

ABSTRACT
Checkpoint kinase 2 (Chk2) is a pivotal effector kinase in the DNA damage response, with an emerging role in mitotic chromosome segregation. In this study, we show that Chk2 interacts with myosin phosphatase targeting subunit 1 (MYPT1), the targeting subunit of protein phosphatase 1cβ (PP1cβ). Previous studies have shown that MYPT1 is phosphorylated by CDK1 at S473 during mitosis, and subsequently docks to the polo-binding domain of PLK1 and dephosphorylates PLK1. Herein we present data that Chk2 phosphorylates MYPT1 at S507 in vitro and in vivo, which antagonizes pS473. Chk2 inhibition results in failure of γ-tubulin recruitment to the centrosomes, phenocopying Plk1 inhibition defects. These aberrancies were also observed in the MYPT1-S507A stable transfectants, suggesting that Chk2 exerts its effect on centrosomes via MYPT1. Collectively, we have identified a Chk2-MYPT1-PLK1 axis in regulating centrosome maturation.
Abbreviations: Chk2: checkpoint kinase 2; MYPT1: myosin phosphatase targeting subunit 1; PP1cβ: protein phosphatase 1c β; Noc: nocodazole; IP: immunoprecipitation; IB: immunoblotting; LC-MS/MS: liquid chromatography-tandem mass spectrometry; Chk2: checkpoint kinase 2; KD: kinase domain; WT: wild type; Ub: ubiquitin; DAPI: 4′,6-diamidino-2-phenylindole; IF: Immunofluorescence; IR: ionizing radiation; siCHK2: siRNA targeting CHK2
KEYWORDS: Chk2, Plk1, MYPT1, Phosphoprotein phosphatase 1β (PP1β), centrosome
Introduction
Checkpoint kinase 2 (Chk2) is a conserved Ser/Thr effector kinase that plays an instrumental role in the DNA damage response (DDR) [1]. Upon recognition of DNA double-strand breaks by the sensor proteins, the transducing kinase Ataxia-telangiectasia mutated (ATM) is activated, which phosphorylates Chk2 at T68, followed by Chk2 oligomerization, auto-phosphorylation and ultimate activation [2,3]. The downstream substrates of Chk2 include p53 and cell cycle regulators, such as Cdc25 [1]. The signaling cascade thus ensures cell cycle arrest to allow ample time for DNA repair or apoptosis if the damage is beyond repair.
Although the role of Chk2 in the DDR has long been documented, its mitotic role is just beginning to unfold. Sporadic evidence has been accumulating. Firstly, a fraction of phosphorylated Chk2 colocalizes with Polo-like kinase 1 (Plk1), a mitotic master kinase, on the centrosome and midbody [4]. Secondly, BRCA1 colocalizes with Chk2 on the centrosome and is also a direct target of Chk2 [5]. Thirdly, Chk2 depletion in colon cancer cells induces aberrant spindle assembly, mitotic delay and chromosome instability [6].
A series of investigations start to shed light on the mechanistic role of Chk2 in cell cycle control. An elegant study in human somatic cells pinpointed BRCA1 as a mitotic target of Chk2 [7]. Importantly, loss of Chk2-mediated BRCA1 phosphorylation results in spindle defects and chromosomal instability [7]. In addition, Chk2 phosphorylates and stabilizes human monopolar spindle 1 (MPS1) [8], which governs the kinetochores localization of MPS1 [9]. More recently, Chk2 has been implicated in regulating mitosis when the majority of kinetochores are unattached [10]. Specifically, Chk2 stabilizes MPS1 and phosphorylates Aurora B kinase at S331 to delay anaphase and prevent mitotic exit [10].
In an effort to identify other binding partners of Chk2, we have performed a large-scale anti-Chk2 immunoprecipitation assay followed by Mass Spectrometry (MS) analysis, and discovered myosin phosphatase targeting subunit 1 (MYPT1). MYPT1 is the targeting subunit of protein phosphatase 1cβ (PP1cβ), and plays a plethora of roles in mitosis [11]. First, cyclin-dependent kinase 1 (CDK1) phosphorylates MYPT1 at S437 to dock MYPT1 to the polo-box domain (PBD) of Plk1, and subsequently MYPT1/PP1cβ dephosphorylates Plk1 at T210, thus inactivating Plk1 [12] and destabilizes kinetochore-microtubule attachments [13]. Secondly, MYPT1 depletion leads to precocious sister chromatid segregation [14]. Thirdly, MYPT1 is required to sustain nocodazole or taxol-induced cell cycle arrest [15].
Herein we show that Chk2 interacts and phosphorylates MYPT1 at S507, which neighbors and antagonizes pS473. Ablation of the phosphorylation compromises MYPT1 stability. Chk2 inhibition leads to failure of γ-tubulin localization to the centrosomes, which is phenocopied by MYPT1-S507A-rescue stable transfected cells. Our study thus delineates a Chk2-MYPT1-Plk1 axis in the centrosome cycle.
Experimental procedures
Cell culture, antibodies and plasmids
HeLa and HEK293 cells were purchased from ATCC. Anti-MYPT1 antibodies, MYPT1 and rescue plasmids were described before [15]. Antibodies against MYPT1 phospho-Ser507 (pS507-Ab) were raised in rabbits using the sequence of CRRLA(pS)TSDIE and manufactured by Beijing B&M Biotech Co., Ltd. Antibodies against MYPT1 phospho-Ser473 (pS473-Ab) were raised as described [12]. MYPT1-S507A mutants were generated using specific primers (sequences available upon request) following the manufacturer’s instructions (QuickChange II, Stratagene). Primer sequences for MYPT1-rescue plasmids were: ACAAAAATTTCTCCTAAGGAGGAGGAACGTAAAGATGAGTCT. siCHK2 oligo: GGT GCC TAT GGA GAA GTTC.
Transfections
HeLa cells were transfected twice with a 24 h interval using Oligofectamine (Invitrogen) according to the manufacturer’s instructions. Transfectants were used for further experiments 24 hours after the second transfection.
For plasmid transfection, cells were seeded at 50–60% confluence/10 cm2 petri dish and transfected with 7.5 μg of plasmid DNA using FuGene 6 according to the manufacturer’s instructions for immunoprecipitation (IP) experiments. Where indicated, CHK2 inhibitor II (EMD Millipore, #516,480–79-8) was used at 10 μM for two hrs, Nocodazole (Noc) was used at 320 nM for 16 hrs.
Immunoblotting (IB) and IP
IB and IP experiments were performed as described before [16]. The following primary antibodies were used for immunoblotting: anti-MYPT1 (Bethyl, BL3866), anti-Chk2 (Bethyl, A300-681A), anti-β-actin, anti-HA, anti-Myc and anti-FLAG M2 (Sigma). Peroxidase-conjugated secondary antibodies were from JacksonImmuno Research. Blotted proteins were visualized using the ECL detection system (Amersham). Signals were detected by a LAS-4000, and analyzed using Multi Gauge (Fujifilm).
In vitro kinase (IVK) assays
Chk2 IVK assays were performed as previously described. Briefly, recombinant Chk2 kinase was purchased from R & D systems (Cat. # 1630-KS), incubated with purified GST-MYPT1 with kinase buffers (20 mM Tris HCl pH 7.5, 10 mM MgCl2, 2 mM MnCl2, 1 mM DTT), and 0.1 mM ATP or 1 μCi of γ-[32P]ATP. After 20 min at 30°C, reactions were stopped by the sample buffer. Protein samples were separated by SDS-PAGE and phosphate incorporation was determined by PhosphorImager. LC-MS/MS analysis were performed as described [17].
Results
Chk2 associates MYPT1
In an effort to identify proteins that interact with Chk2, anti-Chk2 immunoprecipitates from HeLa cells were stained with Coomassie blue (Figure 1(a)), and the corresponding bands were incised and subject to MS analysis. In the immunoprecipitates, we identified MYPT1. We aim to dissect whether the interaction is real, and performed coIP experiments between exogenous and endogenous MYPT1 and Chk2 (Figure 1(b-c)), and the two proteins do coIP. A GST pull-down assay was carried out, and Figure 1(d) indicated that GST-Chk2 could pulldown MYPT1 from cell extracts. To map which domain of Chk2 interacts with MYPT1, we constructed different plasmids that either harbor the kinase domain (KD) of Chk2 or not (ΔKD) (Figure 1(e)). And IP assays showcased that both Chk2-WT and KD interacted with MYPT1, but not the ΔKD proteins (Figure 1(f)), suggesting that the Chk2-KD is indispensible for the interaction. Taken together, the results above indicate that Chk2 interacts with MYPT1, probably via the KD.
Figure 1.
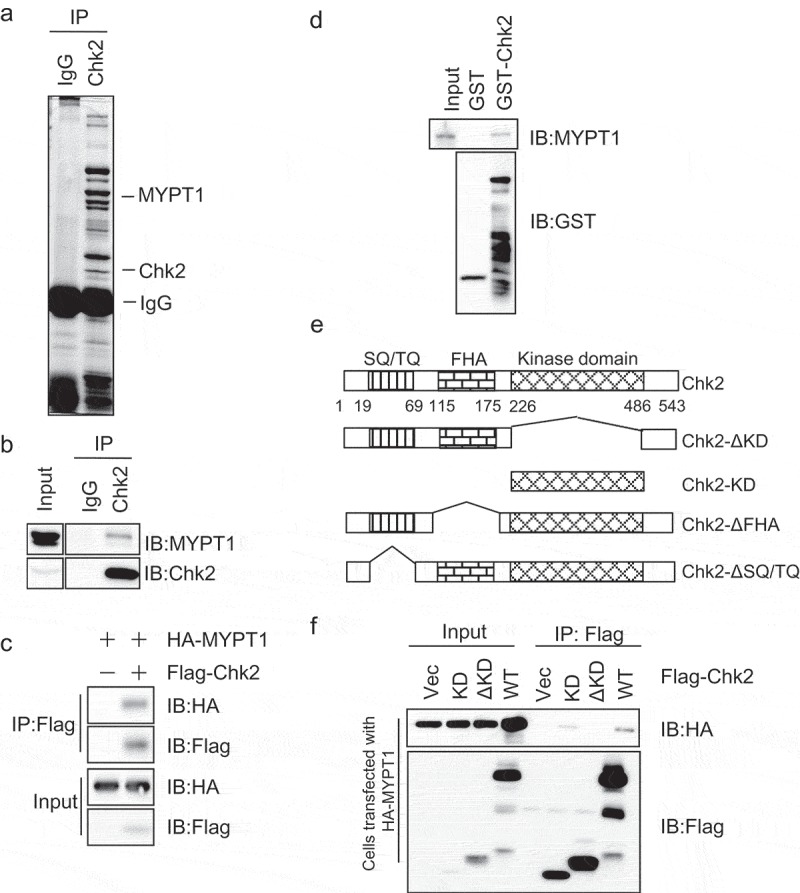
Interaction between Chk2 and MYPT1.
(a) Chk2 was immunoprecipitated from HeLa cells, and samples were analyzed by SDS-PAGE with Coomassie blue staining. Chk2-interacting proteins were identified by mass spectrometry. Indicated are bands identified as Chk2 and MYPT1. (b) CoIP between endogenous proteins. Chk2 immunoprecipitates were blotted with anti-Chk2 and anti-MYPT1 antibodies. (c) CoIP between exogenous Chk2 and MYPT1. HeLa cells transfected with vector or HA-MYPT1 and Flag-Chk2 constructs, and lysates were subjected to IP and IB assays using antibodies indicated. (d) Bacterially expressed Chk2 proteins were incubated with HeLa extracts, and the GST-pulldowns were blotted with anti-MYPT1 and anti-GST antibodies. (e) Domain structure of Chk2. The SQ/TQ motif, the FHA domain, the kinase domain, and the boundaries of Chk2 fragments used in this study are indicated. (f) HeLa cells transfected with HA-MYPT1 and various Chk2 fragments were subject to IP and IB assays using the antibodies indicated.
Chk2 phosphorylates MYPT1 at S507
Given the results aforementioned, we naturally examined whether Chk2 could phosphorylate MYPT1 through an in vitro kinase (IVK) assay. As shown in Figure 2(a), recombinant GST-Chk2 was incubated with GST-MYPT1 in kinase buffers containing γ-[32P]ATP. Chk2 indeed phosphorylates MYPT1 (Figure 2(a)). To identify the phosphorylation site, the IVK products were subject to liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis, and only one site, S507, was pinpointed (Figure 2(b)). A closer look surrounding the S507 residue suggested that it matched a minimum consensus site of Chk2 phosphorylation (Figure 2(c)). When the GST-MYPT1-S507A protein was incubated with GST-Chk2 in an IVK assay, the autoradiography signal was abolished (Figure 2(a)), suggesting that S507 is a phosphorylation site by Chk2 in vitro.
Figure 2.
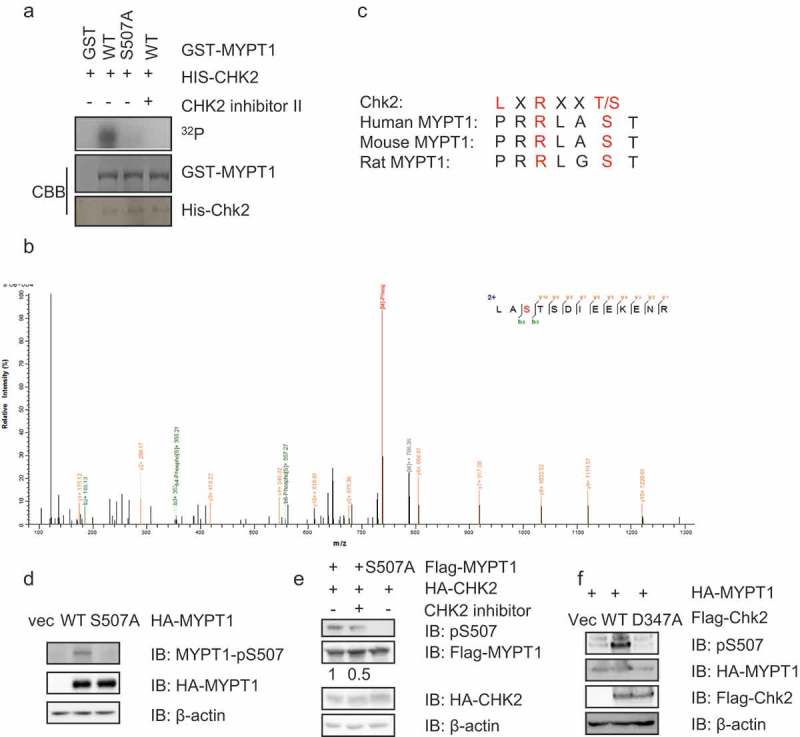
Chk2 phosphorylates MYPT1 at S507.
(a) GST-Chk2 and GST-MYPT1 fragments were purified from E. coli cells and incubated in kinase buffers containg 32P-ATP. Coomassie blue staining showed the amount of the input proteins. Phosphorylated GST-MYPT1 was detected by autoradiography. (b) LC-MS/MS analysis identified S507 of MYPT1 as the only site phosphorylated by Chk2 in vitro. From this collision-induced dissociation spectrum, a phosphorylated peptide LA(pS)TSDIEEKENR of MYPT1 was identified following incubation with Chk2 in an IVK reaction. “b” and “y” ion series represent fragment ions containing the N- and C-termini of the peptide, respectively. (c) S507 is evolutionarily conserved. (d) MYPT1-pS507 phospho-specific antibodies were prepared as described in Experimental Procedures. HeLa cells were transfected with HA-MYPT1-WT or S507A, then the cell extracts were IBed with the indicated antibodies. Numbers indicate quantitation of pS507/Flag-MYPT1 signals. (e) HeLa cells were transfected with Flag-MYPT1 WT or S507A plasmids, together with HA-CHK2 with or without its inhibitor. Cell extracts were then collected and IBed with the antibodies indicated. (f) Cells were transfected with empty vectors, Flag-Chk2-WT, or -D347A (kinase dead) plasmids together with HA-MYPT1, then the lysates were collected for IB assays.
To assess whether the phosphorylation modification also occurs in vivo, we prepared a phosphorylation-specific antibody targeting pS507 as described in Experimental Procedures. As shown in Figure 2(d), the antibody showed a specific band when MYPT1-WT was transfected into HeLa cells, but not when MYPT1-S507A was transfected. When a Chk2 inhibitor (Chk2i) was employed or a kinase-dead allele of Chk2 (D347A) was adopted in the IVK assay, the pS507 signal was abolished (Figure 2(e-f)), suggesting that pS507 occurs in vivo.
pS507 compromised interaction between MYPT1 and Plk1 by antagonizing pS473
As S507 abuts S473, we sought to investigate where there is any interplay between these two phosphorylation events. To this end, we first prepared a specific antibody toward pS473 as described previously [12]. When cells were treated with Noc, the pS473 levels elevated robustly in the cellular lysates (Figure 3(a)), consistent with previous reports [12]. When RO-3306 (CDK1 inhibitor) was used, however, the pS473 signal was abolished (Figure 3(a)), indicating that the antibody is specific.
Figure 3.
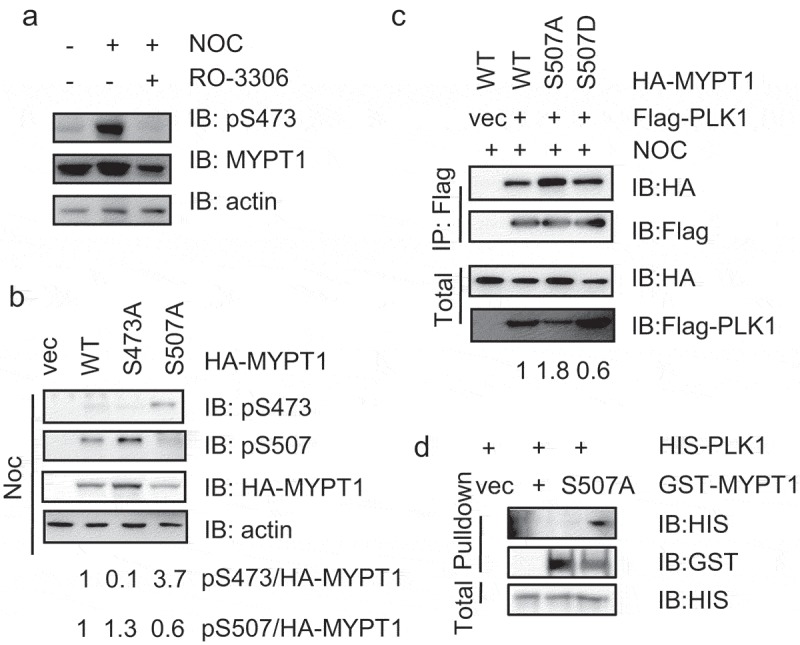
MYPT1-pS507 antagonizes pS473.
(a) MYPT1-pS473 phospho-specific antibodies were prepared as described in Experimental Procedures. HeLa cells were treated with the CDK inhibitor RO-3306 or without. Cell extracts were then collected and IBed with the indicated antibodies. (b) HeLa cells were transfected with HA-MYPT1-WT, -S473A or -S507A plasmids and then the cell extracts were IBed with the antibodies indicated. Numbers are quantitation results as indicated. (c) HeLa cells were transfected with Flag-Plk1 together with HA-MYPT1-WT, -S507A or -S507D plasmids, and then treated with Noc. The cell extracts were IPed with anti-Flag antibodies and IBed with the antibodies indicated. (d) Recombinant His-PLK1 and GST-MYPT1 WT or S507A were purified from E. coli, and then GST pulldown assays were performed.
Then we employed MYPT1-WT, -S473A and -S507A mutants. When these plasmids were transfected into HeLa cells and treated with Noc, pS473 levels were significantly elevated in S507A mutants, and pS507 levels were enhanced in S473A mutants (Figure 3(b)), suggesting that these two adjoining phosphorylation events could be antagonizing against each other.
Since pS473 is a prerequisite for binding between PLK1 and MYPT1 [12], we used MYPT1-WT, S507A and S507D mutants to assess their interaction with PLK1. S507D attenuates PLK1binding, while S507A enhanced PLK1 interaction (Figure 3(c)). This is consistent with Figure 3(b), where S507A promotes pS473 levels. Moreover, we used pulldown assays. GST-MYPT1-WT and S507A were purified from E. coli, and incubated with His-PLK1, and S507A increased PLK1 association (Figure 3(d)). In sum, pS507 antagonizes pS473 and MYPT1-S507A augments PLK1 binding.
CHK2-dependent phosphorylation of pS507 stabilizes MYPT1
Previously we identified that Chk1 phosphorylates MYPT1 at S20 to degrade MYPT1 [18], we wonder whether Chk2-mediated phosphorylation also regulates MYPT1 stability. In CHK2−/- cells, lower levels of MYPT1 is discernable (Figure 4(a)) and elevated ubiquitination levels are detectable (Figure 4(b)), suggesting that Chk2 stabilizes MYPT1. We then used siRNA to deplete Chk2, and again, less MYPT1 is identified (Figure 4(c)). When distinct Chk2 constructs were co-transfected with HA-MYPT1, MYPT1 levels lessened in the kinase-dead CHK2-D347A transfectants (Figure 4(d)), suggesting that MYPT1 levels are dependent on the kinase activity of Chk2.
Figure 4.
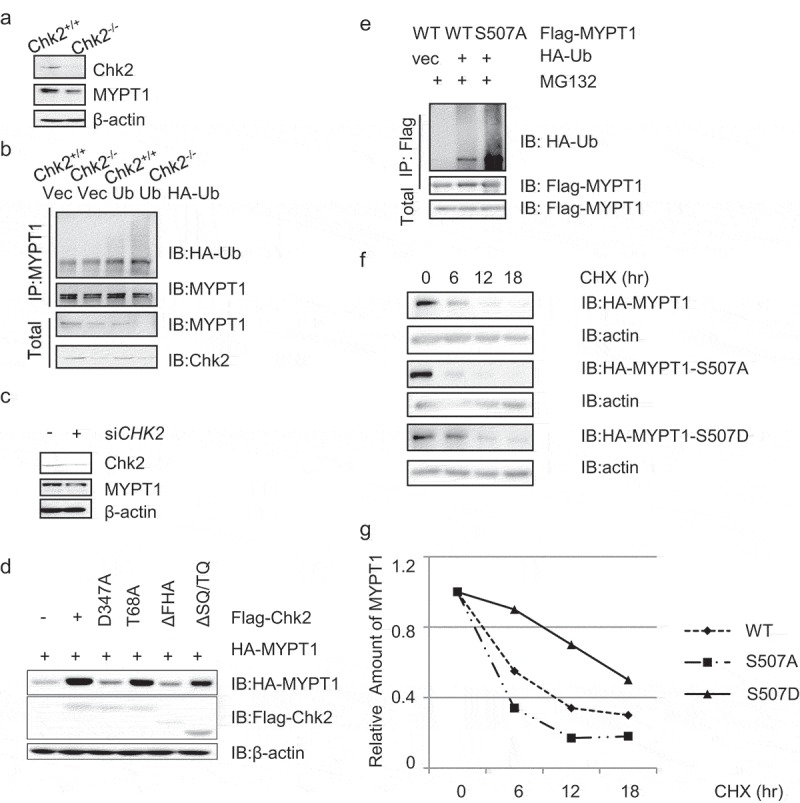
The Chk2 kinase activity stabilizes MYPT1.
(a) HCT116 WT and CHK2−/- cell extracts were collected and IBed with the antibodies indicated. (b) HCT116 WT and CHK2−/- cells were transfected with vector controls or HA-Ub plasmids, and then IPed with anti-MYPT1 antibodies. (c) HeLa cells were treated with control siRNA or siCHK2 oligos, and then subject to IB assays. (d) HeLa cells were transfected with HA-MYPT1 together with Flag-Chk2 WT, D347A (kinase dead), T68A, ΔFHA or ΔSQ/TQ plasmids, and the cell extracts were IBed with the indicated antibodies. (e) Cells were transfected with HA-MYPT1-WT or -S507A plasmids, together with HA-Ub plasmids. The anti-Flag immunoprecipitates were IBed with the antibodies indicated. (f) Cells were transfected with HA-MYPT1-WT or -S507A plasmids, and then treated with Noc together with CHX for different time durations. The cell extracts were collected and subject to IB. (g) Quantitation of the results in (f).
As Chk2 phosphorylates MYPT1 at S507, we examined the ubiquitination and protein levels of S507A. As shown in Figure 4(e), S507A levels increased compared with WT, suggesting that phosphorylation inhibits ubiquitination. Then we used cycloheximide (CHX) to block new protein synthesis, and MYPT1-S507A displayed shorter half-life than WT (Figure 4(f-g)), suggesting that phosphorylation could stabilize MYPT1.
CHK2 inhibition results in failure of γ-tubulin localization
As PLK1 regulates centrosome maturation and γ-tubulin staining becomes absent in PLK1-depleted cells by siRNA [19], we wonder whether Chk2 could function in the same pathway. Cells were first treated with Noc, then incubated with Chk2i or not treated. When the cells were stained with anti-γ-tubulin antibodies, the typical two centrosome localization pattern was discernable in the WT cells, but not in the Chk2i-treated cells (Figure 5(a-b)), suggesting that Chk2 kinase activity is a prerequisite for centrosome maturation.
Figure 5.
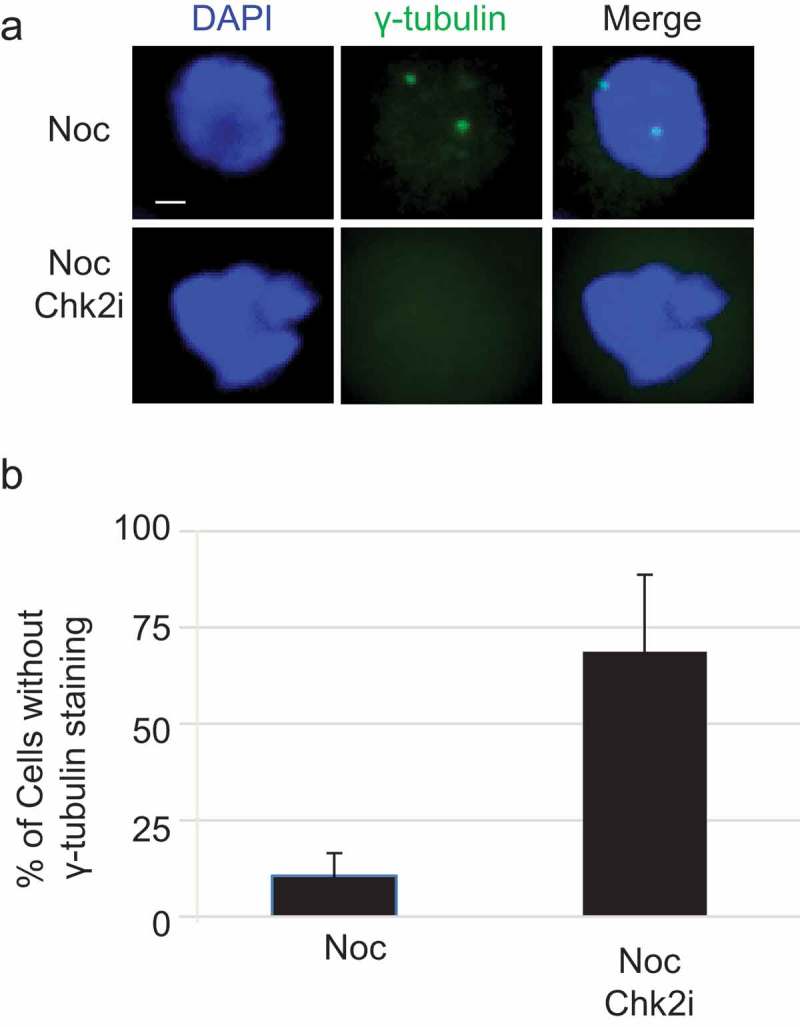
CHK2 inhibition results in failure of γ-tubulin localization.
(a) U2OS cells were treated with Noc, then with or without Chk2i. IF assays were carried out by staining the cells with anti-γ-tubulin antibodies and DAPI. (B) Quantitation of the results in (a). At least 100 cells were measured for centrosome numbers. The bar graph represents mean ± S. D. of three independent experiments. Student’s T-test was performed with p = 0.009. Scale bar, 10 μm.
MYPT1-S507A-rescue constructs induce centrosome maturation defects
We then constructed MYPT1-rescue plasmids as previously described [15], and made corresponding MYPT1-S507A-rescue plasmids. Then MYPT1-WT and -rescue plasmids were transfected into U2OS cells and selected for stable transfectants. When treated with siMYPT1, the endogenous MYPT1 protein was efficiently down-regulated, but not the stably expressed HA-MYPT1 proteins (Figure 6(a)).
Figure 6.
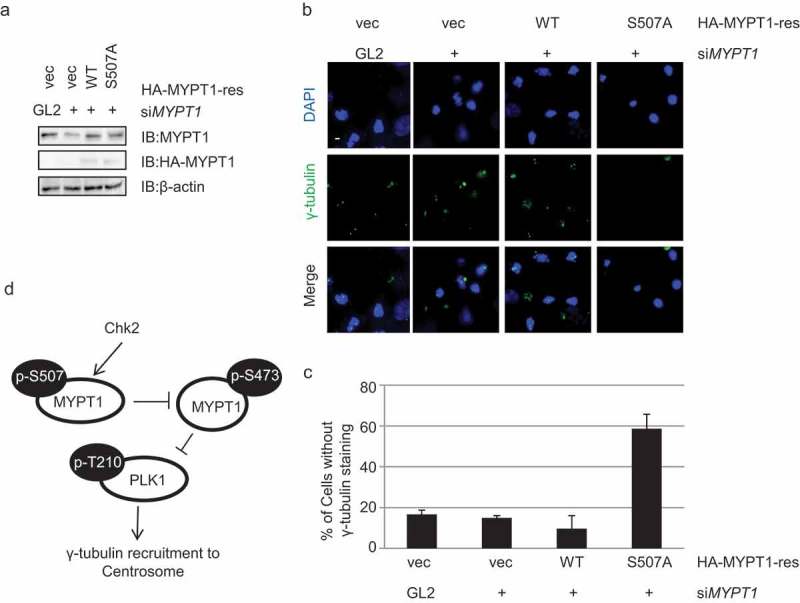
MYPT1-S507A induces centrosome maturation defects.
(a) IB of stable transfectants of Vec, HA-MYPT1-rescue-WT and -S507A plasmids in U2OS cells after treating with siMYPT1, indicative of efficient knock-down of endogenous MYPT1. (b) Cells in (a) were fixed and stained with anti-γ-tubulin antibodies together with DAPI. Scale bar, 10 μm. (c) Quantitation of (b). At least 120 cells were measured for centrosome numbers. The bar graph represents mean ± S. D. of three independent experiments. Student’s T-test was performed with p = 0.015. (d) A model depicting how Chk2 exerts its function during centrosome maturation. Chk2 phosphorylates MYPT1 at S507, which attenuates the neighboring S473 residue that is essential to dock MYPT1 to Plk1. Consequently, Chk2 functions in centrosome maturation through Plk1.
Immunofluorescence (IF) assays were carried out to examine the localization of γ-tubulin. As shown in Figure 6(b), the MYPT1-S507A stable transfectants again displayed the loss of γ-tubulin staining phenotype (Figure 6(b-c)), in the same line as Chk2i-treated cells, indicating that Chk2-dependent phosphorylation of MYPT1 at S507 is indispensable for centrosome maturation.
Discussion
In this report, we identify MYPT1 to be a new mitotic substrate of Chk2, a vital kinase orchestrating not only mitosis but also DDR. We present data that Chk2 phosphorylates MYPT1 at S507, which antagonizes pS473, a known CDK1-phosphorylation site. Chk2 inhibition obliterates γ-tubulin localization during mitosis, thus Chk2 is essential for centrosome maturation.
The link between Chk2 and centrosomes has long been revealed. Chk2-pT68 antibodies as well as HA-Chk2 were found to reside in the centrosomes by cytological studies [4]. Flag-Chk2 and GFP-Chk2 were also shown to localize to centrosomes during mitosis, which is dependent on Plk1 [20]. Recently, a revisit of Chk2-pT68 unveils its localization to the mother centrioles during G2 and M phase [21]. Biochemically, Chk2 co-purifies with centrosomes [22]. In embryonic stem cells, Chk2 localizes exclusively at the centrosomes [23], compromising the ATM-Chk2-p53 pathway. Thus upon ionizing radiation (IR), the damaged cells will undergo apoptosis and hence be removed.
Chk2 also modulates centrosomes during DNA damage. During hydroxyurea-induced replication stress, the centrosomes will amplify and Chk2-pT68 localizes to all amplified centrioles [21]. Depletion of DNA-dependent protein kinase (DNA-PK) will block the centrosome amplification phenotype, probably through inactivating Chk2 [21]. In cisplatin-treated cells, silencing of Xeroderma Pigmentosum Group C (XPC) in human bladder cancers also results in centrosome amplification [24]. The underlying mechanism could be that XPC down-regulation hinders BRCA1 expression, accumulates more DNA double-strand breaks (DSBs) and thus prolongs ATM-Chk2 signaling [24].
On the other side of coin, PLK1 also regulates Chk2. In Plk1-overexpression mouse embryonic fibroblasts (MEFs), IR insults will reduce Chk2 activation and suppress DDR pathways, and lead to tumorigenesis [25]. In line with it, a Plk1 inhibitor, BI6727, will activate the Chk2 pathway and induce DNA damage, hence dampen tumor growth in small cell lung cancer cells [26].
In a recent endeavor to dissect the relationship between DDR during mitosis and chromosome segregation, genetic depletion of ATM and Chk2 culminates in segregation defects prior to DNA damage [27], consistent with previous observations [7]. And it is proposed that the molecular underpinning might be spindle defects not associated with DDR. Our data suggest that besides the Chk2-BRCA1 pathway to ensure chromosomal stability, Chk2 also engages in centrosomal maturation by phosphorylating MYPT1.
Funding Statement
This work was supported by the National Natural Science Foundation of China (NSFC) fund (31872720) and Capacity Building for Sci-Tech Innovation - Fundamental Scientific Research Funds (19530050137) to J. L. X.X. was supported by the National Natural Science Foundation of China (NSFC) grants 31761133012 and 31530016, the 973 projects 2017YFA0503900 and 2015CB910601, the Shenzhen Science and Technology Innovation Commission grants JCYJ20170412113009742 and JCYJ20180507182213033.
Acknowledgments
We thank Dr. Huiqiang Lou (China Agriculture Univ.) for support and members of the Li lab for helpful discussion and comments on the manuscript.
Author contributions
J. L., M-Q. D. and X. X. designed the project and analyzed the data; S. N.,Y. S., H. R., Z. L, Q. G. performed the biochemical assays; Y. D. carried out the MS analysis. J. L. wrote the manuscript; all authors reviewed and approved the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Zannini L, Delia D, Buscemi G.. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol. 2014;6:442–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Xu X, Tsvetkov LM, Stern DF.. Chk2 activation and phosphorylation-dependent oligomerization. Mol Cell Biol. 2002;22:4419–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stracker TH, Usui T, Petrini JH. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst). 2009;8:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tsvetkov L, Xu X, Li J, et al. Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes and the midbody. J Biol Chem. 2003;278:8468–8475. [DOI] [PubMed] [Google Scholar]
- [5].Zhang S, Hemmerich P, Grosse F. Centrosomal localization of DNA damage checkpoint proteins. J Cell Biochem. 2007;101:451–465. [DOI] [PubMed] [Google Scholar]
- [6].Stolz A, Ertych N, Bastians H. Loss of the tumour-suppressor genes CHK2 and BRCA1 results in chromosomal instability. Biochem Soc Trans. 2010;38:1704–1708. [DOI] [PubMed] [Google Scholar]
- [7].Stolz A, Ertych N, Kienitz A, et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nat Cell Biol. 2010;12:492–499. [DOI] [PubMed] [Google Scholar]
- [8].Yeh YH, Huang YF, Lin TY, et al. The cell cycle checkpoint kinase CHK2 mediates DNA damage-induced stabilization of TTK/hMps1. Oncogene. 2009;28:1366–1378. [DOI] [PubMed] [Google Scholar]
- [9].Yeh CW, Yu ZC, Chen PH, et al. Phosphorylation at threonine 288 by cell cycle checkpoint kinase 2 (CHK2) controls human monopolar spindle 1 (Mps1) kinetochore localization. J Biol Chem. 2014;289:15319–15327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Petsalaki E, Zachos G. Chk2 prevents mitotic exit when the majority of kinetochores are unattached. J Cell Biol. 2014;205:339–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Matsumura F, Yamakita Y, Yamashiro S. Myosin light chain kinases and phosphatase in mitosis and cytokinesis. Arch Biochem Biophys. 2011;510:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yamashiro S, Yamakita Y, Totsukawa G, et al. Myosin phosphatase-targeting subunit 1 regulates mitosis by antagonizing polo-like kinase 1. Dev Cell. 2008;14:787–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dumitru AMG, Rusin SF, Clark AEM, et al. Cyclin A/Cdk1 modulates Plk1 activity in prometaphase to regulate kinetochore-microtubule attachment stability. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Matsumura F, Yamakita Y, Yamashiro S. Myosin phosphatase-targeting subunit 1 controls chromatid segregation. J Biol Chem. 2011;286:10825–10833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Li J, Liu X, Liao J, et al. MYPT1 sustains centromeric cohesion and the spindle-assembly checkpoint. J Genet Genomics. 2013;40:575–578. [DOI] [PubMed] [Google Scholar]
- [16].Li J, Wang J, Hou W, et al. Phosphorylation of Ataxin-10 by polo-like kinase 1 is required for cytokinesis. Cell Cycle. 2011;10:2946–2958. [DOI] [PubMed] [Google Scholar]
- [17].Tian J, Tian C, Ding Y, et al. Aurora B-dependent phosphorylation of Ataxin-10 promotes the interaction between Ataxin-10 and Plk1 in cytokinesis. Sci Rep. 2015;5:8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hu X, Li Z, Ding Y, et al. Chk1 modulates the interaction between myosin phosphatase targeting protein 1 (MYPT1) and protein phosphatase 1cbeta (PP1cbeta). Cell Cycle. 2018;17:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sumara I, Gimenez-Abian JF, Gerlich D, et al. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol. 2004;14:1712–1722. [DOI] [PubMed] [Google Scholar]
- [20].Chouinard G, Clement I, Lafontaine J, et al. Cell cycle-dependent localization of CHK2 at centrosomes during mitosis. Cell Div. 2013;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang CY, Huang EY, Huang SC, et al. DNA-PK/Chk2 induces centrosome amplification during prolonged replication stress. Oncogene. 2015;34:1263–1269. [DOI] [PubMed] [Google Scholar]
- [22].Golan A, Pick E, Tsvetkov L, et al. Centrosomal Chk2 in DNA damage responses and cell cycle progression. Cell Cycle. 2010;9:2647–2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hong Y, Stambrook PJ. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc Natl Acad Sci U S A. 2004;101:14443–14448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang H, Huang Y, Shi J, et al. XPC deficiency leads to centrosome amplification by inhibiting BRCA1 expression upon cisplatin-mediated DNA damage in human bladder cancer. Cancer Lett. 2019;444:136–146. [DOI] [PubMed] [Google Scholar]
- [25].Li Z, Liu J, Li J, et al. Polo-like kinase 1 (Plk1) overexpression enhances ionizing radiation-induced cancer formation in mice. J Biol Chem. 2017;292:17461–17472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang Y, Wu L, Yao Y, et al. Polo-like kinase 1 inhibitor BI 6727 induces DNA damage and exerts strong antitumor activity in small cell lung cancer. Cancer Lett. 2018;436:1–9. [DOI] [PubMed] [Google Scholar]
- [27].Bakhoum SF, Kabeche L, Murnane JP, et al. DNA-damage response during mitosis induces whole-chromosome missegregation. Cancer Discov. 2014;4:1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]