

Abstract
The first halonium‐ion‐based helices were designed and synthesized using oligo‐aryl/pyridylene‐ethynylene backbones that fold around reactive iodonium ions. Halogen bonding interactions stabilize the iodonium ions within the helices. Remarkably, the distance between two iodonium ions within a helix is shorter than the sum of their van der Waals radii. The helical conformations were characterized by X‐ray crystallography in the solid state, by NMR spectroscopy in solution and corroborated by DFT calculations. The helical complexes possess potential synthetic utility, as demonstrated by their ability to induce iodocyclization of 4‐penten‐1‐ol.
Keywords: 3c–4e bonds, halocyclization, halogen bonds, helices, iodonium ions
Helices are indispensable secondary structures that serve many functions in biochemistry. Additionally, helices derived from synthetic polymers and oligomers have been the focus of enantioselective catalysis,1 molecular recognition,1a–1c, 2 and selective molecular channels.1a, 3 Helical conformations are typically induced by non‐covalent interactions such as hydrogen bonding,1a–1c, 4 π‐stacking,1a–1c, 5 and host–guest chemistry.1a–1c, 6 Herein, we present synthetically applicable helices stabilized by halogen bonds with halonium ions that occur in both solution and in the solid state.
Halogen bonding is a noncovalent interaction between an electron deficient halogen donor and a Lewis base.7 Halogen bonding has received growing attention in recent years for its use in a wide range of applications including catalysis,8 molecular recognition,8a, 8b, 9 medicinal chemistry,8a, 8b, 10 and self‐assembly.8a, 8b, 11 Halonium ions are strong halogen‐bond donors that tend to form symmetric three‐center, four‐electron (3c–4e) bonds, for example an [N−I−N]+ halogen bond between two nitrogen Lewis bases.12 Such 3c–4e halogen bonds have been previously used to form supramolecular capsules,13 and for stabilization of halonium ions in synthetic reagents applied to halonium transfer and oxidation reactions.14
Polymer and oligomer helices have been widely used in organic synthesis.1 The active functionalities are typically located on the exterior of the helices. One notable exception is the report by Ousaka et al. of a manganese catalyzed epoxidation inside an oligo(phenylene ethynylene) helix.4a Motivated by the scarcity of examples utilizing the interior of helical structures for reactivity, we decided to explore the effects of positioning reactive iodonium ions in the interior of a helix through the use of stabilizing [N−I−N]+ halogen bonds. This approach is unique as helices have not previously been induced by halonium cations.
Our work was inspired by the helical metal complexes reported by Bosch et al.,16 and by our previous work on [N−I−N]+ halogen bonds (Figure 1). Examples of halogen bonding helices are sparse,6b, 17 and most have only been confirmed in the solid state. A noteworthy exception is the work of Massena et al., who have produced solution‐stable, anion‐induced, halogen‐bonding triple helices.6b, 17a
Figure 1.
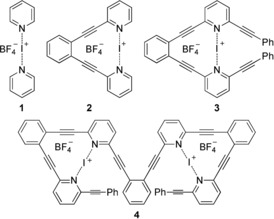
Examples of [N−I−N]+ halogen bond forming systems. 1 and 2 have been previously studied12b, 12c, 15 and are included here for comparison, whereas 3 and 4 are first reported in this study.
Multivalent complex 4 contains two [N−I−N]+ halogen bonds. Complex 4 was predicted to fold into a helix by DFT calculations (see the Supporting Information for details). This geometry is stabilized by the strong halogen bonds and by five aryl–aryl interactions. Partially unfolded conformations of 4 were also examined, but they were found to be at least 6 kcal mol−1 less stable than the helix form.
Motivated by the computational prediction, we synthesized ligands 5 and 6 following a strategy based on Sonogashira couplings and deprotections (Supporting Information). The silver(I) complexes 7 and 8 were formed as precursors to 3 and 4, following a previously described pathway (Scheme 1).18 The formation of the silver(I) and iodonium complexes in CD2Cl2 were associated with large 15N NMR coordination shifts, approximately 60 ppm and 90 ppm, respectively, consistent with previous reports.12a, 15
Scheme 1.
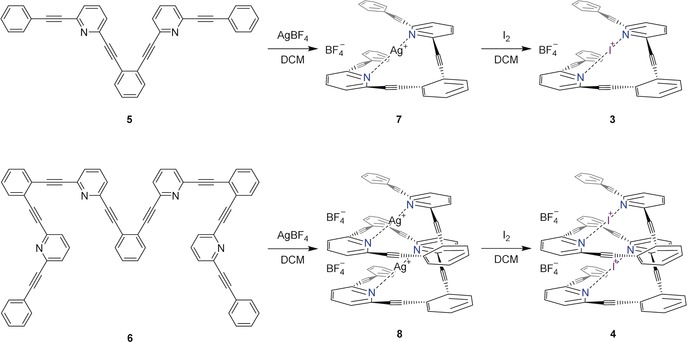
Formation of the helical iodonium complexes.
Single crystals of 3, 4, and 8 were grown by vapor diffusion of hexane into solutions of either dichloromethane or 1,2‐dichloroethane at reduced temperature. Single‐crystal X‐ray analysis corroborated the predicted helical structures (Figure 2, Table 1, and Supporting Information). The crystals of the [N−I−N]+ helical complex 3 and of the [N−Ag−N]+ complex 8 contained both helical enantiomers, whereas the enantiomeric helices of 4 exhibited homochiral self‐sorting. The crystal structure of 4 is similar to the computationally predicted conformation (Figure 2). Bond distances and angles were in agreement with those reported for the related complex 1 (Table 1),19 and notably the observed [N−I−N]+ angles were consistent with the linear geometry of halogen bonds. The range of distances between aromatic centroids deviates only slightly from the generally accepted range for π‐stacking interactions.20 We have also observed an edge‐to‐face type interaction for 8, with one of the terminal phenyl groups showing a 37° angle with the overlying aromatic plane and a short, 2.827 Å, CH–π distance. Such edge‐to‐face type interactions involving the terminal aryl groups might also occur in solution for all of the helices.
Figure 2.
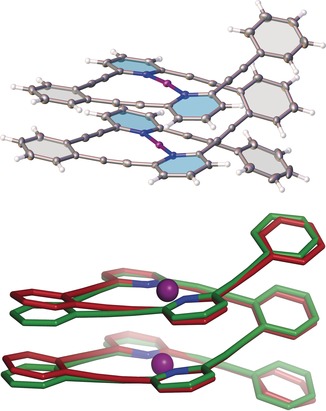
Top: X‐ray crystal structure of complex 4. Bottom: Superimposition of the DFT computed conformation (green) of complex 4 with the X‐ray crystal structure (red).
Table 1.
X‐ray crystallographic data: selected bond lengths and angles.
| Complex | r(N−X) [Å] | r(X−X) [Å] | σ(N‐X‐N) [°] | r(Ar−Ar) [Å][a] |
|---|---|---|---|---|
| 8 [b] (X=Ag) | 2.146(5) to 2.165(5) | 3.5541(8) to 3.6351(8) | 174.9(2) to 178.7(2) | 3.628(4) to 4.067(7)[c] |
| 1 [d][19] (X=I) | 2.255(3) to 2.261(3) | – | 177.66(12) to 180.0 | – |
| 3 [e] (X=I) | 2.256(5) to 2.316(5) | – | 174.1(2) to 176.17(18) | 3.678(4) to 3.972(5) |
| 4 [f] (X=I) | 2.248(5) to 2.302(5) | 3.8621(7) to 3.887(1) | 175.26(18) to 177.03(17) | 3.641(3) to 3.914(4) |
[a] Distances between the centroids of overlying intramolecular aryl groups. [b] Two slightly different helices occur in the crystal structure of 8. [c] The range does not include the distance for the one instance of the CH–π interaction (see the Supporting Information). [d] The unit cell of 1 contains multiple complexes. Data from Ref. 19. [e] The unit cell of 3 contains multiple complexes. [f] The range of values is given for both the M and the P helices, which were measured at temperatures of 150 and 100 K, respectively.
The remarkably short distance between the two charged iodonium ions of 4 is worth noting. The distance between the iodoniums is approximately 0.1 Å less than the sum of the van der Waals radii (r vdW=1.98 Å as proposed by Bondi),21 and also less than the sum of the cationic radii as calculated by Rahm et al. (r=2.21 Å).22 Despite the short distance, the Mayer bond order23 between the iodine atoms was calculated to be small (BO=0.01), suggesting negligible orbital overlap between the iodoniums. The proximity of the iodonium ions could be facilitated by the redistribution of the positive charge throughout the conjugated π‐system. Indeed, natural population analysis revealed the atomic charge on the iodonium ions of 4 to be reduced (Q I=+0.45), however, the electrostatic interaction between the two [N−I−N]+ units is still repulsive (Supporting Information). Computations reveal that the enhanced stability and the compact nature of the helical structure arises from attractive dispersion forces acting between the adjacent aryl and ethynylene groups of 4. Furthermore, the electron deficiency of aromatic rings is known to strengthen π‐stacking interactions,24 which also contribute to the stabilization of the helical conformation.
The helicity of 4 and 8 in solution was confirmed by NOE correlations indicating dipolar relaxation between hydrogens of distinct pyridyl groups (Figure 3 and Supporting Information). Such proximity of the pyridines implies a helical conformation, considering the symmetry of the complexes indicated by 1H, 13C, and 15N NMR. NOE correlations also show evidence for a helix conformation of ligand 6, and DFT calculations found the helix to be a plausible conformation of 6 (Supporting Information).
Figure 3.
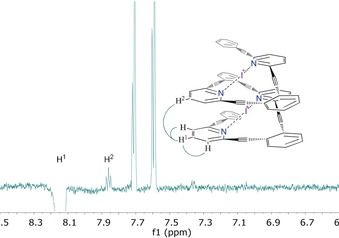
One‐dimensional 1H NOESY NMR spectrum of iodonium complex 4. The triplet at 7.86 ppm is evidence for the close proximity of the two pyridyl groups. The spectrum was obtained with selective excitation centered at 8.14 ppm.
[Bis(pyridine)iodine(I)] complexes are useful reagents for metatheses,25 oxidations,14b, 26 and halofunctionalizations.12c, 14a, 18, 27 Among these, halocyclizations are the most well‐studied and serve as a model reaction for comparing [N−X−N]+ complexes.15a, 18, 27a As a proof of principle, we applied complexes 3 and 4 in iodocyclization reactions with 4‐penten‐1‐ol to produce 2‐(iodomethyl)tetrahydrofuran (Supporting Information). Ligands 5 and 6 were detected post reaction by mass spectrometry (Supporting Information). This indicates that 5 and 6 are not consumed in the reaction and in principle should be recoverable for reuse.
In conclusion, we have synthesized the first halonium‐centered halogen bonding helices. These are stabilized by π–π interactions and 3c–4e [N−I−N]+ bonds. The helices encompass stabilized iodonium ions, which can be transferred to an alkene in a similar fashion to Barluenga's reagent (1). The helical structure of the complexes was demonstrated by X‐ray crystallography in the solid state, NMR spectroscopy in solution and corroborated by computation on the DFT level. The distance between iodonium ions in 4 is remarkably small, less than the sum of the van der Waals radii. Our results extend the possibilities for both the modulation of halonium transfer reagents and for the exploitation of the interior of helices as reaction environments.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to acknowledge funding sources including: the Natural Sciences and Engineering Research Council of Canada's Postdoctoral Fellowship award (516910‐2018), NKFIH (K‐112028) and FORMAS (2017‐01173). This study made use of the NMR Uppsala infrastructure, which is funded by the Department of Chemistry—BMC and the Disciplinary Domain of Medicine and Pharmacy.
A. Vanderkooy, A. K. Gupta, T. Földes, S. Lindblad, A. Orthaber, I. Pápai, M. Erdélyi, Angew. Chem. Int. Ed. 2019, 58, 9012.
References
- 1.
- 1a. Yashima E., Ousaka N., Taura D., Shimomura K., Ikai T., Maeda K., Chem. Rev. 2016, 116, 13752–13990; [DOI] [PubMed] [Google Scholar]
- 1b. Toya M., Ito H., Itami K., Tetrahedron Lett. 2018, 59, 1531–1547; [Google Scholar]
- 1c. Yashima E., Maeda K., Iida H., Furusho Y., Nagai K., Chem. Rev. 2009, 109, 6102–6211; [DOI] [PubMed] [Google Scholar]
- 1d. Yamamoto T., Murakami R., Komatsu S., Suginome M., J. Am. Chem. Soc. 2018, 140, 3867–3870; [DOI] [PubMed] [Google Scholar]
- 1e. Zhou L., Chu B.-F., Xu X.-Y., Xu L., Liu N., Wu Z.-Q., ACS Macro Lett. 2017, 6, 824–829. [Google Scholar]
- 2.
- 2a. Saha S., Kauffmann B., Ferrand Y., Huc I., Angew. Chem. Int. Ed. 2018, 57, 13542–13546; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 13730–13734; [Google Scholar]
- 2b. Prince R. B., Barnes S. A., Moore J. S., J. Am. Chem. Soc. 2000, 122, 2758–2762. [Google Scholar]
- 3.
- 3a. Zhao H., Sheng S., Hong Y., Zeng H., J. Am. Chem. Soc. 2014, 136, 14270–14276; [DOI] [PubMed] [Google Scholar]
- 3b. Xin P., Zhu P., Su P., Hou J.-L., Li Z.-T., J. Am. Chem. Soc. 2014, 136, 13078–13081. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Ousaka N., Yamaguchi T., Yashima E., Chem. Lett. 2014, 43, 512–514; [Google Scholar]
- 4b. Hayashi T., Ohishi Y., Hee-Soo S., Abe H., Matsumoto S., Inouye M., J. Org. Chem. 2018, 83, 8724–8730. [DOI] [PubMed] [Google Scholar]
- 5. Hill D. J., Moore J. S., Proc. Natl. Acad. Sci. USA 2002, 99, 5053–5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Takashima S., Abe H., Inouye M., Chem. Commun. 2012, 48, 3330–3332; [DOI] [PubMed] [Google Scholar]
- 6b. Massena C. J., Decato D. A., Berryman O. B., Angew. Chem. Int. Ed. 2018, 57, 16109–16113; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 16341–16345; [Google Scholar]
- 6c. Suk J.-m., Kim D. A., Jeong K.-S., Org. Lett. 2012, 14, 5018–5021. [DOI] [PubMed] [Google Scholar]
- 7. Desiraju G. R., Ho P. S., Kloo L., Legon A. C., Marquardt R., Metrangolo P., Politzer P., Resnati G., Rissanen K., Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar]
- 8.
- 8a. Cavallo G., Metrangolo P., Milani R., Pilati T., Priimagi A., Resnati G., Terraneo G., Chem. Rev. 2016, 116, 2478–2601; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Gilday L. C., Robinson S. W., Barendt T. A., Langton M. J., Mullaney B. R., Beer P. D., Chem. Rev. 2015, 115, 7118–7195; [DOI] [PubMed] [Google Scholar]
- 8c. Gliese J.-P., Jungbauer S. H., Huber S. M., Chem. Commun. 2017, 53, 12052–12055; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8d. Kniep F., Jungbauer S. H., Zhang Q., Walter S. M., Schindler S., Schnapperelle I., Herdtweck E., Huber S. M., Angew. Chem. Int. Ed. 2013, 52, 7028–7032; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7166–7170. [Google Scholar]
- 9. Sarwar M. G., Dragisic B., Sagoo S., Taylor M. S., Angew. Chem. Int. Ed. 2010, 49, 1674–1677; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1718–1721. [Google Scholar]
- 10. Ford M. C., Ho P. S., J. Med. Chem. 2016, 59, 1655–1670. [DOI] [PubMed] [Google Scholar]
- 11. Vanderkooy A., Pfefferkorn P., Taylor M. S., Macromolecules 2017, 50, 3808–3818. [Google Scholar]
- 12.
- 12a. Lindblad S., Mehmeti K., Veiga A. X., Nekoueishahraki B., Gräfenstein J., Erdélyi M., J. Am. Chem. Soc. 2018, 140, 13503–13513; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Hakkert S. B., Erdelyi M., J. Phys. Org. Chem. 2015, 28, 226–233; [Google Scholar]
- 12c. Barluenga J., González J. M., Campos P. J., Asensio G., Angew. Chem. Int. Ed. Engl. 1985, 24, 319–320; [Google Scholar]; Angew. Chem. 1985, 97, 341–342. [Google Scholar]
- 13.
- 13a. Turunen L., Warzok U., Puttreddy R., Beyeh N. K., Schalley C. A., Rissanen K., Angew. Chem. Int. Ed. 2016, 55, 14033–14036; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 14239–14242; [Google Scholar]
- 13b. Turunen L., Peuronen A., Forsblom S., Kalenius E., Lahtinen M., Rissanen K., Chem. Eur. J. 2017, 23, 11714–11718; [DOI] [PubMed] [Google Scholar]
- 13c. Warzok U., Marianski M., Hoffmann W., Turunen L., Rissanen K., Pagel K., Schalley C. A., Chem. Sci. 2018, 9, 8343–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Okitsu T., Yumitate S., Sato K., In Y., Wada A., Chem. Eur. J. 2013, 19, 4992–4996; [DOI] [PubMed] [Google Scholar]
- 14b. Barluenga J., Gonzalez-Bobes F., Murguia M. C., Ananthoju S. R., Gonzalez J. M., Chem. Eur. J. 2004, 10, 4206–4213. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Carlsson A.-C. C., Mehmeti K., Uhrbom M., Karim A., Bedin M., Puttreddy R., Kleinmaier R., Neverov A. A., Nekoueishahraki B., Gräfenstein J., Rissanen K., Erdélyi M., J. Am. Chem. Soc. 2016, 138, 9853–9863; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Bedin M., Karim A., Reitti M., Carlsson A.-C. C., Topić F., Cetina M., Pan F., Havel V., Al-Ameri F., Sindelar V., Rissanen K., Gräfenstein J., Erdélyi M., Chem. Sci. 2015, 6, 3746–3756; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Carlsson A.-C. C., Gräfenstein J., Budnjo A., Laurila J. L., Bergquist J., Karim A., Kleinmaier R., Brath U., Erdélyi M., J. Am. Chem. Soc. 2012, 134, 5706–5715. [DOI] [PubMed] [Google Scholar]
- 16. Bosch E., Barnes C. L., Brennan N. L., Eakins G. L., Breyfogle B. E., J. Org. Chem. 2008, 73, 3931–3934. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Massena C. J., Wageling N. B., Decato D. A., Martin Rodriguez E., Rose A. M., Berryman O. B., Angew. Chem. Int. Ed. 2016, 55, 12398–12402; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 12586–12590; [Google Scholar]
- 17b. Ng C.-F., Chow H.-F., Mak T. C. W., Angew. Chem. Int. Ed. 2018, 57, 4986–4990; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 5080–5084; [Google Scholar]
- 17c. Casnati A., Liantonio R., Metrangolo P., Resnati G., Ungaro R., Ugozzoli F., Angew. Chem. Int. Ed. 2006, 45, 1915–1918; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1949–1952; [Google Scholar]
- 17d. Cao J., Yan X., He W., Li X., Li Z., Mo Y., Liu M., Jiang Y.-B., J. Am. Chem. Soc. 2017, 139, 6605–6610; [DOI] [PubMed] [Google Scholar]
- 17e. Barry D. E., Hawes C. S., Blasco S., Gunnlaugsson T., Cryst. Growth Des. 2016, 16, 5194–5205; [Google Scholar]
- 17f. Farina A., Meille S. V., Messina M. T., Metrangolo P., Resnati G., Vecchio G., Angew. Chem. Int. Ed. 1999, 38, 2433–2436; [PubMed] [Google Scholar]; Angew. Chem. 1999, 111, 2585–2588; [Google Scholar]
- 17g. Lieffrig J., Niassy A. G., Jeannin O., Fourmigué M., CrystEngComm 2015, 17, 50–57; [Google Scholar]
- 17h. Logothetis T. A., Meyer F., Metrangolo P., Pilati T., Resnati G., New J. Chem. 2004, 28, 760–763; [Google Scholar]
- 17i. Liu C.-Z., Koppireddi S., Wang H., Zhang D.-W., Li Z.-T., Angew. Chem. Int. Ed. 2019, 58, 226–230; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 232–236. [Google Scholar]
- 18. Neverov A. A., Brown R. S., J. Org. Chem. 1998, 63, 5977–5982. [DOI] [PubMed] [Google Scholar]
- 19. Álvarez-Rúa C., García-Granda S., Ballesteros A., González-Bobes F., González J. M., Acta Crystallogr. Sect. E 2002, 58, o1381–o1383. [Google Scholar]
- 20.
- 20a. Janiak C., J. Chem. Soc. Dalton Trans. 2000, 3885–3896; [Google Scholar]
- 20b. Ninković D. B., Janjić G. V., Zarić S. D., Cryst. Growth Des. 2012, 12, 1060–1063; [Google Scholar]
- 20c. Kruszynski R., Sierański T., Cryst. Growth Des. 2016, 16, 587–595. [Google Scholar]
- 21.
- 21a. Bondi A., J. Phys. Chem. 1964, 68, 441–451; [Google Scholar]
- 21b. Batsanov S. S., Inorg. Mater. 2001, 37, 871–885. [Google Scholar]
- 22. Rahm M., Hoffmann R., Ashcroft N. W., Chem. Eur. J. 2016, 22, 14625–14632. [DOI] [PubMed] [Google Scholar]
- 23. Mayer I., Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar]
- 24. Bravin C., Licini G., Hunter C. A., Zonta C., Chem. Sci. 2019, 10, 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Murai K., Tateishi K., Saito A., Org. Biomol. Chem. 2016, 14, 10352–10356. [DOI] [PubMed] [Google Scholar]
- 26. Rousseau G., Robin S., Tetrahedron Lett. 2000, 41, 8881–8885. [Google Scholar]
- 27.
- 27a. Neverov A. A., Feng H. X. M., Hamilton K., Brown R. S., J. Org. Chem. 2003, 68, 3802–3810; [DOI] [PubMed] [Google Scholar]
- 27b. Barluenga J., Vazquez-Villa H., Ballesteros A., Gonzalez J. M., J. Am. Chem. Soc. 2003, 125, 9028–9029; [DOI] [PubMed] [Google Scholar]
- 27c. Brunel Y., Rousseau G., Tetrahedron Lett. 1995, 36, 2619–2622; [Google Scholar]
- 27d. Brunel Y., Rousseau G., Tetrahedron Lett. 1995, 36, 8217–8220; [Google Scholar]
- 27e. Lown J. W., Joshua A. V., Can. J. Chem. 1977, 55, 122–130; [Google Scholar]
- 27f. Homsi F., Rousseau G., J. Org. Chem. 1999, 64, 81–85; [DOI] [PubMed] [Google Scholar]
- 27g. Fujioka H., Nakahara K., Hirose H., Hirano K., Oki T., Kita Y., Chem. Commun. 2011, 47, 1060–1062; [DOI] [PubMed] [Google Scholar]
- 27h. André V., Lahrache H., Robin S., Rousseau G., Tetrahedron 2007, 63, 10059–10066; [Google Scholar]
- 27i. Rousseau G., Robin S., Tetrahedron Lett. 1997, 38, 2467–2470; [Google Scholar]
- 27j. Simonot B., Rousseau G., J. Org. Chem. 1994, 59, 5912–5919. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary