

Abstract
Excessive zinc influx may contribute to neuronal death after certain insults, including transient global ischemia. In light of evidence that levels of intracellular free Zn2+associated with neurotoxicity may be sufficient to inhibit glyceraldehyde-3-phosphate dehydrogenase (GAPDH), experiments were performed looking for reduced glycolysis and energy failure in cultured mouse cortical neurons subjected to lethal Zn2+exposure. As predicted, cultures exposed for 3–22 hr to 40 μm Zn2+ developed an early increase in levels of dihydroxy-acetone phosphate (DHAP) and fructose 1,6-bisphosphate (FBP) and a progressive loss of ATP levels, followed by neuronal cell death; furthermore, addition of the downstream glycolytic substrate pyruvate to the bathing medium attenuated the fall in ATP and neuronal death.
However, an alternative to direct Zn2+ inhibition of GAPDH was raised by the observation that Zn2+exposure also induced an early decrease in nicotinamide-adenine dinucleotide (NAD+) levels, an event itself capable of inhibiting GAPDH. Favoring this indirect mechanism of GAPDH inhibition, the neuroprotective effects of pyruvate addition were associated with normalization of cellular levels of NAD+, DHAP, and FBP. Zn2+-induced neuronal death was also attenuated by addition of the energy substrate oxaloacetate, the activator of pyruvate dehydrogenase, dichloroacetate, or the inhibitors of NAD+ catabolism, niacinamide or benzamide. Acetyl carnitine, α-keto butyrate, lactate, and β-hydroxy-butyrate did not attenuate Zn2+-induced neurotoxicity, perhaps because they could not regenerate NAD+ or be used for energy production in the presence of glucose.
Keywords: pyruvate, niacinamide, energy depletion, PARS, ATP levels, GAPDH inhibition
Zn2+ is an essential ion in mammalian cells. It is incorporated into the active site of many metalloenzymes (Vallee and Falchuk, 1993; Berg and Shi, 1996) and is probably used in the CNS as a neurotransmitter or neuromodulator (Frederickson, 1989). It is stored within vesicles in presynaptic boutons (Frederickson et al., 1983; Danscher et al., 1985) and is released together with glutamate by membrane depolarization in a Ca2+-dependent manner (Assaf and Chung, 1984; Howell et al., 1984; Charton et al., 1985). Although its precise role in neural signaling has not been defined, it may regulate neurotransmission by altering the function of several receptors and channels, including NMDA receptors, GABA receptors, glycine receptors, ATP receptors, and voltage-gated Na+ and Ca2+channels (Harrison and Gibbons, 1994;Smart et al., 1994). Zn2+ may also contribute to neuronal death in disease states, such as transient global ischemia or prolonged seizures (Choi and Koh, 1998). Brain ischemia triggers the translocation of presynaptic Zn2+ (over the ensuing 1–24 hr) into the soma of selectively vulnerable hippocampal CA1 neurons, as well as other vulnerable neurons in cortex, amygdala, striatum, and thalamus that later go on to die (Tonder et al., 1990; Koh et al., 1996). Both this translocation and subsequent selective neuronal cell death can be blocked by the administration of an extracellular chelator CaEDTA (Koh et al., 1996). The extracellular concentration of Zn2+ after intense neuronal activity may reach several hundred micromolar (Assaf and Chung, 1984), concentrations that are neurotoxic to cultured cortical neurons (Yokoyama et al., 1986; Choi and Koh, 1998).
It is presently unknown why exposure to high concentrations of extracellular Zn2+ can induce neuronal death. A critical first step appears to be entry across the plasma membrane, mediated by several routes, including voltage-gated calcium channels, agonist-gated calcium channels, and reverse operation of the sodium-calcium exchanger (Choi and Koh, 1998). We have used mag fura 5 to detect elevations in neuronal intracellular free Zn2+([Zn2+]i) associated with zinc exposure under several conditions (Sensi et al., 1997). Although mag fura 5 is ratiometric and suitable for detecting early increases in [Zn2+]i, its high affinity (KD of ∼10−12) may limit its ability to detect peak concentrations. Therefore we have recently used a low affinity (KD of ∼1 μmZn2+) non-ratiometric Zn2+-selective dye, Newport Green (Haugland, 1996). A 5 min exposure to 300 μmZn2+ under depolarizing conditions - an insult that triggers widespread cortical neuronal death over the next hr (Yokoyama et al., 1986) –resulted in [Zn2+]i reaching 400–600 nm as assessed with Newport Green (Canzoniero et al., 1999; Sensi et al., 1999). At this concentration, Zn2+ can inhibit the key glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in solution, with 400 nm Zn2+ producing 50% inhibition of purified GAPDH (Krotkiewska and Banas, 1992). Zn2+ has also been reported to inhibit phosphofructokinase in solution, with an IC50 of 1.5 μm in the presence of 60 μm fructose 6-phosphate (Ikeda et al., 1980).
The purpose of the present study was to test the hypothesis that neurotoxic Zn2+ exposure leads to inhibition of GAPDH, resulting in a buildup of the upstream substrates dihydroxyacetone phosphate (DHAP) and fructose bisphosphate (FBP) and a fall in neuronal ATP levels. Furthermore, if GAPDH inhibition contributed importantly to the neurotoxic effects of Zn2+, we postulated that the administration of suitable downstream energy substrates might be neuroprotective against Zn2+ exposure.
Parts of this work have been published previously in abstract form (Sheline and Choi, 1997).
MATERIALS AND METHODS
Cell culture and toxicity studies. Near-pure neuronal cultures were prepared from embryonic day 15 (E15) mouse cortices as described previously (Sheline and Choi, 1998). Dissociated cortical neurons were taken from E15 Swiss–Webster mice and plated in Eagle's minimal essential medium (MEM) (Earle's salts, glutamine-free) containing 21 mm glucose, 5% fetal bovine serum, and 5% horse serum at a density of 5 hemispheres per plate onto poly-d-lysine–laminin-coated plates. At 3 din vitro (DIV), cytosine arabinoside was added to 10 μm to inhibit glial growth. Chronic toxicity studies were initiated by washing cultures four times with MEM containing 21 mm glucose, followed by exposure to ZnCl2 in the same media supplemented with 1 μm (+)-5- methyl-10,11-dihydro-5H-dibenzo [a,d] cycloheplen-5,10-imine maleate (MK-801) and 100 ng/ml of neurotrophin-4 (NT-4) or BDNF. MK-801 was included to prevent wash-induced activation of NMDA receptors and was not itself toxic over the ensuing 24 hr, and NT-4 or BDNF were included as a needed survival factor (serum could not be used because it chelates Zn2+). Acute toxicity studies were initiated by washing cultures four times with HEPES-buffered salt solution, followed by exposure to ZnCl2 in the same media supplemented with 1 μm MK-801 and 100 ng/ml NT-4 or BDNF as a survival-promoting activity in the presence or absence of 60 mm KCl for 5 or 15 min. The exposure was terminated by washing three times with MEM containing 21 mm glucose, the cultures were put back into the same media supplemented with 1 μm MK-801 and 100 ng/ml NT-4 or BDNF as a survival-promoting activity, and cell death was assayed 24 hr later. Near-pure neuronal cultures were washed seven times using salt solution (same as in MEM) without glucose but in the presence of 1× amino acids before testing the use of different energy substrates at 6 mm in the same glucose-free solution plus MK-801 and NT-4 for 24 hr. These same substrates were tested against 40 μmZn2+ exposure, as were the effects of late addition of pyruvate. Cell death was estimated by phase-contrast microscopy after staining with 0.01% trypan blue for 60 min at 37°C and assessed quantitatively by measuring lactate dehydrogenase (LDH) efflux (Koh and Choi, 1987) or propidium iodide fluorescence (Sheline and Choi, 1998) and comparing it with the complete neuronal death induced by exposure to 20 μm A23187 for 24 hr.
Determination of dihdroxyacetone phosphate, lactate, and ATP levels. Near-pure neuronal cultures (8–9 DIV) were used for the ATP measurements. Cultures were lysed by addition of 0.1m NaOH–1 mm EDTA at the indicated time points. After centrifugation at 13,000 ×g, the supernatant was neutralized and protein was precipitated by addition of 100 μl of 0.5 mperchloric acid. ATP was measured by the luciferin–luciferase luminescence assay and was normalized to sham-washed controls and to protein content as determined by the bichichonic acid assay (Lust et al., 1981). DHAP, FBP, and extracellular lactate measurements were made on neuronal cell lysates or the bathing medium (for extracellular lactate) prepared in a similar manner, but were lysed by addition of 6% perchloric acid and protein precipitated by addition of potassium carbonate to pH 3.5. DHAP was measured by its enzymatic conversion to glycerol-3-phosphate by glycerol-3-phosphate dehydrogenase, and the concomitant oxidation of nicotinamide-adenine dinucleotide, reduced form (NADH) was measured spectrophotometrically. Glyceraldehyde 3-phosphate was subsequently measured in a linked reaction by its enzymatic conversion to DHAP by triosephosphate isomerase, followed by addition of aldolase to measure FBP levels (Michal, 1974). Extracellular lactate concentration was measured by its conversion to pyruvate in the presence of excess hydrazine, nicotinamide-adenine dinucleotide (NAD+), and LDH to drive the production of NADH (Gutmann and Wahlefeld, 1974). Other metabolites were also measured by their enzymatic conversion and the concomitant oxidation or reduction of NADH, NAD+, or nicotinamide-adenine dinucleotide phosphate (NADP+).
Determination of NAD+ and NADH levels. Neuronal cultures (8–9 DIV) were used for the NAD+ and NADH measurements. For the NAD+ and NADH measurements, cultures were lysed by addition of 75% ethanol–0.05 m K2HPO4 after a 4 hr 40 μmZn2+ exposure. Protein was precipitated by addition of ZnCl2 to 20 mmand centrifuged at 13,000 × g, and the supernatant was assayed for NAD+ and NADH levels (Tilton et al., 1991). NAD+ in the supernatant was measured after its enzymatic conversion to NADH by alcohol dehydrogenase, resulting in an increase in the fluorescence spectrum between 400 and 600 nm after an excitation at 340 nm using a Perkin-Elmer (Emeryville, CA) LS 50B. NADH was measured by the difference in the fluorescence spectrum between 400 and 600 nm before and after treatment of the supernatant with lactate dehydrogenase (Sander et al., 1976).
Whole-cell lysates. Neuronal cultures (8 DIV) were serum-deprived for 1 hr and then exposed as indicated to 100 ng/ml BDNF in the presence or absence of 40 μmZn2+. The cells were then washed twice with ice-cold PBS and resuspended in cold buffer A (1% NP-40, 20 mm Tris-Cl, pH 7.5, 10 mm EGTA, 40 mmβ-glycerophosphate, 2.5 mmMgCl2, 2 mm orthovanadate, 1 mm dithiothreitol, 1 mmphenyl-methyl-sulfonylfluoride, 20 μg/ml aprotinin, and 20 μg/ml leupeptin) for 15 min.
Lysates were centrifuged at 15000 × g for 5 min, and supernatants were retained for analysis. Protein concentrations were determined by the bicinchoninic acid method (Pierce, Rockford, IL) using bovine serum albumin as standard.
Western blotting. For Western blots, 25 μg of total cell protein were resolved in 8% SDS-PAGE gels, transferred to nitrocellulose membranes (MSI, Westboro, MA), and incubated with antibodies specific for phospho-ERKs [anti-active mitogen-activated protein kinase (MAPK) antibodies; New England Biolabs, Beverly, MA]. Bound antibodies were detected by the enhanced chemiluminescence method (Amersham Pharmacia Biotech, Arlington Heights, IL).
Reagents. Most reagents were from Sigma (St. Louis, MO). The neurotrophins NT-4 and BDNF were the kind gift of Amgen (Thousand Oaks, CA).
RESULTS
Zinc exposure did not block neurotrophin signaling
A previous study in PC12 cells suggested that Zn2+ can alter the conformation and biological activities of several neurotrophins (Ross et al., 1997). Arguing against the possibility that Zn2+-induced neuronal death is mediated by an acute loss of neurotrophin influence, ATP depletion and neuronal death occurred more quickly after Zn2+exposure than after serum deprivation (data not shown; see Figs. 3, 5). Furthermore, 40 μm Zn2+ did not prevent 100 ng/ml BDNF or NT-4 from initiating the TrkB-mediated signaling cascade resulting in phosphorylation of the ERK family of protein kinases (Fig. 1).
Fig. 3.
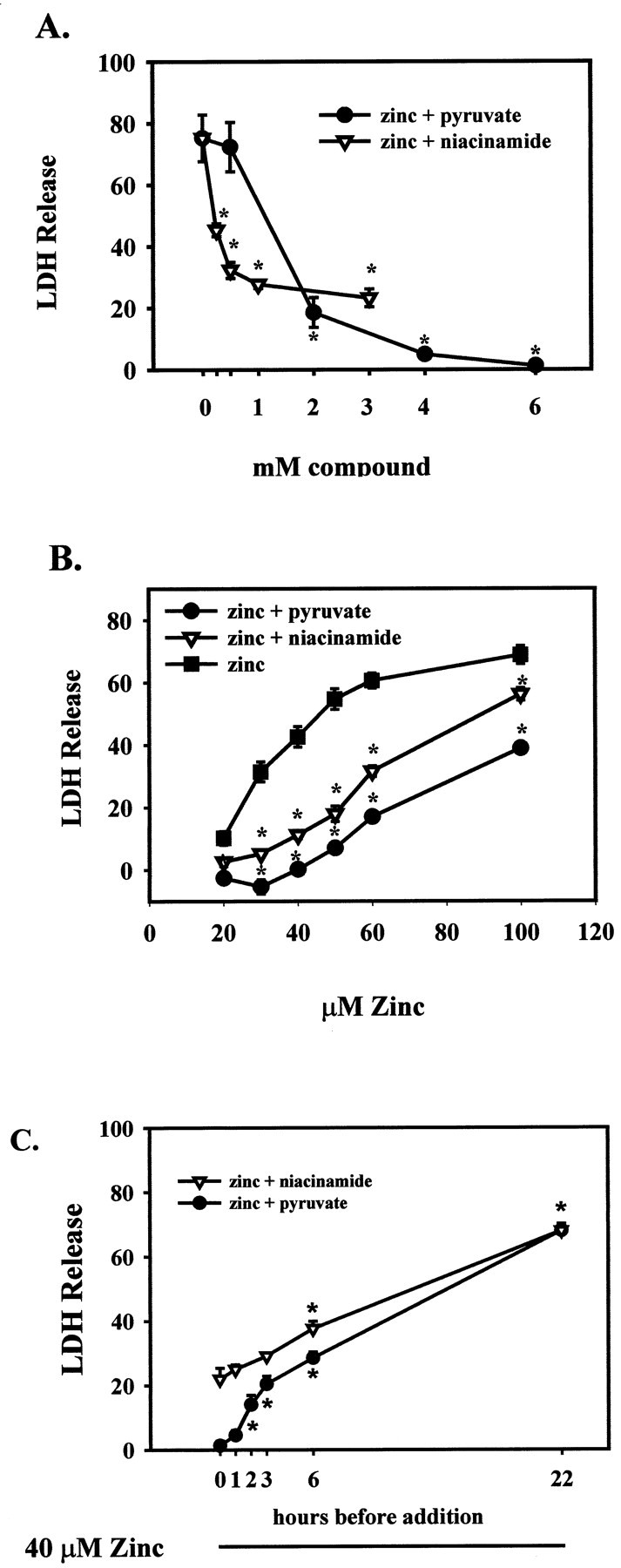
Inhibition of slow zinc toxicity by pyruvate or niacinamide. A, Neuronal cultures were exposed to 40 μm Zn2+ for 24 hr in the presence of the indicated concentrations of pyruvate and niacinamide, and cell death was assessed by LDH release to the bathing medium (mean + SEM,n = 9–12 cultures per condition), scaled to the level associated with near-complete neuronal death (produced by exposure to 20 μm A23187 for 24 hr, 100%). *p < 0.05 indicates difference from Zn2+ exposure alone by one-way ANOVA, followed by a Bonferroni test. B, Neuronal cultures were exposed to the indicated concentrations of Zn2+ for 24 hr in the presence or absence of 4 mm pyruvate or 3 mm niacinamide. *p < 0.05 indicates difference from Zn2+ exposure alone at the same time point. C, Zn2+ (40 μm) was added to the bathing medium at time 0, and afterwards, at the indicated times, 4 mm pyruvate or 3 mmniacinamide were added. Cell death was measured at 22 hr. *p < 0.05 signifies difference from addition at time 0.
Fig. 5.
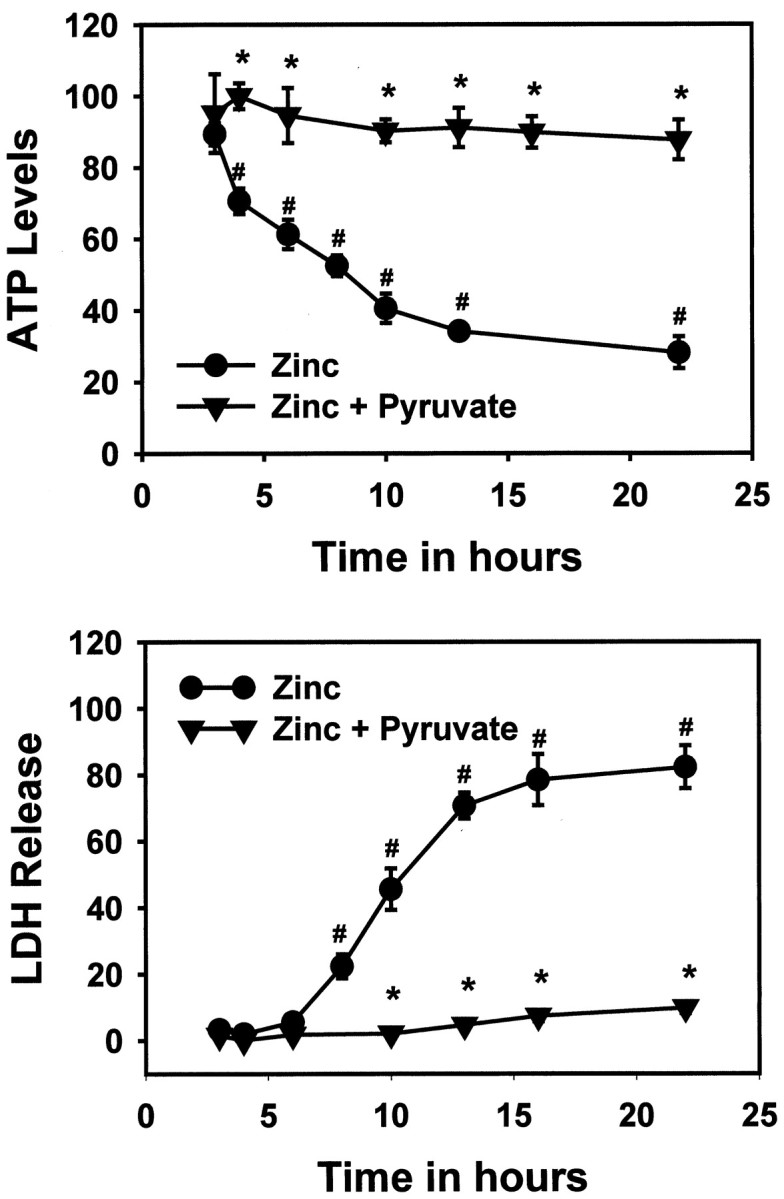
Time course of neuronal cell death and ATP depletion during exposure to zinc with or without pyruvate. Neuronal cultures were exposed continuously to 40 μmZn2+ in the presence or absence of 4 mmpyruvate, and cellular ATP levels (top) or LDH release (bottom) were determined at the indicated times. Cell death was determined by LDH release to the bathing medium (scaled to signal associated with complete neuronal death induced by 24 hr exposure to 20 μm A23187, set as 100). ATP loss was determined by comparison with levels measured in sham-washed controls. Error bars in both parts are SEM; n = 9–12 cultures per condition, pooled from three independent experiments. *p < 0.05 signifies difference from Zn2+ exposure alone by two-way ANOVA, followed by a Bonferroni test. #p < 0.05 signifies difference from sham-washed controls by two-way ANOVA, followed by a Bonferroni test.
Fig. 1.

Toxic levels of extracellular zinc do not block BDNF-induced signaling. Neuronal cultures were deprived of serum and treated in the absence (SD) or presence of 40 μm Zn2+ (Zn) with 100 ng/ml BDNF for the indicated times. Cell extracts were prepared, and activation of the MAP kinase pathway was determined by Western blotting using antibodies that detect the phosphorylated form of ERK1 and ERK2 (p44 and p42, respectively). We have quantified the bands using the 0.5 hr plus BDNF, minus zinc as 100%.
Zinc exposure increased neuronal levels of DHAP and FBP
Near-pure cortical neuronal cultures, 8–9 d in vitro, were exposed to 40 μmZn2+ for 4 hr, an insult duration that did not cause cellmembrane failure as measured by LDH efflux to the bathing medium (see below) or staining with trypan blue or propidium iodide (data not shown). At that time, cells were lysed, and the lysate was assayed for DHAP, FBP, fructose 6-phosphate, glucose 6-phosphate, pyruvate, phosphoenolpyruvate, and glycerate 2-phosphate. Both DHAP and FBP levels were increased several-fold, whereas other measured metabolites (except 2-phospho-glycerate) were unchanged (Table1). Glyceraldehyde 3-phosphate was undetectable in either control cultures or cultures exposed to Zn2+, most likely because it was preferentially converted into DHAP (Lehninger et al., 1993).
Table 1.
Zinc exposure selectively increased DHAP and FBP levels, and this increase was reversed by addition of pyruvate or niacinamide
| (Nanomoles/plate ± SEM) | Control | 40 μm Zinc | Zinc + pyruvate | Zinc + niacinamide | Staurosporine |
|---|---|---|---|---|---|
| Glucose-6-phosphate | 4.2 ± 0.6 | 7.6 ± 1.0 | N.P. | N.P. | N.P. |
| Fructose-6-phosphate | 1.6 ± 0.2 | 3.2 ± 0.4 | N.P. | N.P. | N.P. |
| DHAP | 5.2 ± 0.6 | 22.4 ± 0.8* | 5.2 ± 0.6# | 5.0 ± 0.6# | 3.2 ± 2.2 |
| Fructose bisphosphate | 5.2 ± 0.8 | 51.4 ± 5.0* | 5.0 ± 0.8# | 11.2 ± 1.4# | 5.8 ± 0.6 |
| 2-Phospho-glycerate | 6.25 ± 0.5 | 11.7 ± 0.5* | N.P. | N.P. | N.P. |
| Phospho-enolpyruvate | 5.75 ± 1.7 | 8.2 ± 4.7 | N.P. | N.P. | N.P. |
| Pyruvate | 14.2 ± 2.2 | 20 ± 4.5 | N.P. | N.P. | N.P. |
Near-pure cortical neuronal cultures were sham-washed or exposed to 40 μm Zn2+ in the presence or absence of 4 mm pyruvate or 1 mm niacinamide. The cells were then harvested and assayed for levels of the indicated glycolytic intermediates (results were pooled from three separate experiments; n = 5–8 cultures per condition). Exposure to 100 nm staurosporine for 4–6 hr, sufficient to trigger widespread apoptosis, did not mimic the ability of Zn2+ to elevate DHAP or FBP. N.P. indicates that the experiment was not performed. * signifies difference from sham-washed controls, and
signifies difference from Zn2+-treated cultures at p < 0.05 by one-way ANOVA, followed by a Bonferroni test.
In contrast, when the neuronal cultures were exposed to 100 nm staurosporine for 6 hr, an exposure capable of triggering programmed cell death (Koh et al., 1995; Weil et al., 1996), no changes were seen in the above metabolites (Table 1).
Zinc-induced neuronal death was attenuated by pyruvate
Increasing the duration of 40 μmZn2+ exposure to 24 hr resulted in widespread neuronal degeneration, most marked initially in processes, and later accompanied by trypan blue staining (Fig.2) and release of LDH to the bathing medium (Fig. 3). Addition of 2–6 mm pyruvate to the bathing medium during this toxic Zn2+ exposure resulted in a concentration-dependent reduction in neuronal death, with near-complete preservation of neurons produced by 6 mm pyruvate (Figs. 2,3A). Pyruvate (4 mm) attenuated the neuronal death induced by 24 hr exposure to Zn2+ concentrations between 20 and 100 μm (Fig. 3B), even if added in a delayed manner 3–6 hr after the onset of Zn2+exposure (Fig. 3C). The neuroprotective effect of pyruvate addition was well maintained for at least 48 hr after the onset of Zn2+ exposure.
Fig. 2.
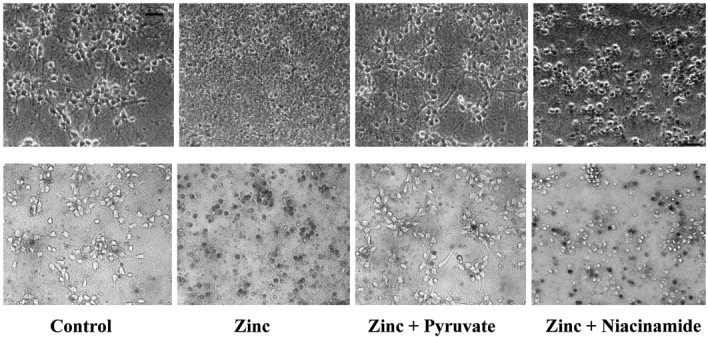
Zinc-induced neuronal death is attenuated by pyruvate. Phase-contrast (top) and matched bright-field fields after staining with trypan blue (bottom) were taken in near-pure neuronal cultures 24 hr after exposure to sham wash (Control), 40 μmZn2+, or 40 μm Zn2+in the presence of 4 mm pyruvate or 1 mmniacinamide. Scale bar, 50 μm.
The more fulminant form of Zn2+ toxicity induced by brief (5–15 min) exposure to high concentrations of Zn2+ (100–400 μm) in the presence of 60 mm K+ [to depolarize cells and enhance entry through voltage-gated calcium channels (Weiss et al., 1993; Sensi et al., 1997)] could also be partially attenuated by 10 mm pyruvate or niacinamide (Fig.4).
Fig. 4.

Inhibition of fast zinc toxicity by pyruvate or niacinamide. Neuronal cultures were exposed to 100 μmZn2+ for 15 min or 400 μmZn2+ for 5 min in the presence or absence of 60 mm KCl, with 10 mm pyruvate or 10 mm niacinamide present 1 hr before, during, and 24 hr afterwards as indicated. Neuronal death was determined 24 hr after the onset of Zn2+ exposure by LDH efflux (n = 9–12 cultures per condition). *p < 0.05 signifies difference from Zn2+ exposure alone. #p < 0.05 signifies difference associated with pyruvate or niacinamide addition on neuronal death induced by Zn2+ plus KCl.
Zinc-induced ATP depletion preceded cell death, and both were sensitive to pyruvate
Neuronal cultures were exposed to 40 μmZn2+ for 3–22 hr, with LDH efflux and ATP levels assessed at intermediate time points. ATP loss was detectable after 4 hr, whereas cell death measured by LDH release was not detectable until after 8 hr (Fig. 5). Cell death assessed by propidium iodide staining was not detectable until after 6 hr of Zn2+ exposure (data not shown). The addition of 4 mm pyruvate to the bathing medium blocked both ATP loss and cell death (Fig. 5).
Zinc-induced decrease in NAD+ was attenuated by pyruvate or niacinamide
Although the above observations were consistent with the hypothesis that direct inhibition of GAPDH by Zn2+ contributed importantly to the neurotoxic effects of the latter, unexpectedly, inclusion of 4 mm pyruvate during Zn2+exposure abolished the buildup of DHAP and FBP found in cultures exposed to Zn2+ alone (Table 1).
Exposure to 40 μm Zn2+ for 4 hr also induced a several-fold decrease in NAD+levels without a compensatory increase in NADH levels, as well as an increase in lactate. The inclusion of 4 mm pyruvate to the Zn2+ exposure abolished the decrease in NAD+, perhaps at the expense of NADH, and further increased lactate (Table 2). In addition, inclusion of 1–3 mm niacinamide, benzamide or 3-aminobenzamide, competitive inhibitors of NAD+-catabolizing enzymes (for review, see Szabo and Dawson, 1998), blocked the drop in NAD+ levels and associated neuronal death (Tables 2, 3; data not shown).
Table 2.
Zinc exposure decreased NAD+, and this effect was reversed by addition of pyruvate or niacinamide
| Condition | NAD+ (nmol/plate) | NADH (nmol/plate) | Lactate (nmol/plate) |
|---|---|---|---|
| Control | 4.63 ± 0.31 | 0.58 ± 0.052 | 2020 ± 130 |
| Control + 4 mm pyruvate | 5.62 ± 0.59* | 0.20 ± 0.016* | 2930 ± 110* |
| Control + 1 mmniacinamide | 6.170 ± 0.36* | 0.66 ± 0.058 | 2650 ± 230* |
| 40 μm Zinc | 1.36 ± 0.16* | 0.83 ± 0.14 | 2470 ± 80* |
| 40 μm Zinc + 4 mm pyruvate | 4.86 ± 0.22# | 0.042 ± 0.027# | 3600 ± 140# |
| 40 μm Zinc + 1 mmniacinamide | 4.38 ± 0.40# | 0.89 ± 0.059 | 3270 ± 170# |
Neuronal cultures (6 × 106 cells per plate) were sham-washed (Control) or exposed to 40 μmZn2+ in the presence or absence of 4 mmpyruvate or 1 mm niacinamide for 4 hr, after which the cells were harvested and assayed for levels of NAD+, NADH, or extracellular lactate (results are pooled from three separate experiments; n = 5–8 cultures per condition). * signifies difference from sham-washed controls at p < 0.05 by two-way ANOVA, followed by a Bonferroni test.
signifies difference from Zn2+-treated cultures at p < 0.05.
Table 3.
Effect of selected energy substates, NAD+catabolism inhibitors, or cofactors for pyruvate dehydrogenase against zinc neurotoxicity or glucose deprivation-induced neuronal death
| % Cell death | 40 μm Zinc | Glucose deprivation |
|---|---|---|
| No addition | 67.7 ± 2.8% | 89.6 ± 4.7% |
| +3 mm Pyruvate | 0.3 ± 1.9%* | 5.2 ± 2.8%* |
| +3 mm Pyruvate + 3 mm CIN | 61.2 ± 4.1%# | 76.5 ± 5.9%# |
| +3 mm Pyruvate + 20 mmoxamate | 47.9 ± 2.7%# | 25.7 ± 2.8%# |
| +3 mmOxaloacetate | 1.6 ± 0.9%* | 17.4 ± 1.7%* |
| +6 mm Malate | 37.3 ± 1.8%* | 67.6 ± 4.3%* |
| +6 mm Succinate | 48.8 ± 2.4%* | 77.4 ± 2.1%* |
| +6 mm Lactate | 67.8 ± 3.5% | 3.4 ± 2.3%* |
| +6 mm β-Hydroxy-butyrate | 73.1 ± 4.9% | 12.9 ± 1.5%* |
| +6 mmα-Keto-butyrate | 61.3 ± 3.4% | 90.3 ± 4.3% |
| +2 mm FBP | 59.1 ± 5.1% | 68.6 ± 1.4%* |
| +2 mm DHAP | 62.3 ± 4.1% | 30.7 ± 4.8%* |
| +6 mm Acetyl-carnitine | 75.0 ± 4.0% | 99.0 ± 2.8% |
| +3 mm Niacinamide | 15.1 ± 1.6%* | 86.9 ± 1.4% |
| +3 mmBenzamide | 21.1 ± 2.9%* | 77.9 ± 3.9% |
| +3 mm 3-Aminobenzamide | 24.0 ± 3.7%* | 83.8 ± 1.7% |
| +6 mm Dichloroacetate | 21.4 ± 1.6%* | 79.0 ± 3.6% |
| +2 mmRiboflavin | 65.4 ± 3.5% | 83.3 ± 2.0% |
| +6 mm Thiamine | 67.0 ± 1.7% | 84.4 ± 1.7% |
| +0.05 mm Lipoic acid (reduced) | 69.4 ± 2.4% | 105.8 ± 1.7% |
| +0.25 mm Lipoic amide | 60.2 ± 2.4% | 89.8 ± 1.4% |
Near-pure neuronal cultures were exposed to 40 μmZn2+ for 24 hr or to glucose deprivation for 24 hr alone or in the presence of the indicated compounds (titrated to optimal concentrations; data not shown), and cell death was determined by LDH efflux or propidium iodide fluorescence. n = 9–12 cultures per condition. * indicates difference from no addition and
indicates difference from exposure plus pyruvate at p < 0.05. CIN, cinnamic acid.
Effect of other energy substrates, NAD+catabolism inhibitors, or pyruvate dehydrogenase cofactors on zinc-induced neuronal death or glucose deprivation-induced death
Other energy substrates, NAD+catabolism inhibitors, and pyruvate dehydrogenase (PDH) cofactors [pyruvate, oxaloacetate, malate, succinate, lactate, β-hydroxy-butyrate, α-keto-butyrate, FBP, DHAP, acetyl-carnitine, niacinamide, benzamide, 3-aminobenzamide, dichloroacetate (DCA), riboflavin, thiamine, lipoic acid, and lipoic amide] were tested at optimal concentrations (concentration titration data not shown) for their ability to reduce Zn2+-induced or glucose deprivation-induced neuronal death. At these concentrations, none of the compounds were found to be toxic or to induce gross changes in cell volume. Oxaloacetate, niacinamide, benzamide, and 3-aminobenzamide were nearly as effective as pyruvate at attenuating Zn2+-induced neuronal death, but of these, only pyruvate and oxaloacetate were used as energy substrates (Table3). Niacinamide, benzamide, and 3-aminobenzamide competitively inhibit all NAD+-catabolizing enzymes; niacinamide is also a precursor for NAD+ synthesis (for review, see Szabo and Dawson, 1998). In addition, DCA, which functions as an activator of the PDH complex (inhibiting the kinase that inhibits the complex), partially attenuated Zn2+ neurotoxicity without serving as an energy substrate. However, the other PDH complex cofactors, thiamine or lipoic acid, were ineffective against Zn2+neurotoxicity. The effect of DCA was synergistic with low levels of pyruvate (data not shown). The protective effect of pyruvate was attenuated by cinnaminic acid, an inhibitor of the monocarboxylate transporter (Schurr et al., 1997), and by oxamate, a competitive inhibitor of lactate dehydrogenase (Wong et al., 1997). These results are summarized in the proposed model for Zn2+-induced neurotoxicity (Fig. 6). Compounds that could not be converted to pyruvate or serve as energy substrates or PDH complex cofactors in neuronal cultures did not attenuate Zn2+-induced neuronal death (Table 3). Two substrates differed in their ability to protect against glucose deprivation-induced death versus Zn2+-induced death. Lactate and β-hydroxy-butyrate protected against the former but not the latter.
Fig. 6.

Summary model for Zn2+-induced neuronal death in vitro. VSCC, Voltage-sensitive calcium channel; G-3-P, glyceraldehyde-3-phosphate; 1,3-BPG, 1,3- bisphosphoglycerate. A, The model for Zn2+-induced changes. B, The protective mechanisms for niacinamide, 3-aminobenzamide, and benzamide involve inhibition of an unknown NAD+-catabolizing enzyme, thereby maintaining NAD+ levels; the protective mechanism for pyruvate involves maintaining NAD+ levels through its conversion to lactate.
DISCUSSION
We observed that neurotoxic levels of Zn2+ exposure induced in cortical neurons an early increase in glycolytic intermediates preceding GAPDH, followed by a progressive loss of ATP levels and neuronal cell death. Addition of the downstream glycolytic substrate pyruvate to the bathing medium attenuated both the fall in ATP and neuronal death. Simple chelation of Zn2+ by pyruvate is unlikely because the log stability constant for Zn2+ pyruvate is 1.3 (Martell and Smith, 1995), and reduction of neuronal death was also achieved by adding another downstream energy substrate, oxaloacetate, or the activator of pyruvate dehydrogenase, dichloroacetate. These findings are consistent with our initial hypothesis that a key mechanism of Zn2+neurotoxicity is energy loss attributable to inhibition of glycolysis at GAPDH, an event that could be mediated directly by Zn2+ (see above). In nondividing bacterial cells, GAPDH has the largest control strength of all the glycolytic enzymes for metabolic regulation (Poolman et al., 1987). We have demonstrated previously that chemical inhibition of GAPDH with α-monochlorohydrin can induce cultured cortical neurons to undergo apoptosis (Sheline and Choi, 1998), an event induced by levels of Zn2+ exposure (20–35 μm) comparable with those used in the present experiments (D. Lobner and D. W. Choi, unpublished observations) (Y. H. Kim et al., 1999a).
However unexpectedly, Zn2+ exposure also induced an early fall in NAD+ levels, an event itself capable of inhibiting GAPDH (Rovetto et al., 1975; Tilton et al., 1991). Favoring indirect inhibition of GAPDH caused by loss of NAD+, the neuroprotective effects of pyruvate addition were associated with normalization of cellular levels of NAD+ and glycolytic intermediates preceding GAPDH. The latter event would not be expected to occur if GAPDH was directly inhibited by Zn2+. Also favoring a central role for Zn2+-induced depression of NAD+ levels in triggering neuronal death, the fall in NAD+ and neuronal death induced by Zn2+ was attenuated by the NAD+ catabolism inhibitors niacinamide, benzamide, or 3-aminobenzamide (for review, seeSzabo and Dawson, 1998).
One postulated mechanism of Zn2+neurotoxicity is inhibition of mitochondrial electron transport (Skulachev et al., 1967; Link and von Jagow, 1995; Manev et al., 1997); this inhibition would decrease neuronal NAD+ levels. However, there was not the expected compensatory increase in NADH levels (Franke et al., 1976;Pryor et al., 1992) (Table 2). Alternative attractive possibilities would be inhibition of NAD+ synthesis or activation of NAD+ catabolism. Zn2+ has been shown to both activate and inhibit different NAD+-catabolizing enzymes depending on the cell type and the conditions (Larsen et al., 1982; Kukimoto et al., 1996; Jorcke et al., 1997). A prominent NAD+-catabolizing enzyme is poly (ADP-ribose) synthetase (PARS), an enzyme likely involved in the detection and repair of single-stranded DNA breaks (Wang et al., 1995). We demonstrated here that three competitive inhibitors of PARS [niacinamide, benzamide, and 3-aminobenzamide (for review, see Szabo and Dawson, 1998)] all attenuate Zn2+neurotoxicity (Fig. 2, Table 3), although these inhibitors also inhibit NAD+ glycohydrylase, another enzyme that breaks down NAD+ (Ziegler et al., 1996). PARS has been implicated as a mediator of neuronal damage after glutamate exposure (Zhang et al., 1994) or hypoxic insults (Eliasson et al., 1997; Endres et al., 1997), consistent with a model in which glutamate receptor overactivation leads to the formation of nitric oxide and other reactive oxygen species (ROS), causing DNA strand breakage and PARS activation (for review, see Szabo and Dawson, 1998).
Cellular Zn2+ overload itself has been suggested to enhance ROS production (E. Y. Kim et al., 1999; Y. H. Kim et al., 1999a,b). We have found that the powerful ROS scavenger C3-polar regioisomer buckministerfullerene (Dugan et al., 1997) was relatively ineffective at attenuating Zn2+-induced cortical neuronal death (L. L. Dugan and D. W. Choi, unpublished observations), and a recent study in cortical cultures did not see a general increase in cytosolic ROS in the first hour after toxic Zn2+ exposure (Sensi et al., 1999). Furthermore α-keto butyrate, which blocks H2O2 hydroxyl radical-induced neuronal death (Desagher et al., 1997) by chemically inactivating H2O2(Holleman, 1904; Desagher et al., 1997), was inactive against Zn2+ neurotoxicity in the present study (Table 3). Regardless of whether Zn2+promotes ROS formation, the possibility that Zn2+ might somehow activate PARS is consistent with the recent observation that the high-affinity Zn2+ chelator N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine inhibits PARS (Virag and Szabo, 1999) (although this observation could simply reflect a baseline requirement of PARS for a Zn2+ cofactor because it has a Zn2+ finger DNA binding domain).
The present proposal that indirect inhibition of GAPDH leading to energy failure is an important mediator of Zn2+-induced neuronal death does not exclude the possibility that Zn2+ may have other death-promoting actions. Direct inactivation of BDNF or NT-4/5 may be considered (Ross et al., 1997) but is unlikely in the present system because Zn2+ exposure did not block the phosphorylation of ERK1 and ERK2, downstream effectors of the TrkB neurotrophic signaling cascade. Activation of an extracellular acid sphingomyelinase producing the apoptotic signaling molecule ceramide (Schissel et al., 1996) is another possible toxic mechanism, although this enzyme is inactive at the physiological pH used in the present experiments.
The observed ability of pyruvate to restore NAD+ to control levels at the expense of NADH provides a plausible explanation for its ability to counteract each of the stated effects of Zn2+exposure on neuronal cultures (Table 2). The increase in extracellular lactate associated with pyruvate addition and the inhibition of pyruvate-induced neuroprotection by oxamate, an inhibitor of lactate dehydrogenase, are consistent with pyruvate generating NAD+ from NADH through its conversion to lactate. However, niacinamide was not as effective as pyruvate in attenuating Zn2+-induced cell death (Fig.2, Table 3), although it also restored NAD+ levels (Table 2). Pyruvate also appeared markedly better than niacinamide in protecting neuronal processes (Fig. 2), raising a possibility that Zn2+ toxicity in processes may differ from that in soma. It is easily possible that pyruvate may have additional beneficial effects on Zn2+-injured neurons, such as enhancement of mitochondrial membrane potential (Kauppinen and Nicholls, 1986) or enhancement of PDH activity, which is reduced after global ischemia in vivo (Kobayashi and Neely, 1983; Zaidan and Sims, 1997).
The inability of lactate and β-hydroxy-butyrate to protect against Zn2+-induced death compared with their ability to substitute for glucose fits with the observation that they are used predominantly after prolonged periods of fasting (Owen et al., 1967; Robinson and Williamson, 1980), whereas Zn2+ may induce too rapid a block in glycolysis to allow the uptake and use of alternative substrates. Also, Zn2+-induced NAD+ deficiency would be expected to inhibit the conversion of lactate to pyruvate by LDH and to inhibit the mitochondrial use of β-hydroxy-butyrate. In contrast, oxaloacetate is effective at preventing Zn2+-induced neuronal death, perhaps because it can be converted to pyruvate (Lehninger et al., 1993).
The present observation that several energy substrates or modulators of energy pathway enzyme activity can attenuate Zn2+-induced neuronal death suggests several new approaches to ameliorating this death in the context of certain disease states.
Footnotes
This work was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke Grant NS 30337 (D.W.C.).
Correspondence should be addressed to Dennis W. Choi, Department of Neurology and Center for the Study of Nervous System Injury, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail: choid@neuro.wustl.edu.
REFERENCES
- 1.Assaf SY, Chung SH. Release of endogenous Zn2+ from brain tissue during activity. Nature. 1984;308:734–736. doi: 10.1038/308734a0. [DOI] [PubMed] [Google Scholar]
- 2.Berg JM, Shi Y. The galvanization of biology: a growing appreciation for the roles of zinc. Science. 1996;271:1081–1085. doi: 10.1126/science.271.5252.1081. [DOI] [PubMed] [Google Scholar]
- 3.Canzoniero LM, Turetsky DM, Choi DW. Measurement of intracellular free zinc concentrations accompanying zinc-induced neuronal death. J Neurosci. 1999;19:RC31. doi: 10.1523/JNEUROSCI.19-19-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charton G, Rovira C, Ben-Ari Y, Leviel V. Spontaneous and evoked release of endogenous Zn2+ in the hippocampal mossy fiber zone of the rat in situ. Exp Brain Res. 1985;58:202–205. doi: 10.1007/BF00238969. [DOI] [PubMed] [Google Scholar]
- 5.Choi DW, Koh JY. Zinc and brain injury. Annu Rev Neurosci. 1998;21:347–375. doi: 10.1146/annurev.neuro.21.1.347. [DOI] [PubMed] [Google Scholar]
- 6.Danscher G, Howell G, Perez-Clausell J, Hertel N. The dithizone, Timm's sulphide silver and the selenium methods demonstrate a chelatable pool of zinc in CNS. A proton activation (PIXE) analysis of carbon tetrachloride extracts from rat brains and spinal cords intravitally treated with dithizone. Histochemistry. 1985;83:419–422. doi: 10.1007/BF00509203. [DOI] [PubMed] [Google Scholar]
- 7.Desagher S, Glowinski J, Premont J. Pyruvate protects neurons against hydrogen peroxide-induced toxicity. J Neurosci. 1997;17:9060–9067. doi: 10.1523/JNEUROSCI.17-23-09060.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dugan LL, Turetsky DM, Du C, Lobner D, Wheeler M, Almli CR, Shen CK, Luh TY, Choi DW, Lin TS. Carboxyfullerenes as neuroprotective agents. Proc Natl Acad Sci USA. 1997;94:9434–9439. doi: 10.1073/pnas.94.17.9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 10.Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose) polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Franke H, Barlow CH, Chance B. Oxygen delivery in perfused rat kidney: NADH fluorescence and renal functional state. Am J Physiol. 1976;231:1082–1089. doi: 10.1152/ajplegacy.1976.231.4.1082. [DOI] [PubMed] [Google Scholar]
- 12.Frederickson CJ. Neurobiology of zinc and zinc-containing neurons. Int Rev Neurobiol. 1989;31:145–238. doi: 10.1016/s0074-7742(08)60279-2. [DOI] [PubMed] [Google Scholar]
- 13.Frederickson CJ, Klitenick MA, Manton WI, Kirkpatrick JB. Cytoarchitectonic distribution of zinc in the hippocampus of man and the rat. Brain Res. 1983;273:335–339. doi: 10.1016/0006-8993(83)90858-2. [DOI] [PubMed] [Google Scholar]
- 14.Gutmann I, Wahlefeld AW. l-(+)-Lactate: determination with lactate dehydrogenase and NAD. In: Bergmeyer HU, editor. Methods of enzymatic analysis. Academic; New York: 1974. pp. 1464–1468. [Google Scholar]
- 15.Harrison NL, Gibbons SJ. Zn2+: an endogenous modulator of ligand- and voltage-gated ion channels. Neuropharmacology. 1994;33:935–952. doi: 10.1016/0028-3908(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 16.Haugland RP. Handbook of fluorescent probes and research chemicals, Ed 6 (Spencer MTZ, ed), pp 530–540. Molecular Probes; Eugene, OR: 1996. [Google Scholar]
- 17.Holleman MAF. Notice sur l'action de l'eau oxygénée sur les acides a-cétoniques et sur les dicétones 1.2. Recl Trav Chim Pays Bas Belg. 1904;23:169–171. [Google Scholar]
- 18.Howell GA, Welch MG, Frederickson CJ. Stimulation-induced uptake and release of zinc in hippocampal slices. Nature. 1984;308:736–738. doi: 10.1038/308736a0. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda T, Kimura K, Morioka S, Tamaki N. Inhibitory effects of Zn2+ on muscle glycolysis and their reversal by histidine. J Nutr Sci Vitaminol. 1980;26:357–366. doi: 10.3177/jnsv.26.357. [DOI] [PubMed] [Google Scholar]
- 20.Jorcke D, Ziegler M, Schweiger M. Characterization of hydrosoluble and detergent-solubilized forms of mitochondrial NAD+ glycohydrolase from bovine liver. Adv Exp Med Biol. 1997;419:447–451. doi: 10.1007/978-1-4419-8632-0_58. [DOI] [PubMed] [Google Scholar]
- 21.Kauppinen RA, Nicholls DG. Synaptosomal bioenergetics. The role of glycolysis, pyruvate oxidation and responses to hypoglycaemia. Eur J Biochem. 1986;158:159–165. doi: 10.1111/j.1432-1033.1986.tb09733.x. [DOI] [PubMed] [Google Scholar]
- 22.Kim EY, Koh JY, Kim YH, Sohn S, Joe E, Gwag BJ. Zn2+ entry produces oxidative neuronal necrosis in cortical cell cultures. Eur J Neurosci. 1999;11:327–334. doi: 10.1046/j.1460-9568.1999.00437.x. [DOI] [PubMed] [Google Scholar]
- 23.Kim YH, Kim EY, Gwag BJ, Sohn S, Koh JY. Zinc-induced cortical neuronal death with features of apoptosis and necrosis: mediation by free radicals. Neuroscience. 1999a;89:175–182. doi: 10.1016/s0306-4522(98)00313-3. [DOI] [PubMed] [Google Scholar]
- 24.Kim YH, Park JH, Hong SH, Koh JY. Nonproteolytic neuroprotection by human recombinant tissue plasminogen activator. Science. 1999b;284:647–650. doi: 10.1126/science.284.5414.647. [DOI] [PubMed] [Google Scholar]
- 25.Kobayashi K, Neely JR. Effects of ischemia and reperfusion on pyruvate dehydrogenase activity in isolated rat hearts. J Mol Cell Cardiol. 1983;15:359–367. doi: 10.1016/0022-2828(83)90320-6. [DOI] [PubMed] [Google Scholar]
- 26.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 27.Koh JY, Wie MB, Gwag BJ, Sensi SL, Canzoniero LM, Demaro J, Csernansky C, Choi DW. Staurosporine-induced neuronal apoptosis. Exp Neurol. 1995;135:153–159. doi: 10.1006/exnr.1995.1074. [DOI] [PubMed] [Google Scholar]
- 28.Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science. 1996;272:1013–1016. doi: 10.1126/science.272.5264.1013. [DOI] [PubMed] [Google Scholar]
- 29.Krotkiewska B, Banas T. Interaction of Zn2+ and Cu2+ ions with glyceraldehyde-3-phosphate dehydrogenase from bovine heart and rabbit muscle. Int J Biochem. 1992;24:1501–1505. doi: 10.1016/0020-711x(92)90078-f. [DOI] [PubMed] [Google Scholar]
- 30.Kukimoto I, Hoshino S, Kontani K, Inageda K, Nishina H, Takahashi K, Katada T. Stimulation of ADP-ribosyl cyclase activity of the cell surface antigen CD38 by zinc ions resulting from inhibition of its NAD+ glycohydrolase activity. Eur J Biochem. 1996;239:177–182. doi: 10.1111/j.1432-1033.1996.0177u.x. [DOI] [PubMed] [Google Scholar]
- 31.Larsen AG, Ostvold AC, Holtlund J, Kristensen T, Laland SG. The inhibitory effect of Zn2+ on poly(ADP-ribose) polymerase activity and its reversal. Biochem J. 1982;203:511–513. doi: 10.1042/bj2030511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehninger AL, Nelson DL, Cox MM. Principles of Biochemistry. Worth; New York: 1993. [Google Scholar]
- 33.Link TA, von Jagow G. Zinc ions inhibit the QP center of bovine heart mitochondrial bc1 complex by blocking a protonatable group. J Biol Chem. 1995;270:25001–25006. doi: 10.1074/jbc.270.42.25001. [DOI] [PubMed] [Google Scholar]
- 34.Lust WD, Feussner GK, Barbehenn EK, Passonneau JV. The enzymatic measurement of adenine nucleotides and P-creatine in picomole amounts. Anal Biochem. 1981;110:258–266. doi: 10.1016/0003-2697(81)90144-5. [DOI] [PubMed] [Google Scholar]
- 35.Manev H, Kharlamov E, Uz T, Mason RP, Cagnoli CM. Characterization of zinc-induced neuronal death in primary cultures of rat cerebellar granule cells. Exp Neurol. 1997;146:171–178. doi: 10.1006/exnr.1997.6510. [DOI] [PubMed] [Google Scholar]
- 36.Martell AM, Smith RM. Critically selected stability constants of metal complexes, p 411. National Institute of Standards and Technology; Gaithersburg, MD: 1995. [Google Scholar]
- 37.Michal GB, Beutler HO. d-Fructose-1,6-P2, dihydroxyacetone phosphate and d-glyceraldehyde-3-phosphate. In: Bergmeyer HU, editor. Methods of enzymatic analysis. Academic; New York: 1974. pp. 1314–1319. [Google Scholar]
- 38.Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF., Jr Brain metabolism during fasting. J Clin Invest. 1967;46:1589–1595. doi: 10.1172/JCI105650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poolman B, Bosman B, Kiers J, Konings WN. Control of glycolysis by glyceraldehyde-3-phosphate dehydrogenase in Streptococcus cremoris and Streptococcus lactis. J Bacteriol. 1987;169:5887–5890. doi: 10.1128/jb.169.12.5887-5890.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pryor WA, Arbour NC, Upham B, Church DF. The inhibitory effect of extracts of cigarette tar on electron transport of mitochondria and submitochondrial particles. Free Radic Biol Med. 1992;12:365–372. doi: 10.1016/0891-5849(92)90085-u. [DOI] [PubMed] [Google Scholar]
- 41.Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. 1980;60:143–187. doi: 10.1152/physrev.1980.60.1.143. [DOI] [PubMed] [Google Scholar]
- 42.Ross GM, Shamovsky IL, Lawrance G, Solc M, Dostaler SM, Jimmo SL, Weaver DF, Riopelle RJ. Zinc alters conformation and inhibits biological activities of nerve growth factor and related neurotrophins. Nat Med. 1997;3:872–878. doi: 10.1038/nm0897-872. [DOI] [PubMed] [Google Scholar]
- 43.Rovetto MJ, Lamberton WF, Neely JR. Mechanisms of glycolytic inhibition in ischemic rat hearts. Circ Res. 1975;37:742–751. doi: 10.1161/01.res.37.6.742. [DOI] [PubMed] [Google Scholar]
- 44.Sander BJ, Oelshlegel FJ, Brewer GJ., Jr Quantitative analysis of pyridine nucleotides in red blood cells: a single-step extraction procedure. Anal Biochem. 1976;71:29–36. doi: 10.1016/0003-2697(76)90006-3. [DOI] [PubMed] [Google Scholar]
- 45.Schissel SL, Schuchman EH, Williams KJ, Tabas I. Zn2+-stimulated sphingomyelinase is secreted by many cell types and is a product of the acid sphingomyelinase gene. J Biol Chem. 1996;271:18431–18436. doi: 10.1074/jbc.271.31.18431. [DOI] [PubMed] [Google Scholar]
- 46.Schurr A, Payne RS, Miller JJ, Rigor BM. Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: an in vitro study. Brain Res. 1997;744:105–111. doi: 10.1016/s0006-8993(96)01106-7. [DOI] [PubMed] [Google Scholar]
- 47.Sensi SL, Canzoniero LM, Yu SP, Ying HS, Koh JY, Kerchner GA, Choi DW. Measurement of intracellular free zinc in living cortical neurons: routes of entry. J Neurosci. 1997;17:9554–9564. doi: 10.1523/JNEUROSCI.17-24-09554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sheline CT, Choi DW. Two strategies for attenuating zinc neurotoxicity on cortical neurons. Soc Neurosci Abstr. 1997;23:2255. [Google Scholar]
- 50.Sheline CT, Choi DW. Neuronal death in cultured murine cortical cells is induced by inhibition of GAPDH and triosephosphate isomerase. Neurobiol Dis. 1998;5:47–54. doi: 10.1006/nbdi.1998.0177. [DOI] [PubMed] [Google Scholar]
- 51.Skulachev VP, Chistyakov VV, Jasaitis AA, Smirnova EG. Inhibition of the respiratory chain by zinc ions. Biochem Biophys Res Commun. 1967;26:1–6. doi: 10.1016/0006-291x(67)90242-2. [DOI] [PubMed] [Google Scholar]
- 52.Smart TG, Xie X, Krishek BJ. Modulation of inhibitory and excitatory amino acid receptor ion channels by zinc. Prog Neurobiol. 1994;42:393–341. doi: 10.1016/0301-0082(94)90082-5. [DOI] [PubMed] [Google Scholar]
- 53.Szabo C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 54.Tilton WM, Seaman C, Carriero D, Piomelli S. Regulation of glycolysis in the erythrocyte: role of the lactate/pyruvate and NAD/NADH ratios. J Lab Clin Med. 1991;118:146–152. [PubMed] [Google Scholar]
- 55.Tonder N, Johansen FF, Frederickson CJ, Zimmer J, Diemer NH. Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci Lett. 1990;109:247–252. doi: 10.1016/0304-3940(90)90002-q. [DOI] [PubMed] [Google Scholar]
- 56.Vallee BL, Falchuk KH. The biochemical basis of zinc physiology. Physiol Rev. 1993;73:79–118. doi: 10.1152/physrev.1993.73.1.79. [DOI] [PubMed] [Google Scholar]
- 57.Virag L, Szabo C. Inhibition of poly(ADP-ribose) synthetase (PARS) and protection against peroxynitrite-induced cytotoxicity by zinc chelation. Br J Pharmacol. 1999;126:769–777. doi: 10.1038/sj.bjp.0702332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang ZQ, Auer B, Stingl L, Berghammer H, Haidacher D, Schweiger M, Wagner EF. Mice lacking ADPRT and poly(ADP-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995;9:509–520. doi: 10.1101/gad.9.5.509. [DOI] [PubMed] [Google Scholar]
- 59.Weil M, Jacobson MD, Coles HS, Davies TJ, Gardner RL, Raff KD, Raff MC. Constitutive expression of the machinery for programmed cell death. J Cell Biol. 1996;133:1053–1059. doi: 10.1083/jcb.133.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weiss JH, Hartley DM, Koh JY, Choi DW. AMPA receptor activation potentiates zinc neurotoxicity. Neuron. 1993;10:43–49. doi: 10.1016/0896-6273(93)90240-r. [DOI] [PubMed] [Google Scholar]
- 61.Wong C, Rodriguez-Paez L, Nogueda B, Perez A, Baeza I. Selective inhibition of the sperm-specific lactate dehydrogenase isozyme-C4 by N-isopropyl oxamate. Biochim Biophys Acta. 1997;1343:16–22. doi: 10.1016/s0167-4838(97)00090-3. [DOI] [PubMed] [Google Scholar]
- 62.Yokoyama M, Koh J, Choi DW. Brief exposure to zinc is toxic to cortical neurons. Neurosci Lett. 1986;71:351–355. doi: 10.1016/0304-3940(86)90646-4. [DOI] [PubMed] [Google Scholar]
- 63.Zaidan E, Sims NR. Reduced activity of the pyruvate dehydrogenase complex but not cytochrome c oxidase is associated with neuronal loss in the striatum following short-term forebrain ischemia. Brain Res. 1997;772:23–28. doi: 10.1016/s0006-8993(97)00833-0. [DOI] [PubMed] [Google Scholar]
- 64.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 65.Ziegler M, Jorcke D, Zhang J, Schneider R, Klocker H, Auer B, Schweiger M. Characterization of detergent-solubilized beef liver mitochondrial NAD+ glycohydrolase and its truncated hydrosoluble form. Biochemistry. 1996;35:5207–5212. doi: 10.1021/bi9527698. [DOI] [PubMed] [Google Scholar]