

Abstract
Long-term depression (LTD) is a form of synaptic plasticity that can be induced either by low-frequency stimulation of presynaptic fibers or in an associative manner by asynchronous pairing of presynaptic and postsynaptic activity. We investigated the induction mechanisms of associative LTD in CA1 pyramidal neurons of the hippocampus using whole-cell patch-clamp recordings and Ca2+ imaging in acute brain slices. Asynchronous pairing of postsynaptic action potentials with EPSPs evoked with a delay of 20 msec induced a robust, long-lasting depression of the EPSP amplitude to 43%. Unlike LTD induced by low-frequency stimulation, associative LTD was resistant to the application ofd-AP-5, indicating that it is independent of NMDA receptors. In contrast, associative LTD was inhibited by (S)-α-methyl-4-carboxyphenyl-glycine, indicating the involvement of metabotropic glutamate receptors. Furthermore, associative LTD is dependent on the activation of voltage-gated Ca2+ channels by postsynaptic action potentials. Both nifedipine, an L-type Ca2+ channel antagonist, and ω-conotoxin GVIA, a selective N-type channel blocker, abolished the induction of associative LTD. 8-hydroxy-2-dipropylaminotetralin (OH-DPAT), a 5-HT1Areceptor agonist, inhibited postsynaptic Ca2+ influx through N-type Ca2+ channels, without affecting presynaptic transmitter release. OH-DPAT also inhibited the induction of associative LTD, suggesting that the involvement of N-type channels makes synaptic plasticity accessible to modulation by neurotransmitters. Thus, the modulation of N-type Ca2+ channels provides a gain control for synaptic depression in hippocampal pyramidal neurons.
Keywords: associative long-term depression, hippocampus, N-type Ca2+ channels, NMDA receptors, metabotropic glutamate receptors, asynchronous pairing
Long-term changes in synaptic strength at glutamatergic synapses are thought to underlie complex functions of neuronal networks, such as learning and memory (Bliss and Collingridge, 1993). The frequency of synaptic stimulation determines both the extent and the direction of the change in synaptic efficacy; high-frequency stimulation (HFS) leads to long-term potentiation (LTP), whereas low-frequency stimulation (LFS) results in long-term depression (LTD) (Dudek and Bear, 1992). Whereas several molecular steps of the induction and expression of LTP have been identified, the mechanisms that lead to LTD are less clear.
In the hippocampus, LFS induces two distinct forms of LTD, which depend either on the Ca2+ influx through NMDA receptors (NMDARs) (Mulkey and Malenka, 1992) or on the activation of metabotropic glutamate receptors (mGluRs) (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Otani and Connor, 1998). The mGluR LTD appears to be the predominant form in young animals (postnatal days 3–8; Bolshakov and Siegelbaum, 1994), whereas NMDAR LTD and mGluR LTD coexist in older animals (Heynen et al., 1996; Oliet et al., 1997;Otani and Connor, 1998).
Although LFS induces robust changes in synaptic strength, the situation that leads to the induction of hippocampal LTD in vivo could be more complex. CA3 and CA1 pyramidal neurons generate action potentials in a precise temporal relationship, depending on the behavioral context (O'Keefe and Reece, 1993), i.e., pyramidal cells in the center of their place field fire action potentials more early in the theta cycle than neurons with adjacent place fields (Skaggs et al., 1996). Thus, the natural paradigm for LTP and LTD induction is likely to be associative, requiring the temporal coincidence of synaptic activation and backpropagating action potentials (for review, seeLinden, 1999). Indeed associative LTD in the hippocampus can be induced by asynchronous pairing of presynaptic and postsynaptic activity (Levy and Steward, 1983; Stanton and Sejnowski, 1989; Stanton et al., 1991). However, the induction mechanisms of associative LTD have remained controversial. Associative LTD in acute slices was reported to be independent of NMDARs (Stanton and Sejnowski, 1989). The opposite was shown for associative LTD in organotypic cell culture (Debanne et al., 1994). Finally, in dissociated hippocampal cell culture, the associative LTD appeared to be dependent on both Ca2+ influx through NMDARs and L-type Ca2+ channels (Bi and Poo, 1998).
Here we investigated the conditions necessary for the induction of associative LTD in acute hippocampal slices by asynchronous paring of presynaptic and postsynaptic activity at the Schaffer collateral–CA1 pyramidal cell synapse. The results suggest that associative LTD is dependent on both mGluRs and Ca2+ influx through voltage-gated L- and N-type Ca2+channels. As N-type Ca2+ channels are preferential targets of G-protein-mediated neuromodulation (Hille, 1994), we have tested whether the modulation of postsynaptic N-type Ca2+ channels could affect LTD induction, which would provide a novel mechanism to regulate activity-dependent synaptic plasticity in the hippocampus.
MATERIALS AND METHODS
Slice preparation. Transverse 300-μm-thick slices were cut from the hippocampus of 11- to 22-d-old Wistar rats with a vibratome (DTK-1000; Dosaka, Kyoto, Japan). For most experiments 14- to 18-d-old animals were used. The animals were killed by decapitation, in accordance with national and institutional guidelines. Slices were kept at 35°C for 30 min after slicing and then at room temperature in physiological extracellular saline containing (in mm): 125 NaCl, 25 NaHCO3, 25 glucose, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCl2, bubbled with carbogene (95% O2 and 5% CO2).
Electrophysiology. The slices were transferred to the recording chamber and continuously superfused with saline at a flow rate of 5–10 ml/min (chamber volume, ∼2 ml). CA1 pyramidal neurons were identified by their location using infrared differential interference contrast video microscopy and their characteristic firing frequency adaptation during long depolarizing current pulses. Patch pipettes were pulled from borosilicate glass tubing (2.0 mm outer diameter, 0.5 mm wall thickness; Hilgenberg, Malsfeld, Germany) and heat-polished immediately before use. An Axopatch 200A amplifier (Axon Instruments, Foster City, CA) or an EPC-9 amplifier (Heka, Lambrecht, Germany) were used for current-clamp (I-clamp fast) and voltage-clamp recordings. The Axopatch amplifier included a bridge-balance circuit for compensation of series resistance in the current-clamp mode, similar to that of the Axopatch 200B. Current and voltage signals were filtered at 5 and 10 kHz, respectively, with a 4-pole lowpass Bessel filter and digitized at 10 or 20 kHz with a 1401plus interface (CED, Cambridge, UK). For data acquisition and analysis we used self-made and commercial programs (EPC, CED; Pulse, Heka).
For current-clamp recordings the patch pipettes were filled with an internal solution containing (in mm): 135 K-gluconate, 20 KCl, 2 MgCl2, 2 Na2ATP, 0.3 NaGTP, 0.2–0.5 EGTA, and 10 HEPES (pH was adjusted to 7.3 with KOH). Dendritic recordings were performed as described previously (Bischofberger and Jonas, 1997). Patch pipette resistance was 5–10 MΩ for somatic and 10–12 MΩ for dendritic recordings. Bridge balance was used to compensate the series resistance of 20–60 MΩ.
Presynaptic Schaffer collateral fibers were stimulated using a stimulus isolator (List, Darmstadt, Germany) and a patch pipette with a resistance of 1–3 MΩ when filled with HEPES-buffered Na+-rich solution. The stimulus pipette was placed in the stratum radiatum of the CA1 region 20–50 μm away from the pyramidal cell layer. Two hundred microsecond voltage pulses of 10–80 V were applied to evoke subthreshold EPSPs at a frequency of 0.1 Hz. Orthodromic stimulation was performed in >90% of the experiments, and antidromic stimulation was performed in <10%, without obvious differences concerning basal transmission and plasticity induction. The latency between the center of the stimulus artifact and the onset of the EPSP was 2.9 ± 0.1 msec (n = 53), indicating monosynaptic transmission. Picrotoxin (20–50 μm) was present in the external solution of all current-clamp experiments. In some experiments 10 μm glycine was added to the bath solution, with no obvious differences in the results.
To record Ba2+ currents, the recording pipettes (mostly 0.8–2 MΩ) were filled with a solution containing (in mm): 140 CsCl, 2 MgCl2, 2 Na2ATP, 0.3 NaGTP, 10 EGTA, and 10 HEPES (pH adjusted to 7.3 with CsOH). The bath solution contained 140 NaCl, 2 tetraethylammonium chloride (TEACl), 2 MgCl2, 2 BaCl2, 1 μm tetrodotoxin (TTX) and 10 HEPES (pH adjusted to 7.4 with NaOH). Recordings were made in the voltage-clamp configuration with series resistance (Rs) compensation (nominally 80–90%, lag 20–100 μsec; Rs before compensation 3–10 MΩ). Leak and capacitive currents were subtracted using a P/-4 protocol. For whole-cell voltage-clamp experiments we used only slices from 11- to 13-d-old animals. Thus the CA1 pyramidal cells had relatively short and thin dendrites, minimizing space-clamp artifacts. Voltage-clamp and current-clamp recordings were made at 22–24°C.
Data analysis and statistics. All values are given as mean ± SEM, error bars in the figures also represent SEM. The decay time course of EPSPs was fit with a single exponential. To calculate the mean EPSP peak amplitude, 10–15 consecutive EPSPs were averaged from each experiment shortly before and 15–20 min after the end of the induction protocol. The changes in the mean EPSP amplitude were analyzed for each experiment, and statistical significance was assessed by a two-tailed Wilcoxon test at the significance level (P) indicated. Traces shown in the figures represent averages of 10–15 consecutive sweeps. Average EPSP amplitudes in amplitude–time plots represent means from seven consecutive EPSPs.
Fluorescence measurements. For the measurement of the intracellular Ca2+ signals we used 0.1 mm fura-2 (Molecular Probes, Eugene, OR) instead of EGTA in the pipette solution. Cells were loaded for at least 15–20 min in the whole-cell configuration before measurements were started. The excitation light source (Polychrome II with 75 W Xenon lamp; TILL Photonics, Munich, Germany) was coupled to the epifluorescent port of the microscope (Axioskop FS2, Zeiss; 60× water immersion objective, Olympus Optical, Tokyo, Japan) via a light guide. To minimize bleaching, the light intensity was reduced to 10%. The filter combination for excitation and emission comprised a beam splitter (BSP410) and emission filters (LWP420, KP600) from Delta Light & Optics (Lyngby, Denmark). Some experiments were done with 0.1 mm Oregon Green (Oregon Green 488 Bapta-1, Molecular Probes) instead of fura-2, using a filter combination from Zeiss(FT510, LP 520) and an excitation light intensity of 5%.
The fluorescence was measured with a backilluminated frame-transfer CCD camera (EBFT 512; Princeton Instruments). Images with full spatial resolution were taken with exposure times of 5 sec. For high-speed Ca2+ measurements (100 Hz repetition rate) we usually defined three rectangular regions of interest (ROIs) of 5 × 5 μm at the soma and 5 × 20 μm at a proximal and distal part of the apical dendrite. The pixels included in the ROIs were binned on-chip and digitized subsequently by the controller (Micromax; 1 MHz; Princeton Instruments). The fluorescence signals were corrected for background, which was obtained from ROIs shifted by 10–15 μm with respect to the original ROIs (Schiller et al., 1995).
Calibration of the Ca2+ measurements with fura-2. To convert the fluorescence signals into Ca2+ concentrations, we used the isosbestic ratioing method (Neher and Augustine, 1992; Schiller et al., 1995). The action potential-induced fluorescence change was recorded at an excitation wavelength of 380 nm. The isosbestic fluorescence was measured immediately before and after this sweep, using an excitation wavelength of 356 nm (the Ca2+-insensitive wavelength in our experimental conditions). The ratio of the background-corrected fluorescence signals R =F356/F380was calculated and converted into the Ca2+concentration using the equation (Grynkiewicz et al., 1985):
where Rmin is the ratio in Ca2+-free solution andRmax the ratio when fura-2 is completely saturated with Ca2+. These values were determined by recording from CA1 pyramidal cells with internal solutions containing either 30 mm EGTA (Rmin = 0.70 ± 0.01;n = 5) or 50 mmCaCl2 (Rmax = 6.97 ± 0.06; n = 5).Keff was calculated according to Neher and Augustine (1992) as Keff =Kd(Rmax/Rmin), with the dissociation constant Kd = 250 nm (Schiller et al., 1995). The resting Ca2+ concentration was on average 47 ± 4 nm (n = 17) and 46 ± 5 nm (n = 17) at the soma and dendrite, respectively. The decay time course of the action potential-induced Ca2+ transients was fitted with the sum of two exponentials and the amplitude-weighted τ was given. Traces in the figures represent averages of 5–10 consecutive sweeps with the fitted curve superimposed.
Ca2+measurements with Oregon Green. To examine the effects of nifedipine on Ca2+ transients, which is a highly light-sensitive substance, we used 0.1 mm Oregon Green instead of fura-2. This dye has two advantages: it can be excited with lower energy light in the visible wavelength range (at 480 nm) and has a higher quantum yield than fura-2. Thus, we could use a lower excitation intensity (5%). In these experiments the relative change in fluorescence intensity ΔF/F was calculated after background subtraction. The decay time constants of the dendritic fluorescence transients (τ = 834 ± 79 msec; 40–100 μm;n = 14) were not significantly different from those measured with fura-2 (τ = 707 ± 41 msec; n= 17; p > 0.2). Although this may suggest that ΔF/F is linearly related to the Ca2+ concentration, we cannot exclude a partial saturation of Oregon Green. This would imply a slight underestimation of the Ca2+ transient and, hence, of the effect of nifedipine.
Chemicals. 8-hydroxy-2-dipropylamino-tetralin (OH-DPAT), (S)-α-methyl-4-carboxyphenyl-glycine (MCPG), andd(−)-2-amino-5-phosphonopentanoic acid (d-AP-5) were obtained from Tocris, and TTX and ω-conotoxin GVIA were obtained from Alomone (Jerusalem, Israel). All other chemicals were from Merck, Sigma, Riedel-de Haen, or Gerbu. Stock solutions were made in distilled water or dimethylsulfoxide (for nifedipine, concentration of dimethylsulfoxide in the final solution, ≤0.1%). ω-conotoxin GVIA was coapplied with 1 mg/ml bovine serum albumin (BSA) to prevent unspecific binding of the peptide toxins. Agonists and antagonists were applied by bath perfusion with the exception of ω-conotoxin GVIA. For this substance, the bath perfusion was interrupted, and the toxin was applied manually in the recording chamber with a pipette. Neither the interruption of the perfusion nor the application of extracellular solution alone with BSA via a pipette had any effect on Ba2+ currents or basal synaptic transmission. Nifedipine was protected from light.
RESULTS
Whole-cell current-clamp recordings from CA1 pyramidal cells were made, and EPSPs were evoked by electrical stimulation of Schaffer collaterals (Fig. 1A). The cells were held at a membrane potential of −68 to −70 mV, near the average resting potential (−68.6 ± 0.3 mV; n= 58). A robust associative LTD was induced by asynchronous pairing of extracellular Schaffer-collateral stimulation with a short postsynaptic current injection generating an action potential 20 msec before the EPSP (Fig. 1A,B). Time intervals of 10–20 msec have been shown to be maximally effective for the induction of associative LTD (Levy and Steward, 1983; Markram et al., 1997; Bi and Poo, 1998). The pairing was repeated 360 times at a frequency of 0.3 or 1 Hz. This asynchronous pairing stimulation (APS) reduced the EPSP amplitude to 42.7 ± 1.8% (n = 17;p < 0.001) of the control value, measured 15–20 min after the induction protocol (Fig. 1C,D). As shown in Figure2, 360 action potentials or EPSPs alone at 1 Hz did not induce significant alterations in EPSP amplitude (action potentials alone: 93.8 ± 5.1%, n = 3;p > 0.5; EPSPs alone: 103.4 ± 4.9% of control EPSP amplitude, n = 3; p > 0.5). Thus, this form of LTD is associative and dependent on the asynchronous activity of both presynaptic and postsynaptic neurons.
Fig. 1.

Associative LTD induced by asynchronous pairing of presynaptic and postsynaptic action potentials in CA1 pyramidal neurons. A, Current-clamp whole-cell recording from a CA1 pyramidal neuron. EPSPs were evoked by extracellular stimulation of the Schaffer collateral pathway with 200 μsec voltage pulses applied through the stimulation pipette (inset). The EPSP amplitude was significantly decreased by asynchronous pairing of presynaptic and postsynaptic activity. Traces shown were taken immediately before and 15 min after asynchronous pairing.B, For the APS, a single action potential was evoked in the postsynaptic CA1 pyramidal neuron by current injection (700 pA, 5 msec). Twenty milliseconds after the onset of the current injection, an EPSP was evoked. This pairing was repeated 360 times at a frequency of 0.3 or 1 Hz. C, The EPSP amplitude during a single representative experiment is plotted against time. D, The mean EPSP amplitude is plotted against time (n= 17). The APS induction paradigm was applied at the time indicated by the arrow.
Fig. 2.
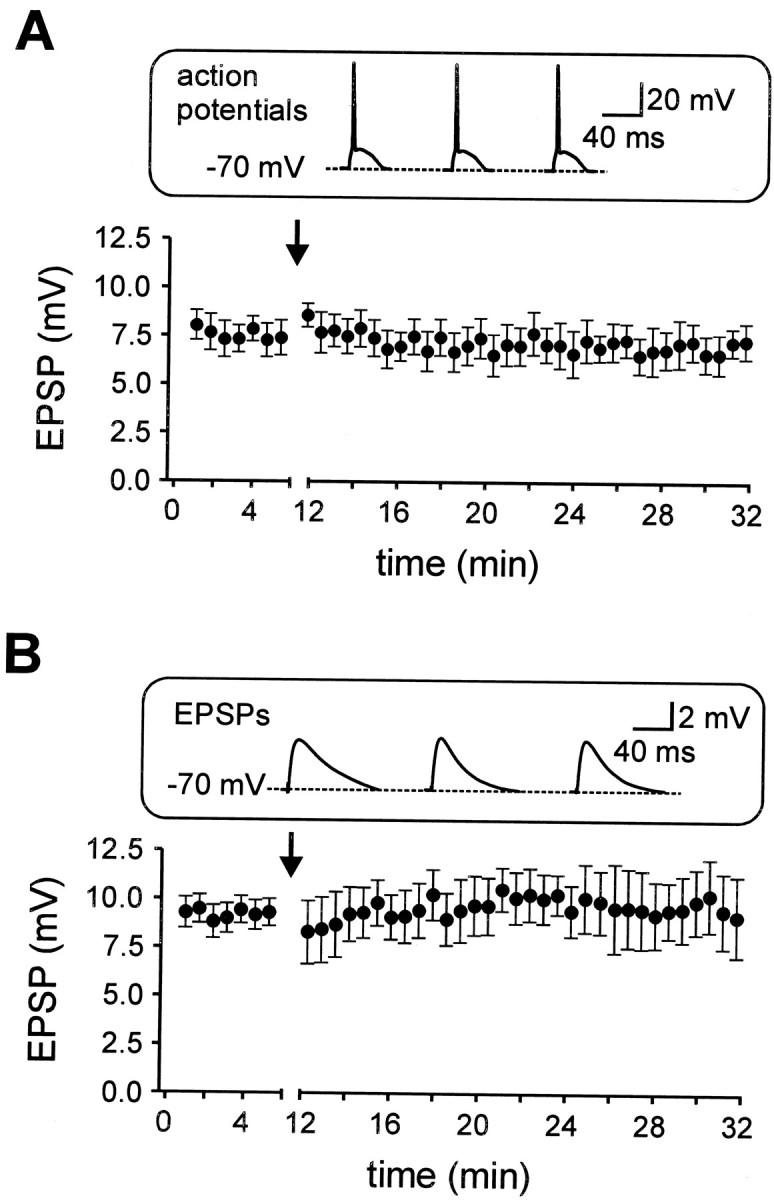
Postsynaptic action potentials or EPSPs alone are not sufficient to induce LTD. A, The mean EPSP amplitude is plotted against time. Three hundred sixty action potentials were evoked in the postsynaptic cell at a frequency of 1 Hz from a membrane potential of −70 mV. This protocol did not change the EPSP amplitude (n = 3). B, Three hundred sixty EPSPs at a membrane potential of −70 mV, as shown in theinset, evoked at a frequency of 1 Hz did not change the EPSP amplitude (n = 3). The induction paradigms were applied at the times indicated by the arrows. Insets represent the first three sweeps of the induction paradigm of a representative experiment.
Associative LTD is dependent on metabotropic glutamate receptors
To investigate the induction mechanisms of associative LTD, we first examined the contribution of metabotropic glutamate receptors (Fig. 3A). In the presence of 500 μm MCPG, an antagonist of metabotropic glutamate receptors, the induction of associative LTD was inhibited (105.3 ± 3.6% of control EPSP amplitude, n = 6;p > 0.1). By contrast, the NMDAR antagonistd-AP-5 (50 μm) was without effect on the synaptic depression (Fig. 3B). In the presence of d-AP-5, the EPSP amplitude was depressed to 45.5 ± 3.5% (n = 6;p < 0.001), similar to the control condition. In addition, d-AP-5 had no significant effects on EPSP peak amplitude (8.6 ± 1.2 mV in control solution vs 8.5 ± 1.2 mV in d-AP-5, n = 11;p > 0.5) and EPSP decay time constant (11.0 ± 11.1% decrease, n = 11; p > 0.05). These results are consistent with a minimal contribution of NMDARs to basal synaptic transmission in CA1 pyramidal cells near the resting membrane potential (Herron et al., 1986; Cash and Yuste, 1999). Thus, under our experimental conditions the NMDARs do not significantly contribute to both basal synaptic transmission and induction of associative LTD.
Fig. 3.

Associative LTD is dependent on activation of metabotropic glutamate receptors. A, The mean EPSP amplitude is plotted against time. In the presence of 500 μm MCPG, an antagonist of metabotropic glutamate receptors, the associative LTD was inhibited (n = 6). B, The NMDAR antagonist d-AP-5 (50 μm) was without effect on both basal synaptic transmission and the induction of associative LTD (n = 6). C, Nine hundred EPSPs at a membrane potential of −62 mV (inset), evoked at a frequency of 1 Hz (indicated by the arrow) reliably depressed the EPSP amplitude (n = 8).D, LFS-induced LTD was blocked by the application of 50 μmd-AP-5 (n = 4).Insets in A and Crepresent the first three sweeps of the induction paradigm of a representative experiment. Horizontal bars indicate the presence of the MCPG and d-AP-5, respectively.
A standard protocol for the induction of LTD is the application of prolonged LFS of presynaptic neurons (e.g., 900 pulses at 1 Hz; Dudek and Bear, 1992). Using this protocol we could only induce long-term depression if the postsynaptic cell was slightly depolarized to −62 mV, but not at the resting membrane potential of −68 to −70 mV. As this potential was closer to firing threshold we used smaller initial EPSP amplitudes (range, 1–4 mV at −70 mV) to avoid postsynaptic spiking during LFS induction (Fig. 3C, inset). The LFS protocol induced a depression of the EPSPs to 40.9 ± 2.5% of the control amplitude (n = 8; p < 0.001; Fig. 3C). Application of d-AP-5 (50 μm) inhibited the induction of LFS-induced LTD (EPSP amplitude was 100.7 ± 10.4% after 15 min,n = 4; p > 0.5, Fig. 3D). Thus, LFS induces an NMDAR-dependent LTD, consistent with previous reports (Dudek and Bear, 1992; Oliet et al., 1997). In addition, the voltage dependence of the LFS-induced NMDAR LTD was similar to that described previously (Debanne et al., 1996; Goda and Stevens, 1996;Oliet et al., 1997; Fitzsimonds et al., 1997). In conclusion, associative pairing selectively induces mGluR-dependent LTD, whereas low-frequency stimulation leads to NMDAR-dependent LTD.
Associative LTD is dependent on activation of voltage-gated Ca2+ channels
Previous studies showed that the induction of both mGluR- and NMDAR-dependent LTD is blocked by the Ca2+chelator BAPTA (Mulkey and Malenka, 1992; Oliet et al., 1997). To examine postsynaptic Ca2+ signaling in associative LTD, we measured Ca2+transients in the soma and apical dendrites of CA1 pyramidal neurons induced by single backpropagating action potentials using 0.1 mm fura-2. After a single action potential, the dendritic Ca2+ concentration increased by 148 ± 14 nm from a resting value of 46 ± 5 nm (distance from soma 40–100 μm; n = 17; Fig. 4A). This transient increase in Ca2+ concentration decayed to initial baseline levels with a time constant of 707 ± 41 msec.
Fig. 4.

Associative LTD is dependent on Ca2+ influx via both, voltage-gated N- and L-type Ca2+ channels. A, Fluorescence image of a CA1 pyramidal neuron filled with 0.1 mm fura-2 (380 nm excitation). Rectangles indicate somatic and dendritic ROIs from where Ca2+ transients were recorded with high time resolution (100 Hz, traces on the right). The action potential-induced Ca2+ transients were reduced in the presence of 1 μm ω-conotoxin GVIA (ω-con, N-type blocker). B, The bar graph summarizes the mean inhibition of the Ca2+ transient by ω-conotoxin GVIA (n = 6) obtained with 0.1 mm fura-2 and 10 μm nifedipine (n = 5; L-type blocker) obtained with 0.1 mm Oregon Green (see Materials and Methods). Dendritic ROIs were located in the stratum radiatum at a distance of ∼0–40 μm or 40–100 μm from the soma. C, Application of 0.5 μm ω-conotoxin GVIA (n = 9) reduced basal synaptic transmission and prevented the induction of LTD by APS.D, A 10 μm concentration of nifedipine did not affect basal transmission but similarly inhibited LTD induction (n = 7). Voltage traces in C andD are EPSPs from a single representative experiment, respectively, at the times indicated by theasterisks.
Application of 1 μm ω-conotoxin GVIA, an irreversible blocker of N-type Ca2+ channels, reduced the dendritic Ca2+ transients by 38.3 ± 4.6% (n = 6; 40–100 μm; Fig.4A,B). This indicates that N-type Ca2+ channels are effectively opened by single backpropagating action potentials. To assess the contribution of L-type Ca2+ channels, we examined the effects of 10 μm nifedipine. Because nifedipine is very light-sensitive, we used 0.1 mm Oregon Green instead of fura-2 (see Materials and Methods). A single action potential evoked a transient fluorescence increase of ΔF/F = 108.5 ± 13% (n = 14). Application of 10 μmnifedipine reduced the dendritic Ca2+transients by 19.4 ± 1.6% (n = 5; 40–100 μm; Fig. 4B). Thus, a single backpropagating action potential induces a reliable Ca2+ influx through voltage-gated Ca2+ channels in the proximal apical dendrite of CA1 pyramidal neurons, with a substantial amount carried by N- and L-type Ca2+channels.
To test the involvement of these channels in LTD induction, we applied the LTD induction protocol in the presence of Ca2+ channel antagonists. When 0.5 μm ω-conotoxin GVIA was applied during basal synaptic transmission, the EPSP amplitude was reduced from 16.8 ± 2.3 mV to 5.8 ± 1.4 mV (n = 9; Fig. 4C), indicating the inhibition of presynaptic N-type Ca2+ channels that mediate neurotransmitter release (Dunlap et al., 1995). As a higher stimulus intensity was used in these experiments, EPSPs in the presence ω-conotoxin were sufficiently large to examine the effects of subsequent pairing. Under these conditions the asynchronous pairing protocol failed to induce significant depression of the EPSP amplitude (102 ± 5.0% of control EPSP amplitude, n = 9;p > 0.5). Although we cannot exclude a contribution of presynaptic N-type Ca2+ channels, these results suggest that Ca2+ influx through postsynaptic N-type channels is required for the induction of associative LTD. Similarly, we tested the involvement of L-type Ca2+ channels in LTD induction (Fig.4D). In contrast to ω-conotoxin, nifedipine did not reduce the initial EPSP amplitude. However, nifedipine markedly reduced the amount of LTD (reduction of EPSP amplitude to 89.6 ± 4.3%,n = 7; p > 0.5). Thus, postsynaptic Ca2+ influx through voltage-gated Ca2+ channels is necessary for the induction of associative LTD.
Modulation of postsynaptic N-type Ca2+ channels inhibits associative LTD
OH-DPAT, a selective agonist of 5-hydroxytryptamine (5-HT)1A receptors, is known to inhibit N-type Ca2+ channels in cortical pyramidal neurons by activation of a Gi/o-protein (Foehring, 1996). Immunocytochemical analysis revealed a high density of 5-HT1A receptors in the hippocampal CA1 region and further suggested an exclusively postsynaptic location (Kia et al., 1996). Thus, we considered OH-DPAT as a selective inhibitor of postsynaptic N-type Ca2+ channels. As a first experimental step, we examined the effect of OH-DPAT on the action potential-induced Ca2+ transient (Fig. 5A). Application of 1 μm OH-DPAT reduced the dendritic Ca2+ transients by 25.6 ± 2.7% (n = 5; 40–100 μm). This is consistent with the previously reported reduction of burst-induced Ca2+ transients by 10 μm 5-HT in CA1 pyramidal neurons (Sandler and Ross, 1999).
Fig. 5.

Selective suppression of N-type Ca2+ channels by 5-HT1A receptors.A, Action potential-induced Ca2+transients recorded from the CA1 pyramidal neuron shown on theleft (0.1 mm fura-2), in control conditions, and in the presence of the 5-HT1A receptor agonist OH-DPAT (1 μm). B, Whole-cell voltage-clamp recordings from CA1 pyramidal neurons. Modulation of Ca2+ channels was analyzed using 30 msec voltage steps from a holding potential of −90 to −10 mV and Ba2+ as a charge carrier in the bath. Thetraces show currents in control and after application of 1 μm OH-DPAT (left traces) or OH-DPAT in the presence of 0.5 μm ω-conotoxin GVIA (right traces), respectively. The bar graph summarizes the mean ofn = 8 (OH-DPAT), n = 4 (ω-conotoxin plus OH-DPAT), and n = 4 (nifedipine plus OH-DPAT) experiments. C, Double recording from the soma and the apical dendrite of a CA1 pyramidal neuron (distance between recording sites was 192 μm; inset). Somatic current injection (900 pA, 5 msec) evoked backpropagating action potentials, which did not significantly change after application of 1 μm OH-DPAT. D, The amplitude of the dendritic action potential is plotted against time (top panel, 6 double recordings, normalized to initial value). The average distance was 110 ± 22 μm. The bar graph (bottom panel) summarizes the effect of 1 μmOH-DPAT on somatic and dendritic AP amplitude, on the half width of the dendritic AP and on the propagation velocity relative to control (n = 6).
The reduction in the dendritic Ca2+transient by OH-DPAT could be attributable to a direct inhibition of Ca2+ channels or a reduction in the amplitude of the backpropagating action potential, or both. For 10 μm 5-HT a slight reduction of the amplitude of the backpropagating action potentials was reported (Sandler and Ross, 1999). To distinguish between these possibilities, we blocked Na+ and K+channels (see Materials and Methods) and examined Ca2+ channels in isolation in the whole-cell voltage-clamp configuration using 2 mmBa2+ as charge carrier (Fig.5B). The application of 1 μm OH-DPAT reduced the Ba2+ currents to 72.9 ± 10.9% (n = 8; p < 0.01). In the presence of ω-conotoxin GVIA the Ba2+currents were reduced to 45.8 ± 6.6% (n = 4). Subsequent to the application of ω-conotoxin, the modulation by OH-DPAT was completely absent, indicating a selective modulation of N-type Ca2+ channels by 5-HT1A receptors. In the presence of 10 μm nifedipine (84.7 ± 7.0% of control;n = 4), however, there was still a substantial reduction of the Ba2+ currents by OH-DPAT (57.0 ± 3.9% of control; p < 0.01).
To examine possible effects of 1 μm OH-DPAT on action potential backpropagation, we made double recordings from the soma and the apical dendrite of CA1 pyramidal cells at distances of 64–192 μm from the soma (Fig. 5C). The shape of the dendritic and somatic action potential in 1 μm OH-DPAT was very similar to control conditions. The resting membrane potential was slightly hyperpolarized by −1.0 ± 0.2 mV at the dendrite and by −0.9 ± 0.2 mV at the soma (p < 0.05; six double recordings). As shown in Figure 5D, the action potential amplitude in the presence of 1 μmOH-DPAT was virtually identical to control conditions (101.2 ± 0.9% of control, n = 6; p > 0.1). Furthermore no significant change in half width of the dendritic AP (102.1 ± 2.1% of control; p > 0.1) or the propagation velocity (98.3 ± 3.2% of control; p> 0.1; Fig. 5D) was observed. In conclusion, these results indicate that the reduction of action potential-induced Ca2+ transients of 1 μm OH-DPAT is attributable to a direct modulation of postsynaptic N-type Ca2+channels and not to an inhibition of dendritic backpropagation.
If postsynaptic N-type Ca2+ channels are necessary for induction of associative LTD (Fig. 4C) and if these channels are selective targets for modulation via 5-HT1A receptors (Fig. 5), then OH-DPAT should affect LTD induction. We first tested the effect of 1 μm OH-DPAT on basal synaptic transmission, and we found that the peak EPSP amplitude remained unchanged (Fig.6A; 95.8 ± 6.4% of control, n = 11; p > 0.5). This allowed us to use OH-DPAT as a tool to inhibit selectively postsynaptic N-type Ca2+ channels. In the presence of 1 μm OH-DPAT, application of the asynchronous pairing paradigm failed to induce associative LTD (102.8 ± 4.9% of control, n = 11; p > 0.5; Fig.6B,C). These results indicate that the Ca2+ influx via postsynaptic N-type channels is necessary for induction of associative LTD and that the G-protein-mediated modulation of these channels strongly controls this form of synaptic plasticity.
Fig. 6.
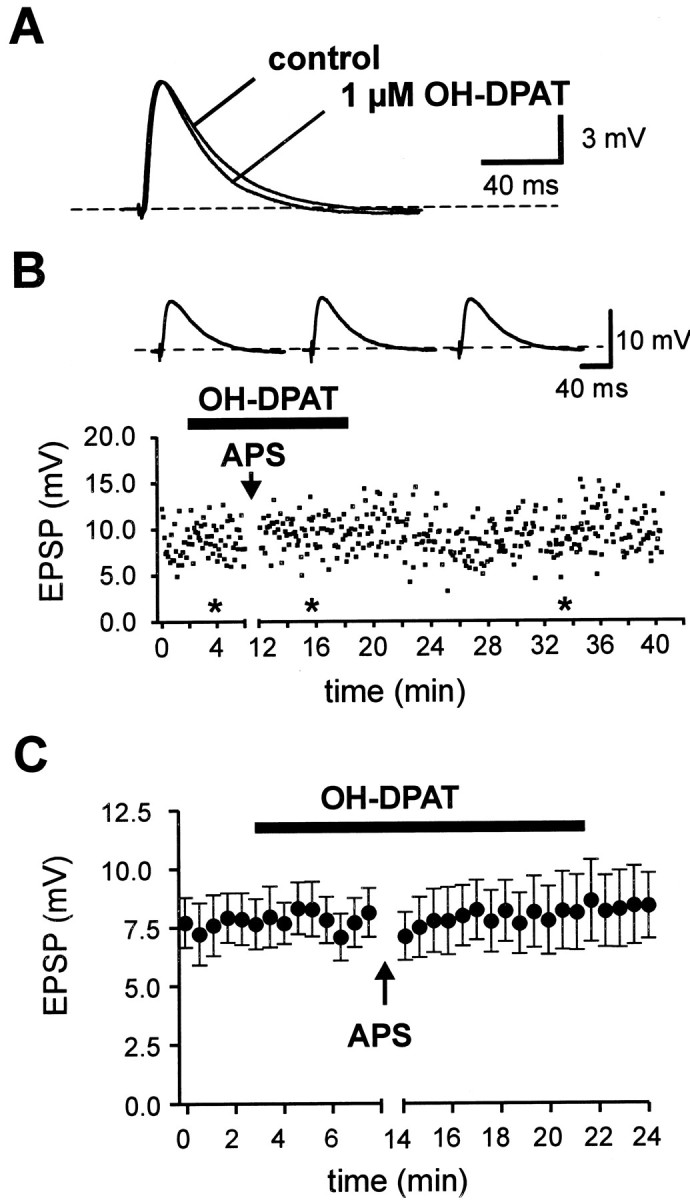
Block of associative LTD by inhibition of postsynaptic N-type Ca2+ channels via 5-HT1A receptors. A, The application of 1 μm OH-DPAT did not affect the EPSP amplitude during basal transmission. B, The EPSP amplitude during a single representative experiment is plotted against time. Voltage traces ontop are EPSPs from a single representative experiment obtained at the times indicated by the asterisks.C, The mean EPSP amplitude is plotted against time (n = 11). Note that the EPSP amplitude remains constant during application of OH-DPAT. The induction of associative LTD by APS, however, was completely blocked.
DISCUSSION
Our results show that associative LTD at the Schaffer collateral–CA1 pyramidal cell synapse can be induced reliably by asynchronous pairing of EPSPs with preceding postsynaptic action potentials. The induction was dependent on both mGluRs and postsynaptic voltage-gated Ca2+ channels. In particular, we show a direct involvement of N-type Ca2+ channels in synaptic plasticity. The modulation of postsynaptic N-type Ca2+channels by 5-HT1A receptors was sufficient to inhibit associative LTD induced by asynchronous pairing.
Associative LTD is dependent on mGluRs
The induction of LTD by the associative paring protocol was blocked by the mGluR antagonist MCPG, similar to the previously described mGluR LTD (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Otani and Connor, 1998). Although we did not use subtype-specific antagonists, it is likely that the depression is mediated by mGluR5. Immunocytochemical evidence indicates that mGluR5 is the most abundant metabotropic glutamate receptor present on the postsynaptic CA1 pyramidal cells (Shigemoto et al., 1997). Furthermore, there is evidence for the involvement of the phospholipase C (PLC) signal transduction pathway in mGluR LTD, consistent with the activation of group 1 mGluRs (Oliet et al., 1997; Otani and Connor, 1998).
The induction mechanism of associative LTD appeared to be different from that of LFS-induced LTD, which was largely blocked by the NMDAR antagonist d-AP-5 (Fig. 3), consistent with previous studies (Dudek and Bear, 1992; Mulkey and Malenka, 1992). The coexistence of two different forms of LTD in hippocampal pyramidal cells was described in detail by Oliet et al. (1997). In bath solutions containing 2.5 mm Ca2+ and 1.3 mmMg2+, LFS primarily induced NMDAR LTD. In 4 mm Ca2+ and 4 mmMg2+, however, an additional NMDAR-independent form of LTD was induced, which was dependent on mGluRs and voltage-gated Ca2+ channels (Oliet et al., 1997). Because associative LTD in acute hippocampal slices is dependent on mGluRs but not on NMDARs, the asynchronous pairing protocol might be the most physiological way to selectively induce the mGluR-dependent LTD.
Thus, two mechanistically distinct forms of LTD coexist in hippocampal pyramidal cells, which can be induced selectively, depending on pyramidal cell firing during network activity (O'Keefe and Reece, 1993). Prolonged presynaptic activity without any postsynaptic spiking may decrease the EPSP amplitude via nonassociative LTD dependent on NMDARs, whereas EPSPs that occur repeatedly at a certain time delay with respect to postsynaptic action potentials will be depressed by associative LTD dependent on mGluRs.
Associative LTD is dependent on activation of postsynaptic Ca2+ channels
The mGluR LTD induced by associative pairing could be blocked by the inhibition of either L- or N-type Ca2+channels (Fig. 4). These channels were reliably activated during single backpropagating action potentials (Fig. 4B). This is consistent with the localization of L- and N-type Ca2+ channels on soma and apical dendrites of CA1 pyramidal neurons (Westenbroek et al., 1992; Kavalali et al., 1997; Magee, 1999) and with cell-attached patch recordings from these dendrites (Magee and Johnston, 1995). In addition, other types of Ca2+ channels are expressed in CA1 pyramidal cells, including P-, R-, and T-type channels (Kavalali et al., 1997). They may be responsible for the ω-conotoxin- and nifedipine-resistant Ca2+-influx.
It may seem surprising that a small (20–40%) reduction of the spatially averaged dendritic Ca2+transient was sufficient to substantially reduce or block the associative LTD. However, the Ca2+concentration at Ca2+-dependent effector molecules may be very different from the measured Ca2+ transients. The peak amplitude of the Ca2+ transient in submembrane cytoplasmic compartments could be much higher because of clustering of Ca2+ channels and local saturation of Ca2+ buffers (Helmchen et al., 1996). If, for example, N-type Ca2+ channels were colocalized with molecules involved in LTD induction, then our data would represent a lower estimate for the contribution of these channels to local Ca2+ signals near these effector molecules. Such a colocalization could occur in dendritic spines, where action potential-induced Ca2+ transients have larger amplitudes than in nearby parent dendrites (Majewska et al., 2000).
Our results and previous reports (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997; Otani and Connor, 1998) converge on the conclusion that postsynaptic Ca2+ influx is essential for LTD induction. However, the target molecules for Ca2+ remain to be identified. A Ca2+-dependent phosphatase is unlikely to be involved, because the phosphatase inhibitor microcystin does not affect mGluR LTD (Oliet et al., 1997). An involvement of Ca2+-dependent isoforms of PKC is more likely, because PKC inhibitory peptide blocks mGluR LTD (Oliet et al., 1997; Otani and Connor, 1998). Because some PKC isoforms are activated by both diacylglycerol and Ca2+(Nishizuka, 1992), they could operate as molecular coincidence detectors, onto which the activation of voltage-gated Ca2+ channels and group 1 mGluRs converge. This would explain the need for both postsynaptic action potentials and the release of glutamate for induction of associative LTD.
Modulation of N-type Ca2+ channels and LTD
The involvement of N-type Ca2+channels in synaptic plasticity is difficult to assess because of the inhibition of basal synaptic transmission by ω-conotoxin GVIA. No selective postsynaptic N-type channel antagonist is available yet. In our study, the selective 5-HT1A agonist OH-DPAT is a valuable tool to distinguish between presynaptic and postsynaptic N-type channels. A 1 μm concentration of OH-DPAT, which selectively inhibits postsynaptic N-type channels by G-protein modulation, was sufficient to block LTD induction. The dependence of associative LTD on N-type Ca2+ channels might be important, because N-type Ca2+channels are preferential targets of neuromodulation by various neurotransmitters like GABA, glutamate, serotonin, somatostatin, and adenosine (Hille, 1994; Kavalali et al., 1997; Magee, 1999). In contrast to the other Ca2+ channel types, which provide a more constant basal Ca2+load per action potential, the Ca2+ influx through N-type channels is highly regulated. Consequently, the recruitment of N-type channels may determine whether the postsynaptic Ca2+ signal is below or above the threshold for LTD induction.
The G-protein-mediated modulation of the N-type channels will therefore enable or disable an activity-dependent depression in synaptic strength. This direct gain control of LTD might be relevant for learning, because learning should occur dependent on behavioral context, which could be signaled by the release of different neuromodulators.
Modulation of associative LTD by backpropagating action potentials
We have shown that N-type Ca2+channels are direct targets of neuromodulation via G-proteins. However, the activation of N-type channels could be also regulated indirectly by modulation of action potential backpropagation. A 1 μmconcentration of OH-DPAT, which is thought to activate selectively 5-HT1A receptors, did not affect the properties of the backpropagated spike within the first 200 μm of the apical dendrite. In contrast, higher concentrations (30 μm) of OH-DPAT and 5-HT induce a marked hyperpolarization of CA1 pyramidal cells by 5 and 14 mV, respectively (Andrade and Nicoll, 1987), which slightly decrease the amplitude of the backpropagated spike (Sandler and Ross, 1999). Furthermore, activation of muscarinic and adrenergic receptors regulates dendritic excitability via modulation of fast dendritic Na+ and K+ channels (Johnston et al., 1999).
Both the amplitude of the backpropagated spike and the evoked dendritic Ca2+ transients decrease with distance from the pyramidal cell soma (Spruston et al., 1995; Magee and Johnston, 1997). Thus, in stratum lacunosum moleculare we would not expect any associative LTD at all, unless backpropagation of action potentials will be enhanced by activation of muscarinic or adrenergic receptors. This will lead to different learning rules for distal and proximal synapses. In general, action potential backpropagation can be very different in different types of neurons (for review, see Magee, 1999). Both CA1 and neocortical pyramidal neurons show decremental spike backpropagation (Magee and Johnston, 1997; Markram et al., 1997). Hippocampal oriens-alveus interneurons and olfactory bulb mitral cells, however, show nondecremental backpropagation of action potentials into the dendrites (Bischofberger and Jonas, 1997; Martina et al., 2000). It would be interesting to know whether glutamatergic synapses on these neurons show LTD, and if so, whether LTD has associative properties over the entire dendritic tree.
Physiological significance of associative LTD
Both associative LTD and LTP in the hippocampus may be important for the dynamical shaping of new place fields during spatial learning and theta-phase associated pyramidal cell firing (O'Keefe and Reece, 1993; Wilson and McNaughton, 1993). In particular, they may contribute to the learning of temporal sequences in the hippocampus (Skaggs and McNaughton, 1996; Mehta et al., 1997). Whereas associative LTP will strengthen the synapses that precede subsequent spike discharge of the postsynaptic cell (Magee and Johnston, 1997), associative LTD will depress EPSPs that occur too late with respect to the postsynaptic spiking, thus leading to temporally asymmetric learning rules. Such rules appeared also to be very effective for the formation of neuronal cell assemblies in artificial neural networks (Sejnowski, 1999), which was shown to be of critical importance for encoding of spatial information in the hippocampus (Wilson and McNaughton, 1993).
In conclusion, we suggest that the induction of associative LTD is a powerful mechanism to depress out-of-phase synaptic input. Thus, it may be important to have a direct gain control of associative synaptic depression, provided by the G-protein-mediated modulation of the voltage-gated N-type Ca2+ channels.
Footnotes
This work was supported by a grant from the Deutsche Forschungsgemeinschaft Bi 642/1–2 and University funds (J.B.) and by the Vada and Theodore Stanley Foundation (J.W.). We thank Drs. M. Bartos, J. R. P. Geiger, and M. Martina for critically reading this manuscript and A. Blomenkamp for technical assistance.
Correspondence should be addressed to Dr. J. Bischofberger, Physiologisches Institut, Universität Freiburg, Hermann-Herder-Strasse 7, D-79104 Freiburg, Germany. E-mail:bischof@uni-freiburg.de.
REFERENCES
- 1.Andrade R, Nicoll RA. Pharmacologically distinct actions of serotonin on single pyramidal neurones of the rat hippocampus recorded in vitro. J Physiol (Lond) 1987;394:99–124. doi: 10.1113/jphysiol.1987.sp016862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bi G, Poo M. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bischofberger J, Jonas P. Action potential propagation into presynaptic dendrites of rat mitral cells. J Physiol (Lond) 1997;504:359–365. doi: 10.1111/j.1469-7793.1997.359be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 5.Bolshakov VY, Siegelbaum SA. Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science. 1994;264:1148–1152. doi: 10.1126/science.7909958. [DOI] [PubMed] [Google Scholar]
- 6.Cash S, Yuste R. Linear summation of excitatory inputs by CA1 pyramidal neurons. Neuron. 1999;22:383–394. doi: 10.1016/s0896-6273(00)81098-3. [DOI] [PubMed] [Google Scholar]
- 7.Debanne D, Gähwiler BH, Thomson SM. Asynchronous pre- and postsynaptic activity induces associative long term depression in area CA1 of the rat hippocampus in vitro. Proc Natl Acad Sci USA. 1994;91:1148–1152. doi: 10.1073/pnas.91.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Debanne D, Gähwiler BH, Thomson SM. Cooperative interaction in the induction of long-term potentiation and depression of synaptic excitation between hippocampal CA3-CA1 cell-pairs in vitro. Proc Natl Acad Sci USA. 1996;93:11225–11230. doi: 10.1073/pnas.93.20.11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dudek S, Bear MF. Homosynaptic long term depression in area CA1 of the hippocampus and effects of NMDA receptor blockade. Proc Natl Acad Sci USA. 1992;89:4363–4367. doi: 10.1073/pnas.89.10.4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 11.Fitzsimonds RM, Song H, Poo M. Propagation of activity dependent synaptic depression in simple neural networks. Nature. 1997;388:439–448. doi: 10.1038/41267. [DOI] [PubMed] [Google Scholar]
- 12.Foehring RC. Serotonin modulates N- and P-type calcium currents in neocortical pyramidal neurons via a membrane-delimited pathway. J Neurophysiol. 1996;75:648–659. doi: 10.1152/jn.1996.75.2.648. [DOI] [PubMed] [Google Scholar]
- 13.Goda Y, Stevens CF. Long-term depression properties in a simple system. Neuron. 1996;16:103–111. doi: 10.1016/s0896-6273(00)80027-6. [DOI] [PubMed] [Google Scholar]
- 14.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 15.Herron CE, Lester RA, Coan EJ, Collingridge GL. Frequency-dependent involvement of NMDA receptors in the hippocampus: a novel synaptic mechanism. Nature. 1986;322:265–268. doi: 10.1038/322265a0. [DOI] [PubMed] [Google Scholar]
- 16.Heynen AJ, Abraham WC, Bear MF. Bidirectional modification of CA1 synapses in the adult hippocampus in vivo. Nature. 1996;381:163–166. doi: 10.1038/381163a0. [DOI] [PubMed] [Google Scholar]
- 17.Helmchen F, Imoto K, Sakmann B. Ca2+ buffering and action potential-evoked Ca2+ signaling in dendrites of pyramidal neurons. Biophys J. 1996;70:1069–1081. doi: 10.1016/S0006-3495(96)79653-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 19.Johnston D, Hoffman DA, Colbert CM, Magee JC. Regulation of backpropagating action potentials in hippocampal neurons. Curr Opin Neurobiol. 1999;9:288–292. doi: 10.1016/s0959-4388(99)80042-7. [DOI] [PubMed] [Google Scholar]
- 20.Kavalali ET, Zhou M, Bito H, Tsien RW. Dendritic Ca2+ channels characterized by recordings from isolated hippocampal dendritic segments. Neuron. 1997;18:651–663. doi: 10.1016/s0896-6273(00)80305-0. [DOI] [PubMed] [Google Scholar]
- 21.Kia HK, Miquel M-C, Brisorgueil MJ, Daval G, Riad M, Mestikawy SE, Hamon M, Verge D. Immunocytochemical localization of serotonin1A receptors in the rat central nervous system. J Comp Neurol. 1996;365:289–305. doi: 10.1002/(SICI)1096-9861(19960205)365:2<289::AID-CNE7>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 22.Levy WB, Steward O. Temporal contiguity requirements for long-term associative potentiation/depression in the hippocampus. Neuroscience. 1983;8:791–797. doi: 10.1016/0306-4522(83)90010-6. [DOI] [PubMed] [Google Scholar]
- 23.Linden DJ. The return of the spike: Postsynaptic action potentials and the induction of LTP and LTD. Neuron. 1999;22:661–666. doi: 10.1016/s0896-6273(00)80726-6. [DOI] [PubMed] [Google Scholar]
- 24.Magee JC. Voltage gated ion channels in dendrites. In: Stuart G, Spruston N, Häusser M, editors. Dendrites. Oxford UP; 1999. pp. 139–160. [Google Scholar]
- 25.Magee JC, Johnston D. Synaptic activation of voltage gated channels in the dendrites of hippocampal pyramidal neurons. Science. 1995;268:301–304. doi: 10.1126/science.7716525. [DOI] [PubMed] [Google Scholar]
- 26.Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- 27.Majewska A, Brown E, Ross J, Yuste R. Mechanisms of calcium decay kinetics in hippocampal spines: role of spine calcium pumps and calcium diffusion through the spine neck in biochemical compartmentalization. J Neurosci. 2000;20:1722–1734. doi: 10.1523/JNEUROSCI.20-05-01722.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- 29.Martina M, Vida I, Jonas P. Distal initiation and active propagation of action potentials in interneuron dendrites. Science. 2000;287:295–300. doi: 10.1126/science.287.5451.295. [DOI] [PubMed] [Google Scholar]
- 30.Mehta MR, Barnes CA, McNaughton BL. Experience-dependent, asymmetric expansion of hippocampal place fields. Proc Natl Acad Sci USA. 1997;94:8918–8921. doi: 10.1073/pnas.94.16.8918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mulkey RM, Malenka RC. Mechanisms underlying induction of homosynaptic long-term depression in the area CA1 in the hippocampus. Neuron. 1992;9:967–975. doi: 10.1016/0896-6273(92)90248-c. [DOI] [PubMed] [Google Scholar]
- 32.Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol (Lond) 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nishizuka Y. Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 34.O'Keefe J, Reece ML. Phase relationship between hippocampal place units and the EEG theta rhythm. Hippocampus. 1993;3:317–330. doi: 10.1002/hipo.450030307. [DOI] [PubMed] [Google Scholar]
- 35.Oliet SH, Malenka RC, Nicoll RA. Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron. 1997;18:969–982. doi: 10.1016/s0896-6273(00)80336-0. [DOI] [PubMed] [Google Scholar]
- 36.Otani S, Connor JA. Requirement of rapid Ca2+ entry and synaptic activation of metabotropic glutamate receptors for the induction of long-term depression in adult rat hippocampus. J Physiol (Lond) 1998;511:761–770. doi: 10.1111/j.1469-7793.1998.761bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sandler VM, Ross WN. Serotonin modulates spike backpropagation and associated Ca2+ changes in the apical dendrites of hippocampal CA1 pyramidal neurons. J Neurophysiol. 1999;81:216–224. doi: 10.1152/jn.1999.81.1.216. [DOI] [PubMed] [Google Scholar]
- 38.Schiller J, Helmchen F, Sakmann B. Spatial profile of dendritic calcium transients evoked by action potentials in rat neocortical pyramidal neurones. J Physiol (Lond) 1995;487:583–600. doi: 10.1113/jphysiol.1995.sp020902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sejnowski TJ. The book of Hebb. Neuron. 1999;24:773–776. doi: 10.1016/s0896-6273(00)81025-9. [DOI] [PubMed] [Google Scholar]
- 40.Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A, Abe T, Nakanishi S, Mizuno N. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci. 1997;17:7503–7522. doi: 10.1523/JNEUROSCI.17-19-07503.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Skaggs WE, McNaughton BL. Replay of neuronal firing sequences in rat hippocampus during sleep following spatial experience. Science. 1996;271:1870–1873. doi: 10.1126/science.271.5257.1870. [DOI] [PubMed] [Google Scholar]
- 42.Skaggs WE, McNaughton BL, Wilson MA, Barnes CA. Theta phase precession in hippocampal neuronal populations and the compression of temporal sequences. Hippocampus. 1996;6:149–172. doi: 10.1002/(SICI)1098-1063(1996)6:2<149::AID-HIPO6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 43.Spruston N, Schiller Y, Stuart G, Sakmann B. Activity-dependent action potential invasion and Ca2+ influx into hippocampal CA1 dendrites. Science. 1995;268:297–300. doi: 10.1126/science.7716524. [DOI] [PubMed] [Google Scholar]
- 44.Stanton PK, Sejnowski TJ. Associative long-term depression in the hippocampus induced by Hebbian covariance. Nature. 1989;339:215–218. doi: 10.1038/339215a0. [DOI] [PubMed] [Google Scholar]
- 45.Stanton PK, Chattarji S, Sejnowski TJ. 2-amino-3-phosphonopropionic acid, an inhibitor of glutamate-stimulated phosphoinositide turnover, blocks induction of homosynaptic long-term depression, but not potentiation, in rat hippocampus. Neurosci Lett. 1991;127:61–66. doi: 10.1016/0304-3940(91)90895-z. [DOI] [PubMed] [Google Scholar]
- 46.Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA. Biochemical properties and subcellular distribution of an N-type calcium channel α1 subunit. Neuron. 1992;9:1099–1115. doi: 10.1016/0896-6273(92)90069-p. [DOI] [PubMed] [Google Scholar]
- 47.Wilson MA, McNaughton BL. Dynamics of hippocampal ensemble code for space. Science. 1993;261:1055–1058. doi: 10.1126/science.8351520. [DOI] [PubMed] [Google Scholar]