

ABSTRACT
DNA repair proteins have been found to localize to the centrosomes and defects in these proteins cause centrosome abnormality. Centrobin is a centriole-associated protein that is required for centriole duplication and microtubule stability. A recent study revealed that centrobin is a candidate substrate for ATM/ATR kinases. However, whether centrobin is involved in DNA damage response (DDR) remains unexplored. Here we show that centrobin is phosphorylated after UV exposure and that the phosphorylation is detected exclusively in the detergent/DNase I-resistant nuclear matrix. UV-induced phosphorylation of centrobin is largely dependent on ATR activity. Centrobin-depleted cells show impaired DNA damage-induced microtubule stabilization and increased sensitivity to UV radiation. Interestingly, depletion of centrobin leads to defective homologous recombination (HR) repair, which is reversed by expression of wild-type centrobin. Taken together, these results strongly suggest that centrobin plays an important role in DDR.
KEYWORDS: Centrobin, DNA damage response, ATR-dependent phosphorylation, microtubule stability, homologous recombination
Introduction
Mirotubules (MTs) are polymers of tubulin that are key components of the eukaryotic cytoskeleton and provide structure and shape to the cytoplasm of the cells [1,2]. Recently, it has been shown that fission yeast with defective DNA repair functions displayed elongated morphology with stabilized microtubules [3]. Another recent study demonstrated that microtubule stabilization is required for intracellular trafficking of DNA repair proteins to the nucleus in response to DNA damage [4], revealing a link between the DNA damage response (DDR) and microtubules networks.
MTs are nucleated and organized by the microtubule organizing center (MTOC), and centrosome serves as the main MTOC in most animal cells [5,6]. Centrobin is a daughter centriole-specific protein which is required for centriole duplication and elongation [7–11]. Centrobin binds to microtubules and this interaction is required for microtubule polymerization and stabilization [8,11,12]. Two different serine/threonine kinase are known to regulate the microtubule stabilizing activity of centrobin. PLK1 (polo-like kinase 1) enhances the microtubule stabilizing activity of centrobin during mitosis, while centrosomal kinase NEK2 (NIMA-related kinase 2) antagonizes the microtubule stabilizing the activity of centrobin during interphase [8,12]. Centrobin is also detected outside the centrosome and is essential for microtubule formation in the cytoplasm [8,13]. Intriguingly, a proteomics analysis for ATM/ATR substrates following IR irradiation identified centrobin as one of 700 putative substrates [14]. Combined, we, therefore, hypothesized that centrobin plays a role in the cellular response to DNA damage, possibly through its ability to organize microtubules.
In the present study, we found that centrobin is phosphorylated following UV radiation and its phosphorylation is largely reduced by ATR inhibition. Centrobin-depleted cells are sensitive to UV exposure and had a defect in UV-induced microtubule stabilization. Centrobin knockdown in DR-GFP/U2OS cells exhibits reduced DNA repair efficiency of homologous recombination (HR) and this HR defect is restored by expression of wild-type centrobin. Taken together, our results suggest that centrobin plays an important role in DDR, leading to DNA damage-induced microtubule stabilization and efficient DNA repair.
Materials and methods
Antibodies
The anti-α-tubulin (T5168), anti-γ-tubulin (T6557), and anti-MCM7 (M7931) antibodies were purchased from Sigma Aldrich; anti-centrobin (ab70448) antibody was purchased from abcam; anti-lamin B1 (sc374015) and anti-centrin 2 (sc27793) antibodies were purchased from Santa Cruz; anti-ATR (#2790), anti-phospho CHK1 (S345) (#2341), anti-phospho CHK2 (T68) (#2661), and anti-K40 acetylated α-tubulin (#5335) antibodies were purchased from Cell signaling; anti-BRCA2 (OP95), anti-BRCA1 (OP92), anti-RPA2 (04-1481), anti-Histone H3 (06-755), and anti-γ-H2AX (05-636) antibodies were purchased from Millipore; anti-ATM (A300-299A), anti-RPA2 (S4/S8) (A300-245A), anti-RPA2 (S33) (A300-246A), and anti-MCM3 (A300-192A) antibodies were purchased from Bethyl Laboratory.
Cell culture and transfection
HeLa and DR-GFP U2OS were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum. Cell transfection was performed using LTX (Thermo Fisher Scientific, Waltham, MA, USA) for plasmids and RNAimax (Thermo Fisher Scientific, Waltham, MA, USA) for siRNAs, respectively, following the manufacturer’s protocol.
siRNAs
siRNAs were synthesized by Qiagen and Bioneer (Korea). The sense sequence of centrobin #1 and #2 siRNAs are GGAUGGUUCUAAGCAUAUCdTdT and AGUGCCAGACUGCAGCAACdTdT, respectively. While both siRNAs efficiently knockdown centrobin, siRNA#1 was used to perform all the knockdown experiments throughout this study. siRNA for centrin-2 (GAGCAAAAGCAGGAGAUCCdTdT), PALB2, and BRCA2 were based on previous report [15]. Negative Control siRNAs were purchased from Qiagen and Bioneer (Korea).
Constructs and retroviral infection
Total RNA isolated from HeLa cells was reverse transcribed and the full-length centrobin cDNA was amplified by PCR. Centrobin cDNA resistant to the siRNA was created by introducing silent mutations. Site-directed mutagenesis was carried out with QuickChange site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) and verified by sequencing. siRNA resistant centrobin cDNA was sub-cloned into a pMSCV-FLAG retroviral vector and then retroviral vectors were cotransfected with pCL-Ampho into 293T cells for virus production. The virus was collected 48 and 72 h after transfection and subsequently used to infect DR-GFP U2OS cells. Two days after the last infection, cells were selected in 1 μg/ml puromycin (Thermo Fisher Scientific, Waltham, MA, USA) for the establishment of stable clones.
Cell fractionation
Cell fractionation was performed as described previously with some modifications [16]. Cell pellets were suspended in modified CSK buffer (10 mM HEPES-KOH (pH 7.5), 100 mM NaCl, 0.3 M sucrose, 0.1% Triton X-100, 0.1 mM EDTA, 1 mM dithiothreitol (DTT) and protease inhibitors. After 10 min of incubation on ice, samples were centrifuged at 1000 × g for 5 min. Supernatants (cytosolic & nucleoplasmic fraction) were saved for S fraction, and remaining pellets (chromatin-enriched fraction; P) were resuspended in the same buffer. Further fractionation of P1 suspension were performed by treatment with high-salt NaCl or DNaseI (New England Biolabs, MA, USA), then supernatants were saved after high-speed centrifugation.
Lambda phosphatase treatment
Cell extracts were incubated with 400 U of λ phosphatase (New England Biolabs, MA, USA) at 30°C for 40 min and then analyzed by Western blotting.
Immunofluorescence
Exponentially growing cells were seeded onto coverslips and then treated (or mock-treated) with the indicated DNA-damaging agent. After treatment, the cells were washed with PBS and fixed in cold-methanol. The cells were then permeabilized with 0.5% Triton X-100 for 5 min and blocked in 5% BSA for 30 min. The cells were incubated with the indicated primary antibodies overnight at 4°C, and Alexa-conjugated secondary antibodies (Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature. After three washing steps, slides were counterstained using DAPI (Sigma-Aldrich, St. Louis, MO, USA), mounted with ProLong Gold antifade (Thermo Fisher Scientific, Waltham, MA, USA), and the results were visualized using a Leica Fluorescence Microscope with a Plan-Apochromat 63x/1.4 oil immersion objective.
Cell survival assay
Cells were seeded onto 12-well plates in triplicate and then transfected with siRNAs. At 48 h after transfection, cells were exposed to UV at the indicated dose. Treated cells were grown for 24 h before being collected and viable cells were counted by methylene blue staining. Growth percentage was calculated as treated cells/untreated cells × 100.
Homologous recombination (HR) assays
Homologous recombination (HR) assays were performed as described previously [17]. DR-GFP U2OS cells were seeded onto 12-well plates in triplicate and then transfected with the indicated siRNAs. Twenty-four hours after transfection, the cells were then transfected with either I-SceI plasmid or GFP plasmid. Forty-eight hours later, the cells were harvested and analyzed for green fluorescent protein (GFP) by fluorescence-activated cell sorting (FACS) analysis. In each experiment, the percentage of green (GFP+) cells was measured in triplicate samples. Values were normalized for the transfection efficiency and were displayed as mean ± SEM GFP+ frequencies relative to that of control siRNA-treated cells.
Nocodazole resistance assay
Nocodazole treatment to image stabilized microtubules was performed as previously described [18]. At 72 hr after siRNAs transfection, HeLa cells were treated with 2 μM of nocodazole (Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 45 min and then pre-extracted with 0.2% triton X-100 at 4°C for 1 min to remove free tubulin. Then, cells were fixed with cold-methanol, permeabilized with 0.5% Triton X-100 for 5 min and blocked in 5% BSA for 30 min. Cells were incubated with α-tubulin and anti-K40 acetylated α-tubulin antibodies for 2 h at room temperature, and Alexa-conjugated secondary antibodies (Invitrogen) for 1 h each at room temperature. After three washing steps, slides were counterstained using DAPI (Sigma-Aldrich), mounted with ProLong Gold antifade (Thermo Fisher Scientific, Waltham, MA, USA), and the results were visualized using a Leica Fluorescence Microscope with a Plan-Apochromat 63x/1.4 oil immersion objective.
Results
Centrobin is phosphorylated after UV radiation
We first examined whether centrobin undergoes post-translational modification in response to DNA damage. HeLa cells were treated with different types of DNA-damaging agents (Figure 1(a)). As shown in Figure 1(a), a slower migrating band of centrobin appeared in UV-radiated cells, while we could not observe any change for other types of DNA-damaging agents. Immunoblots of phosphorylated H2AX (γ-H2AX) and Chk1 (S345) were used to show the extent of DNA damage induced under the indicated conditions. γ-H2AX was induced at approximately similar levels in UV-, CPT- and HU-treated cells, but the centrobin band shift was only detected in UV-irradiated cells. To determine whether the centrobin band shift was due to protein phosphorylation, lysates were treated with λ phosphatase (Figure 1(b)). Lambda phosphatase treatment of centrobin from either untreated or UV-treated cells reverted the shifted band to non-shifted position, indicating that the centrobin band shift seen in UV-treated cells appeared to result from phosphorylation. As a positive control, we confirmed that the phosphorylated form of BRCA1 disappeared in this condition (lower panel in Figure 1(b)). To determine the kinetics of centrobin phosphorylation, HeLa cells were irradiated with 30 J/m2 of UV, and the cells were harvested at indicated time points (Figure 1(c)). γ-H2AX increased progressively during the time course. Centrobin phosphorylation was detectable after 3 h of UV radiation, and the phosphorylation signal lasted until 9 h post-UV radiation then decreased with the appearance of cell death (data not shown).
Figure 1.
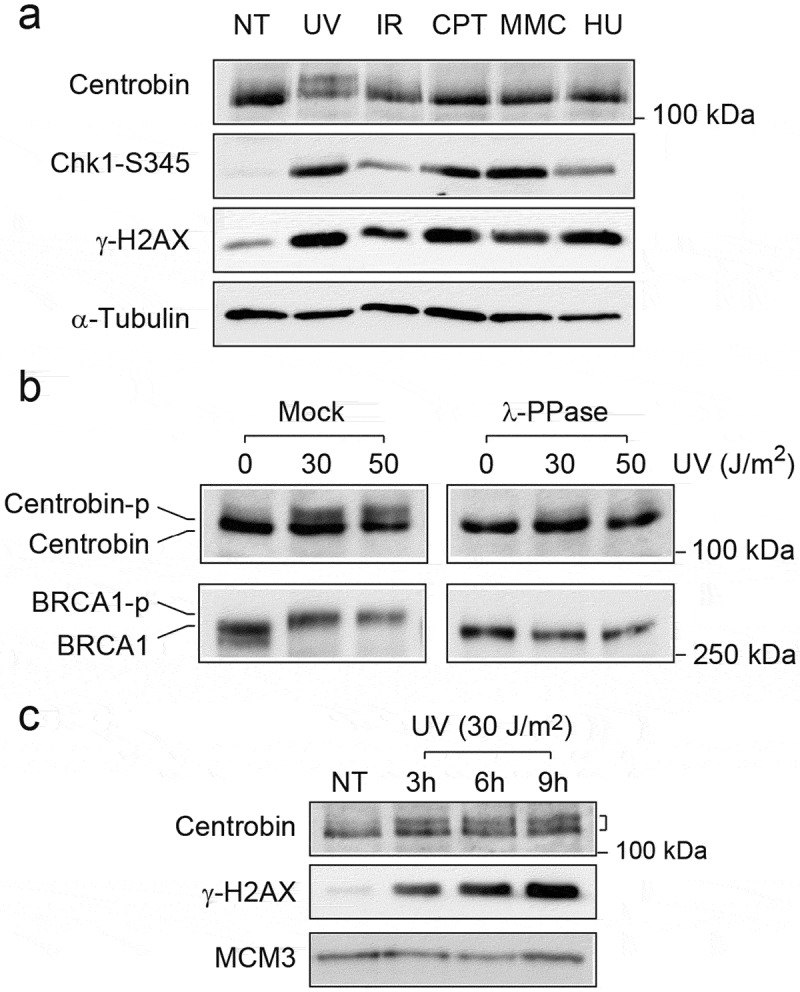
Centrobin is phosphorylated in response to UV radiation. (a) HeLa cells were either left untreated or treated with ultraviolet (UV, 30 J/m2, 6 h), ionizing radiation (IR, 10 Gy, 6 h), mitomycin C (MMC, 50 ng/ml, 16 h), camptothecin (CPT, 100 nM, 16h), or hydroxyurea (HU, 2 mM, 16 h). Cells were lysed and immunoblotted with the indicated antibodies. (b) HeLa cells were either left untreated or treated with UV (30 and 50 J/m2, 6 hr). Cell lysates were incubated with lambda phosphatase and immunoblotted with the indicated antibodies. (c) HeLa cells were exposed to UV (30 J/m2) in a time-course. Cells lysates were immunoblotted with the indicated antibodies.
Centrobin phosphorylation is detected in nuclear matrix-enriched fraction
Many DNA repair and checkpoint proteins change its subcellular localization in response to DNA damage and form intranuclear foci at DNA damage sites [19]. To examine whether subcellular localization of centrobin changes after DNA damage, we initially used immunofluorescence analysis (Figures S1 and S2). However, we did not observe any significant differences in subcellular localization between untreated and UV-treated cells. Alternatively, we used a biochemical fractionation method to examine the subcellular localization of centrobin following DNA damage (Figure 2(a)). Cells were lysed in a low salt and detergent-containing CSK buffer (100 mM NaCl and 0.1% Triton X-100), yielding a low salt-extractable fraction containing cytoplasmic and nucleoplasmic proteins (S) and a remaining pellet containing proteins stably bound to nuclear structures, e.g. chromatin and nuclear matrix (P) (Figure 2(b)). Only non-phosphorylated centrobin was detected in both S and P fractions in untreated cells. Following UV exposure, the phosphorylated form of centrobin was found specifically in P fraction. The whole-cell pellet was also suspended in hypotonic buffer and soluble cytoplasmic proteins (PNL) were separated in the first fraction (Figure 2(c)). By the addition of NaCl (100 mM) and detergent (0.1% Triton X-100) to the remaining pellet, soluble nuclear components were released (NS 100). Subsequent addition of 500 mM NaCl to the resulting pellet released DNA-binding proteins from DNA (NS 500), leaving the complete matrix fraction in the final pellet. Unexpectedly, centrobin was found in PNL at very low levels and ~50% of centrobin was detected in NS 100 fractions, while the majority of α- and γ-tubulin were detected in PNL and NS 100 fractions. After further extraction with 500 mM NaCl, the rest of centrobin (~50%) still remained in the pellet, regardless of its phosphorylation state, suggesting centrobin is stably associated with certain stable nuclear structures such as nuclear envelope or matrix. To further analyze its subcellular localization (Figure 2(d)), cell pellets were lysed with 100 mM NaCl CSK buffer, and the remaining pellets (P) after centrifugation were subsequently incubated with DNase I. As expected, chromatin-bound MCM3 was mostly released into the supernatant, whereas centrobin was not released. Previously, it has been shown that after extraction with 2 M NaCl, matrix proteins that are not part of the core filament network localize in the supernatant, whereas the core matrix fraction remains in the pellet [20]. The remaining pellet after DNase I digestion was further extracted with 2M NaCl. As a result, significant amounts of centrobin were released into the supernatant, but were still present in the final pellet, while Histone H3 extracted with 2M NaCl was mostly detected in the supernatant, further confirming the association of centrobin with the nuclear matrix. Another centrosome proteins, such as γ-tubulin and centrin-2 [21], were still detectable in the final pellet, suggesting that centrosome proteins localize to nuclear-matrix fraction. Collectively, these results suggest that centrobin is part of the nuclear matrix or strongly interacts with its constituents, regardless of centrobin phosphorylation state, and that a portion of nuclear matrix-associated centrobin is selectively phosphorylated after UV radiation.
Figure 2.
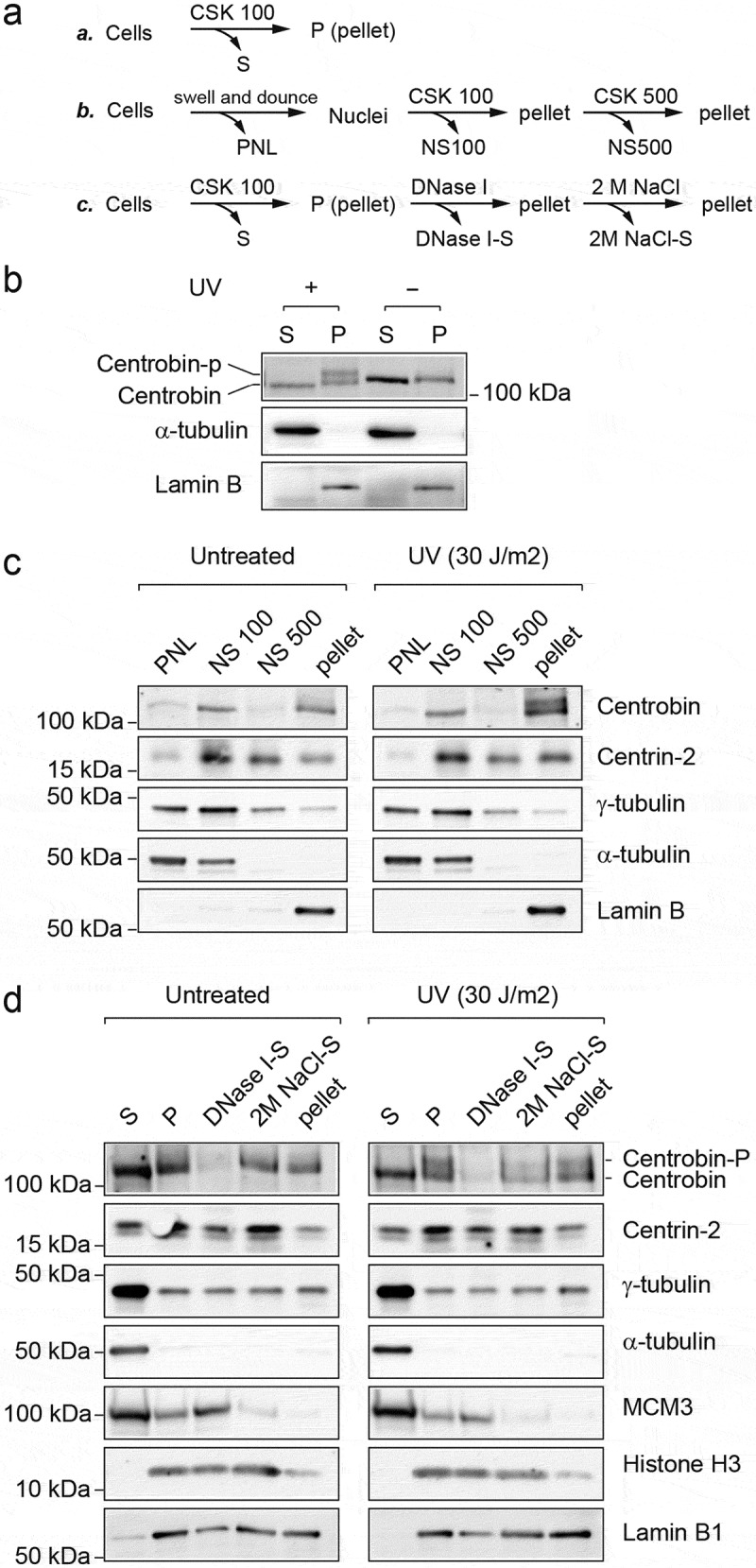
Phosphorylation of centrobin is detected in the nuclear-matrix fraction. (a) Schematic diagrams of the fractionation procedures described in (b–d). (b–d) HeLa cells were either left untreated or treated with UV (30 J/m2, harvested at 6 hr after irradiation). (b) Cells were fractionated into the cytosol and nucleoplasmic fraction (S) and the chromatin-containing fraction (P) with 100 mM CSK buffer. (c) Cells were fractionated into nuclei and postnuclear lysate (PNL). The nuclei were further fractionated into NS100, NS500, and the final pellet. (d) Cells were fractionated into S and P. “P” was incubated with DNase I for 30 min at room temperature and the supernatant was designated DNase I-S. The remaining insoluble material was further extracted with 2 M NaCl CSK buffer, and the supernatant was designated 2M NaCl-S. The final pellet was re-suspended in PBS buffer. The volume of extraction buffers used in all the above steps was kept the same. As a result, each lane represents protein fractions derived from equivalent number of cells, except that final pellets were suspended in x1/2 vol. These fractions were subjected to SDS-PAGE followed by immunoblotting with the indicated antibodies.
ATR is required for UV-induced phosphorylation of centrobin
ATR is activated by UV-induced DNA damage and replication stress, while ATM is activated by IR [19,22–24]. Treating cells with 5 mM caffeine, an inhibitor of ATR/ATM, 2 h prior to UV radiation inhibited the phosphorylation of centrobin (Figure 3(a)), indicating that ATR or ATM is responsible for centrobin phosphorylation. Next, the phosphorylation of centrobin was further analyzed after depletion of ATR and ATM by siRNA (Figure 3(b)). After depletion of either ATR or ATM, the phosphorylation of CHK1 at Ser345 was significantly reduced, indicating an efficient inhibition of ATR and ATM activity. In ATR-depleted cells, there was a significant reduction of centrobin phosphorylation compared to control siRNA-treated cells, whereas depletion of ATM showed little effect on UV-induced centrobin phosphorylation. A similar reduction was observed for combined depletion of ATM and ATR. Taken together, these results indicate that UV-induced phosphorylation of centrobin is dependent on ATR.
Figure 3.
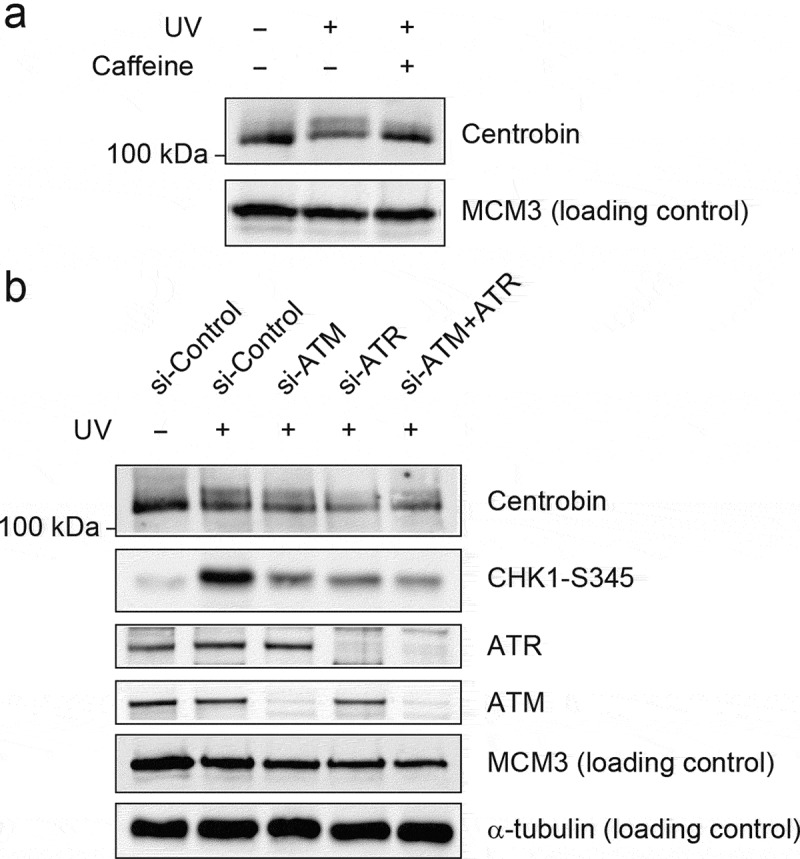
UV-induced centrobin phosphorylation is dependent on ATR activity. (a) HeLa cells pretreated with and without caffeine (5 mM) for 1 h prior to exposing them UV (30 J/m2). Cells were collected at 6 h after UV radiation (30 J/m2) and then fractionated the same as described in Figure 2(b). The chromatin-enriched fractions (P) were immunoblotted with the indicated antibodies. (b) Hela cells were transfected with the indicated siRNAs for 72 hr then exposed to UV (30 J/m2). Cells were collected at 6 h after UV radiation and then fractionated same as described in Figure 2(b). The “P” fractions were immunoblotted with the indicated antibodies.
Centrobin is required for cell survival after UV radiation
Next, to explore the role of centrobin in the DDR, centrobin was depleted using specific siRNAs. As expected, the levels of centrobin were greatly reduced in the centrobin knockdown cells using either siRNA #1 or #2 (Figure S3). To address the impact of reduced expression of centrobin in UV-induced checkpoint activation, immunoblotting was performed (Figure 4(b)). After depletion of centrobin, reduced G1/S population and concomitant increase in S and G2/M population was observed (Figure 4(a)). At 6 h after UV radiation, change in cell cycle distribution was less obvious, but both control and centrobin-depleted cells exhibited reduced G1/S population compared with untreated cells. The levels of CHK1 (Ser345), CHK2 (Thr68), and RPA2 (Ser33 and Ser4/8) phosphorylation induced by UV radiation were comparable between cells treated with control siRNA and centrobin siRNA, indicating that centrobin is dispensable for DNA damage checkpoint activation. Rather, slightly increased levels of CHK1 S345 and RPA2 Ser33 were observed, suggesting that depletion of centrobin may induce a higher amount of DNA damage following UV radiation. Next, we evaluated the relevance of centrobin for cell survival following treatment with UV radiation (Figure 4(c,d)). Centrobin-depleted cells exhibited a significant increase in UV sensitivity compared with control cells (Figure 4(c)). In the absence of UV exposure, the level of γ-H2AX was slightly higher in centrobin-depleted cells compared to control cells (Figure 4(d)). Upon UV radiation, significant increases in γ-H2AX levels were observed at 12 and 24 h in centrobin-depleted cells. These results suggest that DNA damage was not repaired efficiently in centrobin-depleted cells, leading to the accumulation of unrepaired DNA lesion and decreased cell viability.
Figure 4.
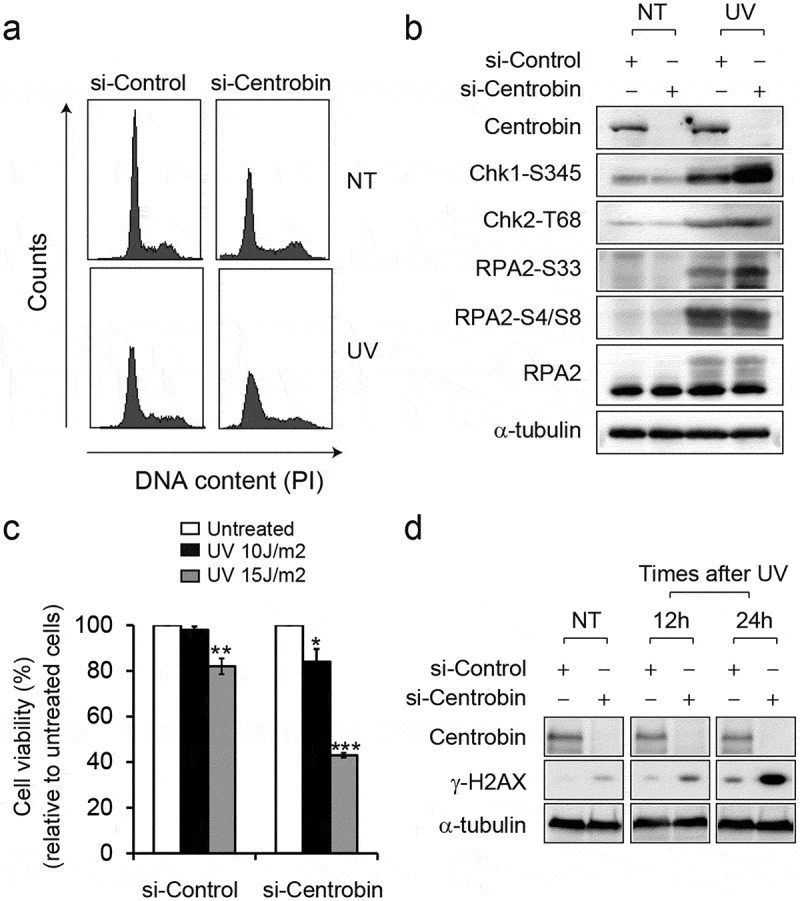
Depletion of centrobin causes the hypersensitivity to UV radiation. (a) HeLa cells were transfected with the indicated siRNAs for 72 hr then either left untreated or treated with UV (30 J/m2). Cells were collected at 6 h after treatment and cell cycle distribution was analyzed by flow cytometry. Representative histograms for cell cycle distribution are presented. (b) HeLa cells were transfected with the indicated siRNAs for 72 hr then either left untreated or treated with UV (30 J/m2). Cells were collected at 6 h after treatment and then analyzed by immunoblotting with the indicated antibodies. (c) HeLa cells were transfected with the indicated siRNAs for 60 hr then either left untreated or treated with UV (10 and 15 J/m2). After 24 h treatment, the viable cells were counted by trypan blue staining. Data are expressed as a percentage of control siRNA. Each experiment was performed in triplicate and repeated in three different sets of tests. *P< 0.05, **P< 0.01, ***P< 0.001 compared to the untreated cells transfected with indicated siRNAs. (d) HeLa cells were transfected with the indicated siRNAs for 60 hr then either left untreated or treated with UV (15 J/m2). Cells were collected 12-hr intervals after UV radiation and analyzed by immunoblotting with the indicated antibodies.
Depletion of centrobin impairs DNA damage-induced microtubule stabilization
Recently it has been reported that microtubule-depolymerizing agents, such as vincristine, increase the toxicity of DNA-damaging agents by disrupt intracellular trafficking of DNA repair proteins [4]. Given that centrobin binds to microtubules and this interaction is required for microtubule polymerization and stabilization [8,11,12], one can speculate that the DNA repair defect by centrobin depletion may be partly due to defective microtubule stability. To test this hypothesis, we examined the microtubule stability in centrobin-depleted cells by analyzing microtubule resistance to nocodazole treatment after UV exposure (Figure 5(a,b)). In the absence of UV radiation, very few nocodazole-resistant microtubules were detected in either control, centrobin or ATR siRNA-treated cells and there were no significant differences between the groups. Following UV radiation, control siRNA-transfected cells displayed an increased number of nocodazole-resistant microtubules and enhanced α-tubulin lysine 40 acetylation (αTu-Ac) around the nucleus, indicative of increased microtubule stability [25]. In contrast, in centrobin-depleted cells, considerably reduced number of nocodazole-resistant microtubules were detected and these were very short and dispersed throughout the cells. Similarly, ATR depletion resulted in defective UV-induced microtubule stabilization, as observed in centrobin-depleted cells. Taken together, these results suggest that centrobin is required for UV-induced microtubule stabilization and this may promote UV damage repair.
Figure 5.
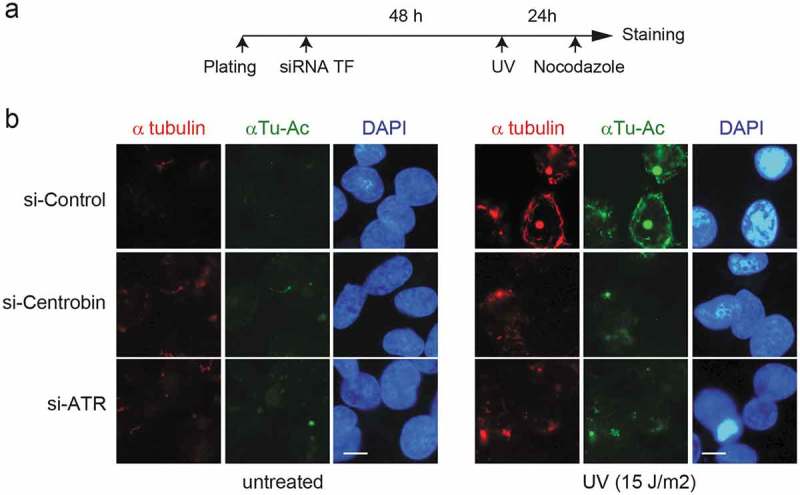
Depletion of centrobin has a defect in UV-induced microtubule stabilization. (a) Experimental scheme. HeLa cells were transfected with the indicated siRNAs. At 48 hr after transfection, cells were either left untreated or treated with 15 J/m2 UV and then incubated for 20 h. Before staining, siRNA-treated HeLa cells were treated with 2 μM nocodazole for 45 min. (b) Representative immunofluorescence (IF) images of siRNA-treated HeLa cells stained for α-tubulin (red), acetylated α-tubulin (green), and DNA (blue). Scale bar, 10 μM.
Centrobin depletion causes an impairment in HR repair
While UV irradiation is not expected to directly cause DNA double-strand breaks (DSBs), replication-associated one-ended DSBs could accumulate when elongating forks encounter UV lesions [26,27]. Moreover, centrobin has been known to interact with BRCA2 tumor suppressor [9,10], a key regulator of DSB-induced homologous recombination (HR) repair. Therefore, we examined the effect of centrobin depletion in HR repair (Figure 6). The direct repeat green fluorescent protein (DR-GFP) assay is a commonly used assay to measure HR in living cells [28,29]. Centrobin was depleted in U2OS cells integrated with DR-GFP reporter substrate in which expression of I-SceI endonuclease generates a DSB that when repaired by HR restores GFP expression, and GFP fluorescence then can be measured by flow cytometry (Figure 6(a,b)). Depletion of either BRCA2 or PALB2 showed an expected reduction in HR frequency. Whereas, DR-GFP/U2OS cells treated with control siRNAs displayed efficient HR repair, approximately 40% of reduction in HR efficiency was observed after centrobin depletion. Depletion of centrin-2, another centriole protein, resulted in a moderate reduction (~20%). For the rescue experiment, we generated DR-GFP U2OS cells that stably expressing siRNA-resistant wild-type centrobin cDNA (Figure 6(c–e)). The diminished centrobin staining in centrobin-depleted cells was completely restored by forced expression of wild-type centrobin (Figure 6(c)) and HR deficiency was efficiently recovered (Figure 6(d,e)). The cell cycle distribution between DR-GFP U2OS cells transfected with control siRNA and centrobin siRNA was comparable (Figure S4), suggesting that the observed effect is not due to an indirect effect of cell cycle change. Taken together, these results indicate that centrobin is required for an efficient HR repair.
Figure 6.
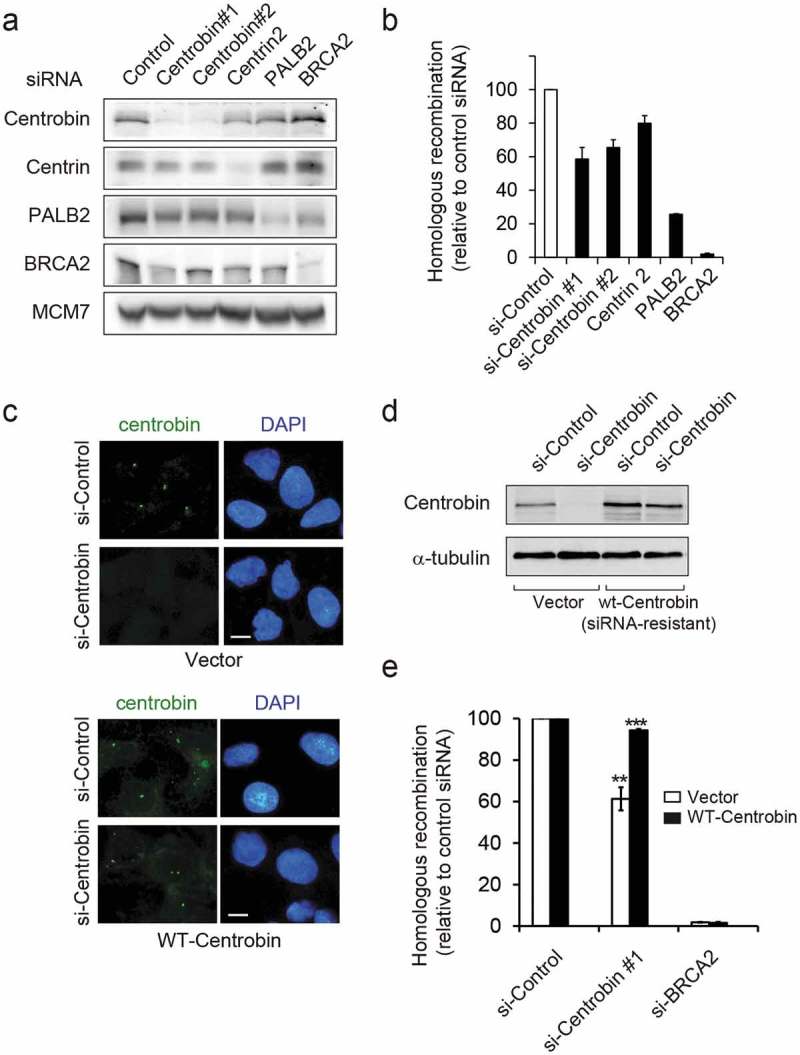
Centrobin-depleted cells have defects in homologous recombination repair. (a,b) DR-GFP U2OS were transfected with the indicated siRNAs for 24 hr and then transfected with an I-SceI expression plasmid to induce double-stranded breaks. Flow cytometric analysis of GFP+ cells was carried out 48 hr after I-SceI transfection. (a) At 48h after I-SceI transfection, cells were collected and immunoblotted with the indicated antibodies. (b) Quantification of the I-SceI -induced HR frequency. HR assay were performed in triplicate and repeated N = 6 times. Values are normalized for the transfection efficiency and are displayed as mean ± SEM GFP+ frequencies relative to that of control siRNA-treated cells. (c–e) DR-GFP U2OS cells stably expressing either empty vector or siRNA-resistant centrobin were transfected with the indicated siRNAs, and then 24hr later transfected with I-SceI plasmid. The cells were grown for 2 days after transfection and then analyzed. (c) Representative IF images of siRNA-treated DR-GFP U2OS cells stained for centrobin (green), and DNA (blue). Scale bar, 10 μM. (d) Cells lysates were immunoblotted with the indicated antibodies. (e) Quantification of the I-SceI -induced HR frequency. HR assay were performed in triplicate and repeated N= 3 times. Values are normalized for the transfection efficiency and are displayed as mean ± SEM GFP+ frequencies relative to that of each control siRNA-treated cell line. **P< 0.01, ***P< 0.001, compared to control siRNA-transfected each cell line.
Discussion
The DDR is a network of cellular pathways that sense, signal, and repair DNA lesions. Many checkpoint or DNA repair proteins including ATR, CHK1, BRCA1, and Rad51 have been shown to localize in the nucleus and the centrosomes [30,31], implicating the existence of a molecular crosstalk between two organelles following DNA damage.
In the present study, we propose that centrobin plays a role during DDR, which is required for genotoxic resistance following DNA damage. Centrobin is phosphorylated in response to UV radiation in an ATR-dependent manner (Figures 1–3). Interestingly, we did not observe phosphorylation-induced centrobin band shifts for other different types of DNA-damaging agents (Figure 1). However, because not all phosphorylation leads to any observable change in mobility shift, we cannot completely rule out the possibility of phosphorylation by other types of DNA-damaging agents without using anti-phospho specific antibodies.
As shown in Figure 2, phosphorylated centrobin was detected in DNAase I and high-salt resistant fraction, suggesting that centrobin phosphorylation may occur in nuclear matrix. Since it has been shown that centrosomes is closely associated with the nuclear envelope (NE) in interphase cells [32,33], we speculate that centrosomal centrobin might be phosphorylated by ATR. How does ATR phosphorylate nuclear matrix-bound centrobin in response to UV radiation? It has been well established that in response to DNA damage, ATR activation is dependent on its binding with ssDNA-RPA complex [22,34]. Recently, however, some studies support the notion of an alternative pathway for ATR activation in response to DNA damage [35]. ATR responds to membrane stress caused by external forces or altered membrane fluidity [36,37]. Under these stress conditions, ATR relocates into the nuclear envelope and phosphorylates Chk1 locally [36]. Following UV radiation, deterioration of the cell membrane can cause increased membrane permeability [38]. In this scenario, we think that ATR may relocate into the nuclear envelope and phosphorylate centrobin.
A recent study in yeast showed that DDR induces microtubule stabilization in an ATR-dependent manner [3]. Consistently, we observed defective microtubule stabilization following ATR depletion (Figure 5). Given that centrobin-depleted cells also displayed impaired microtubule stabilization and increased sensitivity to UV exposure (Figure 4), we speculate that centrobin might function as a DDR target for microtubule reorganization in response to DNA damage. Moreover, we found that centrobin depletion had a significant reduction in HR efficiency. Since centrobin interacts with BRCA2 tumor suppressor [9,10], a key regulator of DNA double-strand break (DSB) repair, it would be interesting to investigate the effect of centrobin depletion on BRCA2 stability or subcellular localization, which may clarify how centrobin deficiency causes HR defect.
Although our results suggest the potential role of centrobin in DDR, it is not clear whether and how centrobin phosphorylation is involved in this process. Nine potential ATR/ATM substrate sites (SQ/TQ) are present in centrobin. The Serine 781 was identified as a putative phosphorylation site of the ATM/ATR following IR irradiation [14], however, its physiological function remains largely unknown. The Serine 36 has been shown to be phosphorylated by NEK2, a cell cycle-regulated serine/threonine kinase, during interphase that suppresses the microtubule-stabilizing activity of centrobin [8,12]. Because NEK2 activity is inhibited by DNA damage such as IR irradiation [39,40], Serine 36 could become available for phosphorylation by another kinase such as ATR following DNA damage. Thus, it will be very interesting to investigate if these phosphorylation sites are directly phosphorylated by ATR or ATM kinases in vivo and to determine their functional significance in DDR-related activities of centrobin. In the present study, some links between centrobin and DDR are proposed, however, additional studies are required to understand the exact role of centrobin in the DDR.
Funding Statement
This work was supported by the Ministry of Education [2017R1D1A1A09]; Ministry of Science (KR) [2011-0030132].
Acknowledgments
We are grateful to Jin Ki Jung and Seok-Won Jang who started this project. We thank I Park for valuable reagents and helpful discussion. This work was supported by Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (2017R1D1A1A09). This work was partly supported by the National Research Foundation of Korea (Medical Research Council for Gene Regulation) funded by the Ministry of Science, ICT and Future Planning (2011-0030132).
Author contributions
N.R. conducted experiments; J.M.K. conceived and designed the experiments: N.R. and J.M.K. wrote the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
References
- [1].Mitchison T, Kirschner M.. Dynamic instability of microtubule growth. Nature. 1984;312:237–242. [DOI] [PubMed] [Google Scholar]
- [2].Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. [DOI] [PubMed] [Google Scholar]
- [3].Graml V, Studera X, Lawson JLD, et al. A genomic Multiprocess survey of machineries that control and link cell shape, microtubule organization, and cell-cycle progression. Dev Cell. 2014;31:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Poruchynsky MS, Komlodi-Pasztor E, Trostel S, et al. Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc Natl Acad Sci U S A. 2015;112:1571–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kimble M, Kuriyama R. Functional components of microtubule-organizing centers. Int Rev Cytol. 1992;136:1–50. [DOI] [PubMed] [Google Scholar]
- [6].Conduit PT, Wainman A, Raff JW. Centrosome function and assembly in animal cells. Nat Rev Mol Cell Biol. 2015;16:611–624. [DOI] [PubMed] [Google Scholar]
- [7].Gudi R, Zou C, Li J, et al. Centrobin-tubulin interaction is required for centriole elongation and stability. J Cell Biol. 2011;193:711–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jeong Y, Lee J, Kim K, et al. Characterization of NIP2/centrobin, a novel substrate of Nek2, and its potential role in microtubule stabilization. J Cell Sci. 2007;120:2106–2116. [DOI] [PubMed] [Google Scholar]
- [9].Zou C, Li J, Bai Y, et al. Centrobin: a novel daughter centriole-associated protein that is required for centriole duplication. J Cell Biol. 2005;171:437–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jeffery JM, Urquhart AJ, Subramaniam VN, et al. Centrobin regulates the assembly of functional mitotic spindles. Oncogene. 2010;29:2649–2658. [DOI] [PubMed] [Google Scholar]
- [11].Lee J, Jeong Y, Jeong S, et al. Centrobin/NIP2 is a microtubule stabilizer whose activity is enhanced by PLK1 phosphorylation during mitosis. J Biol Chem. 2010;285:25476–25484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Park J, Rhee K. NEK2 phosphorylation antagonizes the microtubule stabilizing activity of centrobin. Biochem Biophys Res Commun. 2013;431:302–308. [DOI] [PubMed] [Google Scholar]
- [13].Shin W, Yu NK, Kaang BK, et al. The microtubule nucleation activity of centrobin in both the centrosome and cytoplasm. Cell Cycle. 2015;14:1925–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. [DOI] [PubMed] [Google Scholar]
- [15].Xia B, Sheng Q, Nakanishi K, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–729. [DOI] [PubMed] [Google Scholar]
- [16].Kim JM, Kee Y, Gurtan A, et al. Cell cycle-dependent chromatin loading of the Fanconi anemia core complex by FANCM/FAAP24. Blood. 2008;111:5215–5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Kim JM, Parmar K, Huang M, et al. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell. 2009;16:314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Khawaja S, Gundersen GG, Bulinski JC. Enhanced stability of microtubules enriched in detyrosinated tubulin is not a direct function of detyrosination level. J Cell Biol. 1988;106:141–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, et al. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. [DOI] [PubMed] [Google Scholar]
- [20].He DC, Nickerson JA, Penman S. Core filaments of the nuclear matrix. J Cell Biol. 1990;110:569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Salisbury JL, Suino KM, Busby R, et al. Centrin-2 is required for centriole duplication in mammalian cells. Curr Biol. 2002;12:1287–1292. [DOI] [PubMed] [Google Scholar]
- [22].Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. [DOI] [PubMed] [Google Scholar]
- [24].Melo J, Toczyski D. A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol. 2002;14:237–245. [DOI] [PubMed] [Google Scholar]
- [25].Hammond JW, Cai D, Verhey KJ. Tubulin modifications and their cellular functions. Curr Opin Cell Biol. 2008;20:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shibata A, Jeggo PA. DNA double-strand break repair in a cellular context. Clin Oncol (R Coll Radiol). 2014;26:243–249. [DOI] [PubMed] [Google Scholar]
- [27].Ward JD, Barber LJ, Petalcorin MI, et al. Replication blocking lesions present a unique substrate for homologous recombination. Embo J. 2007;26:3384–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pierce AJ, Hu P, Han M, et al. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Pierce AJ, Johnson RD, Thompson LH, et al. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shimada M, Komatsu K. Emerging connection between centrosome and DNA repair machinery. J Radiat Res. 2009;50:295–301. [DOI] [PubMed] [Google Scholar]
- [31].Zhang S, Hemmerich P, Grosse F. Centrosomal localization of DNA damage checkpoint proteins. J Cell Biochem. 2007;101:451–465. [DOI] [PubMed] [Google Scholar]
- [32].Burakov AV, Nadezhdina ES. Association of nucleus and centrosome: magnet or velcro? Cell Biol Int. 2013;37:95–104. [DOI] [PubMed] [Google Scholar]
- [33].Bolhy S, Bouhlel I, Dultz E, et al. A Nup133-dependent NPC-anchored network tethers centrosomes to the nuclear envelope in prophase. J Cell Biol. 2011;192:855–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. [DOI] [PubMed] [Google Scholar]
- [35].Kidiyoor GR, Kumar A, Foiani M. ATR-mediated regulation of nuclear and cellular plasticity. DNA Repair (Amst). 2016;44:143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kumar A, Mazzanti M, Mistrik M, et al. ATR mediates a checkpoint at the nuclear envelope in response to mechanical stress. Cell. 2014;158:633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhang XH, Zhao C, Ma ZA. The increase of cell-membranous phosphatidylcholines containing polyunsaturated fatty acid residues induces phosphorylation of p53 through activation of ATR. J Cell Sci. 2007;120:4134–4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Bowden GT. Prevention of non-melanoma skin cancer by targeting ultraviolet-B-light signalling. Nat Rev Cancer. 2004;4:23–35. [DOI] [PubMed] [Google Scholar]
- [39].Fry AM, O’Regan L, Sabir SR, et al. Cell cycle regulation by the NEK family of protein kinases. J Cell Sci. 2012;125:4423–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mi J, Guo C, Brautigan DL, et al. Protein phosphatase-1alpha regulates centrosome splitting through Nek2. Cancer Res. 2007;67:1082–1089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.