

Abstract
The ε4 genotype of apolipoprotein E (apoE4) is the most established predisposing factor in Alzheimer's disease (AD); however, it remains unclear how apoE4 contributes to the pathophysiology. Here, we report that the apoE4 protein (ApoE4) evokes apoptosis in neuronal cells through the low-density lipoprotein receptor-related protein (LRP) and heterotrimeric GTPases. We examined neuron/neuroblastoma hybrid F11 cells and found that these cells were killed by 30 μg/ml ApoE4, but not by 30 μg/ml ApoE3. ApoE4-induced death occurred with typical features for apoptosis in time- and dose-dependent manners, and was observed in SH-SY5Y neuroblastomas, but not in glioblastomas or non-neuronal Chinese hamster ovary cells. Activated, but not native, α2-macroglobulin suppressed this ApoE4 toxicity. Suppression by the antisense oligonucleotide to LRP and inhibition by low nanomolar concentrations of LRP-associated protein RAP provided evidence for the involvement of LRP. The involvement of heterotrimeric GTPases was demonstrated by the findings that (1) ApoE4-induced death was suppressed by pertussis toxin (PTX), but not by heat-inactivated PTX; and (2) transfection with PTX-resistant mutant cDNAs of Gαi restored the toxicity of ApoE4 restricted by PTX. We thus conclude that one of the neurotoxic mechanisms triggered by ApoE4 is to activate a cell type-specific apoptogenic program involving LRP and the Gi class of GTPases and that the apoE4 gene may play a direct role in the pathogenesis of AD and other forms of dementia.
Keywords: apolipoprotein E, isoform-specific action, neuronal apoptosis, lipoprotein receptor-related protein, G-proteins, pertussis toxin, Alzheimer's disease
The apoE ε4 allele in chromosome 19q13.2 has been recognized as a susceptibility gene for late-onset Alzheimer's disease (AD) (Saunders et al., 1993; Strittmatter et al., 1993; Ueki et al., 1993; Goedert et al., 1994), AD types of dementia by diffuse Levy body disease (Helisalmi et al., 1996), and non-AD types of dementia (Helisalmi et al., 1996; Ji et al., 1998), including vascular and ischemic dementia. Inheritance of the apoE ε4 allele also seems to influence the pathogenesis of other neurodegenerative diseases, such as amyotrophic lateral sclerosis (Moulard et al., 1996), Pick's disease (Helisalmi et al., 1996; Kalman et al., 2000), and Parkinson's disease (Zareparsi et al., 1997; Kruger et al., 1999), although the ε4 association with some of them is controversial (Mui et al., 1995;Egensperger et al., 1996; The French Parkinson's Disease Genetics Study Group, 1997; Siddique et al., 1998). The gene for apoE is highly polymorphic. The common ε3 and ε4 alleles encode the isoforms of the apoE protein (ApoE): ApoE3 and ApoE4 (Zannis et al., 1982). ApoE mediates the delivery of lipids (Wilson et al., 1991) and also plays a neuron-specific role. ApoE3 stimulates neurite outgrowth, whereas ApoE4 decreases outgrowth (Nathan et al., 1994). This neurite-trophic action is mediated by the low-density lipoprotein receptor-related protein (LRP) (Holtzman et al., 1995; Narita et al., 1997). However, exactly how ApoE4 contributes to the development of AD remains virtually unknown.
An important clue is the finding (Marques et al., 1997; Tolar et al., 1997, 1999; Jordan et al., 1998; Michikawa and Yanagisawa, 1998;DeMattos et al., 1999) that ApoE4 exerts neurotoxicity in culture. Although ApoE4-induced neurodegeneration has not yet been clearly shown in transgenic mice (Raber et al., 1998; Sun et al., 1998), it might result from in vivo suppression of ApoE4 neurotoxicity. Therefore, the molecular mechanism for ApoE4 neurotoxicity deserves investigation. Recently, Buttini et al. (2000) analyzed apoE knock-out mice that express ApoE3 or ApoE4 or both in the brain and found that ApoE4 acts as an inhibitor of neuroprotection by ApoE3.
ApoE4 binds Aβ and facilitates its aggregation (Strittmatter et al., 1993; LaDu et al., 1994, 1995). However, it is unlikely that this action is implicated in the ApoE4 neurotoxicity, because (1) ApoE3 binds Aβ at 20-fold higher levels than does ApoE4 (LaDu et al., 1994); (2) binding of Aβ to rabbit ApoE decreases Aβ toxicity in rat hippocampal neurons (Whitson et al., 1994); and (3) the N-terminal 22 kDa fragment of ApoE4, which lacks the Aβ binding domain (Pillot et al., 1999), exhibits isoform-specific neurotoxicity (Marques et al., 1996, 1997; Tolar et al., 1997, 1999). Also, Demattos et al. (1999)demonstrated that ApoE4 exerts neurotoxicity not through interaction with intracellular Aβ or tau. The present study was conducted to examine whether ApoE4 has a direct action on neuronal death, and if so, with what molecular mechanism. We find that ApoE4 exerts isoform-specific neurotoxicity through LRP and the Gi class of GTPases.
MATERIALS AND METHODS
Materials. F11 cells, described previously (Platika et al., 1985; Yamatsuji et al., 1996), were grown in Ham's F-12 (Life Technologies, Gaithersburg, MD) supplemented with 18% fetal bovine serum (FBS; HyClone, Logan, UT) and antibiotics. Bu695 cells (Hayashi et al., 1992), provided by Dr. K. Yoshikawa (Osaka University, Osaka, Japan), were grown in DMEM (Life Technologies) plus 10% FBS and antibiotics. SH-SY5Y cells were provided by Drs. M. Morishima and Y. Ihara (University of Tokyo, Tokyo, Japan). CHO cells were described previously (Ikezu et al., 1994). ApoE3 and ApoE4 were from Chemicon (Temecula, CA). These recombinant ApoE proteins were >95% pure, forming a single band in SDS-PAGE. Unless otherwise described, they were used as ApoE. Purified native ApoE3 and ApoE4 proteins (Rall et al., 1986) were kindly provided by Dr. K. H. Weisgraber (University of California, San Francisco, CA). Dimyristoylphosphatidylcholine (DMPC) was purchased from Sigma. DMPC reconstitution was performed as described previously (Innerarity et al., 1979). Pertussis toxin (PTX) was from Calbiochem-Novabiochem. For heat inactivation, PTX was incubated at 90°C for 1 hr. Acetyl-l-Aspartyl-l-Glutaminyl-l-Valyl-l-Aspart-1-al (Ac-DEVD-CHO) was purchased from Peptide Institute (Mino, Osaka, Japan). α2-Macroglobulin (α2M) was from Yagai Research Center. For activation, native α2M was treated with 200 mmmethylamine in 50 mm Tris/HCl, pH 8.0, and 150 mm NaCl for 16–18 hr at room temperature in the dark. Unreacted methylamine was removed by dialysis for 48 hr with five changes of 20 mm HEPES/NaOH, pH 7.4, and 150 mmNaCl. The α2M preparation was dialyzed again with serum-free Ham's F-12 for 4 hr. Dialyzed α2M was sterilized by filtering through a 0.22 μm microfilter and then stored at 4°C and used within 2 weeks. Enhanced green fluorescent protein (EGFP) cDNA was purchased from Clontech (Cambridge, UK) (pEGFP-N1). The cDNAs encoding GαiPT and GαoPT, described previously (Taussig et al., 1992), were provided by Dr. T. Kozasa (University of Texas, Southwestern Medical Center, Dallas, TX) and Dr. R. Taussig (University of Michigan, Ann Arbor, MI). RAP and Anti-LRP antibody 8G1 were purchased from PROGEN Biotechnic. The RAP used in this study was a rat recombinant fusion protein with N-terminal His tag and C-terminal c-myc tag, produced inEscherichia coli and purified by affinity beads, and similar to rat GST-fusion RAP described previously (Herz et al., 1991). The purity of the RAP fusion protein was >95%.
Oligonucleotide transfer. Antisense oligonucleotides were transferred into F11 cells using a particle bombardment-mediated gene transfer method. This was performed using the Helios Gene Gun system (Bio-Rad, Hercules, CA), according to the manufacturer's instructions, as described previously in detail (Yoshida et al., 1997). Briefly, antisense oligonucleotide-coated gold particles were constructed by mixing 25 mg gold particles (ø = 0.6 μm) with 100 μg of antisense oligonucleotides. F11 cells were seeded at 105 cells/well in a 12-well plate and incubated for 24 hr in the presence of 18% FBS. After washing, cells then underwent particle bombardment-mediated gene transfer with a gold particle/antisense oligonucleotide mixture. After culturing cells in K-PBS solution (in mm: 30.8 NaCl, 120.7 KCl, 8.1 Na2HPO4 · 12H2O, 5.0 MgCl2, pH adjusted to 7.4 with HCl) for 2 hr, the medium was changed to Ham's F-12 plus 18% FBS, and cells were cultured for another 22 hr. Cells were then treated with ApoE4 in serum-free Ham's F-12 medium. In some experiments, introduction of antisense oligonucleotides was performed using lipofection with similar results. Briefly, F11 cells were seeded at 7 × 104 cells/well in a six-well plate and incubated for 12–18 hr in the presence of 18% FBS. After washing, cells were transfected with antisense oligonucleotides by lipofection (oligonucleotide, 1 μg; Lipofectamine, 2 μl; PLUS reagent, 4 μl) in the absence of serum for 3 hr, and were incubated with Ham's F-12 plus 18% FBS for 2 hr. Next, the culture medium was changed to Ham's F-12 plus 10% FBS. Twenty-four hours after the onset of transfection, cells were treated with 30 μg/ml ApoE and cultured with serum-free Ham's F-12, and cell mortality was measured by Trypan blue exclusion assay 72 hr after the onset of treatment. Purified phosphorothionate-modified oligonucleotides were obtained from Sawady Technology (Tokyo, Japan). The sequence of the antisense oligonucleotide to mouse LRP (AS-LRP) mRNA corresponds to the position from −13 to +11, which includes the ATG initiation codon (5′-GGG GTC AGC ATG GTG TGG GCC GAT-3′). The scrambled oligonucleotide for the control of AS-LRP was 5′-GCG GAG GTG GTC TGG TAG ACG CGT-3′. The sequence of the antisense oligonucleotide to ApoER2 mRNA corresponds to the position from −13 to +11, which includes the ATG codon (5′-GGG AGG CCC ATG GCG GGC CCG GGC-3′). Because this antisense oligonucleotide is for human ApoER2 and the nucleotide sequence of rodent ApoER2 has not yet been determined, it was used as a control oligonucleotide that carries 50% (12/24 base) identity. PTX-resistant mutant cDNAs coding for GαiPT or GαoPT were transfected using Lipofectamine PLUS (Life Technologies). In brief, F11 cells were seeded at 105 cells/well in a six-well plate, incubated for 24 hr in the presence of 18% FBS, and mixed for 3 hr with 10 μg cDNA, 20 μl PLUS reagent, and 25 μl Lipofectamine. Adding an equal volume of Ham's F-12 plus 20% FBS (final FBS concentration 10%) into cultured media, cells were incubated for 24 hr. Then cells were treated with ApoE4 in fresh serum-free Ham's F-12 medium. This condition yielded ∼80% transfection efficiency, as assessed with EGFP cDNA.
Assays. Cell mortality was measured by Trypan blue exclusion assay as follows. Cells were seeded in 12-well plates at a density of 104 cells/well (when transfection was not necessary; see above for transfection experiments). After culturing these cells in complete growth medium, they were washed with serum-free medium once, and the cultured medium was changed to a fresh serum-free medium containing ApoE4 or other reagents. Two different protocols were used for this assay in the present study. (1) At the termination of experiments, cells were suspended by pipetting gently. To ensure the collection of total cells, PBS was added to the well and collected into the cell suspension, using phase-contrast microscopy, to confirm that no cells were left; 0.1% Trypan blue solution (final concentration 0.02%) was then added to the cell suspension and incubated at 37°C for 1–2 min. (2) At the termination of experiments, cells were suspended by pipetting gently, and 50 μl of 0.4% Trypan blue solution was mixed with 200 μl of the cell suspension (final concentration 0.08%) at room temperature. Stained cells were counted within 3 min after mixing with Trypan blue solution. Both protocols yielded similar results. The mortality of cells was then determined as the percentage of Trypan blue-stained cells in total cells. The cell mortality assessed by these methods thus represented the population of dead cells in total cells, including both adhesive and floating cells. The basal death rates without ApoE treatment indicated the actual fraction of dead cells, but not artificial cell death occurring after cell detachment, because in situ staining of Trypan blue-positive cells indicated the presence of similar fractions of dead cells (data not shown). Cell viability was also measured by 2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H- tetrazolium, monosodium salt (WST-8), using Cell Counting kit-8 (Wako Pure Chemicals, Tokyo, Japan). After treatment with ApoE proteins, cells were suspended, and 1/10 volume (100 μl) of the cell suspension was incubated with 10 μl of WST-8 solution in a 96-well plate for 2 hr at 37°C. Absorbance of the samples at 450 nm wavelength was measured by Wallac 1420 ARVOsx Multi Label Counter (Amersham Pharmacia Biotech). Terminal deoxynucleotidyl transferase-mediated fluorescein-deoxy UTP nick end labeling (TUNEL) was performed using a kit (In SituCell Death Detection Kit with Fluorescein; Boehringer Mannheim, Mannheim, Germany), according to the manufacturer's instructions. For this assay, F11 cells were seeded onto slide glasses precoated with poly-d-lysine in Ham's F-12 containing 18% FBS and antibiotics, as described previously (Yamatsuji et al., 1996). Experiments to investigate ApoE4-induced formation of the DNA ladder were performed using a kit (Takara, Japan), according to the manufacturer's instructions. Immunoblot analysis of expressed LRP was performed with 2.5 μg/ml anti-LRP antibody 8G1. Data were analyzed by Student's unpaired t test. All experiments described in this study were repeated at least three times independently.
RESULTS
Cell death effect of ApoE4 in F11 neuronal cells
F11 cells are embryonic neurons immortalized by fusion of E13 rat dorsal root ganglion neurons with a mouse neuroblastoma cell line NTG18. They carry the traits of primary neurons, including the maintenance of neuronal gangliosides and the generation of action potentials (Platika et al., 1985). When F11 cells were treated with 30 μg/ml of recombinant ApoE4 (rApoE4) for 72 hr, 70–80% of treated cells underwent cell death, as assessed by Trypan blue exclusion assay (Fig. 1A). In contrast, the same concentration of rApoE3 failed to kill F11 cells after 72 hr treatment. Similar observations were obtained with seven different batches of rApoE4 preparations and three different batches of rApoE3 preparations. Also, essentially the same observations were obtained from purified native ApoE3 and ApoE4, except that neurotoxicity of ApoE4 occurred approximately three times more rapidly, relative to the same concentrations of rApoE4 (Fig. 1A). The toxic effect of rApoE4 was dose-dependent in a concentration range from 3 to 30 μg/ml. Mortality increased unidirectionally for 72 hr when cells were treated with rApoE4 (Fig. 1B). The data for ApoE4 toxicity were reproduced when cell viability was measured by metabolic activity of viable cells, using tetrazolium salt WST-8, similar to MTT. As shown in Figure 1, C and D, this viability assay revealed that treatment of F11 cells with rApoE4 induced time- and dose-dependent decreases in cell viability, which were inversely proportional to the time- and dose-dependent increases in cell mortality assessed by Trypan blue exclusion assay (Fig.1E).
Fig. 1.

Cell toxicity of ApoE4 in F11 neuronal cells. A, Cell toxicity by ApoE3 and ApoE4. F11 cells were treated with ApoE4 preparations [increasing concentrations of recombinant ApoE4 (rApoE4), 30 μg/ml of DMPC-reconstituted ApoE4 (1) or native ApoE4 (2)] or 30 μg/ml of ApoE3 preparations [native ApoE3 (3), recombinant ApoE3 (4), DMPC-reconstituted ApoE3 (5), or DTT-treated ApoE3 (6)], and cell mortality was measured by Trypan blue exclusion assay. The incubation periods were 24 hr for native ApoE proteins and 72 hr for other cases. The values presented in all figures in this study indicate means ± SD of at least three independent experiments. B, Time course of ApoE4-induced cell death, assessed by Trypan blue exclusion assay. F11 cells were treated with 30 μg/ml rApoE4 for indicated periods, and dead cell numbers were counted in each treatment. C, Time courses of the toxic effects of ApoE proteins, assessed by WST-8 cell viability assay. F11 cells were treated with vehicle (open circles), 30 μg/ml rApoE3 (closed triangles), or rApoE4 (closed circles) for indicated periods, and cell viability was measured by WST-8 assay, as described in Materials and Methods. D, The dose–response relationship for the toxic effect of ApoE4. F11 cells were treated with increasing concentrations of rApoE4 for 72 hr, and cell viability was similarly measured by WST-8 assay. E, The relationship between cell mortality assessed by Trypan blue exclusion assay and cell viability assessed by WST-8 assay. The time course and the dose–response of the rApoE4 toxicity in F11 cells were measured by Trypan blue exclusion assay or by WST-8 assay in independently performed experiments, and corresponding data were plotted. Each value indicates means ± SD of three measurement data (each obtained from one independent experiment, and each experiment was independently repeated three times).
These rApoE proteins are not able to bind to the LDL receptor (LDLR), whereas DMPC-reconstituted rApoE proteins are as potent in binding to LDLR as purified ApoE proteins (Gretch et al., 1991). We thus examined the effects of DMPC-reconstituted rApoE and found that (1) rApoE4 was as active in causing cell death as DMPC-reconstituted rApoE4; (2) DMPC-reconstituted rApoE3 was as nontoxic as native ApoE3 and rApoE3 (Fig. 1A). These data suggest that the action of rApoE can be generalized to native ApoE, as far as the cell death effect is concerned. The failure of DMPC-reconstituted rApoE3 to cause cell death also indicated that lack of toxicity in rApoE3 was not because of disturbed activity or inappropriate folding of rApoE3. This was further supported by the findings that (1) native ApoE3 was not toxic either; and (2) ApoE4-induced cell death was antagonized by rApoE3 (Table 1). As the action of ApoE3 might have been lost by potential oligomerization through Cys, we also tested the effect of DTT. We found that DTT-treated rApoE3 was also as nontoxic as native ApoE3, again indicating that the nontoxic effect of rApoE3 equals that of native ApoE3.
Table 1.
Inhibition of ApoE4-induced cell death by PTX or ApoE3
| Cell mortality (% dead cells of total cells) | |
|---|---|
| ApoE3 | 10.2 ± 5.1 |
| ApoE4 | 74.7 ± 2.2 |
| ApoE4 + PTX | 25.1 ± 6.6* |
| ApoE4 + inactivated PTX | 73.5 ± 11.61-160 |
| ApoE4 + ApoE3 | 30.4 ± 10.0* |
F11 cells were treated with 30 μg/ml ApoE4 with or without several reagents for 72 hr. Cell mortality was measured by Trypan blue exclusion. PTX was used at 1 μg/ml; ApoE3, at 30 μg/ml. The values indicate means ± SE of four independent experiments.
p < 0.01 and
F1-160: not significant versus ApoE4 effect.
Because it has been reported that a high concentration (6 μm, 200 μg/ml) but not 3.2 μm (100 μg/ml) of ApoE3 induces significant toxicity in primary neurons (Marques et al., 1997), we also examined the toxicity of high concentrations of rApoE3. As shown in Table2, treatment with 200 μg/ml rApoE3 resulted in significant induction of cell death, whereas 100 μg/ml rApoE3 caused little toxicity. These data show that the toxicity of rApoE3 was several dozen times weaker than that of rApoE4, consistent with the study of Marques et al. (1997). Because rApoE behaved similarly to native ApoE, we thereafter analyzed the actions of rApoE.
Table 2.
Cell toxicity by ApoE3
| Cell mortality (% dead cells of total cells) | Cell viability (arbitrary unit/well) | |||
|---|---|---|---|---|
| RAP (−) | RAP (+) | RAP (−) | RAP (+) | |
| No treatment | 18.1 ± 1.2 | ND | 1.12 ± 0.02 | ND |
| 100 μg/ml ApoE3 | 18.2 ± 0.3 | 19.7 ± 2.2 | 1.12 ± 0.04 | 1.23 ± 0.04 |
| 200 μg/ml ApoE3 | 31.4 ± 3.2 | 19.4 ± 2.4 | 0.71 ± 0.03 | 1.15 ± 0.09 |
F11 cells were treated with or without 100 μg/ml or 200 μg/ml ApoE3 in the absence (−) or presence (+) of 50 nM RAP for 72 hr. Cell mortality was measured by Trypan blue exclusion assay, and in parallel, cell viability was measured by WST-8 assay. The values indicate means ± SE of three independent experiments. Cell mortality is indicated as a percentage of Trypan blue-stained cells in total cells, and cell viability is indicated as absorbance at 450 nm (arbitrary unit/well).
p < 0.01 versus no treatment, and
F2-160: p < 0.01 versus 200 μg/ml ApoE3.
ND, Not determined.
Characterization of ApoE4-induced cell death
Seventy-two hours after treatment with ApoE4, most dead cells had shrunk and become round, and eventually detached from plates (see Fig.3B, bottom left panel), indicating that ApoE4 caused cells to undergo apoptosis. We thus further characterized the mode of F11 cell death induced by ApoE4. As shown in Figure2A, 24 hr treatment with 30 μg/ml ApoE4 induced a 180 bp ladder formation of DNA, whereas ApoE3 caused as little formation of DNA laddering as no treatment. When the cells were treated with 30 μg/ml ApoE4 in the presence of 10 μm Ac-DEVD-CHO, a specific inhibitor of caspases, DNA ladder formation in the treated cells was suppressed (Fig. 2A, lane 4), suggesting that the oligonucleosomal DNA cleavage induced by ApoE4 is a result of caspase-activated DNase. Note that coexisting ApoE3 protected F11 cells from ApoE4-induced DNA cleavage (Fig. 2A, lane 6), consistent with the ApoE3 action observed by cell mortality assay (Table 1). ApoE4-induced neurotoxicity was associated with staining by TUNEL. As shown in Figure 2B, 24 hr treatment with 30 μg/ml ApoE4 remarkably increased the population of F11 cells stained by TUNEL, whereas ApoE3 treatment resulted in as little TUNEL positivity as no treatment.
Fig. 3.

Inhibition of ApoE4-induced cell death by α2M*. F11 cells were treated with either 100 nm activated (act) or native α2M, 30 μg/ml ApoE4, or 30 μg/ml ApoE4 plus 100 nm activated or native α2M for 72 hr. InA, cell mortality was measured by Trypan blue exclusion assay; in B, representative phase-contrast microscopic images were presented. For both A and B, similar experiments were performed three times with similar results.
Fig. 2.

Characterization of ApoE4 toxicity.A, DNA laddering. F11 cells were treated with 30 μg/ml of ApoE4 (lane 2) or 30 μg/ml of ApoE3 (lane 3) for 24 hr. Cells were also treated with 30 μg/ml of ApoE4 in the presence of 10 μm Ac-DEVD-CHO (lane 4), 1 μg/ml PTX (lane 5), or 30 μg/ml of ApoE3 (lane 6) for 24 hr. Extracted DNA was applied onto a 2% agarose gel (2 μg/lane). As another control, DNA extracted from F11 cells grown in a complete growth medium was examined for DNA ladder formation (lane 1). MK indicates a molecular marker, which is a StyI digestion product of λ (cI857Sam7) DNA. This marker gives a 500–600 bp interval in the bottom half of a gel. The results shown are representative of three similar experiments independently performed.B, TUNEL assay. F11 cells were treated with 30 μg/ml of ApoE3 or ApoE4 for 24 hr; cells were stained with TUNEL. In the bottom panels, cells were treated with 30 μg/ml of ApoE4 in the presence of 1 μg/ml PTX or 10 μmAc-DEVD-CHO for 24 hr. Phase-contrast images were superimposed on the TUNEL fluorescence images. The fragmented DNA was stainedgreen by TdT and TUNEL. Apoptotic cells are known to undergo leakage of DNA from nuclei, which allowed cellular staining of DNA as well as nuclear staining. The results shown are representative of three similar experiments independently performed.C, Effect of ApoE in other cell lines. SH-SY5Y cells, Bu695 glial cells, or CHO cells were treated with or without 30 μg/ml of ApoE3 or ApoE4, and cell mortality was measured by Trypan blue exclusion assay. In SH-SY5Y cells, 30 μg/ml of ApoE4 was also treated in the presence of 30 μg/ml ApoE3 (the extreme right lane). The incubation periods were 48 hr for SH-SY5Y and CHO cells and 72 hr for Bu695 cells. CHO cell mortality at 72 hr after 30 μg/ml ApoE4 treatment was similar to that at 48 hr after treatment (data not shown).
In another attempt at characterization, ApoE4-induced toxicity was briefly tested in other types of cells. We examined two different kinds of neural cells, SH-SY5Y and Bu695, and one non-neuronal cell line, CHO. Whereas both of the former (SH-SY5Y and Bu695) are neural, SH-SY5Y cells are neuroblastic; and Bu695 cells are glial (Hayashi et al., 1992). As shown in Figure 2C, 30 μg/ml of ApoE4 failed to induce death in Bu695 cells, whereas it caused massive death in SH-SY5Y cells more rapidly than in F11 cells. Lack of toxicity by ApoE4 was also the case in non-neuronal CHO cells. Significant toxicity by ApoE4 was observed in SH-SY5Y cells at 24 hr after the start of treatment, whereas ApoE3 was unable to cause death in these cells (data not shown). The inhibitory effect of ApoE3 was also noted in SH-SY5Y cells (Fig. 2C). These data indicate that ApoE4 toxicity may be specific for cells of neuroblast origin.
Effect of α2-macroglobulin (α2M) on toxic action of ApoE4
We next examined the interfering effect of α2M on the toxic effect of ApoE4. α2M is another known ligand for LRP (LRP is also termed the α2M receptor). For α2M to bind LRP, α2M must be treated with and activated by methylamine. The interaction of proteases or methylamine with α2M results in its activation, a conformational change, and exposure of a latent LRP-binding site.
Whether treated with or without methylamine, α2M alone showed little toxicity in F11 cells (Fig. 3). In contrast, methylamine-treated α2M (activated α2M or α2M*) antagonized ApoE4-induced cell death (Fig. 3A) and inhibited apoptotic morphological changes caused by ApoE4 treatment (Fig.3B). Native α2M either failed to suppress ApoE4-induced death or it did not protect cells from apoptotic morphological changes induced by ApoE4. These data suggest that the α2M* binding to LRP may affect ApoE4-induced neurotoxicity.
Involvement of LRP in ApoE4 neurotoxicity
Effect of antisense LRP oligonucleotide on ApoE4 toxicity
To examine whether LRP is involved in ApoE4 toxicity, we sought to disrupt mRNA function using antisense oligonucleotides. Phosphorothionate-modified antisense oligonucleotides complementary to the translation initiation site (position −13 to +11) of LRP mRNA (AS-LRP) or to the translation initiation site (position −13 to +11) of type 2 ApoE receptor (ApoER2) mRNA, were synthesized. These oligonucleotides were designed as each cross-reactivity was minimized. We introduced them by means of a particle bombardment-mediated gene transfer method. In this method, transfection efficiency evaluated by EGFP cDNA, which was introduced into F11 cells in mock-transfection under the same conditions, was at least >60%, and mostly >80% (data not shown). Inclusion of antisense LRP oligonucleotide resulted in >60% inhibition of ApoE4-induced mortality. On the other hand, antisense ApoER2 oligonucleotide had only marginal effects. Figure4A depicts the results of four independent series of experiments, in each of which six, six, six, and four independent transfections using a particle bombardment-mediated gene transfer method were performed under the same conditions on different days. Although the inhibition of F11 cell death by AS-LRP transfer fluctuated within a certain range in quantity, ApoE4-induced cell death was constantly and significantly inhibited by this procedure. The marginal inhibition of the ApoE4 effect by AS-ApoER2 was attributed to weak cross-reactivity of the oligonucleotides, because introduction of a scrambled oligonucleotide resulted in no inhibition of ApoE4-induced cell death (Fig.4B). The inset of Figure 4Bindicates the alteration in the 515 kDa LRP expression after transfection with AS-LRP or the scrambled oligonucleotide. Under the condition in which the cell death experiment results shown in the bottom panel of Figure 4B were obtained, LRP expression decreased by ∼40% at 48 hr after transfection (24 hr after treatment) and by ∼70% at 72 hr after transfection (48 hr after treatment). In contrast, no decrease in LRP expression occurred by transfection with the scrambled oligonucleotide. Therefore, AS-LRP reduced toxicity by ApoE4 to the level of ApoE3 toxicity, whereas the scrambled oligonucleotide exerted no effect. Combined with the fact that the complementary nucleotide region of LRP corresponding to AS-LRP has no homology to any known lipoprotein-binding domain-containing receptors, these data provide evidence that LRP mediates the neurotoxicity of ApoE4.
Fig. 4.
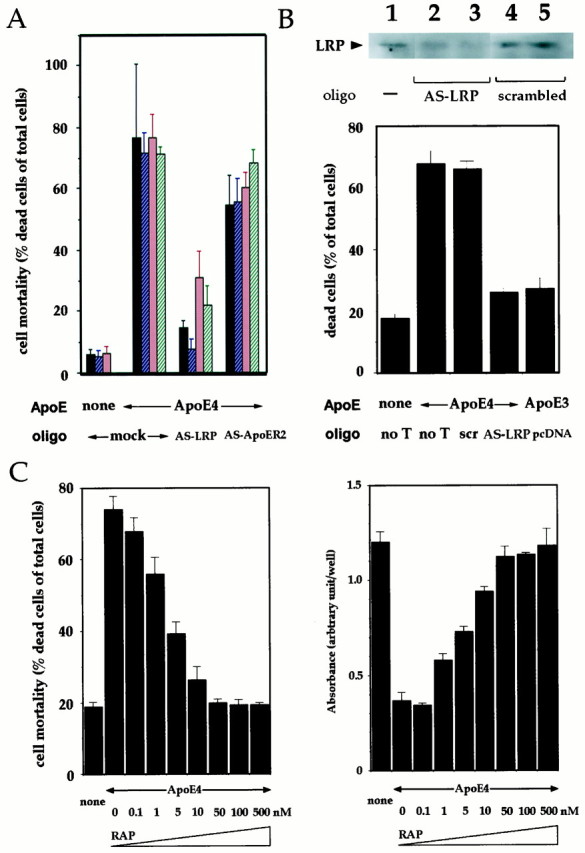
Evidence for the mediation by LRP of ApoE4-induced cell death. A, Effects of the antisense oligonucleotide to LRP or ApoER2. Twenty-four hours after either antisense oligonucleotide for LRP (AS-LRP) or ApoER2 (AS-ApoER2) was transfected into F11 cells with gold particles by the gene gun system, cells were treated with 30 μg/ml ApoE4 for 72 hr, and cell mortality was determined by Trypan blue exclusion assay. As controls, gold particles alone were transferred into F11 cells, and cells were treated with or without 30 μg/ml ApoE4 for 72 hr (mock). Each column indicates means ± SD of four to five independent transfections performed on 1 d. The results indicate four series of similar experiments, represented by different colors, each done on different days. In the fourth experiment, mortality was not measured in mock-transfected cells in the absence of ApoE4. B,Effects of the antisense oligonucleotide to LRP on ApoE4-induced cell mortality. F11 cells were transfected with or without AS-LRP, the scrambled oligonucleotide (scr), or pcDNA plasmid (no T, no transfection) and treated with 30 μg/ml ApoE3 or ApoE4, as described in Materials and Methods, and cell mortality was measured 72 hr after ApoE treatment.Inset, As performed in the panel, F11 cells were transfected with AS-LRP (lanes 2, 3) or a scrambled oligonucleotide (lanes 4, 5) for 12 hr (lane 1), 48 hr (lanes 2,4), or 72 hr (lanes 3,5). Cell lysates were submitted to immunoblot analysis with anti-LRP antibody. Because ApoE4 treatment was started 24 hr after transfection, the time point of each lane corresponds to before (lane 1), 24 hr (lanes 2,4), and 48 hr (lanes 3,5) after treatment. C, The dose–response relationship for the RAP suppression of ApoE4 toxicity. F11 cells were treated with or without 30 μg/ml ApoE4 in the presence or absence of increasing concentrations of recombinant RAP. Cell mortality (assessed by Trypan blue exclusion assay, left panel) and cell viability (assessed by WST-8 assay, right panel) were measured 72 hr after the onset of ApoE4 treatment.
Effect of LRP-associated protein RAP
RAP is a cell surface-associated protein that inhibits the delivery of ApoE to LRP. To confirm the involvement of LRP in the action of ApoE4, we examined the effect of RAP on ApoE4-induced death in neuronal cells. F11 cell death by ApoE4 was almost completely blocked by 50 nm recombinant RAP (Fig. 4C), whereas 1 μm RAP alone had no effect (data not shown). In addition, both Trypan blue exclusion assay and WST-8 cell viability assay consistently revealed that RAP dose-dependently suppressed ApoE4-induced neurotoxicity with an IC50 value of ∼5 nm. Table 2 indicates that the weak toxicity by high concentrations of ApoE3 was also suppressed by 50 nm RAP. Given that the IC50 value is 1–5 nm for RAP to specifically inhibit the function of LRP (Herz et al., 1991), it was highly likely that the toxic action of ApoE4 (and probably that of high concentrations of ApoE3) is mediated by LRP.
Involvement of PTX-sensitive GTPases in ApoE4 toxicity
Effect of PTX on the actions of ApoE4
AD-linked V642 mutants of the amyloid precursor protein (APP) cause apoptosis through the Go class of PTX-sensitive GTPases in neuronal cells (Yamatsuji et al., 1996;Giambarella et al., 1997). APP can directly interact with Go (Nishimoto et al., 1993), and V642I-APP can directly activate this G-protein in vitro (Okamoto et al., 1996). Wolozin et al. (1996) found that presenilin (PS)-2 induces apoptosis in PC12 cells in a PTX-sensitive manner, and Smine et al. (1998) showed that PS-1 activates Go through the C-terminal 39 residues. PS-1 and -2 are implicated in certain types of early onset familial AD. We therefore examined whether G-proteins are involved in ApoE4-induced cell death. F11 cells were treated with 30 μg/ml ApoE4 in the presence of 1 μg/ml PTX. Seventy-two hour incubation resulted in remarkable inhibition of ApoE4 action (Table 1). In contrast, heat-inactivated PTX failed to inhibit ApoE4-induced death, suggesting that the inhibitory effect of PTX was because of its enzymatic activity, not to chemicals or other contaminations in the PTX solution. Consistent with the PTX inhibition of ApoE4-induced cell mortality, the induction of DNA laddering was drastically attenuated when the cells were treated with 30 μg/ml ApoE4 in the presence of 1 μg/ml PTX (Fig. 2A,lane 5). Furthermore, PTX treatment appreciably inhibited TUNEL staining of F11 cells stimulated by 30 μg/ml ApoE4 (Fig.2B). These data provide evidence that PTX-sensitive G-proteins are involved in ApoE4-induced cell death.
Transfection of PTX-resistant mutants of Gi family Gα cDNAs
To confirm the involvement of PTX-sensitive G-proteins, we transfected PTX-resistant mutants of Gi family Gα cDNA into F11 cells, treated the transfected cells with ApoE4 in the presence of PTX, and examined whether ApoE4 induced cell death in a manner resistant to PTX. Resistance to PTX is conferred on the four members of the Gi family GTPases (Gαi1, Gαi2, Gαi3, Gαo) by the substitution of the Cys residue at the fourth position in the extreme C terminus (Taussig et al., 1992). The PTX-resistant mutants were termed GαiPT and GαoPT. With or without PTX, transfection of each PTX-resistant mutant cDNA did not significantly increase mortality in the absence of ApoE4, as compared with mock transfection (data not shown). In the presence of PTX, however, ApoE4 treatment killed cells transfected with either Gαi1PT, Gαi2PT, or Gαi3PT, whereas ApoE4 could not do so in cells transfected with an empty vector or GαoPT (Fig.5). Either transfection resulted in similar expression of the PTX-resistant mutants (data not shown). These data clearly indicate that ApoE4 causes cell death mediated by the PTX-sensitive Gi class of GTPases.
Fig. 5.

Recovery of ApoE4 toxicity in the presence of PTX by the transfection of PTX-resistant mutants of Gαi.A, Lack of the effect of GαPT transfection on ApoE4-induced cell death in the absence of PTX, but not in the presence of PTX. B, ApoE4 killed cells transfected with either GαiPT in the presence of PTX. Twenty-four hours after F11 cells were transfected with each PTX-resistant mutant of Gαi (Gαi1, Gαi2, or Gαi3) or Gαo or empty plasmid (vec), cells were treated with or without 30 μg/ml ApoE4 in the presence or absence of 500 ng/ml PTX for 72 hr, and mortality was measured by Trypan blue exclusion assay. The values indicate means ± SD of six independent transfections. Similar experiments were repeated three times in total, each with essentially the same results.
DISCUSSION
We have herein shown that ApoE causes death in neuronal cells in an isoform-specific manner and that at least one mechanism for neurotoxic actions of ApoE4 is apoptosis mediated by LRP. The toxicity of ApoE4 was observed in cells of neuroblast origin, but not in glial cells or non-neuronal CHO cells. The observed resistance of glial cells is consistent not only with the study of Crutcher et al. (1994), indicating that glial cells are resistant to neurotoxic ApoE peptides, but also with the well established finding that LRP is found abundantly in neurons but not in glial cells (Wolf et al., 1992; Lopes et al., 1994; Tooyama et al., 1995; Fabrizi et al., 1997). Whereas this study provides additional evidence that ApoE4 is toxic in neuronal cells, discrepancies have existed in the literature. In some studies, ApoE4 causes neurotoxic effects (Marques et al., 1997; Tolar et al., 1997,1999; Jordan et al., 1998; Michikawa and Yanagisawa, 1998; DeMattos et al., 1999), whereas in others, no toxicity has been found (Bellosta et al., 1995; Nathan et al., 1995; DeMattos et al., 1998). This variability could be attributed to several possibilities. One is that ApoE4 sensitivity of the neuronal cells used may be different not only in cell preparations but in cell conditions. Michikawa and Yanagisawa (1998) found that same neurons exhibit different responses to ApoE4 toxicity, in the presence or absence of compactin. Also, the tissue distribution (Bu et al., 1994; Zheng et al., 1994) suggests that RAP expression is differentially regulated from LRP expression, whereas their expression is mutually related (Willnow et al., 1995). It is thus conceivable that the ratio of cellular expression of RAP versus LRP, which could vary among neuronal cells and by cellular conditions, influences the toxic effects of ApoE4. Another possibility is that the culture conditions may affect ApoE4 neurotoxicity. The aforementioned studies reporting negative effects of ApoE4 were performed under conditions with serum or serum supplements including 5 μg/ml of insulin, which could suppress apoptosis. In contrast, our study was performed in the complete absence of serum or other supplements.
We also found that α2M* suppresses ApoE4-induced neuronal death. As this suppression was observed for α2M*, but not native α2M, this effect is highly likely mediated by LRP. However, it is unlikely that this suppression occurs only through inhibition by α2M* of ApoE4 binding to LRP, because Hussain et al. (1991) demonstrated only partial cross-competition between α2M* and ApoE-activated β-migrating very low-density lipoproteins for binding to LRP. Another possibility is that α2M* binding evokes internalization of LRP (Gliemann, 1998) and decreases the amount of cell surface LRP, resulting in impaired toxicity of ApoE4. The third possibility is that α2M* binding to LRP may suppress the function of LRP stimulated by another ligand ApoE4, without inhibiting the binding of ApoE4 to LRP. Such a phenomenon has been observed for another multiligand receptor, the mannose 6-phosphate/insulin-like growth factor-II receptor (Murayama et al., 1990; Takahashi et al., 1993; Ikezu et al., 1995). These possibilities are not mutually exclusive and could help to explain the nearly compete suppression of the ApoE4 effect, if they occur in combination. Whereas this is the first report that α2M negatively interferes with neuronal cell death caused by AD gene products, this α2M antagonism against ApoE4 concurs with recent reports (Blacker et al., 1998; Liao et al., 1998; Alvarez et al., 1999; Dodel et al., 2000; Romas et al., 2000) that polymorphisms of α2M are genetically associated with AD, although this association is controversial (Kovacs et al., 1999; Gibson et al., 2000; Higuchi et al., 2000; Sodeyama et al., 2000).
The receptors responsible for the reported neurotoxicity of ApoE have not been fully determined (Crutcher et al., 1994; Tolar et al., 1997,1999; Jordan et al., 1998; Moulder et al., 1999). Jordan et al. (1998) argued against the mediation of ApoE4-induced neurotoxicity by LDLR family members, based mainly on their finding that RAP treatment did not inhibit the toxicity of ApoE4 in rat hippocampal neurons. In contrast, Tolar et al. (1997, 1999) indicated that RAP suppresses the toxicity of 22 kDa N-terminal fragments of ApoE4, as well as full-length ApoE4, in chick lumbar sympathetic ganglions and rat hippocampal neurons, suggesting that LRP is involved in the neurotoxicity of ApoE4. Whereas the reported discrepancies might have been caused by different experimental conditions, our study provides different lines of evidence that ApoE4-bound LRP can cause neuronal cell apoptosis, although the possibility still exists that a hitherto unidentified LRP-like receptor, whose function is inhibitable by RAP and antisense LRP oligonucleotides, is responsible. Multiple groups (Lendon et al., 1997; Wavrant-DeVrieze et al., 1997, 1999; Kang et al., 1997; Kamboh et al., 1998; Hollenbach et al., 1998; Lambert et al., 1998; Beffert et al., 1999) observed the genetic association of LRP polymorphisms with AD, although this association is controversial (Clatworthy et al., 1997; Fallin et al., 1997; Baum et al., 1998; Scott et al., 1998). The involvement of LRP in ApoE4 neurotoxicity is consistent with the notion that LRP could be a risk factor for AD.
Because binding of ApoE to its receptor is usually thought to require lipid, the toxic action of rApoE4 in the absence of exogenous lipid was unexpected. However, Marques et al. (1997) reported that ApoE could exhibit neurotoxic effects in the absence of exogenous lipoproteins. In fact, in the present study, both rApoE3 and rApoE4 behaved similarly to DMPC-reconstituted rApoE as well as native ApoE proteins, regarding cell death. Recently, Tolar et al. (1999) reported that truncated ApoE4, which does not contain the lipid-binding domain, exerts cellular responses and toxicity from neurons through LRP, indicating that neurotoxicity by ApoE4 does not depend on lipoprotein interactions. Yu et al. (1998) also reported that lipid-free ApoE proteins are degraded by LRP. Therefore, there could be a mechanism that allows LRP to bind rApoE. In support, Demattos et al. (1998) reported that a minimally lipidated form of ApoE exhibits isoform-specific stimulation of neurite outgrowth, which has been confirmed to be mediated by LRP (Holtzman et al., 1995; Narita et al., 1997).
The molecular basis for the observed complicated actions of ApoE3 also deserves investigation. Because it is unlikely that ApoE3 binds to LRP in a manner different from that of ApoE4 binding to LRP, the binding of ApoE to LRP may not sufficiently explain the basis for the striking difference in the cytotoxic effects of these ApoE isoforms. Consider the following: (1) ApoE3 and ApoE4 share an identical LRP-binding domain; (2) low concentrations (≤30 μg/ml) of ApoE3 inhibited the toxic action of ApoE4; (3) higher concentrations (≥200 μg/ml) of ApoE3 exerted a toxic effect, probably through LRP; and (4) low concentrations (≤30 μg/ml) of ApoE3, but not ApoE4, suppress neurotoxicity not mediated by LRP (Jordan et al., 1998). Given these facts, it is highly likely that ApoE3 may exert two opposite effects, one a neurotoxic effect through LRP, and the other a neuroprotective effect through unknown mechanisms; and that ApoE4 may only exert its neurotoxic action through LRP. Although the mechanistic basis for the neuroprotective action of ApoE3 remains unknown, both in vitro and in vivo neuroprotections by ApoE3 have been reported in the literature (Puttfarcken et al., 1997; Jordan et al., 1998; Buttini et al., 1999, 2000; Pedersen et al., 2000). Pedersen et al. (2000) argued that the neuroprotective action of ApoE3 may be through a direct lipid peroxidation-detoxifying effect allowed by the presence of one Cys residue in ApoE3, a residue absent in ApoE4.
The present study also indicates, for the first time, that ApoE4-induced neurotoxicity occurs through the Giclass of GTPases. PTX-sensitive G-proteins have been implicated in the apoptotic death of neuronal cells (Yan et al., 1995; Yamatsuji et al., 1996; Wolozin et al., 1996; Yin et al., 1997; Farkas et al., 1998;Okazawa et al., 1998). Given that transfected Gβ2γ2, but not α subunits of PTX-sensitive G-proteins, induces DNA fragmentation in cultured cells (Giambarella et al., 1997), neurotoxicity of ApoE4 may occur through the Gβγ subunit released from activated Gi. A functional linkage between LRP and PTX-sensitive GTPases has so far been postulated (Misra et al., 1994,1999; Wang and Gruenstein, 1997). Goretzki and Mueller (1998) reported that RAP-precipitated LRP associates with several G-proteins, mainly Gs but also including Gi to some extent. Because, in their study, LRP was probably unbound to ApoE by virtue of RAP being used for precipitation, it is tempting to examine the LRP interaction with Gi in the absence of RAP and the presence of ApoE proteins. Recent studies (Yamatsuji et al., 1996; Wolozin et al., 1996; Hashimoto et al., 2000) have suggested that PTX-sensitive G-proteins act as a common target of multiple AD genes. The characterization of their downstream mechanisms would open a new avenue for the understanding and treatment of AD.
Footnotes
This work was supported in part by grants from the Naito Foundation, Brain Science Foundation, Takeda Science Foundation, the Ministry of Health and Welfare of Japan, the Ministry of Education, Science, and Culture of Japan and the Organization for Pharmaceutical Safety and Research.
We thank K. H. Weisgraber for his kind cooperation and native ApoE proteins; M. C. Fishman for F11 neuronal hybrid cells; T. Kozasa and R. Taussig for PTX-resistant Gα cDNAs; J. T. Potts Jr, E. Ogata, and Y. & Y. Tamai for indispensable encouragement; K. Yoshikawa for Bu695 cells; M. Morishima and Y. Ihara for SH-SY5Y cells; and E. Arakawa, D. Wylie, and K. Nishihara for expert technical assistance. We are especially indebted to T. Hiraki for cooperation in this study.
Y.H. and H.J. contributed equally to this study.
Correspondence should be addressed to Dr. Nishimoto or Dr. Murayama at the above addresses. E-mail: nisimoto@mc.med.keio.ac.jp.
REFERENCES
- 1.Alvarez V, Alvarez R, Lahoz CH, Martinez C, Pena J, Guisasola LM, Slas-Puig J, Moris G, Uria D, Menes BB, Ribacoba R, Vidal JA, Sanchez JM, Coto E. Association between an alpha(2) macroglobulin DNA polymorphism and late-onset Alzheimer's disease. Biochem Biophys Res Commun. 1999;264:48–50. doi: 10.1006/bbrc.1999.1295. [DOI] [PubMed] [Google Scholar]
- 2.Blacker D, Wilcox MA, Laird NM, Rodes L, Horvath SM, Go RCP, Perry R, Watson B, Jr, Bassett SS, McLnnis MG, Albert MS, Hyman BT, Tanzi RE. α-2 Macroglobulin is genetically associated with Alzheimer disease. Nat Genet. 1998;19:357–360. doi: 10.1038/1243. [DOI] [PubMed] [Google Scholar]
- 3.Baum L, Chen L, Ng HK, Chan YS, Mak YT, Woo J, Chiu HF, Pang CP. Low density lipoprotein receptor related protein gene exon 3 polymorphism association with Alzheimer's disease in Chinese. Neurosci Lett. 1998;247:33–36. doi: 10.1016/s0304-3940(98)00294-8. [DOI] [PubMed] [Google Scholar]
- 4.Beffert U, Arguin C, Poirier J. The polymorphism in exon 3 of the low density lipoprotein receptor-related protein gene is weakly associated with Alzheimer's disease. Neurosci Lett. 1999;259:29–32. doi: 10.1016/s0304-3940(98)00888-x. [DOI] [PubMed] [Google Scholar]
- 5.Bellosta S, Nathan BP, Orth M, Dong L-M, Mahley RW, Pitas RE. Stable expression and secretion of apolipoproteins E3 and E4 in mouse neuroblastoma cells produces differential effects on neurite outgrowth. J Biol Chem. 1995;270:27063–27071. doi: 10.1074/jbc.270.45.27063. [DOI] [PubMed] [Google Scholar]
- 6.Bu G, Maksymovitch EA, Nerbonne JM, Schwartz AL. Expression and function of the low density lipoprotein receptor-related protein (LRP) in mammalian central neurons. J Biol Chem. 1994;269:18521–18528. [PubMed] [Google Scholar]
- 7.Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW. Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: isoform-specific effects on neurodegeneration. J Neurosci. 1999;19:4867–4880. doi: 10.1523/JNEUROSCI.19-12-04867.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buttini M, Akeefe H, Lin C, Mahley RW, Pitas RE, Wyss-Coray T, Mucke L. Dominant negative effects of apolipoprotein E4 revealed in transgenic models of neurodegenerative disease. Neuroscience. 2000;97:207–210. doi: 10.1016/s0306-4522(00)00069-5. [DOI] [PubMed] [Google Scholar]
- 9.Clatworthy AE, Gomez-Isla T, Rebeck GW, Wallace RB, Hyman BT. Lack of association of a polymorphism in the low-density lipoprotein receptor-related protein gene with Alzheimer disease. Arch Neurol. 1997;54:1289–1292. doi: 10.1001/archneur.1997.00550220087019. [DOI] [PubMed] [Google Scholar]
- 10.Crutcher KA, Clay MA, Scott SA, Tian X, Tolar M, Harmony JAK. Neurite degeneration elicited by apolipoprotein E peptides. Exp Neurol. 1994;130:120–126. doi: 10.1006/exnr.1994.1191. [DOI] [PubMed] [Google Scholar]
- 11.DeMattos RB, Curtiss LK, Williams DL. A minimally lipidated form of cell-derived apolipoprotein E exhibits isoform-specific stimulation of neurite outgrowth in the absence of exogenous lipids or lipoproteins. J Biol Chem. 1998;273:4206–4212. doi: 10.1074/jbc.273.7.4206. [DOI] [PubMed] [Google Scholar]
- 12.DeMattos RB, Thorngate FE, Williams DL. A test of the cytosolic apolipoprotein E hypothesis fails to detect the escape of apolipoprotein E from the endocytic pathway into the cytosol and shows that direct expression of apolipoprotein E in the cytosol is cytotoxic. J Neurosci. 1999;19:2464–2473. doi: 10.1523/JNEUROSCI.19-07-02464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dodel RC, Du Y, Bales KR, Gao F, Eastwood B, Glazier B, Zimmer R, Cordell B, Hake A, Evans R, Gallagher-Thompson D, Thompson LW, Tinklenberg JR, Pfefferbaum A, Sullivan EV, Yesavage J, Alstiel L, Gasser T, Farlow MR, Murphy GM, Jr, Paul SM. α2-Macroglobulin and the risk of Alzheimer's disease. Neurology. 2000;54:438–442. doi: 10.1212/wnl.54.2.438. [DOI] [PubMed] [Google Scholar]
- 14.Egensperger R, Bancher C, Kosel S, Jellinger K, Mehraein P, Graeber MB. The apolipoprotein E epsilon 4 allele in Parkinson's disease with Alzheimer lesions. Biochem Biophys Res Commun. 1996;224:484–486. doi: 10.1006/bbrc.1996.1053. [DOI] [PubMed] [Google Scholar]
- 15.Fabrizi C, Businaro R, Persichini T, Fumagalli L, Lauro GM. The expression of the LDL receptor-related protein (LRP) correlates with the differentiation of human neuroblastoma cells. Brain Res. 1997;776:154–161. doi: 10.1016/s0006-8993(97)01035-4. [DOI] [PubMed] [Google Scholar]
- 16.Fallin D, Kundtz A, Town T, Gauntlett AC, Duara R, Barker W, Crawford F, Mullan M. No association between the low density lipoprotein receptor-related protein (LRP) gene and late-onset Alzheimer's disease in a community-based sample. Neurosci Lett. 1997;233:145–147. doi: 10.1016/s0304-3940(97)00634-4. [DOI] [PubMed] [Google Scholar]
- 17.Farkas I, Baranyi L, Liposits ZS, Yamamoto T, Okada H. Complement C5a anaphylatoxin fragment causes apoptosis in TGW neuroblastoma cells. Neuroscience. 1998;86:903–911. doi: 10.1016/s0306-4522(98)00108-0. [DOI] [PubMed] [Google Scholar]
- 18.Giambarella U, Yamatsuji T, Okamoto T, Matsui T, Ikezu T, Murayama Y, Levine MA, Katz A, Gautam N, Nishimoto I. G protein βγ complex-mediated apoptosis by familial Alzheimer's disease mutant of APP. EMBO J. 1997;16:4897–4907. doi: 10.1093/emboj/16.16.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibson AM, Singleton AB, Smith G, Woodward R, McKeith IG, Perry RH, Ince PG, Ballard CG, Edwardson JA, Morris CM. Lack of association of the α2-macroglobulin locus on chromosome 12 in AD. Neurology. 2000;54:433–438. doi: 10.1212/wnl.54.2.433. [DOI] [PubMed] [Google Scholar]
- 20.Gliemann J. Receptors of the low density lipoprotein (LDL) receptor family in man. Multiple functions of the large family members via interaction with complex ligands. Biol Chem. 1998;379:951–964. [PubMed] [Google Scholar]
- 21.Goedert M, Strittmatter WJ, Roses A. Risky apolipoprotein in brain. Nature. 1994;372:45–46. doi: 10.1038/372045a0. [DOI] [PubMed] [Google Scholar]
- 22.Goretzki L, Mueller BM. Low-density-lipoprotein-receptor-related protein (LRP) interacts with a GTP-binding protein. Biochem J. 1998;336:381–386. doi: 10.1042/bj3360381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gretch DG, Sturley SL, Friesen PD, Beckage NE, Attie AD. Baculovirus-mediated expression of human apolipoprotein E in Manduca sexta larvae generates particles that bind to the low density lipoprotein receptor. Proc Natl Acad Sci USA. 1991;88:8530–8533. doi: 10.1073/pnas.88.19.8530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hashimoto Y, Niikura T, Ito Y, Nishimoto I (2000) Multiple mechanisms underlie neurotoxicity by different types of Alzheimer's disease mutations of amyloid precursor protein. J Biol Chem, in press. [DOI] [PubMed]
- 25.Hayashi Y, Kashiwagi K, Yoshikawa K. Protease inhibitors generate cytotoxic fragments from Alzheimer amyloid protein precursor in cDNA-transfected glioma cells. Biochem Biophys Res Commun. 1992;187:1249–1255. doi: 10.1016/0006-291x(92)90437-p. [DOI] [PubMed] [Google Scholar]
- 26.Helisalmi S, Linnaranta K, Lehtovirta M, Mannermaa A, Heinonen O, Ryynanen M, Riekkinen Sr P, Soininen H. Apolipoprotein E polymorphism in patients with different neurodegenerative disorders. Neurosci Lett. 1996;205:61–64. doi: 10.1016/0304-3940(96)12373-9. [DOI] [PubMed] [Google Scholar]
- 27.Herz J, Goldstein JL, Strickland DK, Ho YK, Brown MS. 39-kDa protein modulates binding of ligands to low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J Biol Chem. 1991;266:21232–21238. [PubMed] [Google Scholar]
- 28.Higuchi S, Matsushita S, Nakane J, Arai H, Matsui T, Urakami K, Yuzuriha T, Takeda A. α2-Macroglobulin gene polymorphisms show racial diversity and are not associated with Alzheimer's disease. NeuroReport. 2000;11:1167–1171. doi: 10.1097/00001756-200004270-00005. [DOI] [PubMed] [Google Scholar]
- 29.Hollenbach E, Ackermann S, Hyman BT, Rebeck GW. Confirmation of an association between a polymorphism in exon 3 of the low-density lipoprotein receptor-related protein gene and Alzheimer's disease. Neurology. 1998;50:1905–1907. doi: 10.1212/wnl.50.6.1905. [DOI] [PubMed] [Google Scholar]
- 30.Holtzman DM, Pitas RE, Kilbridge J, Nathan B, Mahley RW, Bu G, Schwartz AL. Low density lipoprotein receptor-related protein mediates apolipoprotein E-dependent neurite outgrowth in a central nervous system-derived neuronal cell line. Proc Natl Acad Sci USA. 1995;92:9480–9484. doi: 10.1073/pnas.92.21.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hussain MM, Maxfield FR, Mas-Oliva J, Tabas I, Ji Z-S, Innerarity TL, Mahley RW. Clearance of chylomicron remnants by the low density lipoprotein receptor-related protein/α2-macroglobulin receptor. J Biol Chem. 1991;266:13936–13940. [PubMed] [Google Scholar]
- 32.Ikezu T, Okamoto T, Murayama Y, Okamoto T, Homma Y, Ogata E, Nishimoto I. Bidirectional regulation of c-fos promoter by an oncogenic gip2 mutant of Gαi2: a novel implication of retinoblastoma gene product. J Biol Chem. 1994;269:31955–31961. [PubMed] [Google Scholar]
- 33.Ikezu T, Okamoto T, Giambarella U, Yokota T, Nishimoto I. In vivo coupling of IGF-II/M6P receptor to heteromeric G proteins: distinct roles of cytoplasmic domains and signal sequestration by the receptor. J Biol Chem. 1995;270:29224–29228. doi: 10.1074/jbc.270.49.29224. [DOI] [PubMed] [Google Scholar]
- 34.Innerarity TL, Pitas RE, Mahley RW. Binding of arginine-rich (E) apoprotein after recombination with phospholipid vesicles to the low density lipoprotein receptors of fibroblasts. J Biol Chem. 1979;254:4186–4190. [PubMed] [Google Scholar]
- 35.Ji Y, Urakami K, Adachi Y, Maeda M, Isoe K, Nakashima K. Apolipoprotein E polymorphism in patients with Alzheimer's disease, vascular dementia and ischemic cerebrovascular disease. Dement Geriatr Cogn Disord. 1998;9:243–245. doi: 10.1159/000017068. [DOI] [PubMed] [Google Scholar]
- 36.Jordan J, Galindo MF, Miller RJ, Reardon CA, Getz GS, LaDu MJ. Isoform-specific effect of apolipoprotein E on cell survival and β-amyloid-induced toxicity in rat hippocampal pyramidal neuronal cultures. J Neurosci. 1998;18:195–204. doi: 10.1523/JNEUROSCI.18-01-00195.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalman J, Juhasz A, Majtenyi K, Rimanoczy A, Jakab K, Gardian G, Rasko I, Janka Z. Apolipoprotein E polymorphism in Pick's disease in Huntington's disease. Neurobiol Aging. 2000;21:555–558. doi: 10.1016/s0197-4580(00)00150-0. [DOI] [PubMed] [Google Scholar]
- 38.Kamboh MI, Ferrell RE, DeKosky ST. Genetic association studies between Alzheimer's disease and two polymorphisms in the low density lipoprotein receptor-related protein gene. Neurosci Lett. 1998;244:65–68. doi: 10.1016/s0304-3940(98)00141-4. [DOI] [PubMed] [Google Scholar]
- 39.Kang DE, Saitoh T, Chen X, Xia Y, Masliah E, Hansen LA, Thomas RG, Thal LJ, Katzman R. Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer's disease. Neurology. 1997;49:56–61. doi: 10.1212/wnl.49.1.56. [DOI] [PubMed] [Google Scholar]
- 40.Kovacs T, Cairns NJ, Lantos PL. α2-Macroglobulin intronic polymorphism is not associated with autopsy-confirmed late-onset Alzheimer's disease. Neurosci Lett. 1999;273:61–83. doi: 10.1016/s0304-3940(99)00604-7. [DOI] [PubMed] [Google Scholar]
- 41.Kruger R, Vieira-Saecker AM, Kuhn W, Berg D, Muller T, Kuhnl N, Fuchs GA, Storch A, Hungs M, Woitalla D, Przuntek H, Epplen JT, Schols L, Riess O. Increased susceptibility to sporadic Parkinson's disease by a certain combined alpha-synuclein/apolipoprotein E genotype. Ann Neurol. 1999;45:611–617. doi: 10.1002/1531-8249(199905)45:5<611::aid-ana9>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 42.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to β-amyloid. J Biol Chem. 1994;269:23403–23406. [PubMed] [Google Scholar]
- 43.LaDu MJ, Pederson TM, Frail DE, Reardon CA, Getz GS, Falduto MT. Purification of apolipoprotein E attenuates isoform-specific binding to β-amyloid. J Biol Chem. 1995;270:9039–9042. doi: 10.1074/jbc.270.16.9039. [DOI] [PubMed] [Google Scholar]
- 44.Lambert JC, Wavrant-DeVrieze F, Amouyel P, Chartier-Harlin MC. Association at LRP gene locus with sporadic late-onset Alzheimer's disease. Lancet. 1998;351:1787–1788. doi: 10.1016/s0140-6736(05)78749-3. [DOI] [PubMed] [Google Scholar]
- 45.Lendon CL, Talbot CJ, Craddock NJ, Han SW, Wragg M, Morris JC, Goate AM. Genetic association studies between dementia of the Alzheimer's type and three receptors for apolipoprotein E in a Caucasian population. Neurosci Lett. 1997;222:187–190. doi: 10.1016/s0304-3940(97)13381-x. [DOI] [PubMed] [Google Scholar]
- 46.Liao A, Nitsch RM, Greenberg SM, Finckh U, Blacker D, Albert M, Rebeck GW, Gomez-Isla T, Clatworthy A, Binetti G, Hock C, Mueller-Thomsen T, Mann U, Zuchowski K, Beisiegel U, Staehelin H, Growdon JH, Tanzi R, Hyman BT. Genetic association of an α2-macroglobulin (Val1000Ile) polymorphism and Alzheimer's disease. Hum Mol Genet. 1998;7:1953–1956. doi: 10.1093/hmg/7.12.1953. [DOI] [PubMed] [Google Scholar]
- 47.Lopes MBS, Bogaev CA, Gonias SL, VandenBerg SR. Expression of α2-macroglobulin is increased in reactive and neoplastic glial cells. FEBS Lett. 1994;338:301–305. doi: 10.1016/0014-5793(94)80288-2. [DOI] [PubMed] [Google Scholar]
- 48.Marques MA, Tolar M, Harmony JA, Crutcher KA. A thrombin cleavage fragment of apolipoprotein E exhibits isoform-specific neurotoxicity. NeuroReport. 1996;7:2529–2532. doi: 10.1097/00001756-199611040-00025. [DOI] [PubMed] [Google Scholar]
- 49.Marques MA, Tolar M, Crutcher KA. Apolipoprotein E exhibits isoform-specific neurotoxicity. Alzheimer Res. 1997;3:1–6. doi: 10.1097/00001756-199611040-00025. [DOI] [PubMed] [Google Scholar]
- 50.Michikawa M, Yanagisawa K. Apolipoprotein E4 induces neuronal cell death under conditions of suppressed de novo cholesterol synthesis. J Neurosci Res. 1998;54:58–67. doi: 10.1002/(SICI)1097-4547(19981001)54:1<58::AID-JNR7>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 51.Misra UK, Chu CT, Gawdi G, Pizzo SV. The relationship between low density lipoprotein-related protein/α2-macroglobulin (α2M) receptors and the newly described α2M signaling receptor. J Biol Chem. 1994;269:18303–18306. [PubMed] [Google Scholar]
- 52.Misra UK, Gawdi G, Pizzo SV. Ligation of low-density lipoprotein receptor-related protein with antibodies elevates intracellular calcium and inositol 1,4,5-trisphosphate in macrophages. Arch Biochem Biophys. 1999;372:238–247. doi: 10.1006/abbi.1999.1521. [DOI] [PubMed] [Google Scholar]
- 53.Moulard B, Sefiani A, Laamri A, Malafosse A, Camu W. Apolipoprotein E genotyping in sporadic amyotrophic lateral sclerosis: evidence for a major influence on the clinical presentation and prognosis. J Neurol Sci [Suppl] 1996;139:34–37. doi: 10.1016/0022-510x(96)00085-8. [DOI] [PubMed] [Google Scholar]
- 54.Moulder KL, Narita M, Chang LK, Bu G, Johnson EM., Jr Analysis of a novel mechanism of neuronal toxicity produced by an apolipoprotein E-derived peptide. J Neurochem. 1999;72:1069–1080. doi: 10.1046/j.1471-4159.1999.0721069.x. [DOI] [PubMed] [Google Scholar]
- 55.Mui S, Rebeck GW, McKenna-Yasek D, Hyman BT, Brown RH., Jr Apolipoprotein E epsilon 4 allele is not associated with earlier age at onset in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:460–463. doi: 10.1002/ana.410380318. [DOI] [PubMed] [Google Scholar]
- 56.Murayama Y, Okamoto T, Ogata E, Asano T, Iiri T, Katada T, Ui M, Grubb JH, Sly WS, Nishimoto I. Distinctive regulation of the functional linkage between the human cation-independent mannose 6-phosphate receptor and GTP-binding proteins by insulin-like growth factor II and mannose 6-phosphate. J Biol Chem. 1990;265:17456–17462. [PubMed] [Google Scholar]
- 57.Narita M, Bu G, Holtzman DM, Schwartz AL. The low-density lipoprotein receptor-related protein, a multifunctional apolipoprotein E receptor, modulates hippocampal neurite development. J Neurochem. 1997;68:587–595. doi: 10.1046/j.1471-4159.1997.68020587.x. [DOI] [PubMed] [Google Scholar]
- 58.Nathan BP, Bellosata S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE. Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science. 1994;264:850–852. doi: 10.1126/science.8171342. [DOI] [PubMed] [Google Scholar]
- 59.Nathan BP, Chang K-C, Bellosta S, Brisch E, Ge N, Mahley RW, Pitas RE. The inhibitory effect of apolipoprotein E4 on neurite outgrowth is associated with microtubule depolymerization. J Biol Chem. 1995;270:19791–19799. doi: 10.1074/jbc.270.34.19791. [DOI] [PubMed] [Google Scholar]
- 60.Nishimoto I, Okamoto T, Matsuura Y, Okamoto T, Murayama Y, Ogata E. Alzheimer amyloid protein precursor forms a complex with brain GTP binding protein Go. Nature. 1993;362:75–79. doi: 10.1038/362075a0. [DOI] [PubMed] [Google Scholar]
- 61.Okamoto T, Takeda S, Giambarella U, Matsuura Y, Katada T, Nishimoto I. Intrinsic G-coupling function of amyloid precursor protein as a novel target of V642 mutations linked to familial Alzheimer disease. EMBO J. 1996;15:3769–3777. [PMC free article] [PubMed] [Google Scholar]
- 62.Okazawa M, Shiraki T, Ninomiya H, Kobayashi S, Masaki T. Endothelin-induced apoptosis of A375 human melanoma cells. J Biol Chem. 1998;273:12584–12592. doi: 10.1074/jbc.273.20.12584. [DOI] [PubMed] [Google Scholar]
- 63.Pedersen WA, Chan SL, Mattson MP. A mechanism for the neuroprotective effect of apolipoprotein E: isoform-specific modification by the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 2000;74:1426–1433. doi: 10.1046/j.1471-4159.2000.0741426.x. [DOI] [PubMed] [Google Scholar]
- 64.Pillot T, Goethals M, Najib J, Labeur C, Lins L, Chambaz J, Brasseur R, Vandekerckhove J, Rosseneu M. Beta-amyloid peptide interacts specifically with the carboxy-terminal domain of human apolipoprotein E: relevance to Alzheimer's disease. J Neurochem. 1999;72:230–237. doi: 10.1046/j.1471-4159.1999.0720230.x. [DOI] [PubMed] [Google Scholar]
- 65.Platika D, Boulos MH, Braizer L, Fishman MC. Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc Natl Acad Sci USA. 1985;82:3499–3503. doi: 10.1073/pnas.82.10.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Puttfarcken PS, Manelli AM, Falduto MT, Getz GS, LaDu MJ. Effect of apolipoprotein E on neurite outgrowth and beta-amyloid-induced toxicity in developing rat primary hippocampal cultures. J Neurochem. 1997;68:760–769. doi: 10.1046/j.1471-4159.1997.68020760.x. [DOI] [PubMed] [Google Scholar]
- 67.Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW, Mucke L. Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci USA. 1998;95:10914–10919. doi: 10.1073/pnas.95.18.10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rall SC, Jr, Weisgraber KH, Mahley RW. Isolation and characterization of apolipoprotein E. Methods Enzymol. 1986;128:273–287. doi: 10.1016/0076-6879(86)28073-8. [DOI] [PubMed] [Google Scholar]
- 69.Romas SN, Mayeux R, Rabinowitz D, Tang MX, Zadroga HR, Lantigua R, Medrano M, Tycko B, Knowles JA. The deletion polymorphism and Val1000Ile in α2-macroglobulin and Alzheimer disease in Caribbean Hispanics. Neurosci Lett. 2000;279:133–136. doi: 10.1016/s0304-3940(99)00972-6. [DOI] [PubMed] [Google Scholar]
- 70.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Association of apolipoprotein E allele ε4 with late-onset familial and sporadic Alzheimer's disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 71.Scott WK, Yamaoka LH, Bass MP, Gaskell PC, Conneally PM, Small GW, Farrer LA, Auerbach SA, Saunders AM, Roses AD, Haines JL, Pericak-Vance MA. No genetic association between the LRP receptor and sporadic or late-onset familial Alzheimer disease. Neurogenetics. 1998;1:179–183. doi: 10.1007/s100480050026. [DOI] [PubMed] [Google Scholar]
- 72.Siddique T, Pericak-Vance MA, Caliendo J, Hong ST, Hung WY, Kaplan J, McKenna-Yasek D, Rimmler JB, Sapp P, Saunders AM, Scott WK, Siddique N, Haines JL, Brown RH. Lack of association between apolipoprotein E genotype and sporadic amyotrophic lateral sclerosis. Neurogenetics. 1998;1:213–216. doi: 10.1007/s100480050031. [DOI] [PubMed] [Google Scholar]
- 73.Smine A, Xu X, Nishiyama K, Katada T, Gambetti P, Yadav SP, Wu X, Shi YC, Yasuhara S, Homburger V, Okamoto T. Regulation of brain G-protein Go by Alzheimer's disease gene presenilin-1. J Biol Chem. 1998;273:16281–16288. doi: 10.1074/jbc.273.26.16281. [DOI] [PubMed] [Google Scholar]
- 74.Sodeyama N, Yamada M, Itoh Y, Suematsu N, Matsushita M, Otomo E, Mizusawa H. α2-Macroglobulin polymorphism is not associated with AD or AD-type neuropathology in the Japanese. Neurology. 2000;54:443–446. doi: 10.1212/wnl.54.2.443. [DOI] [PubMed] [Google Scholar]
- 75.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sun Y, Wu S, Bu G, Onifade MK, Patel SN, LaDu MJ, Fagan AM, Holtzman DM. Glial fibrillary acidic protein-apolipoprotein E (apoE) transgenic mice: astrocyte-specific expression and differing biological effects of astrocyte-secreted apoE3 and apoE4 lipoproteins. J Neurosci. 1998;18:3261–3272. doi: 10.1523/JNEUROSCI.18-09-03261.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Takahashi K, Murayama Y, Okamoto T, Yokota T, Ikezu T, Takahashi S, Giambarella U, Ogata E, Nishimoto I. Conversion of G-protein specificity of insulin-like growth factor II/mannose 6-phosphate receptor by exchanging of a short region with b-adrenergic receptor. Proc Natl Acad Sci USA. 1993;90:11772–11776. doi: 10.1073/pnas.90.24.11772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taussig R, Sanchez S, Rifo M, Gilman AG, Belardetti F. Inhibition of the ω-conotoxin-sensitive calcium current by distinct G proteins. Neuron. 1992;8:799–809. doi: 10.1016/0896-6273(92)90100-r. [DOI] [PubMed] [Google Scholar]
- 79.The French Parkinson's Disease Genetics Study Group. Apolipoprotein E genotype in familial Parkinson's disease. J Neurol Neurosurg Psychiatry. 1997;63:394–395. [PMC free article] [PubMed] [Google Scholar]
- 80.Tolar M, Marques MA, Harmony JAK, Crutcher KA. Neurotoxicity of the 22 kDa thrombin-cleavage fragment of apolipoprotein E and related synthetic peptides is receptor-mediated. J Neurosci. 1997;17:5678–5686. doi: 10.1523/JNEUROSCI.17-15-05678.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tolar M, Keller JN, Chan S, Mattson MP, Marques MA, Crutcher KA. Truncated apolipoprotein E (apoE) causes increased intracellular calcium and may mediate apoE neurotoxicity. J Neurosci. 1999;19:7100–7110. doi: 10.1523/JNEUROSCI.19-16-07100.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tooyama I, Kawamata T, Akiyama H, Kimura H, Moestrup SK, Gliemann J, Matsuo A, McGeer PL. Subcellular localization of the low density lipoprotein receptor-related protein (α2-macroglobulin receptor) in human brain. Brain Res. 1995;691:235–238. doi: 10.1016/0006-8993(95)00735-9. [DOI] [PubMed] [Google Scholar]
- 83.Ueki A, Kawano M, Namba Y, Kawakami M, Ikeda K. A high frequency of apolipoprotein E4 isoprotein in Japanese patients with late-onset nonfamilial Alzheimer's disease. Neurosci Lett. 1993;163:166–168. doi: 10.1016/0304-3940(93)90373-s. [DOI] [PubMed] [Google Scholar]
- 84.Yamatsuji T, Okamoto T, Takeda S, Fukumoto H, Iwatsubo T, Suzuki N, Asami-Odaka A, Ireland S, Kinane TB, Nishimoto I. Neuronal DNA fragmentation by familial Alzheimer's V642 mutants of APP via heteromeric G proteins. Science. 1996;272:1349–1352. doi: 10.1126/science.272.5266.1349. [DOI] [PubMed] [Google Scholar]
- 85.Yan GM, Lin SZ, Irwin RP, Paul SM. Activation of G proteins bidirectionally affects apoptosis of cultured cerebellar granule neurons. J Neurochem. 1995;65:2425–2431. doi: 10.1046/j.1471-4159.1995.65062425.x. [DOI] [PubMed] [Google Scholar]
- 86.Yin DL, Ren XH, Zheng ZL, Pu L, Jiang LZ, Ma L, Pei G. Etorphine inhibits cell growth and induces apoptosis in SK-N-SH cells: involvement of pertussis toxin-sensitive G proteins. Neurosci Res. 1997;29:121–127. doi: 10.1016/s0168-0102(97)00080-1. [DOI] [PubMed] [Google Scholar]
- 87.Yoshida Y, Kobayashi E, Endo H, Hamamoto T, Yamanaka T, Fujimura A, Kagawa Y. Introduction of DNA into rat liver with a hand-held gene gun: distribution of the expressed enzyme, [32P]DNA, and Ca2+ flux. Biochem Biophys Res Commun. 1997;234:695–700. doi: 10.1006/bbrc.1997.6682. [DOI] [PubMed] [Google Scholar]
- 88.Yu L, Narita M, Bu G, Schwartz A, Holtzman D. Lipid free apoE3 and apoE4 are differentially degraded via cellular LRP. Soc Neurosci Abstr. 1998;24:1712. [Google Scholar]
- 89.Wang XS, Gruenstein E. Rapid elevation of neuronal cytoplasmic calcium by apolipoprotein E peptide. J Cell Physiol. 1997;173:73–83. doi: 10.1002/(SICI)1097-4652(199710)173:1<73::AID-JCP9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 90.Wavrant-DeVrieze F, Perez-Tur J, Lambert JC, Frigard B, Pasquier F, Delacourte A, Amouyel P, Hardy J, Chartier-Harlin MC. Association between the low density lipoprotein receptor-related protein (LRP) and Alzheimer's disease. Neurosci Lett. 1997;227:68–70. doi: 10.1016/s0304-3940(97)00304-2. [DOI] [PubMed] [Google Scholar]
- 91.Wavrant-DeVrieze F, Lambert JC, Stas L, Crook R, Cottel D, Pasquier F, Frigard B, Lambrechts M, Thiry E, Amouyel P, Tur JP, Chartier-Harlin MC, Hardy J, Van Leuven F. Association between coding variability in the LRP gene and the risk of late-onset Alzheimer's disease. Hum Genet. 1999;104:432–434. doi: 10.1007/s004390050980. [DOI] [PubMed] [Google Scholar]
- 92.Whitson JS, Mims MP, Strittmatter WJ, Yamaki T, Morrisett JD, Appel SH. Attenuation of the neurotoxic effect of Aβ amyloid peptide by apolipoprotein E. Biochem Biophys Res Commun. 1994;199:163–170. doi: 10.1006/bbrc.1994.1209. [DOI] [PubMed] [Google Scholar]
- 93.Willnow TE, Armstrong SA, Hammer RE, Herz J. Functional expression of low density lipoprotein receptor-related protein is controlled by receptor-associated protein in vivo. Proc Natl Acad Sci USA. 1995;92:4537–4541. doi: 10.1073/pnas.92.10.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wilson C, Wardell MR, Weisgraber KH, Mahley RW, Agard DA. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science. 1991;252:1817–1822. doi: 10.1126/science.2063194. [DOI] [PubMed] [Google Scholar]
- 95.Wolf BB, Lopes MBS, VandenBerg SR, Gonias SL. Characterization and immunohistochemical localization of α2-macroglobulin receptor (low-density lipoprotein receptor-related protein) in human brain. Am J Pathol. 1992;141:37–42. [PMC free article] [PubMed] [Google Scholar]
- 96.Wolozin B, Iwasaki K, Vito P, Ganjei JK, Lacana E, Sunderland T, Zhao B, Kusiak JW, Wasco W, D'Adamio L. Participation of presenilin 2 in apoptosis: enhanced basal activity conferred by an Alzheimer mutation. Science. 1996;274:1710–1713. doi: 10.1126/science.274.5293.1710. [DOI] [PubMed] [Google Scholar]
- 97.Zannis VI, Breslow JL, Utermann G, Mahley RW, Weisgraber KH, Havel RJ, Goldstein JL, Brown MS, Schonfeld G, Hazzard WR, Blum C. Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes. J Lipid Res. 1982;23:911–914. [PubMed] [Google Scholar]
- 98.Zareparsi S, Kaye J, Camicioli R, Grimslid H, Oken B, Litt M, Nutt J, Bird T, Schellenberg G, Payami H. Modulation of the age at onset of Parkinson's disease by apolipoprotein E genotypes. Ann Neurol. 1997;42:655–658. doi: 10.1002/ana.410420417. [DOI] [PubMed] [Google Scholar]
- 99.Zheng G, Bachinsky DR, Stamenkovic I, Strickland DK, Brown D, Andres G, McCluskey RT. Organ distribution in rats of two members of the low-density lipoprotein receptor gene family, gp330 and LRP/alpha 2MR, and the receptor-associated protein (RAP). J Histochem Cytochem. 1994;42:531–542. doi: 10.1177/42.4.7510321. [DOI] [PubMed] [Google Scholar]