

Abstract
Although norepinephrine (NE) has been implicated in animal models of ethanol consumption for many years, the exact nature of its influence is not clear. Lesioning and pharmacological studies examining the role of NE in ethanol consumption have yielded conflicting results. We took a genetic approach to determine the effect of NE depletion on ethanol-mediated behaviors by using dopamine β-hydroxylase knockout (Dbh −/−) mice that specifically lack the ability to synthesize NE. Dbh −/− males have reduced ethanol preference in a two-bottle choice paradigm and show a delay in extinguishing an ethanol-conditioned taste aversion, suggesting that they drink less ethanol in part because they find its effects more aversive. Both male and female Dbh −/− mice are hypersensitive to the sedative and hypothermic effects of systemic ethanol administration, and the sedation phenotype can be rescued pharmacologically by acute replacement of central NE. Neither the decreased body temperature nor changes in ethanol metabolism can explain the differences in consumption and sedation. These results demonstrate a significant role for NE in modulating ethanol-related behaviors and physiological responses.
Keywords: norepinephrine, dopamine β-hydroxylase, mice, ethanol, conditioned taste aversion, sedation, hypothermia
Although previous lesioning and pharmacological studies have suggested a role for norepinephrine (NE) in ethanol-mediated behaviors, they have, taken together, failed to reach solid conclusions. Acute administration of ethanol can modulate the synthesis, turnover, and release of central NE (Corrodi et al., 1966; Carlsson and Lindqvist, 1973; Hunt and Majchrowicz, 1974;Pohorecky and Jaffe, 1975; Karoum et al., 1976) and the activity of noradrenergic neurons (Aston-Jones et al., 1982; Pohorecky and Brick, 1987; Verbanck et al., 1990). However, the function of these changes in noradrenergic transmission in response to ethanol is controversial because of conflicting behavioral results. Depending on site of administration and strain of rat used, chemical lesions of noradrenergic neurons with 6-hydroxydopamine (6-OHDA) increase (Kiianmaa et al., 1975; Melchior and Myers, 1976; Kiianmaa, 1980), decrease (Myers and Melchior, 1975; Melchior and Myers, 1976), or have no effect (Melchior and Myers, 1976; Richardson and Novakovski, 1978) on voluntary ethanol consumption. There are a number of caveats associated with this technique. For example, 6-OHDA ablates entire neurons, resulting in the loss of neuropeptides such as galanin and neuropeptide Y (NPY) that are colocalized with NE in noradrenergic neurons (Everitt et al., 1984; Melander et al., 1986; Xu et al., 1998). 6-OHDA can also affect dopaminergic function. Last, 6-OHDA does not completely eliminate NE, and both presynaptic and postsynaptic compensation can occur (Segal and Geyer, 1976; De Montigny et al., 1980). There are also conflicting findings based on dopamine β-hydroxylase (DBH) inhibitors (Amit et al., 1977; Daoust et al., 1990) and adrenergic agonists (Andreas et al., 1983; Grupp et al., 1989).
As a consequence of this confusion, NE has been largely discounted as being an important mediator of ethanol-associated behaviors. We took a genetic approach to this question by measuring ethanol consumption, intoxication, and hypothermia in mice with a targeted disruption of the dopamine β-hydroxylase gene, which is required for NE synthesis.Dbh −/− mice completely lack NE and have been useful in determining many critical functions of NE in vivo. These include roles predicted by pharmacology such as cardiovascular function (Cho et al., 1999), smooth muscle contraction, and brown fat thermogenesis (Thomas and Palmiter, 1997a), and novel roles such as embryonic development (Thomas et al., 1995), maternal behavior (Thomas et al., 1997b), and immune function (Alaniz et al., 1999).
MATERIALS AND METHODS
Animals. Dbh knockout mice, maintained on a 129/SvEv and C57BL/6J hybrid background, were developed and generated as described (Thomas et al., 1995, 1998). Dbh −/− males that were rescued for their fertility defect byl-threo-3,4-dihydroxyphenylserine (DOPS) were bred to Dbh +/− females, and 9th-12th generation littermates between 3 and 6 months of age were used in all experiments. Results were similar across generations.
Dbh −/− mice were identified by the delayed growth and ptosis phenotype, which perfectly correlates with the Dbh−/− genotype (data not shown). A subset of genotypes was confirmed by PCR. Dbh +/− mice have normal levels of epinephrine and NE, and they are indistinguishable from wild-type littermates for all previously tested behaviors (Tafari et al., 1997; Thomas and Palmiter, 1997a,b; Thomas et al., 1998; Alaniz et al., 1999; our unpublished data). Therefore, heterozygous (Dbh +/−) littermates were used as controls for all experiments in this study.
Experimental protocols were approved by the Animal Care Committee at the University of Washington and meet the guidelines of the American Association for Accreditation of Laboratory Animal Care.
Ethanol-induced hypothermia. Dbh +/− and Dbh−/− mice of both sexes were anesthetized with 425 mg/kg of a 2.5% tribromoethanol solution (Sigma, St. Louis, MO), and transmitters (Mini-Mitter Company, Sunriver, OR) were surgically implanted into the peritoneal cavity. Mice were allowed to recover for 7 d before body temperature measurement. Body temperature was recorded every 5 min by an ER-4000 Energizer/Receiver via radio telemetry. Experiments were separated by at least 7 d. To measure hypothermia to acute ethanol administration, body temperature was measured before and after a 3 gm/kg ethanol injection, at either 22 or 30°C. To measure changes in body temperature during oral consumption of ethanol, it was recorded over a 12 hr light/dark cycle at room temperature (22°C) in response to ingestion of a 3% (w/v) ethanol solution available ad libitum as the only fluid source.
Ethanol-induced sedation. Naïve male and female mice were either untreated or injected with 2 mg/ml ascorbic acid. Five hours later, mice were injected with 3 gm/kg (i.p.) of a filter-sterilized solution containing 20% (w/v) ethanol and 0.9% NaCl. Mice were placed on their backs in a plastic U-shaped trough until they lost their righting reflex. The number of minutes it took for a mouse to right itself on all four paws, three times in 30 sec, was recorded as latency to regain righting reflex. There were no sex differences or differences between untreated and ascorbic acid-injected animals, and these groups were combined. This experiment was performed at both 22 and 30°C. For the DOPS rescue, mice received 1 mg/gm DOPS, 0.125 mg/gm S-(−)-carbidopa (ICN Biomedicals, Aurora, OH), and 2 mg/ml ascorbic acid subcutaneously 5 hr before ethanol injection. Solutions were made in 0.2 m HCl and neutralized with NaOH just before injection. Data were analyzed with the Wilcoxon–Mann–Whitney U test using a Bonferroni correction for multiple pairwise comparisons.
Ethanol preference test. Throughout the experiment, fluid intake, food intake, and body weight were measured daily. Because of the increased metabolic rate of Dbh −/− mice (Thomas and Palmiter, 1997a), breeder chow (Purina 5015 Irradiated Mouse Diet, Animal Specialties, Hubbard, OR) was provided ad libitum to discourage calorically driven ethanol intake during this study. TwelveDbh +/− and 12 Dbh −/− naïve male mice were housed individually and given continuous access to two water bottles. The first bottle always contained water, and the second bottle contained the following solutions in the order listed: water (8 d), 1.7% sucrose (2 d), 4.25% sucrose (2 d), 0.03 mm quinine (2 d), 0.1 mmquinine (2 d), water (6 d), 3% ethanol (8 d), 6% ethanol (8 d), and 10% ethanol (8 d). The positions of the bottles were changed every day to control for side preferences. In a separate study, six maleDbh +/−, six male Dbh −/−, six femaleDbh +/−, and six female Dbh −/− naïve mice were subjected to the same paradigm without the sucrose and quinine. No differences were found between the male mice in the two experiments, and results were combined. Data were analyzed by comparing the average daily intake of Dbh +/− with Dbh−/− animals at each ethanol concentration with the Wilcoxon–Mann–Whitney U test.
Ethanol metabolism. Male and female mice were injected intraperitoneally with ethanol [3 gm/kg; 20% (w/v) in 0.9% NaCl solution and filtered]. Trunk blood was collected 1 or 3 hr later in heparinized tubes and centrifuged, and plasma was frozen on dry ice and stored at −80° until assayed. Blood ethanol levels were measured spectrophotometrically with an alcohol (ethanol) kit (Sigma). Two measurements were taken for each sample, and the results were averaged. No sex differences were found, and results were combined. Data were analyzed by Student's t tests.
Conditioned taste aversion. Dbh −/− and Dbh +/−males were put on a limited access drinking schedule, where water was available only from 8:00–8:30 A.M. and 4:00–5:00 P.M. Food was available ad libitum. On the first conditioning day, a 0.15% saccharin solution was given instead of water during the morning drinking session. Saccharin consumption was measured after 30 min, and mice in the “paired” groups were injected with either ethanol (2 gm/kg, i.p.) or 0.15 m LiCl (20 ml/kg, i.p.). Mice in the “unpaired” groups were injected with saline. The next day, water was given during the morning drinking session, and unpaired mice were injected with ethanol or LiCl, and paired groups were injected with saline (“reverse injections”). Thus, all mice received identical treatments, except that only the paired groups experienced the pairing of the novel taste of saccharin with the ethanol or LiCl. Water was given during the morning drinking session on days 3 and 4. On day 5, the first test day, saccharin was given during the morning session and consumption was measured and compared with preconditioning saccharin consumption. Because no differences were found for either the paired or unpaired groups, the paired groups were injected with ethanol or LiCl again, and the first test day also served as the second conditioning day. Water and reverse injections were given on day 6. Saccharin consumption was measured on day 7, the second test day, and all paired groups showed similar and significant aversions. However, because the unpaired ethanol groups also showed aversions, ethanol and LiCl were administered to the paired groups for a third time. On day 8, water and reverse injections were given. All groups were given 3 d of rest before the final test on day 12. At this time, all paired groups showed aversions, whereas none of the unpaired groups did.
To measure aversion extinction, all mice were given access to both water and saccharin ad libitum. Intake of each fluid was measured daily, and saccharin preference ratios were calculated by dividing the amount of saccharin solution consumed by the total amount of fluid consumed. After 10 d, the water was removed, and saccharin was available ad libitum as the only fluid source to force aversion extinction. Data were analyzed by ANOVA followed by Student Newman–Keuls post hoc tests.
RESULTS
Ethanol-induced hypothermia is more severe in Dbh−/− mice
To determine the effect of NE deficiency on ethanol-induced hypothermia, Dbh −/− mice and littermate controls were implanted with temperature monitors, and core body temperature was measured in response to an injection of ethanol (3 gm/kg, i.p.). Body temperature averaged over 60 min before ethanol injection inDbh −/− mice was slightly higher than controls at 22°C (Dbh −/−, 37.11 ± 0.07°; Dbh +/−, 36.21° ± 0.05°) and 30°C (Dbh −/−, 37.43° ± 0.82°; Dbh +/−, 36.47° ± 0.27°). When ethanol was administered at 22°C, Dbh −/− mice experienced prolonged hypothermia (∼6 hr) and an approximately sixfold greater decrease in body temperature than controls (Fig.1A,C). Presumably as a direct result of the decreased core body temperature, ethanol was metabolized more slowly in Dbh −/− mice (Fig.2A). When this experiment was repeated at thermoneutrality (30°C), there were no gross differences between genotypes in the average hypothermic effect of ethanol (Fig. 1B,C) or ethanol metabolism (Fig. 2B). These results demonstrate that the increased ethanol-induced hypothermia and lower rate of ethanol metabolism in Dbh −/− mice are dependent on ambient temperature.
Fig. 1.
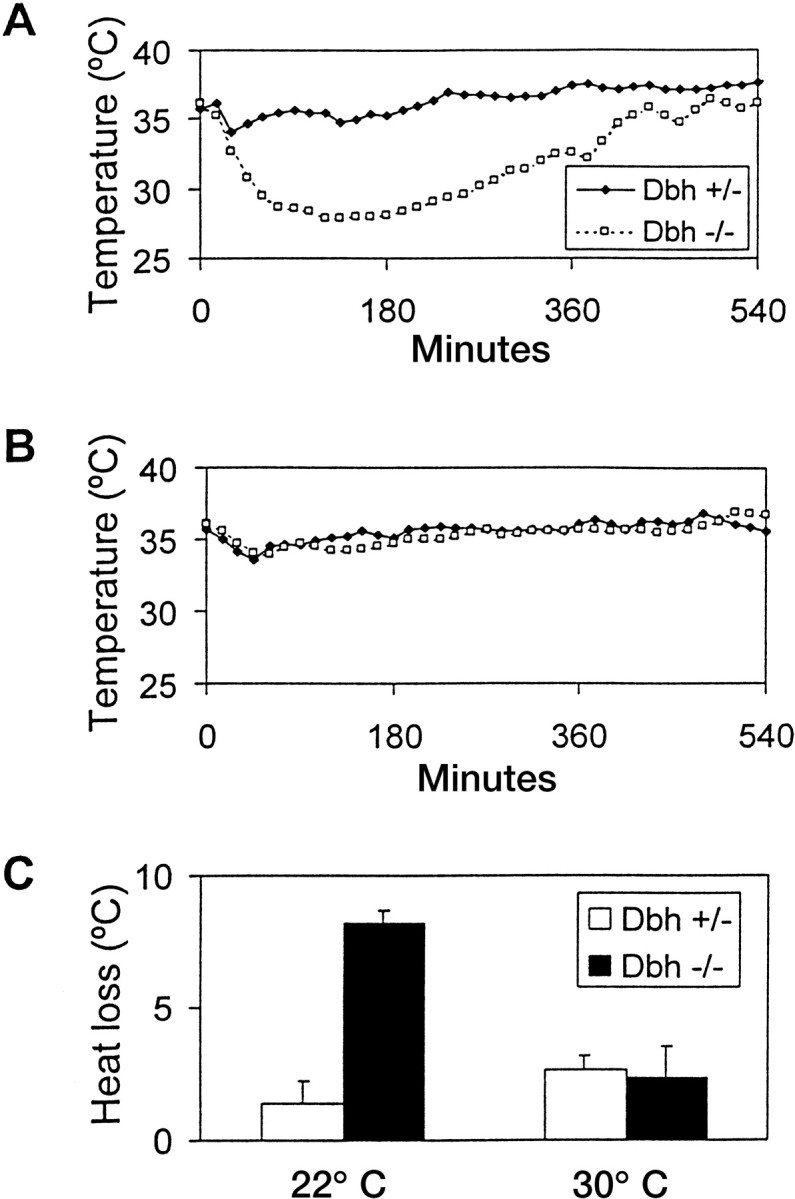
Ethanol-induced changes in body temperature. Representative hypothermic response to acute administration of 3 gm/kg ethanol at (A) 22° ambient temperature and (B) 30° ambient temperature. Each data point is an average of three 5 min measurements. C, Mean hypothermic response at 22° (Dbh +/−,n = 4; Dbh −/−,n = 3) and 30° (Dbh +/−,n = 3; Dbh −/−,n = 3). No sex differences were found, and results were combined.
Fig. 2.

Ethanol metabolism. Serum ethanol concentration 1 and 3 hr after an acute injection of 3 gm/kg ethanol at (A) 22° ambient temperature and (B) 30° ambient temperature (n = 6 for each genotype at each time point). *p < 0.05.
Dbh −/− mice are hypersensitive to ethanol-induced sedation
The role of NE in modulating ethanol sedation was tested by measuring the latency to regain righting reflex after an acute intraperitoneal injection of 3 gm/kg ethanol. At 22°C, Dbh−/− mice took more than three times as long to regain righting reflex than controls (Fig. 3A). To rule out the possibility that this phenotype was a secondary effect of increased hypothermia and decreased metabolism, the experiment was repeated at 30°C. Despite no difference in heat loss or ethanol metabolism at this temperature (Figs. 1B,2B), Dbh −/− mice were still unconscious almost three times as long as controls (Fig. 3B), demonstrating that NE is critical for antagonizing ethanol-induced sedation independent of its role in temperature homeostasis and metabolic rate. No sex differences were observed (data not shown).
Fig. 3.
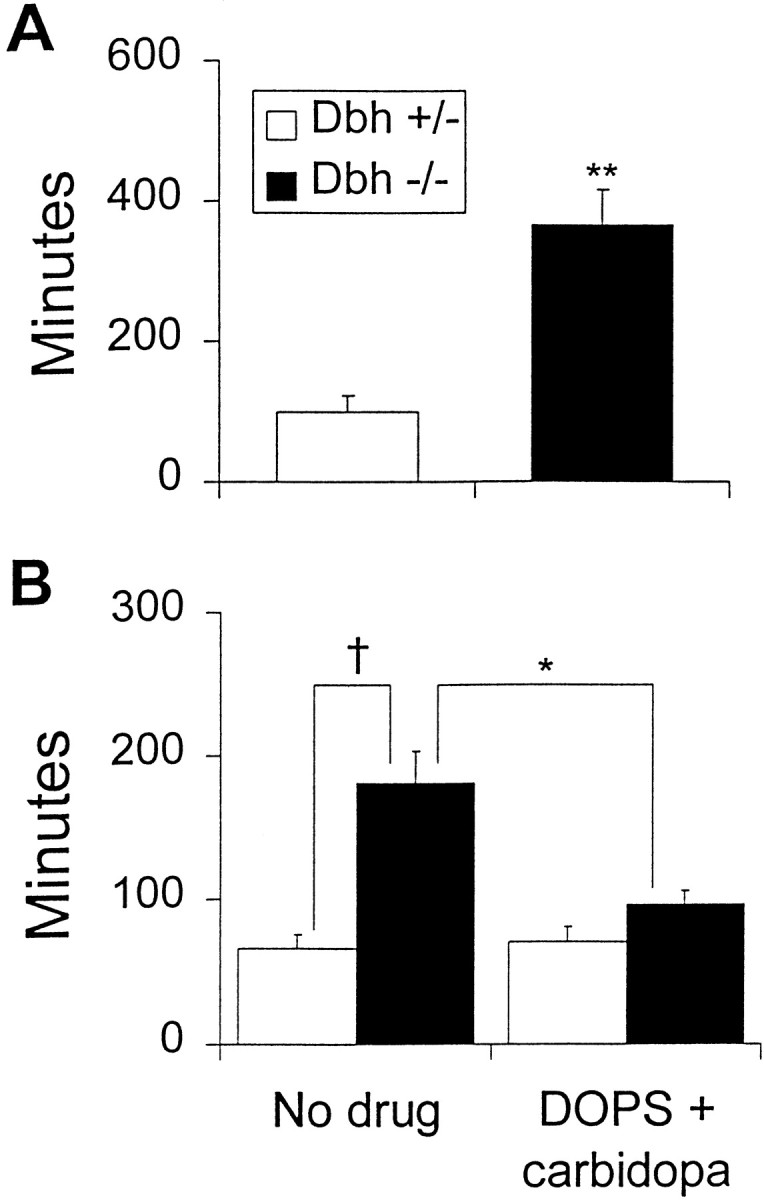
Ethanol-induced sedation. Latency to regain righting reflex after an acute injection of 3 gm/kg ethanol at (A) 22° ambient temperature (Dbh+/−, n = 6; Dbh −/−,n = 6) and (B) 30° ambient temperature (Dbh +/−, n = 18;Dbh −/−, n = 20). DOPS (1 mg/gm) + carbidopa (0.125 mg/gm) (Dbh +/−, n= 13; Dbh −/−, n = 12) was administered 5 hr before ethanol injection. *p < 0.05; **p < 0.01; † p < 0.0001.
To determine whether restoring NE centrally to Dbh −/− mice could reduce the sedation hypersensitivity, DOPS and carbidopa were administered 5 hr before ethanol injection. DOPS can be converted to NE by l-aromatic amino acid decarboxylase (AADC), thus bypassing the requirement for DBH, and carbidopa is an inhibitor of AADC that cannot cross the blood–brain barrier. Similar paradigms restore central but not peripheral NE levels to between 11 and 26% of wild-type levels (Thomas and Palmiter, 1997b; Thomas et al., 1998). The increased latency to regain righting reflex in knockouts was rescued by DOPS + carbidopa treatment (Fig.2B). This result demonstrates that a central NE deficiency in Dbh −/− mice is responsible for the hypersensitivity to ethanol-induced sedation.
Dbh −/− males have reduced ethanol preference
We measured ethanol consumption in males using a two-bottle preference paradigm. At the beginning of the experiment, control mice were larger than Dbh −/− mice. However, during the course of the experiment, Dbh −/− mice ate more food, drank more water and total fluid, and gained more weight than controls and were of comparable weight by the end of the study (Fig.4). Male Dbh −/− mice had a reduced ethanol preference at all ethanol concentrations tested (Fig.5A). Despite having an increase in other ingestive behaviors (food and fluid consumption),Dbh −/− males consumed less ethanol at the 6 and 10% concentrations (Fig. 5B). Preliminary results with a small group of Dbh −/− females suggest that they have no differences in ethanol preference compared with controls (data not shown).
Fig. 4.

Body weight and consumption measurements during preference study. A, Body weight before start of preference study. B, Body weight after conclusion of preference study. C, Weight gain during preference study calculated as a percentage of starting weight. D, Average food consumption over course of preference study.E, Average water consumption over course of preference study. F, Average total fluid (water + ethanol) over course of preference study. G, Sucrose preference ratio (weight of sucrose solution consumed/weight of total fluid consumed).H, Quinine preference ratio (weight of quinine solution consumed/weight of total fluid consumed). *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
Fig. 5.
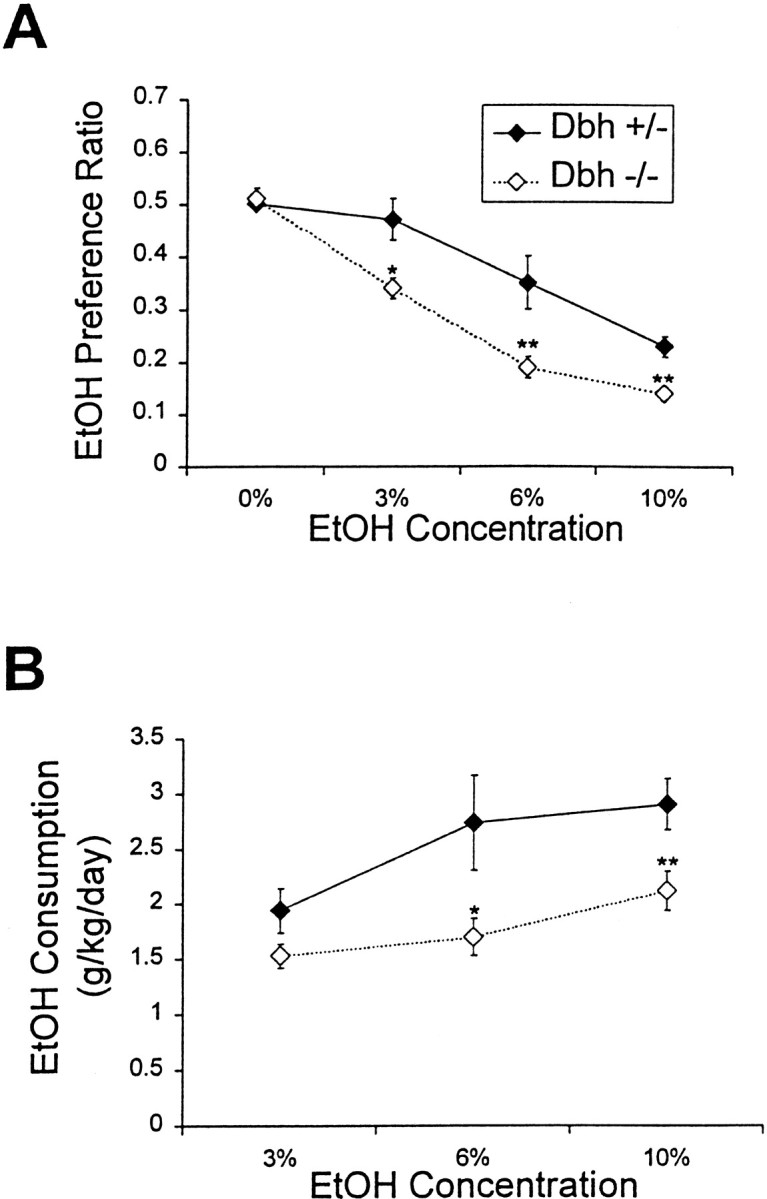
Ethanol preference and consumption.A, Ethanol preference ratio (weight of ethanol consumed/weight of total fluid consumed) of males (Dbh+/−, n = 18; Dbh −/−,n = 18). B, Male ethanol consumption calculated as grams of ethanol per kilogram of body weight. *p < 0.05; **p < 0.01.
To rule out differences in taste preference, mice were tested for their preference for sweet (sucrose) and bitter (quinine) solutions (Fig. 4). There were no differences between genotypes in preference for either compound, demonstrating that the reduced ethanol preference and consumption in Dbh −/− males is not generalized to all tastants or related to caloric value.
Because ethanol-induced hypothermia was more severe in Dbh−/− mice, it was possible that the decreased consumption was secondary to a reduction in body temperature. We tested this hypothesis by measuring body temperature over 12 hr when the mice were given only ethanol to drink. Dbh −/− mice experienced no hypothermia despite consuming more ethanol than animals drank during the preference study (Fig. 6) (data not shown). We conclude that the decreased voluntary ethanol consumption inDbh −/− males is independent of the hypothermic effect.
Fig. 6.

Body temperature during voluntary ethanol consumption. Shown is measurement of body temperature over 12 hr with 3% ethanol the only fluid available (Dbh +/−,n = 2; Dbh −/−,n = 2).
Extinction of a conditioned taste aversion to ethanol is delayed inDbh −/− males
Because ethanol has both rewarding and aversive effects, we reasoned that Dbh −/− males drink less ethanol either because they find it less rewarding or because they find it more aversive. We used a conditioned taste aversion paradigm to test the latter hypothesis. During the conditioning sessions, a novel taste (saccharin) was paired with either a 2 gm/kg ethanol injection (paired groups) or a saline injection (unpaired groups). LiCl was also paired with saccharin. If the aversive effects of the ethanol or LiCl injection are associated with the taste of saccharin, animals should drink less saccharin during subsequent sessions. Saccharin consumption was measured after the conditioning sessions and was compared with preconditioning saccharin consumption levels. Dbh +/− andDbh −/− males in the ethanol- and LiCl-paired groups required two conditioning sessions to establish an aversion and displayed a comparable reduction in saccharin consumption after three conditioning sessions (Fig.7A) (data not shown). No differences in preconditioning or postconditioning saccharin consumption were manifested in the unpaired groups. To assess extinction of taste aversion, saccharin and water consumption were measured in a two-bottle preference paradigm. Dbh +/− males in the paired ethanol group reached a steady-state extinction after 3 d (i.e., showed no preference for water over saccharin).Dbh −/− males took 6 d to achieve similar amounts of extinction (Fig. 7B). No differences were found between genotypes in the paired LiCl groups (Fig. 7C). After 10 d, some paired mice of each genotype fully or partially extinguished the aversion, whereas others failed to do so at all. The unpaired groups of either genotype manifested a preference for saccharin, in that ∼80% of fluid consumed was from the saccharin bottle throughout the experiment. To force extinction in the paired animals so that saccharin consumption was comparable to that of the unpaired groups, the water bottle was removed and saccharin was the only fluid available. Within 1 d, all paired groups were consuming amounts of saccharin comparable to the unpaired groups, demonstrating that all mice were capable of extinguishing the taste aversion (data not shown).
Fig. 7.

Conditioned taste aversion. A, Percentage of preconditioning saccharin solution consumed after the third conditioning trial (n = 6 for each group except for EtOH unpaired Dbh −/−,n = 4). Saccharin preference ratio (weight of saccharin solution consumed/weight of total fluid consumed) after the third conditioning trial for (B) ethanol-paired mice and (C) LiCl-paired mice. *p < 0.05.·
DISCUSSION
Ethanol-induced hypothermia
A decrease in body temperature in response to acute administration of ethanol has been well documented in rodents (Nikki et al., 1971;Lomax et al., 1980) and is evident in humans under conditions of low ambient temperature and strenuous activity (for review, see Pohorecky and Brick, 1987). There is also support for the notion that ethanol plays a role in deaths resulting from accidental hypothermia (Weyman et al., 1974; Hirvonen, 1979). We show that the magnitude and duration of ethanol-induced hypothermia are augmented in mice lacking NE. Previous studies have described the inability of Dbh −/− mice to regulate their body temperature in cold environments because of impaired vasoconstriction and inability to activate brown fat thermogenesis (Thomas and Palmiter, 1997b). Because brown fat thermogenesis is not involved in ethanol-induced hypothermia (Huttunen et al., 1998) and ethanol facilitates vasodilation (Gillespie, 1967;Ritchie, 1980), we hypothesize that the hypersensitivity ofDbh −/− mice to ethanol-induced hypothermia is primarily a result of defective heat conservation rather than an absence of heat production. It is also possible that depletion of hypothalamic NE in the Dbh −/− mice has compromised central temperature regulation and contributes to the increased hypothermia.
Previous studies have shown that Dbh −/− mice have a slightly lower body temperature than controls (Thomas and Palmiter, 1997a), whereas we observed a slightly elevated body temperature. A difference in temperature measurement (single rectal measurements vs continuous measurements from the intraperitoneal cavity) could account for this difference.
Ethanol-induced sedation
To our knowledge, the nearly threefold difference betweenDbh −/− and Dbh +/− animals in sedation sensitivity is the largest described to date. For example, rats bred for ethanol preference regain their righting reflex about twice as quickly as rats bred for nonpreference (Kurtz et al., 1996), and NPY −/− mice differ from controls by only ∼25% (Thiele et al., 1998). There exists a close parallel between the effects of ethanol on neurons of the locus coeruleus (LC), the primary noradrenergic brain nucleus, and on behavior. NE turnover and locomotor activity are increased after low doses of ethanol, whereas high doses of ethanol decrease NE turnover and are sedative (for review, see Pohorecky, 1977; Pohorecky and Brick, 1987). Because activation of the LC is associated with consciousness and arousal (Jouvet, 1969; Hobson et al., 1975;Aston-Jones and Bloom, 1981; Robbins, 1984), it is possible that the behavioral effects of ethanol are mediated in part by its effects on LC activity and NE release. Our result that Dbh −/− mice that lack NE are hypersensitive to ethanol-induced sedation is consistent with this idea.
Evidence from both Drosophila and vertebrates has implicated cAMP signaling in modulating ethanol intoxication (for review, seeBellen, 1998). The β1, β2, and α2adrenoreceptors are G-protein-coupled receptors that regulate cAMP production through adenylate cyclase. The sedation phenotype ofDbh −/− mice could be explained by a deficiency in this signaling pathway, suggesting that ethanol sedation is similarly controlled by cAMP in this model.
DOPS rescue of sedation hypersensitivity
The ability of DOPS to restore normal sedation sensitivity toDbh −/− mice is critical because it addresses four caveats of our experimental design. Dbh is required to convert dopamine (DA) to NE. Consequently, the Dbh −/− mice produce DA instead of NE in noradrenergic neurons, albeit in very small quantities (Thomas et al., 1998); thus, any phenotype we observe could be the result of ectopic DA production. DOPS is converted to NE by AADC, thus bypassing DBH and leaving the ectopic DA unaffected (Thomas et al., 1998). The efficacy of DOPS in this experiment demonstrates that the sedation hypersensitivity is a result of NE deficiency rather than ectopic DA production. Because our mice are a hybrid of two strains with different ethanol sensitivities, it is possible that strain background effects or Dbh-linked genes cause the altered ethanol response rather than NE. This hypothesis is ruled out by the observation that DOPS rescues sedation hypersensitivity inDbh −/− mice without affecting the performance of controls in this assay. The ability of a single injection of DOPS to rescue the sedation phenotype of Dbh −/− adults also demonstrates that this phenotype is caused by an acute requirement for NE and not a defect with a developmental basis. Carbidopa, an inhibitor of AADC that cannot cross the blood–brain barrier, was included to show that the rescue, and therefore the sedation hypersensitivity, is a central effect of ethanol. Dbh −/− mice lack epinephrine in addition to norepinephrine (Thomas et al., 1995). Because DOPS + carbidopa treatment partially restores both catecholamines (Thomas et al., 1998), our experiments cannot distinguish between a requirement for either transmitter in ethanol-related behaviors. However, the amount of epinephrine produced in the CNS is very small relative to NE. Because of technical constraints, we were unable to test whether DOPS + carbidopa treatment could rescue the reduced ethanol preference inDbh −/− mice. However, because nearly all previously described phenotypes of Dbh −/− mice can be rescued by DOPS treatment (Thomas et al., 1995; Thomas and Palmiter, 1997a; Thomas et al., 1998), we suspect that the ethanol consumption phenotype is also caused by an acute lack of central NE.
Ethanol preference and consumption
Despite dozens of studies over the last 30 years suggesting a role for NE in ethanol consumption, NE has been conspicuously absent from recent reviews on ethanol neurobiology (Diamond and Gordon, 1997;McBride and Li, 1998). Perhaps this is because of a lack of consensus produced by studies with conflicting results (see introductory remarks). Our results clearly demonstrate that NE influences normal ethanol consumption in male mice. The reduced ethanol consumption in male Dbh −/− mice is striking considering that other ingestive behaviors (food and water intake) were increased by the lack of NE. Previous work demonstrated that Dbh −/− mice have an increased metabolic rate, which could account for the increased food and water intake (Thomas and Palmiter, 1997b).
It has recently been shown that NPY levels are inversely related to ethanol consumption (Thiele et al., 1998). Because a subset of noradrenergic neurons uses NPY as a cotransmitter, it is possible that the decreased ethanol intake in Dbh −/− mice is a direct result of a compensatory upregulation of NPY in noradrenergic neurons. However, an examination of NPY levels in Dbh −/− mice using mRNA in situ hybridization and RT-PCR argues against this hypothesis (data not shown).
Sensitivity to and preference for ethanol are inversely related (Kurtz et al., 1996; Thiele et al., 1998), and this is also true for Dbh −/− males. Therefore, it is surprising that Dbh −/− females show no decrease in ethanol preference yet still display the sedation hypersensitivity, although more animals will need to be tested to confirm this result. Genetic analyses of mouse strains that differ in ethanol preference (Melo et al., 1996; Peirce et al., 1998) and ethanol sedation (Bennett and Johnson, 1998) have revealed a number of sex-specific loci.
Ethanol aversion
One reason that Dbh −/− males consume less ethanol may be that NE is important for attenuating the aversive effects of ethanol. The locus coeruleus is activated in response to aversive stimuli, and it was the only brain region that showed a consistent difference in c-fos immunoreactivity between genetically high and low ethanol-preferring rats after acute ethanol administration (Thiele et al., 1997). Ethanol-induced sedation and hypothermia could be considered aversive, and the Dbh −/− mice are exquisitely sensitive to these effects. NE-deficient mice were delayed in the extinction of an ethanol-paired conditioned taste aversion compared with controls. No differences between genotypes were found with a LiCl-paired conditioned taste aversion, demonstrating that Dbh −/− mice do not have a general extinction defect using this paradigm. The number of trials required to establish the ethanol-paired aversion and the extent of the aversion were similar between genotypes. The delayed extinction of the conditioned taste aversion supports the idea that Dbh −/− males drink less ethanol because they find it more aversive. However, this interpretation must be made with caution because the phenotype is relatively subtle and the doses of ethanol administered for the conditioned taste aversion are much greater than the amount consumed voluntarily.
NE and ethanol-associated reward
Another possible explanation for the decreased ethanol consumption by Dbh −/− males is that NE is important for mediating the rewarding effects of ethanol. DA has received the bulk of the attention regarding the rewarding effects of drugs of abuse, including ethanol (Wise, 1980; Koob et al., 1998). Electrophysiological, pharmacological, and genetic experiments have established a clear role for DA in voluntary ethanol consumption (Koob et al., 1994; El-Ghundi et al., 1998; Phillips et al., 1998). However, it has been suggested that NE, and not DA, is critical for ethanol reinforcement (Amit and Brown, 1982). Support for this hypothesis comes from studies demonstrating that (1) ethanol has a greater effect on NE turnover and release than DA (Corrodi et al., 1966; Hunt and Majchrowicz, 1974); (2) chemical lesioning of the NE system but not the DA system modulates voluntary ethanol intake (Myers and Melchior, 1975; Kiianmaa et al., 1979;Rassnick et al., 1993); and (3) blocking the conversion of DA to NE via DBH inhibitors attenuates the positive reinforcing effects of ethanol (Brown et al., 1977). One way to reconcile these data is to emphasize that the NE and DA systems need not be disassociated (Wise and Bozarth, 1985). NE modulates the activity of dopaminergic cells in the substantia nigra and ventral tegmental area (Grenhoff et al., 1993) and DA release in the nucleus accumbens (Lategan et al., 1990). Thus, NE could influence ethanol intake via modulation of DA release or in parallel to DA.
Footnotes
D.W., N.C.R., and N.S.M. were supported by the Howard Hughes Medical Institute. We thank Todd Thiele, Don Marsh, Linda Ste. Marie, William Alynick, Suzanne Weinshenker, and Michelle Brot for technical and intellectual input, and Doug Kim and Mark Szczypka for critical reading of this manuscript. We thank Ted Young for the use of his 30°C room and Sumitomo Pharmaceuticals for the generous donation of DOPS.
Correspondence should be addressed to Dr. Richard D. Palmiter, Howard Hughes Medical Institute, UW Biochemistry, Box 357370, University of Washington, Seattle, WA 98195. E-mail:palmiter@u.washington.edu.
REFERENCES
- 1.Alaniz RC, Thomas SA, Perez-Melgosa M, Mueller K, Farr AG, Palmiter RD, Wilson CB. Dopamine β-hydroxylase deficiency impairs cellular immunity. Proc Natl Acad Sci USA. 1999;96:2274–2278. doi: 10.1073/pnas.96.5.2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amit Z, Brown ZW. Actions of drugs of abuse on brain reward systems: a reconsideration with specific attention to alcohol. Pharmacol Biochem Behav. 1982;17:233–238. doi: 10.1016/0091-3057(82)90075-2. [DOI] [PubMed] [Google Scholar]
- 3.Amit Z, Brown ZW, Levitan DE, Ogren SO. Noradrenergic mediation of the positive reinforcing properties of ethanol: I. Suppression of ethanol consumption in laboratory rats following dopamine-beta-hydroxylase inhibition. Arch Int Pharmacodyn Ther. 1977;230:65–75. [PubMed] [Google Scholar]
- 4.Andreas K, Fischer HD, Schmidt J. Effect of central effective substances on alcohol preference. Biomed Biochim Acta. 1983;42:391–398. [PubMed] [Google Scholar]
- 5.Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aston-Jones G, Foote SL, Bloom FE. Low doses of ethanol disrupt sensory responses of brain noradrenergic neurones. Nature. 1982;296:857–860. doi: 10.1038/296857a0. [DOI] [PubMed] [Google Scholar]
- 7.Bellen HJ. The fruit fly: a model organism to study the genetics of alcohol abuse and addiction? Cell. 1998;93:909–912. doi: 10.1016/s0092-8674(00)81195-2. [DOI] [PubMed] [Google Scholar]
- 8.Bennett B, Johnson TE. Development of congenics for hypnotic sensitivity to ethanol by QTL-marker-assisted counter selection. Mamm Genome. 1998;9:969–974. doi: 10.1007/s003359900908. [DOI] [PubMed] [Google Scholar]
- 9.Brown ZW, Amit Z, Levitan DE, Ogren SO, Sutherland EA. Noradrenergic mediation of the positive reinforcing properties of ethanol: II. Extinction of ethanol-drinking behavior in laboratory rats by inhibition of dopamine-beta-hydroxylase. Implications for treatment procedures in human alcoholics. Arch Int Pharmacodyn Ther. 1977;230:76–82. [PubMed] [Google Scholar]
- 10.Carlsson A, Lindqvist M. Effect of ethanol on the hydroxylation of tyrosine and tryptophan in rat brain in vivo. J Pharm Pharmacol. 1973;25:437–440. doi: 10.1111/j.2042-7158.1973.tb09129.x. [DOI] [PubMed] [Google Scholar]
- 11.Cho M-C, Rao M, Kock WJ, Thomas SA, Palmiter RD, Rockman HA. Enhanced contractility and decreased β-adrenergic receptor kinase-1 in mice lacking endogenous norepinephrine and epinephrine. Circulation. 1999;99:2702–2707. doi: 10.1161/01.cir.99.20.2702. [DOI] [PubMed] [Google Scholar]
- 12.Corrodi H, Fuxe K, Hokfelt T. The effect of ethanol on the activity of central catecholamine neurones in rat brain. J Pharm Pharmacol. 1966;18:821–823. doi: 10.1111/j.2042-7158.1966.tb07817.x. [DOI] [PubMed] [Google Scholar]
- 13.Daoust M, Protais P, Ladure P. Noradrenergic system: effect of DSP4 and FLA-57 on ethanol intake in ethanol preferring rats. Pharmacol Biochem Behav. 1990;36:133–137. doi: 10.1016/0091-3057(90)90138-8. [DOI] [PubMed] [Google Scholar]
- 14.De Montigny C, Wang RY, Reader TA, Aghajanian GK. Monoaminergic denervation of the rat hippocampus: microiontophoretic studies on pre- and postsynaptic supersensitivity to norepinephrine and serotonin. Brain Res. 1980;200:363–376. doi: 10.1016/0006-8993(80)90927-0. [DOI] [PubMed] [Google Scholar]
- 15.Diamond I, Gordon AS. Cellular and molecular neuroscience of alcoholism. Physiol Rev. 1997;77:1–20. doi: 10.1152/physrev.1997.77.1.1. [DOI] [PubMed] [Google Scholar]
- 16.El-Ghundi M, George SR, Drago J, Fletcher PJ, Fan T, Nguyen T, Liu C, Sibley DR, Westphal H, O'Dowd BF. Disruption of dopamine D1 receptor gene expression attenuates alcohol-seeking behavior. Eur J Pharmacol. 1998;353:149–158. doi: 10.1016/s0014-2999(98)00414-2. [DOI] [PubMed] [Google Scholar]
- 17.Everitt BJ, Hokfelt T, Terenius L, Tatemoto K, Mutt V, Goldstein M. Differential co-existence of neuropeptide Y (NPY)-like immunoreactivity with catecholamines in the central nervous system of the rat. Neuroscience. 1984;11:443–462. doi: 10.1016/0306-4522(84)90036-8. [DOI] [PubMed] [Google Scholar]
- 18.Gillespie JA. Vasodilator properties of alcohol. Br Med J. 1967;2:274–277. doi: 10.1136/bmj.2.5547.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grenhoff J, Nisell M, Ferre S, Aston-Jones G, Svensson TH. Noradrenergic modulation of midbrain dopamine cell firing elicited by stimulation of the locus coeruleus in the rat. J Neural Transm Gen Sect. 1993;93:11–25. doi: 10.1007/BF01244934. [DOI] [PubMed] [Google Scholar]
- 20.Grupp LA, Sneddon B, Solway E, Perlanski E, Stewart RB. The beta adrenergic agonist isoproterenol suppresses voluntary alcohol intake in rats. Pharmacol Biochem Behav. 1989;33:493–495. doi: 10.1016/0091-3057(89)90537-6. [DOI] [PubMed] [Google Scholar]
- 21.Hirvonen J. Accidental hypothermia. In: Lomax, Schonbaum, editors. Body temperature: regulation, drug effects and therapeutic implications. Dekker; New York: 1979. pp. 561–585. [Google Scholar]
- 22.Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. 1975;189:55–58. doi: 10.1126/science.1094539. [DOI] [PubMed] [Google Scholar]
- 23.Hunt WA, Majchrowicz E. Alterations in the turnover of brain norepinephrine and dopamine in alcohol-dependent rats. J Neurochem. 1974;23:549–552. doi: 10.1111/j.1471-4159.1974.tb06058.x. [DOI] [PubMed] [Google Scholar]
- 24.Huttunen P, Sampi M, Myllyla R. Ethanol-induced hypothermia and thermogenesis of brown adipose tissue in the rat. Alcohol. 1998;15:315–318. doi: 10.1016/s0741-8329(97)00136-5. [DOI] [PubMed] [Google Scholar]
- 25.Jouvet M. Biogenic amines and the states of sleep. Science. 1969;163:32–41. doi: 10.1126/science.163.3862.32. [DOI] [PubMed] [Google Scholar]
- 26.Karoum F, Wyatt RJ, Majchrowicz E. Brain concentrations of biogenic amine metabolites in acutely treated and ethanol-dependent rats. Br J Pharmacol. 1976;56:403–411. doi: 10.1111/j.1476-5381.1976.tb07450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiianmaa K. Alcohol intake and ethanol intoxication in the rat: effect of a 6-OHDA-induced lesion of the ascending noradrenaline pathways. Eur J Pharmacol. 1980;64:9–19. doi: 10.1016/0014-2999(80)90364-7. [DOI] [PubMed] [Google Scholar]
- 28.Kiianmaa K, Fuxe K, Jonsson G, Ahtee L. Evidence for involvement of central NA neurones in alcohol intake. Increased alcohol consumption after degeneration of the NA pathway to the cortex cerebri. Neurosci Lett. 1975;1:41–45. doi: 10.1016/0304-3940(75)90010-5. [DOI] [PubMed] [Google Scholar]
- 29.Kiianmaa K, Andersson K, Fuxe K. On the role of ascending dopamine systems in the control of voluntary ethanol intake and ethanol intoxication. Pharmacol Biochem Behav. 1979;10:603–608. doi: 10.1016/0091-3057(79)90240-5. [DOI] [PubMed] [Google Scholar]
- 30.Koob GF, Rassnick S, Heinrichs S, Weiss F. Alcohol, the reward system and dependence. Experientia [Suppl] 1994;71:103–114. doi: 10.1007/978-3-0348-7330-7_11. [DOI] [PubMed] [Google Scholar]
- 31.Koob GF, Sanna PP, Bloom FE. Neuroscience of addiction. Neuron. 1998;21:467–476. doi: 10.1016/s0896-6273(00)80557-7. [DOI] [PubMed] [Google Scholar]
- 32.Kurtz DL, Stewart RB, Zweifel M, Li TK, Froehlich JC. Genetic differences in tolerance and sensitization to the sedative/hypnotic effects of alcohol. Pharmacol Biochem Behav. 1996;53:585–591. doi: 10.1016/0091-3057(95)02055-1. [DOI] [PubMed] [Google Scholar]
- 33.Lategan AJ, Marien MR, Colpaert FC. Effects of locus coeruleus lesions on the release of endogenous dopamine in the rat nucleus accumbens and caudate nucleus as determined by intracerebral microdialysis. Brain Res. 1990;523:134–138. doi: 10.1016/0006-8993(90)91646-x. [DOI] [PubMed] [Google Scholar]
- 34.Lomax P, Bajorek JG, Chesarek WA, Chaffee RR. Ethanol-induced hypothermia in the rat. Pharmacology. 1980;21:288–294. doi: 10.1159/000137443. [DOI] [PubMed] [Google Scholar]
- 35.McBride WJ, Li TK. Animal models of alcoholism: neurobiology of high alcohol-drinking behavior in rodents. Crit Rev Neurobiol. 1998;12:339–369. doi: 10.1615/critrevneurobiol.v12.i4.40. [DOI] [PubMed] [Google Scholar]
- 36.Melander T, Hokfelt T, Rokaeus A. Distribution of galanin-like immunoreactivity in the rat central nervous system. J Comp Neurol. 1986;248:475–517. doi: 10.1002/cne.902480404. [DOI] [PubMed] [Google Scholar]
- 37.Melchior CL, Myers RD. Genetic differences in ethanol drinking of the rat following injection of 6-OHDA, 5,6-DHT or 5,7-DHT into the cerebral ventricles. Pharmacol Biochem Behav. 1976;5:63–72. doi: 10.1016/0091-3057(76)90289-6. [DOI] [PubMed] [Google Scholar]
- 38.Melo JA, Shendure J, Pociask K, Silver LM. Identification of sex-specific quantitative trait loci controlling alcohol preference in C57BL/6 mice. Nat Genet. 1996;13:147–153. doi: 10.1038/ng0696-147. [DOI] [PubMed] [Google Scholar]
- 39.Myers RD, Melchior CL. Alcohol drinking in the rat after destruction of serotonergic and catecholaminergic neurons in the brain. Res Commun Chem Pathol Pharmacol. 1975;10:363–378. [PubMed] [Google Scholar]
- 40.Nikki P, Vapaatalo H, Karppanen H. Effect of ethanol on body temperature, postanaesthetic shivering and tissue monoamines in halothane-anaesthetized rats. Ann Med Exp Biol Fenn. 1971;49:157–161. [PubMed] [Google Scholar]
- 41.Peirce JL, Derr R, Shendure J, Kolata T, Silver LM. A major influence of sex-specific loci on alcohol preference in C57Bl/6 and DBA/2 inbred mice. Mamm Genome. 1998;9:942–948. doi: 10.1007/s003359900904. [DOI] [PubMed] [Google Scholar]
- 42.Phillips TJ, Brown KJ, Burkhart-Kasch S, Wenger CD, Kelly MA, Rubinstein M, Grandy DK, Low MJ. Alcohol preference and sensitivity are markedly reduced in mice lacking dopamine D2 receptors. Nat Neurosci. 1998;1:610–615. doi: 10.1038/2843. [DOI] [PubMed] [Google Scholar]
- 43.Pohorecky LA. Biphasic action of ethanol. Biobehav Rec. 1977;1:231–240. [Google Scholar]
- 44.Pohorecky LA, Brick J. Pharmacology of ethanol. Pharmacol Ther. 1987;36:335–427. doi: 10.1016/0163-7258(88)90109-x. [DOI] [PubMed] [Google Scholar]
- 45.Pohorecky LA, Jaffe LS. Noradrenergic involvement in the acute effects of ethanol. Res Commun Chem Pathol Pharmacol. 1975;12:433–447. [PubMed] [Google Scholar]
- 46.Rassnick S, Stinus L, Koob GF. The effects of 6-hydroxydopamine lesions of the nucleus accumbens and the mesolimbic dopamine system on oral self-administration of ethanol in the rat. Brain Res. 1993;623:16–24. doi: 10.1016/0006-8993(93)90004-7. [DOI] [PubMed] [Google Scholar]
- 47.Richardson JS, Novakovski DM. Brain monoamines and free choice ethanol consumption in rats. Drug Alcohol Depend. 1978;3:253–264. doi: 10.1016/0376-8716(78)90079-0. [DOI] [PubMed] [Google Scholar]
- 48.Ritchie JM. In: The pharmacological basis of therapeutics (Gilman AG, Goodman LS, Gilman A, eds), pp 376–380. Macmillan; New York: 1980. [Google Scholar]
- 49.Robbins TW. Cortical noradrenaline, attention and arousal. Psychol Med. 1984;14:13–21. doi: 10.1017/s0033291700003032. [DOI] [PubMed] [Google Scholar]
- 50.Segal DS, Geyer MA. Pre- and post-junctional supersensitivity: differentiation by intraventricular infusions of norepinephrine and methoxamine. Psychopharmacology. 1976;50:145–148. doi: 10.1007/BF00430483. [DOI] [PubMed] [Google Scholar]
- 51.Tafari AT, Thomas SA, Palmiter RD. Norepinephrine facilitates the development of the murine sweat response but is not essential. J Neurosci. 1997;17:4275–4281. doi: 10.1523/JNEUROSCI.17-11-04275.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thiele TE, van Dijk G, Bernstein IL. Ethanol-induced c-Fos expression in rat lines selected for low and high alcohol consumption. Brain Res. 1997;756:278–282. doi: 10.1016/s0006-8993(97)00228-x. [DOI] [PubMed] [Google Scholar]
- 53.Thiele TE, Marsh DJ, Ste. Marie L, Bernstein IL, Palmiter RD. Ethanol consumption and resistance are inversely related to neuropeptide Y levels. Nature. 1998;396:366–369. doi: 10.1038/24614. [DOI] [PubMed] [Google Scholar]
- 54.Thomas SA, Palmiter RD. Thermoregulatory and metabolic phenotypes of mice lacking noradrenaline and adrenaline. Nature. 1997a;387:94–97. doi: 10.1038/387094a0. [DOI] [PubMed] [Google Scholar]
- 55.Thomas SA, Palmiter RD. Impaired maternal behavior in mice lacking norepinephrine and epinephrine. Cell. 1997b;91:583–592. doi: 10.1016/s0092-8674(00)80446-8. [DOI] [PubMed] [Google Scholar]
- 56.Thomas SA, Matsumoto AM, Palmiter RD. Noradrenaline is essential for mouse fetal development. Nature. 1995;374:643–646. doi: 10.1038/374643a0. [DOI] [PubMed] [Google Scholar]
- 57.Thomas SA, Marck BT, Palmiter RD, Matsumoto AM. Restoration of norepinephrine and reversal of phenotypes in mice lacking dopamine β-hydroxylase. J Neurochem. 1998;70:2468–2476. doi: 10.1046/j.1471-4159.1998.70062468.x. [DOI] [PubMed] [Google Scholar]
- 58.Verbanck P, Seutin V, Dresse A, Scuvee J, Massotte L, Giesbers I, Kornreich C. Electrophysiological effects of ethanol on monoaminergic neurons: an in vivo and in vitro study. Alcohol Clin Exp Res. 1990;14:728–735. doi: 10.1111/j.1530-0277.1990.tb01235.x. [DOI] [PubMed] [Google Scholar]
- 59.Weyman AE, Greenbaum DM, Grace WJ. Accidental hypothermia in an alcoholic population. Am J Med. 1974;56:13–21. doi: 10.1016/0002-9343(74)90746-3. [DOI] [PubMed] [Google Scholar]
- 60.Wise RA. Action of drugs of abuse on brain reward systems. Pharmacol Biochem Behav [Suppl] 1980;13:213–223. doi: 10.1016/s0091-3057(80)80033-5. [DOI] [PubMed] [Google Scholar]
- 61.Wise RA, Bozarth MA. Actions of abused drugs on reward systems in the brain. In: Blum K, Manzo L, editors. Neurotoxicology. Dekker; New York: 1985. pp. 111–133. [Google Scholar]
- 62.Xu ZQ, Shi TJ, Hokfelt T. Galanin/GMAP- and NPY-like immunoreactivities in locus coeruleus and noradrenergic nerve terminals in the hippocampal formation and cortex with notes on the galanin-R1 and-R2 receptors. J Comp Neurol. 1998;392:227–251. doi: 10.1002/(sici)1096-9861(19980309)392:2<227::aid-cne6>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]