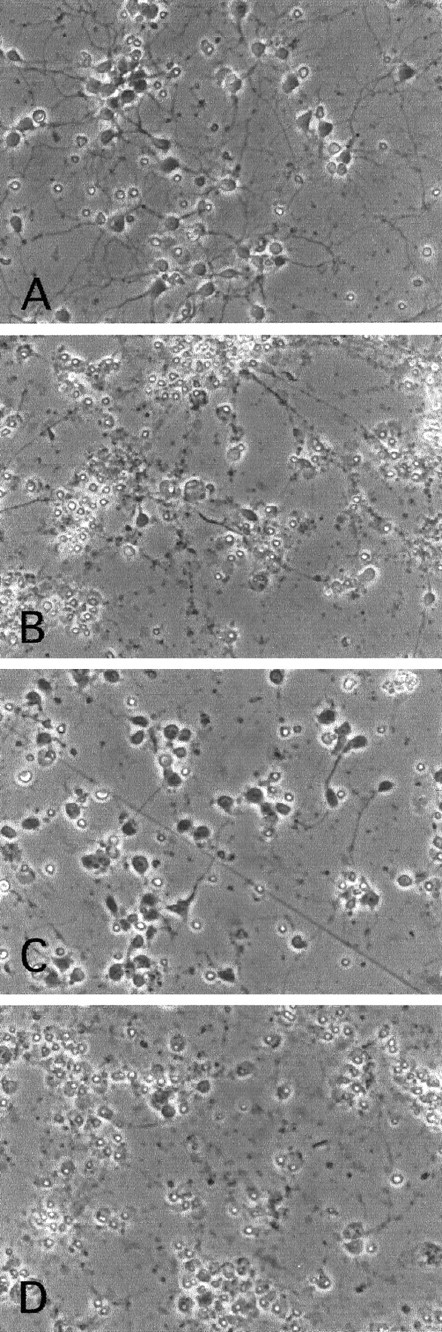
Phase-contrast micrographs of cortical neurons treated for 1 d with the following: no additives (A), camptothecin (10 μm;B), (Δ1–126) DN DP1 virus + camptothecin (C), and (Δ1–126) DN DP1 control virus + camptothecin (D).
We next examined whether death in a another model of DNA damage could also be blocked by expression of DN mutants of DP1. As shown in Figure9, expression of either (Δ1–126) DN DP1 or (Δ103–126) DN DP1 inhibits the death of sympathetic neurons evoked by UV irradiation (∼85% survival with DN DP1 expression vs 50% without). Expression of a mutant of DP1 [(Δ233–272) DP1] bearing the E2F-binding domain deletion or of WT DP1 or infection with stop control viruses that contain deletions of the initiation codon to prevent expression of DP1 proteins had no significant effects on the kinetics of neuronal death (Fig. 9; data not shown). Sympathetic neurons protected by DN DP1 expression showed the typical phase-bright morphology and relatively intact neuritic processes of viable neurons. In contrast, sympathetic neurons treated with UV irradiation alone or after infection with control viruses displayed shrunken, phase-dark somas and degenerating processes (Fig.10). Expression of the FLAG-tagged DP1 constructs was confirmed by immunofluorescence analysis (Fig. 11).
Fig. 9.
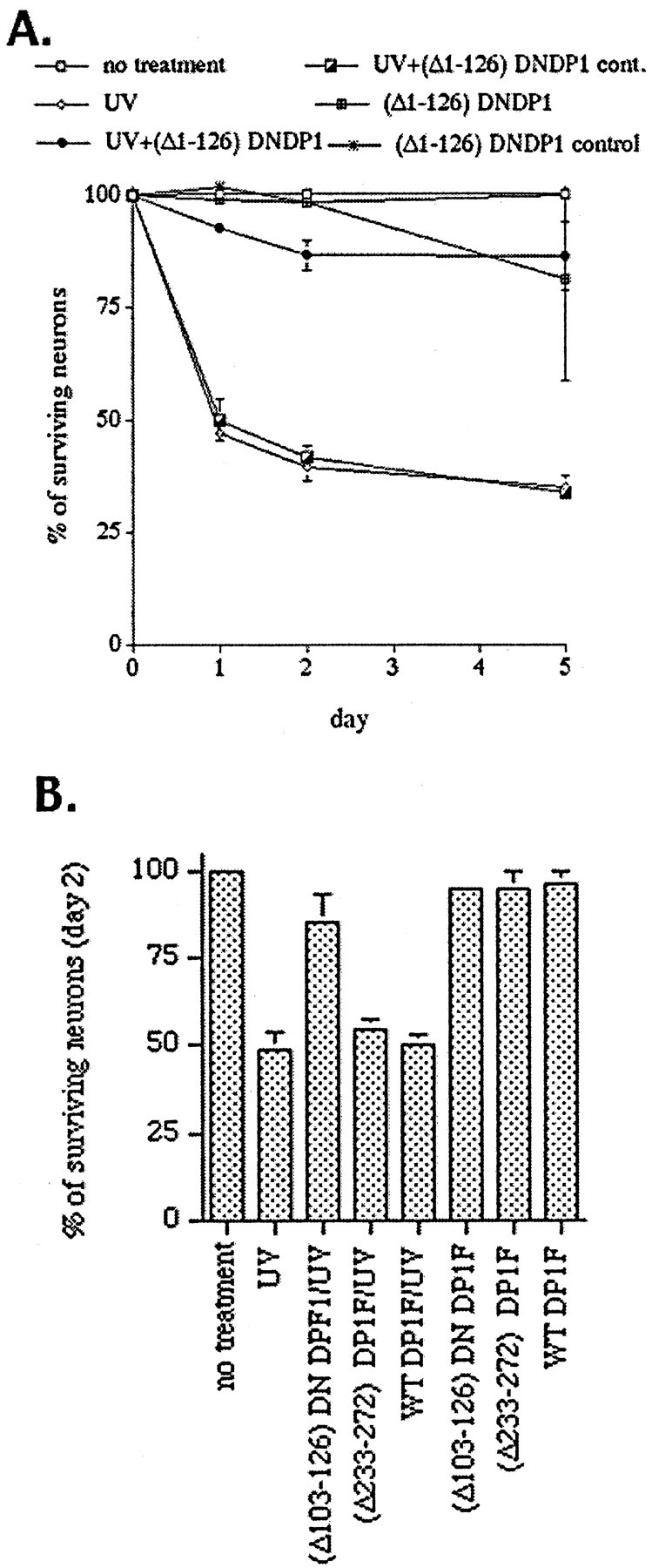
Expression of DN DP1 suppresses the death of cultured sympathetic neurons evoked by UV irradiation. Control viruses for each vector were generated by removal of the start codon from the insert. F denotes that the protein has a FLAG epitope attached to the C terminal. Effects of expression of (Δ1–126) DN DP1, a dominant-negative mutant containing an N-terminal truncation from amino acids 1–126 (A), and of (Δ103–126) DN DP1F, a FLAG-tagged dominant-negative mutant containing a DNA-binding domain deletion of amino acids 103–126, (Δ233–272) DP1F, a FLAG-tagged mutant containing the E2F-binding domain deletion of amino acids (233–272), and wild type (WT) DP1 (B) on the survival of sympathetic neurons after UV irradiation. A, Graph in which each point is the mean ± SEM of combined data from three independent experiments (each independent experiment performed in triplicate) and is expressed relative to the number of neurons present in each culture at the initial time of UV treatment. cont, Control.B, Representative experiment in which each point is the mean ± SEM of three cultures from the same experimental trial. Similar results were obtained in two independent experiments.
Fig. 10.
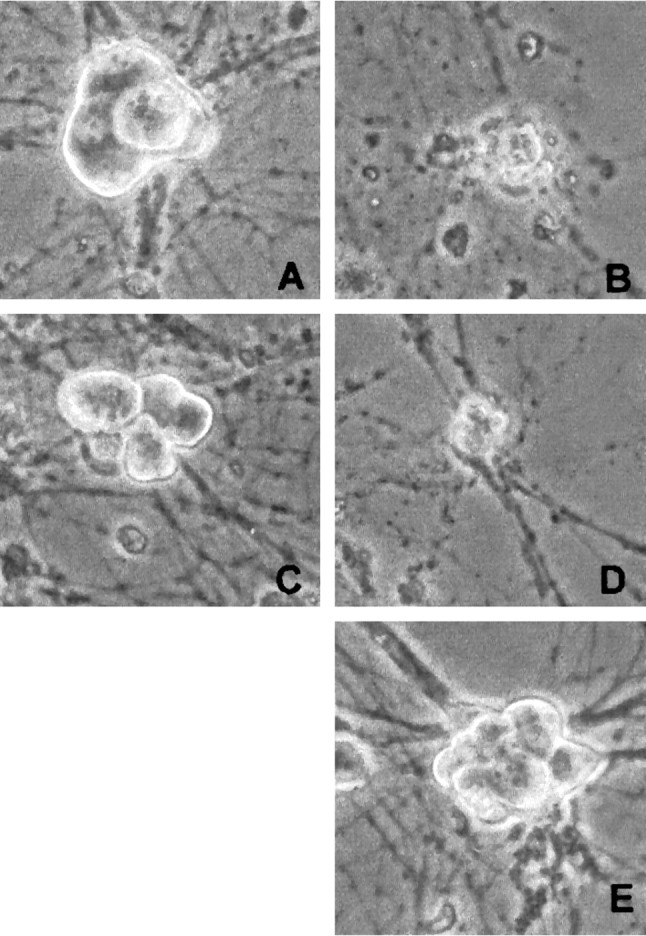
Phase-contrast micrographs of rat sympathetic neurons maintained in NGF-containing medium and exposed to and/or infected with the following: no treatment (A), UV (B), (Δ1–126) DN DP1 virus + UV (C), (Δ1–126) DN DP1 control virus + UV (D), or (Δ1–126) DN DP1 virus alone (E). The photos were taken 2 d after irradiation and/or infection.
Fig. 11.

Immunofluorescence (A–C) staining with an antibody directed against the FLAG epitope or corresponding light micrographs (D–F) of sympathetic neurons in culture infected with Sindbis virus expressing (Δ233–272) DP1F (A, D) or (Δ103–126) DN DP1F (B, E) or containing a nonexpressing control virus (C, F). Neurons were stained 2 d after infection.
DISCUSSION
The potential role of cell cycle elements in neuronal apoptosis is perhaps paradoxical considering the terminally differentiated state of neurons. However, growing evidence has suggested that elements that normally control cell cycle progression may modulate death in a cellular environment not conducive to proliferation. Our previous evidence indicated a role for Cdk4/6 in the mechanism of neuronal death evoked by DNA damage and trophic factor deprivation (Park et al., 1997b, 1998b). Accordingly, we examined (1) the involvement of pRb/p107, substrates for Cdk4/6, in neuronal death evoked by DNA damage and (2) whether E2F/DP complexes play a required role in the death signal downstream of Cdk4/6.
Phosphorylation and loss of pRb/p107
We have shown recently that camptothecin induces rapid (within 10 min) DNA strand breaks in cortical neurons (Morris et al., 1997) and is a convenient model for examining the mechanisms that control DNA damage-induced neuronal death. We examined here the status of key cell cycle regulatory proteins during apoptosis of cortical neurons evoked by camptothecin and demonstrate that pRb/p107 is phosphorylated during such treatment. pRb phosphorylation has also been observed in cultures of cis-platinum-treated sensory neurons (Gill and Windebank, 1998) and in cerebellar granule neurons deprived of K+ (Padmanabhan et al., 1999). We also observed here that phosphorylation of pRb/p107 is blocked by the CDK inhibitor flavopiridol that is known to block Cdk4/6 activity (Filgueira de Azevedo et al., 1996). Moreover, this phosphorylation occurs early (within 1 hr) after the start of camptothecin treatment and correlates well with the induction of cyclin D1-associated kinase activity demonstrated previously (Park et al., 1998b). In addition, we find that levels of pRb and p107 are greatly diminished 3–4 hr after camptothecin treatment. This loss is inhibited by flavopiridol, suggesting a relationship between pRb/p107 phosphorylation and loss. In addition, zVAD and BAF also block the loss, raising the possibility that caspases may mediate pRb and p107 degradation. pRb is a substrate of caspases and contains a consensus caspase cleavage site at its C terminal (Tan et al., 1997). However, it is unclear whether the observed loss of pRb/p107 is directly or indirectly mediated by caspases.
To test whether pRb and p107 are important for maintaining survival, we forced expression of a variant of Rb that contains mutations in several phosphorylation sites including the Cdk4/6 consensus site and found that this delays the death of neurons evoked by camptothecin. In other studies, expression of a caspase cleavage-resistant mutant of pRb abrogated TNF-α-induced apoptosis of Rb−/− fibroblasts (Tan et al., 1997). The importance of Rb in cell death and survival is underscored by observations that its overexpression in various contexts is protective against apoptosis (Berry et al., 1996; Fan et al., 1996), whereas loss of Rb or of Rb activity facilitates death of both non-neuronal (Liu and Kitsis, 1996;Shan et al., 1996) and neuronal cells (Jacks et al., 1992; Lee et al., 1992; Macleod et al., 1996). Taken together, these results suggest that the maintenance of the levels and/or activity of Rb members plays an important role in neuronal death evoked by DNA damage.
Levels of cyclins, CDKs, and CKIs
As discussed above, we found previously that Cdk4/6 increases within 1 hr of camptothecin treatment. How does this activation occur? In certain systems, it has been suggested that this regulation occurs via modulation of levels of cell cycle regulators such as the cyclins and CDKs. However, regulation of CDK activity can occur via multiple mechanisms (Pines, 1993). These include (1) binding to an obligate activating cyclin partner, (2) inhibition by endogenous CKIs, and (3) phosphorylation at sites that are either activating or inhibitory. Accordingly, the observed increase in cyclin D1-associated kinase activity and the consequent pRb/p107 phosphorylation could have occurred as a result of increased cyclin or CDK levels or by a decrease in levels of CKIs. However, no such modulation of cyclin, CDK, or CKI levels was detected during the early period of camptothecin treatment. In only a few cases, for example, with p21, did the levels diminish significantly, and these were too late to account for the early induction of cyclin D1-associated kinase activity. Accordingly, these observations, along with the rapid induction of cyclin D1-associated activity (Park et al., 1998b) and pRb/p107 phosphorylation, suggest that activation of Cdk4/6 during the death of cortical neurons evoked by DNA damage could be caused by post-translational modifications such as phosphorylation/dephosphorylation events and the translocation of cyclins as we have described during the death of cerebellar granule neurons deprived of K+ (Padmanabhan et al., 1999). In addition, our observations differ from that observed in dying neurons of adult brains where upregulation of cell cycle markers has been reported (McShea et al., 1997; Nagy et al., 1997; Vincent et al., 1997; Busser et al., 1998). This may be caused by the downregulation of cell cycle molecules that occur in more mature neurons.
Role of E2F/DP complexes in neuronal death evoked by DNA damage
What are possible consequences of pRb/p107 phosphorylation and loss of pRb and p107 protein? pRb binds E2F and negatively regulates its transcriptional activity; when hyperphosphorylated by Cdk4/6, pRb releases E2F, resulting in the derepression of E2F-responsive genes (Chellappan et al., 1991). This raised the possibility that E2F plays a causal role in the death of camptothecin-treated cortical neurons. E2F members 1–5 are present in embryonic cultured cortical neurons (D. S. Park, unpublished data). Of additional relevance, overexpression of E2F in a variety of non-neuronal cells triggers apoptotic death (Qin et al., 1994; Hiebert et al., 1995), and we have observed that overexpression of E2F1 induces death of neurons (D. S. Park, D. Liu, and L. A. Greene, unpublished data). One means we used to test the possible role of E2F in death was overexpression of ΔK11 Rb. This protein has been shown to suppress E2F-mediated transcription (Brown et al., 1999). We observed that this construct protected neurons from camptothecin-evoked death, consistent with a role for E2F in the apoptotic mechanism.
As a second test of our hypothesis, we used Sindbis virus to express several different DN constructs of DP1 (a member of a family of obligate partners for E2F-transactivating activity) (Bandara et al., 1993; Girling et al., 1993; Wu et al., 1996) in cultures of cortical neurons exposed to camptothecin. These DN forms, but neither WT DP1 nor a variety of control constructs, provided significant protection from death evoked by camptothecin. Similar results were obtained with sympathetic neurons exposed to UV irradiation. The findings provide further support for a role of E2F/DP1 complexes in neuronal death evoked by camptothecin and suggest that this extends to multiple neuronal types and to additional paradigms of DNA damage.
It must be noted that there are alternative means by which DN DP1 may provide neuronal protection. The DN versions of DP1 used in these studies contain a deletion of the DNA-binding domain that is required for mediating E2F activity and cell cycle progression in proliferating cell systems (Wu et al., 1996). The apparent requirement for the DNA-binding domain in death signaling suggests the importance of transcriptionally mediated events. In support of this possibility, neuronal death attributable to DNA damage requires transcription (Morris and Geller, 1996; Park et al., 1998a). A recent report, however, suggests that E2F may mediate death in at least certain proliferating systems via a transcriptionally independent mechanism and that this requires the E2F DNA-binding domain (Hsieh et al., 1997). Accordingly, it is unclear whether transcriptional regulation mediated by E2F/DP complexes is important in neuronal apoptosis. It is important to note that it is unclear which E2F complexes may be inactivated by DN DP1 expression in neurons. Curiously, we find that DN DP1 expression is not exclusively nuclear. Wu et al. (1996) have suggested that DN DP1 is able to inhibit the transcriptional activity of multiple E2F members (1–5). Because several of the E2F members are known to also reside in the cytoplasm [for example, E2F4 (Verona et al., 1997)], it is possible that DN DP1 may act (partially or fully) in the cytoplasm to inhibit death.
Finally, E2F and DP are also reported to interact physically with other proteins including p53 both in vitro and in proliferating cells (O'Connor et al., 1995; Sorensen et al., 1996). The existence of such interactions in neurons and/or their potential functions are unknown. Accordingly, we cannot exclude the possibility that expression of DN DP1 may protect neurons via a mechanism that includes p53 or other proteins. In this regard, it has been reported that p53 expression is required for the death of neurons evoked by DNA damage (Enokido et al., 1996; Johnson et al., 1998; Anderson and Tolkovsky, 1999).
The findings presented here provide a plausible model as to why a variety of inhibitors of the cell cycle are effective in rescuing neurons from the death evoked by DNA damage. Our data suggest that a post-translational mechanism is activated in camptothecin-treated cortical neurons that elevates Cdk4/6 activity and that this leads to enhanced phosphorylation of pRb/p107. Phosphorylation of these proteins in turn contributes to their loss by a mechanism requiring caspase activity. Our findings that expression of ΔK11 Rb and DN DP1 protects neurons from DNA damage further suggest that loss of pRb and p107 permit dysregulated E2F transactivational activity that results in synthesis of gene products that contribute to and are required for the death process. Elements of additional pathways such as p53 (Enokido et al., 1996; Johnson et al., 1998) and c-Jun/JNK (Estus et al., 1994; Ham et al., 1995; Xia et al., 1995; Maroney et al., 1999) have been implicated in neuronal death. It remains to be seen whether these act in parallel, or intersect, with the pathway described here.
Footnotes
This work was supported in part by grants from the National Institutes of Health–National Institute of Neurological Disorders and Stroke and the Blanchette Rockefeller Foundation (L.A.G.), the National Institutes of Health (H.M.G. and M.L.S.), the National Cancer Institute of Canada with funds from the Canadian Cancer Society (R.B.), and the Medical Research Council of Canada and Glaxo Wellcome (D.S.P.) We would like to thank Jie Wu for technical support.
D.S.P. and E.J.M. contributed equally to this manuscript.
Correspondence should be addressed to Dr. David S. Park, Neuroscience Research Institute, University of Ottawa, 451 Smyth Road, Ottawa, Ontario, K1H 8M5, Canada. E-mail: dpark@uottawa.ca.
Dr. Morris's present address: Massachusetts General Hospital Cancer Center, Laboratory of Molecular Oncology Building 149, 13th Street Mailcode 149-7330, Charlestown, MA 02129.
REFERENCES
- 1.Anderson AJ, Su JH, Cotman CW. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J Neurosci. 1996;16:1710–1719. doi: 10.1523/JNEUROSCI.16-05-01710.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson CNG, Tolkovsky AM. A role for MAPK/ERK in sympathetic neuron survival: protection against a p53-dependent, JNK-independent induction of apoptosis by cytosine arabinoside. J Neurosci. 1999;19:664–673. doi: 10.1523/JNEUROSCI.19-02-00664.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bandara LR, Buck VM, Zamanian M, Johnston LH, La Thangue NB. Functional synergy between DP-1 and E2F-1 in the cell cycle-regulating transcription factor DRTF1/E2F. EMBO J. 1993;12:4317–4324. doi: 10.1002/j.1460-2075.1993.tb06116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beijersbergen RL, Carlee L, Kerkhoven RM, Bernards R. Regulation of the retinoblastoma protein-related p107 by G1 cyclin complexes. Genes Dev. 1995;9:1340–1350. doi: 10.1101/gad.9.11.1340. [DOI] [PubMed] [Google Scholar]
- 5.Berry DE, Lu Y, Schimdt B, Falon PG, O'Connell C, Hu S-X, Xu H-J, Blanck G. Retinoblastoma protein inhibits IFN induced apoptosis. Oncogene. 1996;12:1809–1819. [PubMed] [Google Scholar]
- 6.Brown VD, Phillips RA, Gallie BL. Cumulative effect of phosphorylation of pRB on regulation of E2F activity. Mol Cell Biol. 1999;19:3246–3256. doi: 10.1128/mcb.19.5.3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Busser J, Gelmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, Nevins JR. The E2F transcription factor is a cellular target for the RB protein. Cell. 1991;65:1053–1061. doi: 10.1016/0092-8674(91)90557-f. [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Nagayama T, Jin K, Stetler A, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cotman CW. Apoptosis decision cascades and neuronal degeneration in Alzheimer's disease. Neurobiol Aging. 1998;19:S29–S32. doi: 10.1016/s0197-4580(98)00042-6. [DOI] [PubMed] [Google Scholar]
- 11.Enokido Y, Araki T, Tanaka K, Aizawa S, Hatanaka H. Involvement of p53 in DNA strand break-induced apoptosis in postmitotic CNS neurons. Eur J Neurosci. 1996;8:1812–1821. doi: 10.1111/j.1460-9568.1996.tb01325.x. [DOI] [PubMed] [Google Scholar]
- 12.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan G, Ma X, Kren BT, Steer CJ. The retinoblastoma gene product inhibits TGF-b1 induced apoptosis in rat hepatocyte and human HuH-7 hepatoma cells. Oncogene. 1996;12:1909–1919. [PubMed] [Google Scholar]
- 14.Farinelli SE, Greene LA. Cell cycle blockers mimosine, ciclopirox, and deferoxamine prevent the death of PC12 cells and postmitotic sympathetic neurons after removal of trophic support. J Neurosci. 1996;16:1150–1162. doi: 10.1523/JNEUROSCI.16-03-01150.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Filgueira de Azevedo W, Mueller-Dieckmann H-J, Schulze-Gahmen U, Worland PJ, Sausville E, Kim S-H. Structural basis for specificity and potency of a flavanoid inhibitor of human CDK2, a cell cycle kinase. Proc Natl Acad Sci USA. 1996;93:2735–2740. doi: 10.1073/pnas.93.7.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freeman RF, Estus S, Johnson EM. Analysis of cell-related gene expression in postmitotic neurons: selective induction of Cyclin D1 during programmed cell death. Neuron. 1994;12:343–355. doi: 10.1016/0896-6273(94)90276-3. [DOI] [PubMed] [Google Scholar]
- 17.Gao CY, Zelenka PS. Induction of cyclin B and H1 kinase activity in apoptotic PC12 cells. Exp Cell Res. 1995;219:612–618. doi: 10.1006/excr.1995.1271. [DOI] [PubMed] [Google Scholar]
- 18.Gill JS, Windebank AJ. Cisplatin-induced apoptosis in rat dorsal root ganglion neurons is associated with attempted entry into the cell cycle. J Clin Invest. 1998;101:2842–2850. doi: 10.1172/JCI1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Girling R, Partridge JF, Bandara LR, Burden N, Totty NF, Hsuan JJ, La Thangue NB. A new component of the transcription factor DRTF1/E2F. Nature. 1993;362:83–87. doi: 10.1038/362083a0. [DOI] [PubMed] [Google Scholar]
- 20.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 21.Hamel PA, Gill RM, Phillips RA, Gallie BL. Regions controlling hyperphosphorylation and conformation of the retinoblastoma gene product are independent of domains required for transcriptional repression. Oncogene. 1992;7:693–701. [PubMed] [Google Scholar]
- 22.Hardy S, Kitamura M, Hariis-Stansil T, Dai Y, Phillips ML. Construction of adenovirus vectors through Cre-lox recombination. J Virol. 1997;71:1842–1849. doi: 10.1128/jvi.71.3.1842-1849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hiebert SW, Packham G, Strom DK, Haffner R, Oren M, Zambetti G, Cleveland JL. E2F-1: DP-1 induces p53 and overrides survival factors to trigger apoptosis. Mol Cell Biol. 1995;15:6864–6874. doi: 10.1128/mcb.15.12.6864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh JK, Fredersdorf S, Kouzarides T, Martin K, Lu X. E2F1-induced apoptosis requires DNA binding but not transactivation and is inhibited by the retinoblastoma protein through direct interaction. Genes Dev. 1997;11:1840–1852. doi: 10.1101/gad.11.14.1840. [DOI] [PubMed] [Google Scholar]
- 25.Jacks T, Fazeli A, Schitt EM, Bronson RT, Goodell MA, Weinberg RA. Effects of an Rb mutation in the mouse. Nature. 1992;359:295–300. doi: 10.1038/359295a0. [DOI] [PubMed] [Google Scholar]
- 26.Joe AK, Ferrari G, Jiang HH, Liang XH, Levine B. Dominant inhibitory Ras delays sindbis virus-induced apoptosis in neuronal cells. J Virol. 1996;70:7744–7751. doi: 10.1128/jvi.70.11.7744-7751.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson MD, Xiang H, London S, Kinoshita Y, Knudson M, Mayberg M, Korsmeyer SJ, Morrison RS. Evidence for involvement of BAX and p53, but not caspases, in radiation-induced cell death of cultured postnatal hippocampal neurons. J Neurosci Res. 1998;54:721–733. doi: 10.1002/(SICI)1097-4547(19981215)54:6<721::AID-JNR1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 28.Kitagawa M, Higashi H, Jung HK, Suzuki-Takahashi I, Ikeda M, Tamai K, Kato J, Segawa K, Yoshida E, Nishimura S, Taya Y. The consensus motif for phosphorylation by cyclin D1-Cdk4 is different from that for phosphorylation by cyclin A/E-Cdk2. EMBO J. 1996;15:7060–7069. [PMC free article] [PubMed] [Google Scholar]
- 29.La Thangue NB. DTRF1/E2F: an extended family of heterodimeric factors implicated in cell cycle control. Trends Biochem Sci. 1994;19:108–114. doi: 10.1016/0968-0004(94)90202-x. [DOI] [PubMed] [Google Scholar]
- 30.Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Kitsis RN. Induction of DNA synthesis and apoptosis in cardiac myocytes by E1A oncoprotein. J Cell Biol. 1996;133:325–334. doi: 10.1083/jcb.133.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Macleod KF, Hu Y, Jacks T. Loss of RB activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996;15:6178–6188. [PMC free article] [PubMed] [Google Scholar]
- 33.Maroney A, Finn JP, Bozyczko-Coyne D, Kane T, Neff NT, Tolkovsky AM, Park DS, Yan CYI, Troy CM, Greene LA. CEP-1347 (KT7515), an inhibitor of JNK activation, rescues sympathetic neurons and neuronally differentiated PC12 cells from death evoked by three distinct insults. J Neurochem. 1999;73:1901–1912. [PubMed] [Google Scholar]
- 34.Martin K, Trouche D, Hagemeier C, Sorensen TS, La Thangue NB, Kouzarides T. Stimulation of E2F1/DP1 transcriptional activity by MDM2 oncoprotein. Nature. 1995;375:691–694. doi: 10.1038/375691a0. [DOI] [PubMed] [Google Scholar]
- 35.McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am J Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 36.Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase-I: evidence for cell-cycle independent toxicity. J Cell Biol. 1996;134:757–770. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris EJ, Park DS, Dreixler JC, Greene LA, Geller HM. Signal transduction of DNA damage-induced neuronal apoptosis. Soc Neurosci Abstr. 1997;23:8815. [Google Scholar]
- 38.Nagy Z, Esiri MM, Cato AM, Smith AD. Cell cycle markers in the hippocampus in Alzheimer's disease. Acta Neuropathol (Berl) 1997;94:6–15. doi: 10.1007/s004010050665. [DOI] [PubMed] [Google Scholar]
- 39.Nevins JR. E2F, a link between the Rb tumour suppressor protein and viral oncoproteins. Science. 1992;258:424–429. doi: 10.1126/science.1411535. [DOI] [PubMed] [Google Scholar]
- 40.O'Connor DJ, Lam E, Griffin S, Zhong S, Leighton LC, Burbidge SA, Lu X. Physical and functional interactions between p53 and cell cycle co-operating factors, E2F1 and DP1. EMBO J. 1995;14:6184–6192. doi: 10.1002/j.1460-2075.1995.tb00309.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 42.Padmanabhan J, Park DS, Greene LA, Shelanski ML. Role of cell cycle regulatory proteins in cerebellar granule neuron apoptosis. J Neurosci. 1999;19:8747–8756. doi: 10.1523/JNEUROSCI.19-20-08747.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Park DS, Farinelli SE, Greene LA. Inhibitors of cyclin-dependent kinases promote survival of post-mitotic neuronally differentiated PC12 cells and sympathetic neurons. J Biol Chem. 1996a;271:8161–8170. doi: 10.1074/jbc.271.14.8161. [DOI] [PubMed] [Google Scholar]
- 44.Park DS, Stefanis L, Yan CYI, Farinelli SE, Greene LA. Ordering the cell death pathway: differential effects of Bcl-2, an interleukin-1-converting enzyme family protease inhibitor, and other survival agents on JNK activation in serum/nerve growth factor-deprived PC12 cells. J Biol Chem. 1996b;271:21898–21905. doi: 10.1074/jbc.271.36.21898. [DOI] [PubMed] [Google Scholar]
- 45.Park DS, Morris EJ, Greene LA, Geller HM. G1/S cell cycle blockers and inhibitors of cyclin dependent kinases suppress camptothecin-induced apoptosis. J Neurosci. 1997a;17:1256–1270. doi: 10.1523/JNEUROSCI.17-04-01256.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park DS, Levine B, Ferrari G, Greene LA. Cyclin dependent kinase inhibitors and dominant negative cyclin dependent kinase 4 and 6 promote survival of NGF-deprived sympathetic neurons. J Neurosci. 1997b;17:8975–8983. doi: 10.1523/JNEUROSCI.17-23-08975.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park DS, Morris EJ, Stefanis L, Troy CM, Shelanski ML, Geller HM, Greene LA. Multiple pathways of neuronal death induced by DNA damaging agents, NGF deprivation, and oxidative stress. J Neurosci. 1998a;18:830–840. doi: 10.1523/JNEUROSCI.18-03-00830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park DS, Morris EJ, Padmanabhan J, Shelanski ML, Geller HM, Greene LA. Cyclin dependent kinases participate in death of neurons evoked by DNA damaging agents. J Cell Biol. 1998b;143:457–467. doi: 10.1083/jcb.143.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pines J. Cyclins and cyclin-dependent kinases: take your partners. Trends Biochem Sci. 1993;18:195–197. doi: 10.1016/0968-0004(93)90185-p. [DOI] [PubMed] [Google Scholar]
- 50.Qin XQ, Livingston DM, Kaelin WG, Jr, Adams PD. Deregulated transcription factor E2F-1 expression leads to S-phase entry and p53-mediated apoptosis. Proc Natl Acad Sci USA. 1994;91:10918–10922. doi: 10.1073/pnas.91.23.10918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shan B, Durfee T, Lee W-H. Disruption of RB/E2f-1 interaction by a single point mutation in E2f-1 enhances s-phase entry and apoptosis. Proc Natl Acad Sci USA. 1996;93:679–684. doi: 10.1073/pnas.93.2.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sorensen TS, Girling R, Lee CW, Gannon J, Bandara LR, La Thangue NB. Functional interactions between DP-1 and p53. Mol Cell Biol. 1996;16:5888–5895. doi: 10.1128/mcb.16.10.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stefanis L, Burke RE, Greene LA. Apoptosis in neurodegenerative disorders. Curr Opin Neurol. 1997;10:299–305. doi: 10.1097/00019052-199708000-00004. [DOI] [PubMed] [Google Scholar]
- 54.Stefanis L, Park DS, Friedman WJ, Greene LA. Caspase-dependent and -independent death of camptothecin-treated embryonic cortical neurons. J Neurosci. 1999;19:6235–6247. doi: 10.1523/JNEUROSCI.19-15-06235.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suzuki-Takahashi I, Kitagawa M, Saijo M, Higashi H, Ogino H, Matsumoto H, Taya Y, Nishimura S, Okuyama A. The interactions of E2F with pRB and with p107 are regulated via the phosphorylation of pRB and p107 by a cyclin-dependent kinase. Oncogene. 1995;10:1691–1698. [PubMed] [Google Scholar]
- 56.Tan X, Martin SJ, Green DR, Wang JY. Degradation of retinoblastoma protein in tumor necrosis factor- and CD95-induced cell death. J Biol Chem. 1997;272:9613–9616. doi: 10.1074/jbc.272.15.9613. [DOI] [PubMed] [Google Scholar]
- 57.Verona R, Moberg K, Estes S, Starz M, Vernon JP, Lees JA. E2F activity is regulated by cell cycle-dependent changes in subcellular localization. Mol Cell Biol. 1997;17:7268–7282. doi: 10.1128/mcb.17.12.7268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weinberg RA. The retinoblastoma protein and cell cycle control. Cell. 1995;81:323–330. doi: 10.1016/0092-8674(95)90385-2. [DOI] [PubMed] [Google Scholar]
- 60.Wu C-L, Classon M, Dyson N, Harlow E. Expression of dominant negative mutant DP-1 block cell cycle progression in G1. Mol Cell Biol. 1996;16:3698–3706. doi: 10.1128/mcb.16.7.3698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]