

Abstract
Systemic injection of the endotoxin lipopolysaccharide (LPS) upregulates the gene encoding CD14 early in the circumventricular organs (CVOs) and later in the brain parenchyma. The present study tested the hypothesis that the parenchymal production of the proinflammatory cytokine tumor necrosis factor α (TNF-α) by microglial cells is responsible for triggering CD14 transcription in an autocrine/paracrine loop-like manner. In a first set of experiments, Sprague Dawley rats were killed 1, 3, 6, and 12 hr after an intracerebroventricular administration of recombinant rat TNF-α or vehicle solution. Second, anti-rat TNF-α-neutralizing antibody or vehicle solution was administrated into the lateral ventricle 10 hr before an intraperitoneal injection of LPS. Central administration of the cytokine caused a strong induction of IκBα, TNF-α, and CD14 mRNA in parenchymal microglial cells. The hybridization signal for these transcripts was localized to the edge of the ventricles and the site of infusion. The time-related expression of each mRNA suggested that TNF-α has the ability to trigger its own production followed by the transcription of the LPS receptor; the signal for IκBα, TNF-α, and CD14 peaked at 1, 3, and 6 hr, respectively. The genes encoding TNF-α and mCD14 were also induced in the CVOs and within microglial cells across the brain parenchyma in response to intraperitoneal LPS administration. This induction in parenchymal cells of the brain was prevented in animals that received the anti-TNF-antisera intracerebroventricularly 10 hr before the systemic treatment with the endotoxin. The present data provide the evidence that microglial-derived TNF-α is responsible for the production of the LPS receptor CD14 during endotoxemia. This autocrine/paracrine stimulatory loop may be of great importance in controlling the inflammatory events that take place in the CNS during innate immune response as well as under pathological conditions.
Keywords: CD14 receptor, microglia, astrocytes, in situhybridization, histochemistry, inflammation, lipopolysaccharide, cytokines, blood vessels, septic shock
The clinical manifestations of endotoxemia are characterized by hyperventilation, hypercoagulation, pain, fever, cachexia, tachycardia, hypotension, somnolence, change in oxygen consumption, and multiple organ failure (Sweet and Hume, 1996). Most of these symptoms can be mimicked by the stimulation of host monocytes or macrophages in presence of the endotoxin lipopolysaccharide (LPS) (Haziot et al., 1996) or prevented with anti-CD14 antibody (Wright et al., 1990, 1991; Dentener et al., 1993). The endotoxin is released by the outer membrane of the gram-negative bacteria during sepsis and is detected by cells of myeloid origin, which bear the LPS receptor CD14 at their membrane surfaces (Pugin et al., 1994). CD14 is considered as the key player in the induction of the septic shock provoked by gram-negative bacteria.
Two forms of the CD14 receptor can be found. The first one is present at the surface of myeloid cells (mCD14) and acts as a glycosylphosphatidylinositol (GPI)-anchored membrane glycoprotein. The other form is soluble in the serum (sCD14) and lacks the GPI properties, although it can bind LPS to activate cells devoid of mCD14, such as endothelial, epithelial cells and vascular smooth muscle cells (Andersson et al., 1992). It is not well known how cell activation is triggered after binding between the endotoxin and the GPI-anchored mCD14, although there is now evidence that activation of tyrosine kinase leads to transduction signal and cytokine gene transcription through nuclear factor kappa B (NF-κB). The recent characterization of human homologs of Toll, especially the Toll-like receptor 4, may be the missing link for the transduction events leading to NF-κB activity in response to the LPS–mCD14 interaction (for review, seeUlevitch, 1999; Wright, 1999). NF-κB is normally present in the cytoplasm, forming an inactive complex with an inhibitor known as IκBα, which must be phosphorylated and degraded to allow NF-κB nuclear translocation and transcriptional activation of target genes (Baeuerle and Baltimore, 1996; Baeuerle, 1998; Delhase et al., 1999). After its degradation, IκBα is rapidly resynthesized to act as an endogenous inhibitory signal for NF-κB, and monitoring IκBα mRNA expression is a powerful tool to investigate the activity of the transcription factor within the CNS during immune stimuli.
We have recently reported that circulating LPS causes a rapid expression of CD14 mRNA within the circumventricular organs (CVOs), brain regions that contain a rich vascular plexus with specialized arrangements of the blood vessels (Lacroix et al., 1998). The tight junctions normally present between the endothelial cells are shifted in part to the ventricular surface and partly to the boundary between the CVOs and the adjacent structures explaining the diffusion of large molecules into the perivascular region (Oldfield and Mckinley, 1995). Parenchymal cells located in the anatomical boundaries of the CVOs exhibited a delayed response, which was followed by a positive signal for CD14 transcript in microglia across the brain parenchyma (Lacroix et al., 1998). These results strongly suggest that the endotoxin first reaches organs devoid of the blood–brain barrier (BBB) to induce the transcription of its own receptor and thereafter increased CD14 biosynthesis within parenchymal structures surrounding the CVOs and then the entire brain of severely challenged animals. Brain CD14 expression is likely to be a key step in the transcription of proinflammatory cytokines (Quan et al., 1997; Vallières and Rivest, 1997; Nadeau and Rivest, 1999) first within accessible structures from the blood and thereafter through scattered parenchymal cells during severe sepsis.
Like systemic phagocytes (Fearns et al., 1995), CD14 synthesis in parenchymal cells of the brain may be dependent on the production of proinflammatory cytokines. Of interest is the data that systemic injection of the bacterial endotoxin induced strong expression of CD14 (Lacroix et al., 1998) mRNA in a pattern that was closely related to the induction of tumor necrosis factor α (TNF-α) (Nadeau and Rivest, 1999) transcript with a rapid and delayed response. Although there is a large body of evidence that CD14 is necessary for the role of LPS on the induction of cytokine transcription from different myeloid cells, the possibility remains that the cytokine itself acts as an autocrine and paracrine factor to upregulate the LPS receptor. The binding of TNF to its type I receptor (p55) leads to the activation and translocation of p50/65 NF-κB into the nucleus (Beg et al., 1993), an event that has been reported to modulate CD14 expression. Indeed, TNF-α is able to induce a transient increase in plasma CD14 levels with a peak at 6–8 hr, and this elevation in levels of CD14 antigen was shown to be accompanied by increased levels of CD14 mRNA in lung, liver, and kidney (Fearns and Loskutoff, 1997). Pretreatment of mice with an anti-TNF-α antibody significantly prevented LPS-induced mCD14 transcription (Fearns and Loskutoff, 1997; Fearns and Ulevitch, 1998). Whether such mechanism is indeed taking place in the brain has yet to be investigated, but it is possible to believe that the cytokine is one of the paracrine factor that contributes to activate adjacent cells within the parenchymal brain.
The purpose of the present study was therefore to investigate the role of microglial-derived TNF in the regulation of CD14 in the brain during endotoxemia. To ascertain that TNF is capable of triggering its own production and CD14 transcription in specific cellular populations of the brain, animals received a single bolus of recombinant rat TNF-α (rrTNF-α) and were killed at different times thereafter. A second experiment consisted of neutralizing the biological activity of the cytokine in infusing an anti-rat TNF-α antibody in the lateral ventricle 10 hr before the systemic challenge with the endotoxin LPS. The results confirmed the hypothesis that TNF-α has the ability to trigger IκBα, TNF-α, and CD14 transcripts in microglia across the brain parenchyma, and the endogenous release of the proinflammatory molecule is responsible for the paracrine autostimulation of the phagocytic population of cells in the brain during blood sepsis.
MATERIALS AND METHODS
Animals. Adult male Sprague Dawley rats (Charles River Canada, St. Constant, Quebec, Canada; ∼265–300 gm) were acclimated to standard laboratory conditions (14:10 light/dark cycle; lights on at 6:00 A.M. and off at 8:00 P.M.) with ad libitumaccess to rat chow and water. Each rat was only used once for experimentation, and all protocols were approved by the Laval University's Animal Welfare Committee. A total of 116 rats were assigned to different protocols divided among the treatment and route of administration as described in Table1.
Table 1.
Treatment, dose, and number of rats used in each condition
| Treatment | Time (hr) | |||
|---|---|---|---|---|
| 1 3 6 12 | ||||
| Veh/treated | Veh/treated | Veh/treated | Veh/treated | |
| rrTNF-α, i.c.v. (150 ng/10 μl for 2 min) | 3 /5 | 3 /5 | 3 /5 | 3 /5 |
| LPS, i.p. (2.5 mg/kg, b.w.) | 3 /4 | 3 /4 | 3 /4 | 3 /3 |
| rrTNF-α, i.v. (4.1 μg/kg, b.w.) | 3 /4 | 3 /4 | 3 /4 | – |
| Time (hr) | ||||
|---|---|---|---|---|
| 2 | 6 | |||
| Veh, i.c.v. (5 μl for 2 min)/Veh, i.p. (300 μl) | 3 | 3 | ||
| Veh, i.c.v. (5 μl for 2 min)/LPS, i.p. (1.5 mg/kg, b.w.) | 5 | 5 | ||
| Ab, i.c.v. (2 μg/5 μl for 2 min)/Veh, i.p. (300 μl) | 5 | 5 | ||
| Ab, i.c.v. (2 μg/5 μl for 2 min)/LPS, i.p. (1.5 mg/kg, b.w.) | 5 | 5 | ||
| Goat IgG (5 μl for 2 min)/Veh, i.p. (300 μl) | 2 | 2 | ||
| Goat IgG (5 μl for 2 min)/LPS, i.p. (1.5 mg/kg, b.w.) | 2 | 2 | ||
rrTNF-α: catalog
510 RT, lot number AGM017071, R & D Systems (Minneapolis, MN); LPS, bacterial endotoxin from Escherichia coli, serotype 055:B5, Sigma (St. Louis, MO) L-2880, lot number 114H4022; Veh, sterile pyrogen-free saline; Ab, anti-rTNF-α neutralizing antibody, catalog
AF-510-NA, lot number X1028051, R & D Systems; Goat IgG: normal goat serum, catalog
S-1000, Vector Laboratories Burlingame, CA.
Intracerebroventricular infusions. Animals receiving intracerebroventricular injections were anesthetized with an intraperitoneal injection of a mixture [1 ml/kg body weight (b.w.)] of ketamine hydrochloride (91 mg/kg) and xylazine (9 mg/kg). The right lateral ventricle was reached stereotaxically (David Kopf Instruments, Tujunga, CA) using the Paxinos and Watson's atlas (1986). With the incisor bar placed at 3.3 mm below the interaural line (horizontal zero), the coordinates from bregma for the guide cannula were −0.6 mm anteroposterior, −1.4 mm lateral, and −2.8 mm dorsoventral. A 22 gauge stainless steel guide cannula was implanted close to the right lateral ventricle and secured with screws and cranioplastic cement (cranioplastic powder, Plastic One, Roanoke, VA; repair material, Dentsply International, York, PA). The rats were then housed individually for a 7 d recuperation period. During the first 3 d after the surgery, rats received once daily an subcutaneous injection of 8 ml of Ringer's lactate (Abbott Laboratories, Saint-Laurent, Quebec, Canada; catalog #7953(150), lot #48–142-NA at 37°C) and 150 μl of ketoprofen (Rhône Mérieux Canada, Victoriaville, Quebec, Canada; lot #M02008).
On the day of the experiment (∼8:30 A.M.), an internal cannula (28 gauge, 1 mm projection beyond the tip of the guide cannula) was connected to the guide cannula. Thereafter, either 10 μl of rrTNF-α or 10 μl of vehicle solution (pyrogen-free sterile distilled water) was injected into the right lateral ventricle over 2 min by means of a microinjection pump (model A-99; Razel Scientific Instruments, Stanford, CN). The animals were conscious and freely moving at all times throughout the procedure and killed 1, 3, 6, and 12 hr after the intracerebroventricular administration.
In another group of animals, anti-rTNF-α-neutralizing antibody (2 μg/5 μl/rat; catalog #AF-510-NA, lot #X1028051; R & D Systems, Minneapolis, MN) or normal goat serum (5 μl/rat; catalog #S-1000; Vector Laboratories, Burlingame, CA) was injected into the lateral ventricle 10 hr before an intraperitoneal injection of either LPS (1.5 mg/kg, b.w.) or the vehicle solution (300 μl of sterile pyrogen-free saline). This dose was selected to induce mCD14 and TNF-α expression in microglial cells across the brain parenchyma (Lacroix et al., 1998;Nadeau and Rivest, 1999). The animals were conscious and freely moving at all times throughout the intracerebroventricular injections.
Intravenous injection of rrTNF-α. Animals receiving intravenous injections were implanted with sterile cannulas containing sterile pyrogen-free heparin-saline (5.0 U/ml). Catheters were made from a piece of SILASTIC tubing [SILASTIC medical grade tubing, inner diameter (i.d.) 0.020 inch, outer diameter (o.d.) 0.037 inch; Dow Corning, Midland, MI] connected to an Intramedic polyethylene tubing (PE-50; i.d. 0.023 inch, o.d. 0.038 inch; Caly Adams, Parsippany, NJ). Outlets of cannulas were placed at the level of the neck, and rats were housed individually for a recuperation period of ∼5 d. On the day of the experiment (∼8:30 A.M.), the outlet portion of the catheter was fixed to a truncated 27 gauge needle that was attached to a PE-50 tubing. These connectors were then fixed to a 1 ml syringe, and rats were housed individually in a quiet room for at least 2 hr before the intravenous injections. Recombinant rat TNF-α (4.1 μg/kg, b.w.; catalog #510 RT, lot #AGM017071; R & D Systems) or the vehicle solution (300 μl of sterile pyrogen-free saline) was injected through the right jugular vein. The animals were killed 1, 3, and 6 hr after the intravenous injection of rrTNF-α.
Rats were deeply anesthetized with an intravenous (100 μl) or intraperitoneal (500 μl) injection of a mixture of ketamine hydrochloride and xylazine, then rapidly perfused transcardially with 0.9% saline, followed by 4% paraformaldehyde in 100 mmborax buffer, pH 9.5, at 4°C. The brains were removed from the skull, post-fixed for 1–4 d, and placed in 10% sucrose in a solution of 4% paraformaldehyde–borax buffer overnight at 4°C. For the combination of immunocytochemistry (especially for OX-42-immunoreactive cells) toin situ hybridization, rats were perfused with saline followed by 4% paraformaldehyde in 0.1 m sodium phosphate, pH 7.2. Brains were removed from the skull, post-fixed for 2 hr, and then placed in 20% sucrose diluted in 4% paraformaldehyde–sodium phosphate buffer for 12–15 hr. The brains were mounted onto a sliding microtome (SM2000R; Leica Instruments GmbH, Nussloch, Germany), frozen with dry ice, and cut into 30 μm coronal sections from the olfactory bulb to the caudal medulla. The slices were collected in a cold cryoprotectant solution (0.05m sodium phosphate buffer, pH 7.3, 30% ethylene glycol, 20% glycerol) and stored at −20°C.
Single in situ hybridization. Hybridization histochemical localization of CD14, TNF-α, and IκBα mRNA was performed on every sixth section of the whole rostrocaudal extent of each brain using 35S-labeled cRNA probes. All solutions were treated with diethylpyrocarbonate (DEPC) and sterilized to prevent RNA degradation. Tissue sections mounted onto poly-l-lysine-coated slides were desiccated overnight under vacuum, fixed in 4% paraformaldehyde for 30 min, and digested with proteinase K (10 μg/ml in 0.1 m Tris HCl, pH 8.0, and 50 mm EDTA, pH 8.0, at 37°C for 25 min). Thereafter, the brain sections were rinsed in sterile DEPC water followed by a solution of 0.1 m triethanolamine (TEA), pH 8.0, acetylated in 0.25% acetic anhydride in 0.1 m TEA, and dehydrated through graded concentrations of alcohol (50, 70, 95, and 100%). After vacuum drying for a minimum of 2 hr, 90 μl of hybridization mixture (107 cpm/ml) was spotted on each slide, sealed under a coverslip, and incubated at 60°C overnight (∼15–20 hr) in a slide warmer. Coverslips were then removed, and the slides were rinsed in 4× SSC at room temperature. Sections were digested by RNase A (20 μg/ml, 37°C, 30 min), rinsed in descending concentrations of SSC (2×, 1×, 0.5× SSC), washed in 0.1× SSC for 30 min at 60°C (1× SSC: 0.15 m NaCl and 15 mmtrisodium citrate buffer, pH 7.0) and dehydrated through graded concentrations of alcohol. After being dried for 2 hr under vacuum, the sections were exposed at 4°C to x-ray films (Biomax; Eastman Kodak, Rochester, NY) for 1–3 d. The slides were thereafter defatted in xylene, dipped in NTB2 nuclear emulsion (Kodak; diluted 1:1 with distilled water), exposed for 7 d (IκBα transcript) or 14 d (CD14 and TNF-α transcript), developed in D19 developer (Kodak) for 3.5 min at 14–15°C, washed 15 sec in water, and fixed in rapid fixer (Kodak) for 5 min. Tissues were then rinsed in running distilled water for 1–2 hr, counterstained with thionin (0.25%), dehydrated through graded concentrations of alcohol, cleared in xylene, and coverslipped with distrene plasticizer xylene (DPX) mounting medium.
cRNA probe synthesis and preparation. The pBluescript SK plasmids containing the full-length coding sequence of the rat CD14 cDNA (kindly provided by Dr. Doug Feinstein; Cornell University Medical College, New York, NY; Lacroix et al., 1998), rat TNF-α cDNA (Nadeau and Rivest, 1999), or the mouse IκBα cDNA (kindly provided by Dr. Alain Israël, Institut Pasteur, Paris, France; Laflamme and Rivest, 1999) were linearized with SacI and KpnI (CD14), EcoRI and BamHI (TNF-α), orBamHI and HindIII (IκBα) for the antisense and sense riboprobes, respectively. Radioactive cRNA copies were synthesized by incubation of 250 ng of linearized plasmid in (in mm): 6 MgCl2, 40 Tris, pH 7.9, 2 spermidine, 10 NaCl, 10 dithiothreitol, and 0.2 ATP/GTP/CTP, and 200 μCi of α-35S-UTP (DuPont NEN, Boston, MA; #NEG 039H), 40 U of RNAsin (Promega, Madison, WI), and 20 U of either T7 (CD14 and IκBα antisense probe, TNF-α sense probe) or T3 (TNF-α antisense probe, CD14 and IκBα sense probe) RNA polymerase for 60 min at 37°C. Unincorporated nucleotides were removed using ammonium–acetate method; 100 μl of DNase solution (1 μl of DNase, 5 μl of 5 mg/ml tRNA, 94 μl of 10 mm Tris–10 mmMgCl2) was added, and 10 min later, a phenol–chloroform extraction was performed. The cRNA was precipitated with 80 μl of 5 m ammonium acetate and 500 μl of 100% ethanol for 20 min on dry ice. The pellet was washed with 500 μl of 70% ethanol, dried, and resuspended in 100 μl of 10 mm Tris/1 mm EDTA. A concentration of 107 cpm probe was mixed into 1 ml of hybridization solution [500 μl of formamide, 60 μl of 5 m NaCl, 10 μl of 1 mTris, pH 8.0, 2 μl of 0.5 m EDTA, pH 8.0, 50 μl of 20× Denhardt's solution, 200 μl of 50% dextran sulfate, 50 μl of 10 mg/ml tRNA, and 10 μl of 1 m DTT, (118 μl of DEPC water − volume of probe used)]. This solution was mixed and heated for 5 min at 65°C before being spotted on slides.
Combination of immunocytochemistry with in situhybridization. Immunocytochemistry was combined with the in situ hybridization histochemistry protocol to determine the type of cells that express CD14, IκBα, and TNF-α mRNA transcript in the rat brain after intracerebroventricular administration of TNF-α. The anti-rat CD11b/c monoclonal antibody (OX-42; Cedarlane Laboratories, Hornby, Ontario, Canada; catalog #CL042B, lot #4241) and the mouse anti-glial GFAP monoclonal antibody (Chemicon, Temecula, CA; catalog #MAB360, lot #19020465) were used to stain microglia and astrocytes, respectively. Every sixth brain section was processed by using the avidin–biotin bridge method with peroxidase as a substrate. Briefly, slices were washed in sterile DEPC-treated 50 mm potassium PBS (KPBS) and incubated 2 hr at room temperature with anti-CD11b/c or anti-GFAP antibody diluted in sterile KPBS plus 0.4% Triton X-100 plus 1% bovine serum albumin (fraction V; Sigma, St. Louis, MO) plus 0.25% heparin sodium salt USP (ICN Biomedicals, Aurora, OH). Monoclonal anti-CD11b/c or anti-GFAP antibodies were diluted at 1:1500 or 1:2500, respectively. After incubation with the primary antibody, brain slices were rinsed in sterile KPBS and incubated with a mixture of KPBS plus 0.4% Triton X-100 plus 1% BSA plus 0.25% heparin plus biotinylated secondary antibody (anti-mouse IgG; catalog #BA-2001; Vector Laboratories, 1:1500) for 60 min. Sections were then rinsed with KPBS and incubated at room temperature for 60 min with an avidin–biotin–peroxidase complex (Vectastain ABC Elite kit; Vector Laboratories). After several rinses in sterile KPBS, the brain slices were reacted in a mixture containing sterile KPBS, the chromagen 3,3′-diaminobenzidine tetrahydrochloride (DAB; 0.05%), and 0.003% hydrogen peroxide (H2O2).
Thereafter, tissues were rinsed in sterile KPBS, immediately mounted onto gelatin and poly-l-lysine-coated slides, desiccated under vacuum for 30 min, fixed in 4% paraformaldehyde, pH 7.2, for 30 min, and digested by proteinase K (10 μg/ml in 100 mm Tris HCl, pH 8.0, and 50 mm EDTA, pH 8.0) at 37°C for 25 min. Prehybridization, hybridization, and posthybridization steps were performed according to the above description with the difference of dehydration (alcohol 50, 70, 95, 100%), which was shortened to avoid decoloration of immunoreactive cells (brown staining). After being dried for 2 hr under the vacuum, sections were exposed at 4°C to x-ray film (Kodak) for 17 hr, defatted in xylene, and dipped in NTB2 nuclear emulsion (Kodak; diluted 1:1 with distilled water). Slides were exposed for 10 d (IκBα) or 21 d (CD14, TNF-α), developed in D19 developer (Kodak) for 3.5 min at 15°C, and fixed in rapid fixer (Kodak) for 5 min. Tissues were then rinsed in running distilled water for 1–2 hr, rapidly dehydrated through graded concentrations of alcohol, cleared in xylene, and coverslipped with DPX. The presence of the different transcripts was detected by the agglomeration of silver grains in perikarya, whereas GFAP or OX-42 immunoreactivity within the cell cytoplasm and ramifications was indicated by brown homogeneous coloration.
Quantitative analysis. Quantitative analysis of hybridization signal for the IκBα, TNF-α, and CD14 mRNA signal was performed on x-ray films over numerous regions ipsilateral to the site of injection. The selected structures were the isocortex (layers 1–4), septum, ependymal lining of the right lateral ventricle (infusion site), paraventricular nucleus, geniculate nucleus, periaqueductal gray, locus coeruleus, and the ventrolateral medulla. These regions were chosen to facilitate the analysis among animals, although the hybridization signal was not limited to specific nuclei but through the parenchymal elements surrounding the cerebroventricular system (see Results). Transmittance values (referred here as optical density) of the hybridization signals was measured under a Northern Light Desktop Illuminator (Imaging Research) using a Sony (Tokyo, Japan) camera video system attached to a Micro-Nikkor 55 mm Vivitar extension tube set for Nikon lens and coupled to a Macintosh computer (Power Macintosh 7100/66) and NIH Image software, version 1.59/ppc (written by W. Rasband at the United States National Institutes of Health and available from the Internet by anonymous ftp fromzippy.nih.gov). Optical density (OD) values for each pixel were calculated using a known standard of intensity and distance measurements from a logarithmic specter adapted from Bioimage Visage 110s (Millipore, Ann Arbor, MI). Sections from intracerebroventricular vehicle- and TNF-challenged animals were digitized and subjected to densitometric analysis, yielding measurements of mean density per area. The OD of each region was then corrected for the average background signal by subtracting the OD of areas without positive signal located in the contralateral site. All measurements were performed in triplicate. Data are reported as mean OD values (± SEM), and statistical analysis was performed by ANOVA for each transcript, followed by a Bonferroni–Dunn test procedure as post hoccomparisons with Statview software (version 4.01, Macintosh). Factors were identified as follows: intracerebral treatment, which was composed of two levels (i.c.v. vehicle and i.c.v. rrTNF-α) and time after injection that comprised 4 levels (1, 3, 6, and 12 hr).
The number of hybridized IκBα, TNF-α, and CD14 cells was performed on nuclear emulsion-dipped slides with an Olympus Optical System (BX-50, Bmax) coupled to a Macintosh computer (Power PC 7100/66) and Image software (version 1.59, non-FPU; W. Rasband, National Institutes of Health). Positive cells were counted in three different areas (1.0 mm2) of the caudoputamen under dark-field illumination at a magnification of 10×. The three transcripts were strongly induced in this region in response to circulating LPS, and the number of cells per square millimeter was the selected dependent variable for an objective analysis among animals. Data are reported as mean values of cells/1.0 mm2 (± SEM) for experimental and control animals. Statistical analysis was performed by ANOVA followed by a Bonferroni–Dunn test procedure as post hoc comparisons with the Statview program. Six different groups were included in the analysis; intracerebroventricular vehicle and intraperitoneal vehicle solution, intracerebroventricular vehicle and intraperitoneal LPS, intracerebroventricular anti-TNF and intraperitoneal vehicle solution, intracerebroventricular anti-TNF and intraperitoneal LPS, intracerebroventricular goat IgG and intraperitoneal vehicle solution, and intracerebroventricular goat IgG and intraperitoneal LPS.
RESULTS
TNF-α-induced cerebral gene expression
Figure 1 depicts representative examples of hydridization signal on x-ray films for CD14 mRNA in the brain of control (unmanipulated), intracerebroventricular vehicle and intracerebroventricular TNF-α-injected rats. The brain of the naive rat exhibited a low to moderate expression of CD14 transcript in various structures including the leptomeninges, choroid plexus (chp), and the CVOs, vascular organ of the lamina terminalis (OVLT), subfornical organ (SFO), median eminence (ME), and area postrema (AP) (Fig. 1, left column). A strong but localized hybridization signal for the LPS receptor was found in cells lining the cortical tract (placement of the guide cannula) and the ependymal cells of the lateral (site of injection) and the third ventricle (Fig. 1,middle column). This localized mCD14 message contrasts with the profound induction of the transcript across the brain parenchyma in response to intracerebroventricular TNF-α infusion (Fig. 1,right column). Indeed, the cytokine triggered CD14 transcription in regions ipsilateral to the site of injection, i.e., the right caudoputamen, cerebral cortex, hippocampal and septal formation, and parenchymal elements around the third ventricle, beneath the leptomeninges, and near the central canal and the fourth ventricle. The expression profile seemed to follow the diffusion pattern of the intracerebroventricularly injected TNF-α solution, although we have no direct evidence of such phenomenon (see Discussion). The intensity of the hydridization signal in regions in circumference to the ventricle system gradually increased to reach a maximal level at 6 hr after TNF infusion (Fig.2). A slight decrease was detected 6 hr later, although numerous positive CD14 cells were still found throughout the parenchymal brain 12 hr after the single intracerebroventricular TNF-α bolus. Hybridized tissues with sense probe did not exhibit detectable signal in any of the regions that displayed positive message with antisense CD14 probe (data not shown).
Fig. 1.
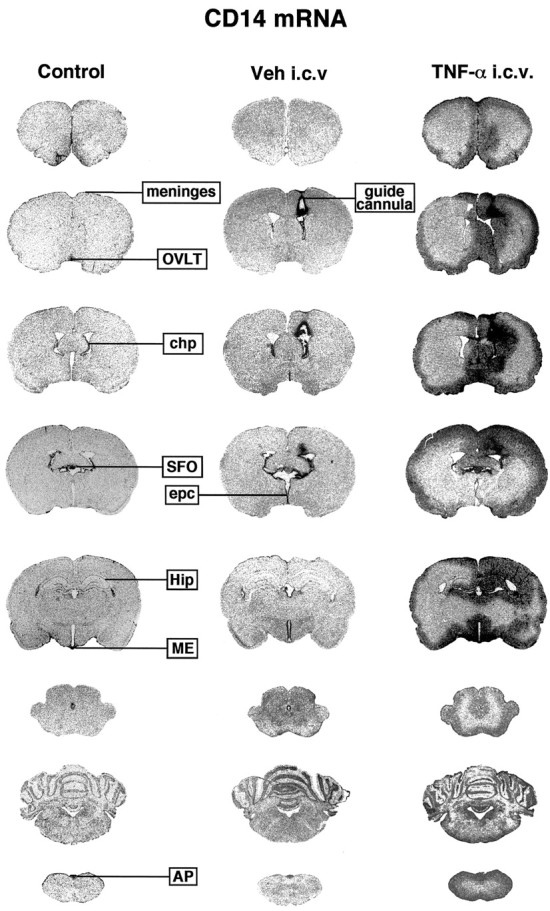
Expression of the gene encoding the LPS receptor CD14 across the brain of rats that received an intracerebroventricular infusion of recombinant rat tumor necrosis factor α (rrTNF-α). These rostrocaudal sections (30 μm) depict positive signal on x-ray films (Biomax) in the brain of an intact animal (Control) and 6 hr after the intracerebroventricular administration of either vehicle (Veh) or the proinflammatory cytokine.AP, Area postrema; chp, choroid plexus;epc, ependymal cells; Hip, hippocampus;ME, median eminence; OVLT, organum vasculosum of the lamina terminalis; SFO, subfornical organ.
Fig. 2.

Time-related induction of IκBα, TNF-α, and CD14 mRNA after intracerebroventricular rrTNF-α administration. The OD was measured in the same areas (isocortex, septum, ependymal lining of the lateral ventricle, paraventricular nucleus, geniculate nucleus, periaqueductal gray, locus coeruleus, and the ventrolateral medulla) for each transcript. The areas were digitized and subjected to densitometric analysis using a logarithmic specter of standardized OD adapted from Bioimage Visage 110s (Millipore), yielding measurements of mean density per area (referred here to as OD). For more information on image analysis, see Materials and Methods. Results represent means ± SEM for five animals per group. *Significantly different (p < 0.05) from their corresponding vehicle-treated groups.
Injection of the proinflammatory cytokine into the lateral ventricle caused a rapid transcriptional activation of the gene encoding IκBα and TNF-α in regions near the ventricles (Fig. 2). A low to moderate signal for IκBα mRNA was observed in the leptomeninges, chp, hippocampus, OVLT, ME, SFO, AP, supraoptic nucleus (SON), and the paraventricular nucleus (PVN) of vehicle-administered rats (Fig.3, left column). The signal increased in these structures and over the regions neighboring the ventricle walls, the leptomeninges, and the central canal after intracerebroventricular rrTNF-α administration (Fig. 3). The IκBα mRNA signal was particularly intense along the periventricular zone of the third ventricle. Although TNF-α transcript was not detected in the brain of naive rats (data not shown), few positive cells were found along the cerebral tract as well as the walls of the lateral and third ventricle (Fig. 3). Central TNF-α infusion caused a robust transcriptional activation of its own gene in regions adjacent to the ventricles and the isocortex underneath the leptomeninges at time 1–6 hr. The IκBα hydridization labeling reached its maximal level 1 hr after the intracerebroventricular rrTNF-α administration and gradually returned to basal levels 12 hr later, whereas TNF-α mRNA signal peaked at 3 hr and decreased slowly thereafter (Fig. 2).
Fig. 3.

Representative examples of the influence of intracerebroventricular administration of rrTNF-α on IκBα and TNF-α gene expression in the rat brain. These rostrocaudal sections (30 μm) depict the hybridization signal on x-ray films (Biomax) from animals killed 1 (IκBα mRNA) or 3 (TNF-α mRNA) hr after intracerebroventricular TNF-α infusion. For abbreviations, see legend for Figure 1.
Despite the different time-related induction of IκBα, CD14, and TNF-α by intracerebroventricular injection with the proinflammatory cytokine, the pattern of expression of the three tran-scripts was quite comparable in the regions adjacent to the site of injection, the periventricular zone of the third ventricle and underneath the leptomeninges. However, the intensity of the signal and the spread of positive cells differed among genes. The Figure4 shows examples of IκBα, TNF-α, and CD14-expressing cells in various regions of the brain at 1, 3, and 6 hr after TNF-α injection, respectively. Blood vessels and ependymal cells of the ventricle walls exhibited positive signal for IκBα and CD14, but not TNF-α. TNF-α- and IκBα-expressing cells were present in a restricted surface lining the periventricular area, whereas a more diffused distribution of the mRNA encoding the LPS receptor was observed in response to the central TNF challenge (Fig.4C). It is of interest to note the well-defined lining distribution of TNF-α-positive cells in circumference to the ventricles and around the medulla at ∼0.3 mm beneath the meningeal formation (Fig. 4C–E). Adjacent sections hybridized with IκBα and TNF-α sense probes did not exhibit any positive signal (data not shown).
Fig. 4.
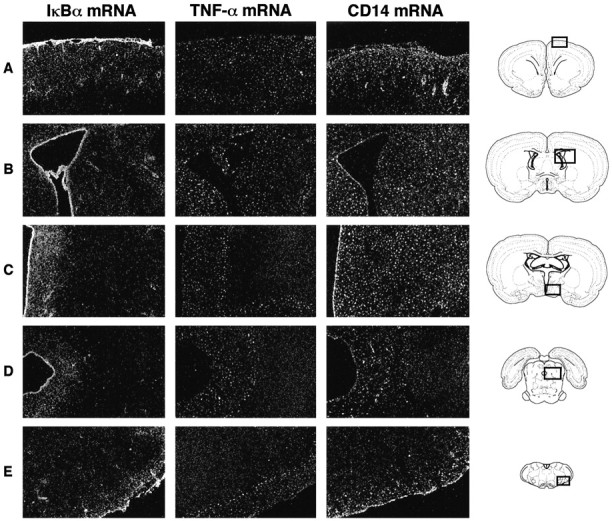
Comparative pattern of IκBα-, TNF-α-, and CD14-expressing cells in different areas of the rat brain in response to centrally injected TNF-α. These dark-field photomicrographs of nuclear emulsion-dipped sections show scattered small positive cells in the brain of rats killed 1 (IκBα), 3 (TNF-α), or 6 (CD14) hr after the intracerebroventricular infusion of the proinflammatory cytokine. These times correspond to the maximal expression levels of each transcript after the intracerebroventricular infusion.A, Leptomeninges and isocortex; B, ependymal lining cells and areas around the lateral ventricle;C, region of the third ventricle; D, central canal; E, the ventrolateral medulla. The schemes of the right column were taken from the atlas of Swanson (1992). Magnification, 10×.
Dual-labeling data
To determine the phenotype of IκBα-, TNF-α-, and CD14-expressing cells in the brain of intracerebroventricular TNF-α-infused rats, immunocytochemistry was combined with in situ hybridization histochemistry together on the same coronal sections. Antisera against OX-42 and GFAP was used to stain microglia and astrocytes, respectively. Positive hybridization signal for the three transcripts was detected in numerous OX-42-IR cells in the brain of TNF-injected rats (Fig. 5, top panels, filled arrowheads), but not all microglial cells displayed agglomeration of silver grains (open arrowheads). In contrast, astrocytes failed to show positive signal for the mRNA encoding either IκBα, TNF-α, or CD14 (Fig. 5, bottom panels). Indeed, hybridized cells (white arrowheads) did not overlap with GFAP immunoreactivity at all the postinjection times evaluated in this study (black open arrowheads, bottom panels).
Fig. 5.

Central injection of TNF-α caused upregulation of the mRNA encoding IκBα, TNF-α, and CD14 in the parenchymal microglia. The anti-rat OX-42 (CD11b/c) and the mouse anti-glial GFAP monoclonal antibodies were used to stain microglia and astrocytes, respectively. Note that numerous microglia contain positive hybridization signal for the three transcripts, whereas no convincing mRNA signal was found over the astrocytes. Filled black arrowheads, Double-labeled cells (OX-42/IκBα mRNA,top left; OX-42/TNF-α mRNA, top middle; OX-42/CD14 mRNA, top right); open arrowheads, single OX-42-IR (top) and GFAP-IR (bottom panels) cells; white arrowheads, single mRNA-expressing cells. Magnification, 150×.
CD14 expression during endotoxemia and in response to circulating TNF-α
Figure 6 depicts representative expression of CD14 transcript in the AP and surrounding areas after a single intraperitoneal administration of the endotoxin LPS or intravenous rrTNF-α injection. As mentioned, a low to moderate hybridization signal was found in the AP under basal conditions (left column). The labeling for mCD14 mRNA increased gradually to reach a maximum level of intensity 6 hr after the intraperitoneal LPS bolus. At that time, numerous scattered small CD14-expressing cells were detected across the brain parenchyma of endotoxin-treated rats (Fig. 6, bottom, middle column). These cells are of myeloid origin because they were immunoreactive to the rat complement receptor type 3 (Lacroix et al., 1998). Intravenous rrTNF-α injection also caused an increase in the expression of the LPS receptor in the CVOs, but not within parenchymal elements of the brain (Fig. 6, right column). These results indicate that centrally produced TNF-α only may be responsible for triggering CD14 transcription in microglial cells of the brain parenchyma during blood endotoxemia.
Fig. 6.
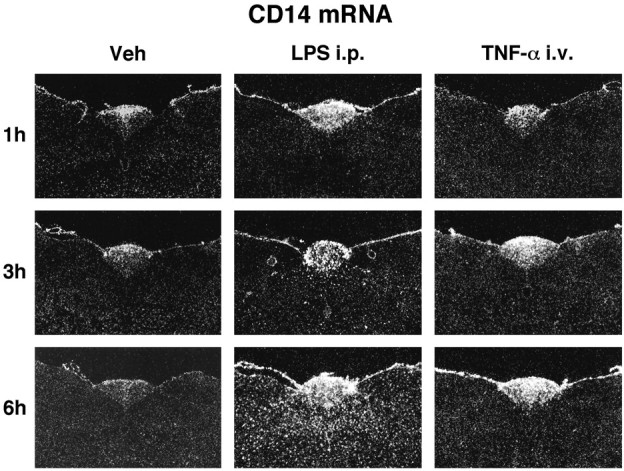
Time-related expression of CD14 mRNA in the AP and its adjacent regions in response to a single intraperitoneal injection of the endotoxin LPS or intravenous rrTNF-α administration. These dark-field photomicrographs show the constitutive expression of the LPS receptor in the circumventricular organ and the robust induction in the parenchymal elements by the endotoxin. Although intravenous injection of recombinant rat TNF-α increased the relative CD14 mRNA levels in the AP, this treatment failed to activate the LPS receptor in the parenchymal brain. Magnification, 10×.
Neutralization of centrally produced TNF-α
To confirm such mechanism, the biological activity of TNF-α was neutralized by the intracerebroventricular administration of an anti-rTNF-α antibody 10 hr before the systemic treatment with the endotoxin. Figure 7 shows the effect of anti-rTNF-α-neutralizing antibody on LPS-induced IκBα, TNF-α, and CD14 expression in rat cerebral tissues. The signal for these genes was low to undetectable in the parenchymal brain of vehicle-injected rats, but the endotoxin caused a strong increase in the cerebral capillaries (IκBα mRNA) and across the brain (IκBα, TNF-α, and CD14 transcripts) at 6 hr after injection (Fig. 7, second row). The intracerebroventricular administration of anti-rTNF-α-neutralizing antibody 10 hr before an intraperitoneal injection of the vehicle solution had no effect on these transcripts (Fig. 7, third row), but it significantly prevented LPS-induced transcription of IκBα, TNF-α, and CD14 mRNA (Fig. 7,bottom panels). The influence of the anti-TNF in interrupting IκBα expression was however only partial and localized to parenchymal cells, because the cerebral blood vessels still exhibited strong hybridization signal in response to circulating LPS (Fig. 7, bottom left). Scattered small positive IκBα cells were still present in the brain of rats that received both intracerebroventricular anti-TNF and intraperitoneal LPS administration. Pretreatment with the anti-TNF also failed to prevent LPS-induced genetic expression of the proinflammatory molecules in the CVOs, chp, and the leptomeninges, although it was effective in blocking completely CD14 and TNF biosynthesis by parenchymal microglial cells. Indeed, the number of CD14 and TNF-α cells was comparable in rats that received an intraperitoneal vehicle administration and those pretreated with the anti-TNF and challenged with the endotoxin (Fig.8).
Fig. 7.
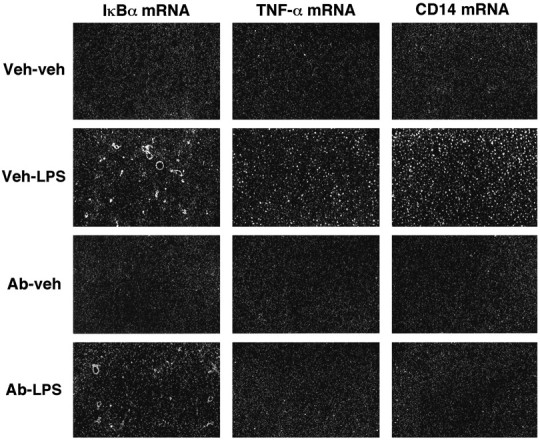
Inhibition of centrally produced TNF-α prevents the LPS-induced transcription of IκBα, TNF-α, and CD14 in the brain parenchyma. The rats received an intracerebroventricular injection of vehicle (Veh; 5 μl for 2 min) or anti-rTNF-α-neutralizing antibody (Ab; 2 μg/5 μl for 2 min) 10 hr before an intraperitoneal injection of either LPS or the vehicle solution. These dark-field photomicrographs of 30 μm coronal sections dipped into NTB2 emulsion depict the numerous scattered positive cells in the caudate putamen of animals that received the intracerebroventricular vehicle solution before the systemic LPS injection (second row). Very few positive cells were found in parenchymal elements of the CNS of rats pretreated with the anti-TNF and challenged with the endotoxin 10 hr later (bottom panels). Note that LPS-induced IκBα expression in the cerebral capillaries was not prevented by the anti-TNF antisera (bottom left). Magnification, 10×.
Fig. 8.
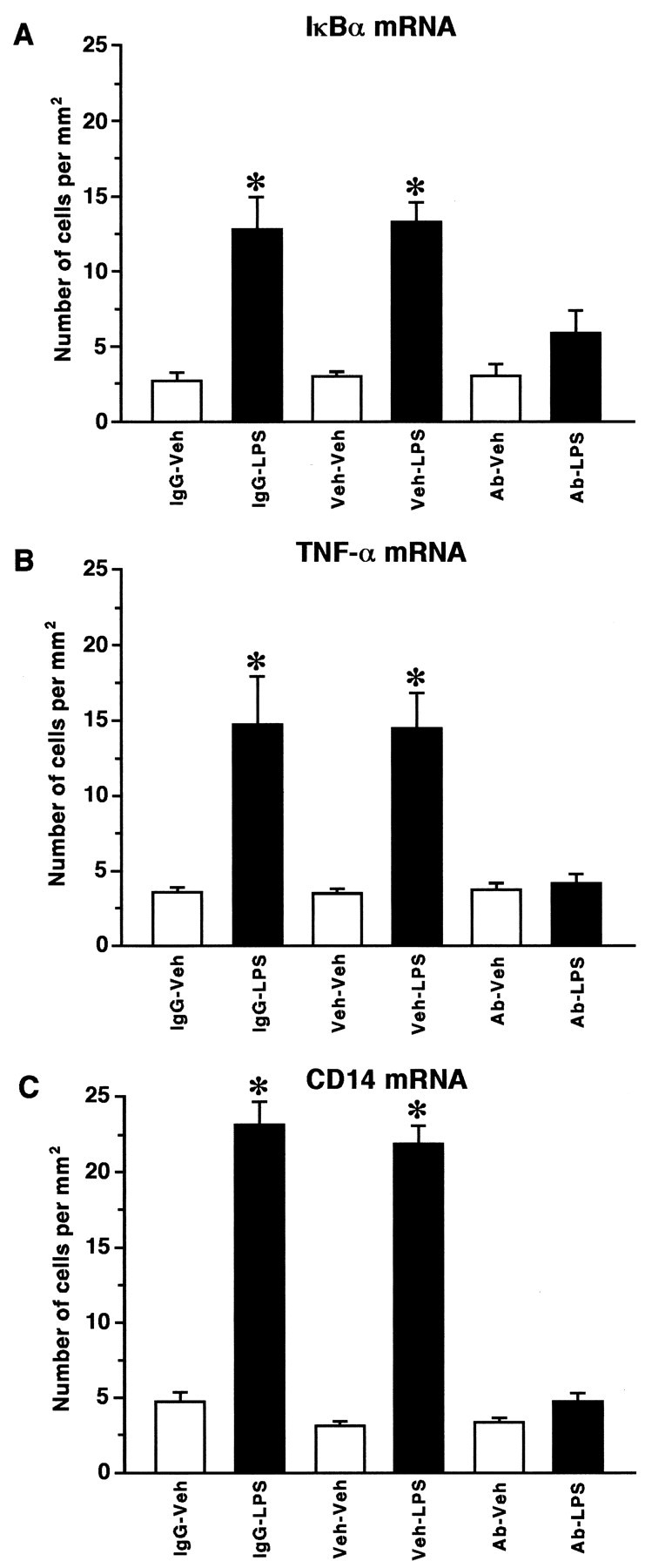
Number of IκBα-, TNF-α-, and CD14-positive cells in the caudate putamen of animals pretreated or not with an anti-rTNF-α-neutralizing antibody before the systemic challenge with the endotoxin LPS. Rats received an intracerebroventricular injection of vehicle (Veh; 5 μl for 2 min), anti-rTNF-α-neutralizing antibody (Ab; 2 μg/5 μl for 2 min), or goat serum as a control IgG (5 μl/2 min) 10 hr before an intraperitoneal injection of either a vehicle solution or LPS (1.5 mg/kg, b.w.). Positive cells were counted in three different areas (1.0 mm2) of the caudoputamen under dark-field illumination at a magnification of 10×. Data are reported as mean values of cells/1.0 mm2 (± SEM) for experimental and control animals killed 6 hr after the intraperitoneal injection of the endotoxin or its vehicle solution. *Significantly different (p < 0.05) from their corresponding vehicle-treated groups.
DISCUSSION
Central injection of the recombinant rat TNF-α caused a robust expression of the genes encoding IκBα, TNF-α, and CD14 in microglial cells of the brain parenchyma. The time-related induction of these transcripts suggested a potential role of NF-κB in mediating TNF-induced transcriptional activation of the LPS receptor. Systemic injection with the endotoxin LPS provoked a similar microglial activation that was prevented in inhibiting the biological activity of the proinflammatory cytokine in the CNS. Together these data provide the evidence that centrally produced TNF-α plays an essential autocrine/paracrine role in triggering parenchymal microglial cells during severe endotoxemia. These events may be determinant for orchestrating the neuroinflammatory responses that take place in a well coordinated manner to activate the resident phagocytic population of cells in the brain. The physiological outcomes of this innate immune response of the CNS are likely to include a rapid elimination of LPS particles via an increased opsonic activity of the transmembrane CD14 receptor to prevent potential detrimental consequences on neuronal elements during blood sepsis.
As previously reported, systemic LPS injection induced mCD14 expression first within structures devoid of BBB and thereafter throughout the brain parenchyma of animals that received high doses of LPS (Lacroix et al., 1998). Microscopic analysis of emulsion-dipped slides revealed that CD14-positive cells spread over the anatomical boundaries of the CVOs in a migratory-like pattern during the course of the endotoxemia. The direct action of LPS on myeloid-derived cells expressing mCD14 and present in structures that are accessible from the systemic circulation may allow a rapid production of proinflammatory cytokines within these organs. The rapid induction of TNF-α mRNA in the CVOs by intraperitoneal LPS clearly indicates that such events take place in specific populations of cells in the brain. Like CD14, scattered small TNF-expressing cells can be found across the brain parenchyma in response to LPS, although this depends on the dose of the endotoxin and the route of administration (Nadeau and Rivest, 1999). Indeed, the CVOs displayed low but positive signal as soon as 1 hr after the LPS challenge and increased to reach maximal levels at 6 hr, but positive cells gradually became apparent in the boundary of these organs and spread over the entire brain from 6 to 24 hr during severe endotoxemia. However, this phenomenon is only provoked by a high dose of the bacterial LPS (2.5 mg/kg, i.p.); a lower dose (250 μg/kg, i.p.) caused a more restricted transcriptional activation of the gene encoding the proinflammatory cytokine within the CVOs and the adjacent areas (Nadeau and Rivest, 1999). The same dose injected intravenously is capable of inducing CD14 and TNF across the brain parenchyma, whereas these transcripts are quite localized to the CVOs and chp in response to 5 μg of LPS intravenously (N. Laflamme and S. Rivest, unpublished data). This clearly indicates that the endotoxin first reaches available targets devoid of BBB, which in return, depending on the severity of the challenge, prime adjacent cells within the parenchymal brain to stimulate TNF-α transcription.
The bacterial endotoxins are among the most powerful agents known to stimulate circulating monocytes and tissue macrophages, which lead to the synthesis and release of a variety of proinflammatory cytokines (Andersson et al., 1992). The most important target of macrophage-derived secretory products is the macrophage itself (Sweet and Hume, 1996). The early production of TNF may be an essential step in these autocrine and paracrine loops, because this cytokine is able to induce its own production by an autocrine stimulation that is followed by the synthesis of other proinflammatory cytokines, such as IL-1β and IL-6 (Andersson et al., 1992). TNF has also been found to be a major factor inducing shock, and passive immunization against the cytokine can attenuate the appearance of IL-1β and IL-6 (Fong et al., 1989). Surprisingly however, cytokine production and circulating IL-6 and IL-1β induced by intraperitoneal LPS injection is intact in TNF-deficient mice (Marino et al., 1997), which suggests that TNF is certainly not the sole primary event that leads to production of subsequent cytokines in response to the endotoxin and other models of inflammation. In the brain, the cytokine seems to act as a key ligand to activate parenchymal microglia in a paracrine manner during endotoxemia. It is suggested here that circulating LPS targets its transmembrane receptor in CVO/chp macrophages and microglia, which may stimulate the NF-κB signaling events and trigger TNF transcription. The cytokine may in return bind to its cognate p55 receptor and lead to the formation of the TNF-R1-associated death domain (TRADD)/TNF receptor-associated factor 2 (TRAF2)/receptor-interacting protein (RIP) complex, which may activate the NF-κB signaling kinases in adjacent microglia. TNF-α is actually one of the most potent effectors of NF-κB activity through the 55 kDa TNF type I receptor (Baeuerle and Baltimore, 1996; Baeuerle, 1998). Such events are likely to contribute to the transcriptional activation of both CD14 and TNF genes in the brain of endotoxin-treated animals.
Central production of TNF-α is a key mechanism controlling CD14 expression in the brain parenchyma, but not in the CVOs and chp. The anti-TNF did not significantly change the relative CD14 mRNA levels in these organs, but prevented quite specifically the parenchymal expression of the LPS receptor during endotoxemia. The constitutive expression of CD14 in regions that can be reached by the bloodstream may allow a rapid production of TNF that in return acts as the endogenous ligand to activate adjacent cells of myeloid lineage. An important question however is what mechanisms control the spreading of the message from regions close to the CVOs to deep parenchymal elements. As mentioned, the spreading of CD14- and TNF-α-expressing cells depends on the severity of the endotoxin challenge (Lacroix et al., 1998; Nadeau and Rivest, 1999). It is therefore possible that the paracrine influence of the cytokine remains localized in response to low circulating levels of LPS, but takes place across the cerebral tissue during severe endotoxemia. Such effects of TNF in activating CD14 expression have previously been reported in different systemic organs and like the present study, pretreatment of mice with anti-TNF-α antibody significantly prevented LPS-induced mCD14 transcription in the lung, liver, and kidney (Fearns and Loskutoff, 1997; Fearns and Ulevitch, 1998).
IL-1β has also been reported to stimulate CD14 expression in different organs, and anti-IL-1β antibody attenuated the induction of the LPS receptor in response to the endotoxin (Fearns and Loskutoff, 1997; Fearns and Ulevitch, 1998). IL-1β and TNF-α are known to have numerous overlapping activities, and inhibiting one cytokine may frequently be associated with redundant mechanisms because of the presence of the other cytokine (Baumann et al., 1993). Although IL-1β may have the ability to stimulate CD14 in the brain microglia, its involvement depends most likely on the previous production of TNF as the anti-TNF completely inhibited LPS-induced CD14 transcription. On the other hand, IL-1β is the key inflammatory signal in the brain to stimulate the production of growth factors by astrocytes during brain trauma, whereas TNF is not essential for such response. Indeed, we have recently observed a rapid production of numerous proinflammatory molecules in cells lining the lesion site that was followed by a robust increase in ciliary neurotrophic factor (CNTF) biosynthesis (Herx et al., 1999). The release of CNTF was completely inhibited in IL-1β-deficient mice notwithstanding the ongoing TNF-α production by microglial cells lining the corticectomy (Herx et al., 1999). Circulating IL-1β has also been recently shown to be the key mediator of the NF-κB activity and COX-2 transcription in cells of the BBB during a systemic model of inflammation (Laflamme et al., 1999). These data, together with the present study, support the concept that despite the recognized overlapping activities of both NF-κB-signaling cytokines in the systemic immune system, IL-1β and TNF-α seem to have a distinct role in orchestrating the inflammatory events that take place in the brain.
The possibility that circulating TNF may influence CNS CD14 expression was also investigated, because the cytokine is rapidly detected into the bloodstream during endotoxemia (Basu et al., 1997). Intravenous injection of rrTNF-α caused upregulation of mCD14 transcript quite selectively in the CVOs and not within parenchymal cells adjacent to these organs. Therefore, circulating TNF may not contribute to the robust transcriptional activation of the LPS receptor in the parenchymal microglia of endotoxin-challenged rats. The positive autoregulatory loop is therefore not always associated with a paracrine influence of the cytokine to trigger CD14 in surrounding cerebral tissues. This unexpected result provides the evidence that microglial-derived TNF-α only is responsible to activate the biosynthesis of the LPS receptor in deep parenchymal cells of the brain in response to high circulating levels of the bacterial endotoxin.
Microscopic analysis of emulsion-dipped slides revealed some differences in the pattern of expression for the three transcripts studied. The intensity of positive signal also decreased in distant structures from the site of injection, which may be attributed to the high molecular weight of TNF-α and the slow diffusion of the cytokine into the CSF, a known phenomenon for intracerebroventricularly injected products (Anderson et al., 1995). It is therefore quite surprising that anti-TNF was capable of preventing CD14 expression across the brain parenchyma during endotoxemia. In this particular case however, the neutralizing antibody was infused 10 hr before the LPS challenge, and the CVOs and their adjacent areas initiate the response. Because these sensorial CVOs are the direct extension of the cerebroventricular system (Oldfield and Mckinley, 1995), the antisera may have access to these organs and their surrounding structures. Inhibition of the migratory-like pattern of CD14/TNF production may then prevent the paracrine influence of the cytokine within deep regions of the brain.
The depth of the hybridization signal in the parenchyma was different among transcripts; CD14-positive signal was wider than IκBα and TNF-α transcripts in response to centrally injected TNF. It is possible that the signaling molecules that control the transcription of the gene encoding CD14 are highly sensitive to small quantities of TNF-α in regions that can be difficult to reach by the cytokine after the intracerebroventricular injection. TNF-α may also not be the only mediator responsible for the induction of mCD14, although it may act as the first signal responsible for the induction of a series of events in cascade leading to the stimulation of the LPS receptor. This concept is further supported by the fact that CD14-positive cells were clearly more abundant than those expressing either IκBα or TNF-α. The suppression of mCD14 synthesis by the anti-TNF-neutralizing antibody indicates that the proinflammatory cytokine has a leading role in initiating these events during endotoxemia.
The role of CD14 in the CNS is unknown. The constitutive expression of CD14 in the CVOs and its upregulation in the brain parenchyma suggest a potential role in protecting the neural elements against the LPS particles. The macrophages and microglia in the CVOs are strategically well positioned for responding rapidly to circulating endotoxin or bacteria, whereas parenchymal microglia are the phagocytic population of cells in the brain in case of invasion. There is alteration of the BBB during severe endotoxemia (Mayhan, 1998), which may allow diffusion of molecules that normally have no access to the parenchymal elements and be detrimental for neurons. Activation of microglial cells across the CNS may rapidly eliminate this foreign material, although a sustained activity of these cells is not suitable because it may have opposite effects and be associated with neurodegenerative disorders (Gonzalez-Scarano and Baltuch, 1999).
Footnotes
This work was supported by the Medical Research Council of Canada (MRCC). S.N. holds a Studentship from the MRCC, and S.R. is an MRCC Scientist. We thank Dr. Alain Israel (Institut Pasteur, Paris, France) and Dr. Doug Feinstein (Cornell University Medical College, New York, NY) for the gift of the plasmid containing the IκBα cDNA and CD14 cDNA, respectively. We also thank Ms. Nathalie Laflamme for invaluable technical assistance.
Correspondence should be addressed to Dr. Serge Rivest, Laboratory of Molecular Endocrinology, CHUL Research Center and Department of Anatomy and Physiology, Laval University, 2705, boulevard Laurier, Québec, Canada G1V 4G2. E-mail: Serge.Rivest@crchul.ulaval.ca.
REFERENCES
- 1.Anderson KD, Alderson RF, Altar CA, DiStefano PS, Corcoran TL, Lindsay RM, Wiegand SJ. Differential distribution of exogenous BDNF, NGF, and NT-3 in the brain corresponds to the relative abundance and distribution of high-affinity and low-affinity neurotrophin receptors. J Comp Neurol. 1995;357:296–317. doi: 10.1002/cne.903570209. [DOI] [PubMed] [Google Scholar]
- 2.Andersson J, Nagy S, Björk L, Abrams J, Holm S, Andersson U. Bacterial toxin-induced cytokine production studied at the single-cell level. Immunol Rev. 1992;127:69–96. doi: 10.1111/j.1600-065x.1992.tb01409.x. [DOI] [PubMed] [Google Scholar]
- 3.Baeuerle PA. Pro-inflammatory signaling: last pieces in the NF-kappa B puzzle. Curr Biol. 1998;8:R19–R22. doi: 10.1016/s0960-9822(98)70010-7. [DOI] [PubMed] [Google Scholar]
- 4.Baeuerle PA, Baltimore D. NF kappa B: ten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- 5.Basu S, Dunn AR, Marino MW, Savoia H, Hodgson G, Lieschke GJ, Cebon J. Increased tolerance to endotoxin by granulocyte-macrophage colony- stimulating factor-deficient mice. J Immunol. 1997;159:1412–1417. [PubMed] [Google Scholar]
- 6.Baumann H, Morella KK, Wong GH. TNF-alpha, IL-1 beta, and hepatocyte growth factor cooperate in stimulating specific acute phase plasma protein genes in rat hepatoma cells. J Immunol. 1993;151:4248–4257. [PubMed] [Google Scholar]
- 7.Beg AA, Finco TS, Nantermet PV, Baldwin AS. Tumor necrosis factor and interleukin-1 lead to phosphorylation and loss of IκBα: a mechanism for NF-κB activation. Mol Cell Biol. 1993;13:3301–3310. doi: 10.1128/mcb.13.6.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science. 1999;284:309–13. doi: 10.1126/science.284.5412.309. [DOI] [PubMed] [Google Scholar]
- 9.Dentener MA, Bazil V, Von Asmuth EJ, Ceska M, Buurman WA. Involvement of CD14 in lipopolysaccharide-induced tumor necrosis factor- alpha, IL-6 and IL-8 release by human monocytes and alveolar macrophages. J Immunol. 1993;150:2885–91. [PubMed] [Google Scholar]
- 10.Fearns C, Kravchenko VV, Ulevitch RJ, Loskutoff DJ. Murine CD14 gene expression in vivo: extramyeloid synthesis and regulation by lipopolysaccharide. J Exp Med. 1995;181:857–866. doi: 10.1084/jem.181.3.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fearns C, Loskutoff DJ. Role of tumor necrosis factor alpha in induction of murine CD14 gene expression by lipopolysaccharide. Infect Immun. 1997;65:4822–4831. doi: 10.1128/iai.65.11.4822-4831.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fearns C, Ulevitch RJ. Effect of recombinant interleukin-1beta on murine CD14 gene expression in vivo. Shock. 1998;9:157–163. doi: 10.1097/00024382-199803000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Fong Y, Tracey KJ, Moldawer LL, Hesse DG, Manogue KB, Kenney JS, Lee AT, Kuo GC, Allison AC, Lowry SF, Cerami A. Antibodies to cachectin/tumor necrosis factor reduce interleukin 1 beta and interleukin 6 appearance during lethal bacteremia. J Exp Med. 1989;170:1627–1633. doi: 10.1084/jem.170.5.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–240. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 15.Haziot A, Ferrero E, Kontgen F, Hijiya N, Yamamoto S, Silver J, Stewart CL, Goyert SM. Resistance to endotoxin shock and reduced dissemination of gram- negative bacteria in CD14-deficient mice. Immunity. 1996;4:407–414. doi: 10.1016/s1074-7613(00)80254-x. [DOI] [PubMed] [Google Scholar]
- 16.Herx L, Rivest S, Yong W. Inflammatory cytokine regulation of neurotrophism following CNS trauma. Soc Neurosci Abstr. 1999;25:1535. [Google Scholar]
- 17.Lacroix S, Feinstein D, Rivest S. The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol. 1998;8:625–640. doi: 10.1111/j.1750-3639.1998.tb00189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laflamme N, Rivest S. Effects of systemic immunogenic insults and circulating proinflammatory cytokines on the transcription of the inhibitory factor kappa B alpha within specific cellular populations of the rat brain. J Neurochem. 1999;73:309–321. doi: 10.1046/j.1471-4159.1999.0730309.x. [DOI] [PubMed] [Google Scholar]
- 19.Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1β in mediating NF-κB activity and COX-2 transcription in cells of the blood-brain barrier in response to systemic and localized inflammation, but not during endotoxemia. J Neurosci. 1999;19:10923–10930. doi: 10.1523/JNEUROSCI.19-24-10923.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, Jungbluth A, Wada H, Moore M, Williamson B, Basu S, Old LJ. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci USA. 1997;94:8093–8098. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mayhan WG. Effect of lipopolysaccharide on the permeability and reactivity of the cerebral microcirculation: role of inducible nitric oxide synthase. Brain Res. 1998;792:353–357. doi: 10.1016/s0006-8993(98)00259-5. [DOI] [PubMed] [Google Scholar]
- 22.Nadeau S, Rivest S. Regulation of the gene encoding tumor necrosis factor alpha in the rat brain and pituitary in response to different models of systemic immune challenge. J Neuropathol Exp Neurol. 1999;58:61–77. doi: 10.1097/00005072-199901000-00008. [DOI] [PubMed] [Google Scholar]
- 23.Oldfield BJ, Mckinley MJ. Circumventricular organs. In: Paxinos G, editor. The rat nervous system. Academic; San Diego: 1995. pp. 391–403. [Google Scholar]
- 24.Paxinos G, Watson C. The rat brain in sterotaxic coordinates. Academic; San Diego: 1986. [Google Scholar]
- 25.Pugin J, Heumann ID, Tomasz A, Kravchenko VV, Akamatsu Y, Nishijima M, Glauser MP, Tobias PS, Ulevitch RJ. CD14 is a pattern recognition receptor. Immunity. 1994;1:509–516. doi: 10.1016/1074-7613(94)90093-0. [DOI] [PubMed] [Google Scholar]
- 26.Quan N, Whiteside M, Herkenham M. Time course and localization patterns of interleukin-1β mRNA expression in the brain and pituitary after peripheral administration of lipopolysaccharide. Neuroscience. 1997;83:281–293. doi: 10.1016/s0306-4522(97)00350-3. [DOI] [PubMed] [Google Scholar]
- 27.Swanson LW. Brain maps: structure of the rat brain. Elsevier; New York: 1992. [Google Scholar]
- 28.Sweet MJ, Hume DA. Endotoxin signal transduction in macrophages. J Leukoc Biol. 1996;60:8–26. doi: 10.1002/jlb.60.1.8. [DOI] [PubMed] [Google Scholar]
- 29.Ulevitch RJ. Toll gates for pathogen selection. Nature. 1999;401:755–756. doi: 10.1038/44490. [DOI] [PubMed] [Google Scholar]
- 30.Vallières L, Rivest S. Regulation of the genes encoding interleukin-6, its receptor, and gp130 in the rat brain in response to the immune activator lipopolysaccharide and the proinflammatory cytokine interleukin-1β. J Neurochem. 1997;69:1668–1683. doi: 10.1046/j.1471-4159.1997.69041668.x. [DOI] [PubMed] [Google Scholar]
- 31.Wright SD. Toll, a new piece in the puzzle of innate immunity. J Exp Med. 1999;189:605–609. doi: 10.1084/jem.189.4.605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wright SD, Ramos RA, Tobias PS, Ulevitch RJ, Mathison JC. CD14, a receptor for complexes of lipolysaccharide (LPS) and LPS binding protein. Science. 1990;249:1431–1433. doi: 10.1126/science.1698311. [DOI] [PubMed] [Google Scholar]
- 33.Wright SD, Ramos R, Hermanowski-Vosatka A, Rockwell P, Detmers PA. Activation of the adhesive capacity of CR3 on neutrophils by endotoxin: dependence on lipopolysaccharide binding protein and CD14. J Exp Med. 1991;173:1281–1286. doi: 10.1084/jem.173.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]