

Graphical Abstract
1. INTRODUCTION
Bioanalytical sensors capable of accurately measuring in vivo chemical dynamics of important physiological analytes have been transformative on several fronts, including disease/health management and improved understanding of biological processes. The glucose biosensor, which over the past 35 years has evolved from initial proof-of-concept to an indispensable technology in diabetes management, serves as a paragon of the utility of implantable chemical sensors.1 Care for critically-ill patients (i.e., those in the intensive care unit; ICU) has benefited from monitoring of several analyte classes that hold prognostic/diagnostic value, including ions (e.g., Na+, K+), gases (O2, CO2), and metabolites (glucose, lactate).2 A large number of electrochemical sensors have been used to study in vivo dynamics of neurotransmitters, biological gases, and metabolites for more than 40 years.3 The vast majority of sensors across these areas rely on miniaturized electrochemical or, to a lesser extent, optical probes that meet strict benchtop analytical performance merits required for such measurements (e.g., dynamic range, response time, analyte selectivity). Unfortunately, sensor function is often impeded partially or completely for in vivo devices due to the implant-initiated host response.
The in vivo environment that a sensor encounters changes with protein adsorption and subsequent cell adhesion. Although the specific progression of biochemical events that occur in the various physiological fluids and tissues is diverse, a shared trait is the ability to recognize the in vivo sensor as foreign and mount a prolonged cellular attack. The end-stage of the host response is remarkably similar, regardless of the location (e.g., blood, tissue), culminating in isolation of the implant by formation of thrombi or scar tissue. For many inactive implants (e.g., fixation devices), moderate degrees of cellular infiltration and isolation are tolerated without severe detriment to the implant’s intended purpose, and they are thus deemed biocompatible.4 In contrast, chemical sensors must persist in vivo under a changing physiological environment while providing stable signals that accurately follow the concentration of an analyte. Even moderate cellular presence or isolation will alter analyte transport in the region surrounding the sensor such that the surface concentration of an analyte may not reflect bulk blood/tissue levels.5 As such, this seemingly insurmountable host response compromises both the accuracy and usable lifetime of all in vivo sensors.
Ratner previously defined biocompatibility as the ability of a material to guide a more passive response in which the sensor is not wholly recognized as a foreign body.6 Many researchers have worked to develop strategies that might reduce the various biological reactions to an in vivo sensor with the goal of improving analytical performance. Unfortunately, a critical void that has not been addressed in the literature is whether mitigating the host response generates perceptible improvements to the in vivo accuracy or usable lifetime of such devices. In this regard, the “analytical biocompatibility” of new chemical strategies should be used to describe improvements to in vivo sensor performance directed at reducing the host response. In this review article, we highlight the individual host responses, as they relate to in vivo chemical sensor performance, and discuss recent approaches for improving sensor analytical biocompatibility.
2. APPLICATIONS OF IN VIVO CHEMICAL SENSORS
Many scientists and clinicians would welcome the ability to accurately monitor analytes and biochemical reactions in vivo. Implantable sensors capable of continuous measurement for biological gases (e.g., O2, CO2), ions (Na+, K+, Ca2+), glucose, and lactate represent impactful technologies for use in bedside and at-home disease management. Sensors for glucose, lactate, glutamate, and catecholamines are routinely implanted and operated in central nervous system tissue for understanding the roles of metabolite and neurotransmitter dynamics in physiological processes. Given that the field of chemical sensors is vast and this review article is focused on recent advances in analytical biocompatibility, coverage of novel sensors designs/applications will inevitably be incomplete. General sensing strategies including neurotransmitter detection via fast-scan cyclic voltammetry,7 use of microdialysis probes,8–15 and aptamer-based biosensors16 will not be covered in great detail. Nevertheless, these methods similarly experience degradation of in vivo performance associated with the host response, and notable examples are highlighted in the biocompatibility strategies section.
2.1. Ion sensors
Electrolytes are involved in multiple physiological functions. Abnormal blood ion concentrations are commonly associated with increased morbidity and mortality in the intensive-care unit.17 The most widely-utilized ion sensors are potentiometric devices that have been modified with recognition chemistries (ionophores) that selectively complex with target ions to generate an electromotive force.18 The various recognition agents (e.g., tridodecyl amine for H+, calcium ionophore II, valinomycin for K+) are typically immobilized in an inert, viscous liquid polymer matrix (polyvinylchloride) mixed with water-immiscible plasticizers (bis(2-ethylhexyl)sebacatate, bis(2-ethylhexyl)phthalate) and a suitable lipophilic counterion (e.g., potassium tetrakis(chlorophenyl)borate as an anionic site).19–20 Optical ion sensors have also been described that possess inherent advantages over potentiometric sensors, including ease of miniaturization and ratiometric detection.21 Optical detection schemes rely on co-immobilization of an ionophore (the recognition agent for the ion of interest) and a suitable pH indicator in an inert polymer. Migration of the analyte into the polymer either induces expulsion of protons from the membrane (for cations) or co-migration of protons into the polymer, which in turn will trigger a change in the absorbance or fluorescence properties of the pH indicator.21 Disadvantages of the optical systems include photobleaching of the pH indicator and slower response to changing analyte concentrations than their potentiometric sensor counterparts.
2.2. Biological gas sensors
2.2.1. Oxygen
Sensors for molecular oxygen (O2) have been fabricated in both electrochemical (amperometric) and optical (fluorescence) detection configurations22–25 and integrated within intravascular (IV) catheters for detection in blood.2,23,25 Electrochemical O2 sensors generally rely on O2 reduction on platinum working electrodes that are immersed in an electrolyte solution and separated from the external medium by a thin, O2 permeable polymer membrane (e.g., silicone rubber).23–24 Although housing the electrode in an electrolyte solution places constraints on the minimum size of the resulting device (>0.3 mm), larger dimensions are generally tolerable for IV use. A large reduction potential is required for O2 detection (−0.65 or −0.70 V vs. silver-silver chloride (Ag|AgCl, 3 M KCl)), yet electrochemical O2 sensors do not generally suffer from response to interferents due to low permeability of most species through the hydrophobic polymer membrane. As is the case with the optical ion sensors, optical O2 sensors display certain advantages over traditional amperometric sensors including ratiometric detection.2,21,25 Adopting an optical detection scheme also eliminates the need for the electrode filling solution and may be advantageous for minimizing the physical dimensions of the sensor. Optical sensor designs rely on fluorescence quenching of immobilized indicators (e.g., pyrene/perylene) by paramagnetic oxygen, resulting in a decrease in fluorescence emission intensity.25
2.2.2. Carbon dioxide
Patients with poor lung function or those experiencing respiratory failure are unable to efficiently remove CO2 from the blood, thereby resulting in an increase in blood CO2 levels.26 Electrochemical sensors for CO2 monitoring are almost exclusively variants of the Stow-Severinghaus electrode.27 The device itself is a pH sensor immobilized in a bicarbonate buffer solution that is isolated from the external (sample) solution by a CO2-permeable polymeric membrane (e.g., Teflon). Equilibrium partitioning of CO2 into the bicarbonate buffer increases the proton activity in solution, which is measured by the pH sensor.2,27 Despite wide use, analytical performance issues associated with the Stow-Severinghaus electrode include slow response times (>1 min), logarithmic response/poor sensitivity at low pCO2, and susceptibility to interference from other gases (e.g., hydrogen sulfide).28 Xie and Bakker recently reported an alternative CO2 electrode design that alleviates several of these issues.28 The device makes use of dual potentiometric H+ and carbonate ion sensors to determine the activities of both ions, which are used to calculate the pCO2 in the sample solution. However, this electrode design has yet to be adapted for IV use.
2.2.3. Nitric oxide
Nitric oxide (NO) is a blood-soluble, free radical gas produced endogenously from a group of isoenzymes called nitric oxide synthases (NOS).29 Since the discovery that NO was the primary factor regulating endothelial relaxation/vascular tone in 1987,30 NO has been implicated as a key molecule in many physiological processes including inflammation31–33 and neurotransmission.34 Burgeoning evidence suggests NO measurement may be useful in the ICU for sepsis monitoring.35 Thus, demand for analytical technologies that facilitate accurate, real-time NO detection is increasing.
The majority of NO-selective sensors are electrochemical devices that rely on oxidation of NO at common electrode materials (e.g., platinum, gold, glassy carbon, carbon fiber).36 Although NO detection is also possible via electroreduction, interference by O2 reduction precludes reliable NO determination in biological media.36 Nevertheless, the NO oxidation method is also susceptible to interference by a number of endogenous molecules (e.g., l-ascorbate, nitrite, dopamine) due to the high required working potential (+0.7–0.9 V vs. Ag|AgCl). Polymer membranes such as Nafion, Teflon, nitrocellulose, chloroprene, xerogels, and electropolymerized films are thus used to differentially permit NO permeation to the electrode surface over interfering species via size-exclusion and coulombic repulsion mechanisms.36–37 Alternatively, electrocatalysts have been used to shift the oxidation potential of NO to less positive values, thereby circumventing the oxidation of interfering analytes. To date, the most promising catalysts are metalloporphyrins and metallophthalocyanines that allow for ~0.15 V negative shifts in the NO oxidation peak potential and simultaneously amplify the current response up to 3-fold.36
In addition to selectivity, the sensor sensitivity, LOD, and dynamic range are critical analytical merits that must be carefully selected for the specific context of NO measurement as physiological concentrations vary greatly (i.e., picomolar to micromolar).38 The short half-life of NO (<10 s) and abundance of natural scavengers (O2, peroxides, superoxide, metalloproteins, etc.) further complicate accurate measurement and demand fast sensor response.39 As such, advances in NO detection technologies are still needed as the significance of NO and suitable measurement locations are further revealed.
2.3. Enzyme-based biosensors
2.3.1. Glucose
The ability to facilitate self-management of diabetes has resulted in extensive investigation of blood glucose monitoring strategies. Indeed, diabetics must self-monitor BG using discrete, non-implantable electrochemical glucometer technologies in order to maintain normoglycemic levels (4–7 mM). Unfortunately, infrequent sampling via discrete blood glucose measurement has not resulted in adequate disease management.40 Implantable continuous glucose monitoring (CGM) technologies have thus been proposed and developed for improved glucose management. CGM devices generally measure glucose levels in interstitial fluid of sub-dermal tissues. More recently, impetus for continuous glucose monitoring in the blood (i.e., in the critical-care setting) has increased as tight glycemic control has been shown to improve outcomes for ICU patients, even in the absence of diabetes.2
The most successful glucose sensor technologies involve electrochemical sensors that rely on immobilized glucose oxidase (GOx; generally isolated from the fungus Aspergillus Niger) which serves as a recognition element and transducer for glucose.41 Oxidation of glucose by GOx both decreases O2 (the enzyme co-substrate) concentrations and elevates hydrogen peroxide (product) levels, both of which may be monitored electrochemically. Peroxide oxidation is generally the more practical sensor configuration because electrochemical O2 reduction requires a separate electrode for monitoring background oxygen concentrations.41–50 However, a fully-implantable subcutaneous sensor has been developed for glucose detection via differential oxygen consumption.22 The majority of the hydrogen peroxide-detecting sensors are percutaneous devices that resemble either the “needle-type” sensor initially described by Shichiri et al43 or the variant coil-type sensor.45 Both sensors are of course suitable for in vivo use. Peroxide is oxidized amperometrically at platinum/iridum alloy working electrodes (+0.6 to +0.7 V vs. Ag|AgCl, 3 M KCl).
A drawback to hydrogen peroxide-detecting sensors is poor linear dynamic range due to limited availability of the enzyme co-substrate (O2), which is ~15 fold lower in concentration than glucose in tissue.51 This obstacle is generally overcome by application of an outer permselective polymer layer that differentially reduces glucose and oxygen diffusion to the immobilized enzyme, effectively balancing their concentrations at the electrode. The permselective layer is used to extend the sensor linear range (at the expense of sensor response time) to cover physiological blood/interstitial fluid glucose concentrations (1–18 mM). An additional disadvantage of hydrogen peroxide detection is the high required working potentials at which other, endogenous species (e.g., ascorbate) can also be oxidized. The issue of glucose selectivity has been largely addressed by deposition of additional polymeric layers (e.g., electropolymerized films, Nafion) that obstruct the fluxes of interferents to the sensor surface via charge or size exclusion mechanisms.52–53 Redox mediators (e.g., [Os(N,N’-dialkylated-2,2’-biimidazole)3]2+/3+, carbon nanotubes)52,54 have also been employed to directly shuttle electrons from GOx to the electrode surface, greatly reducing the required working potentials (−0.1 to +0.1 V vs. Ag|AgCl, 3 M KCl) and thereby obviating the need for additional permselective membranes.
Optical approaches to in vivo glucose sensing have also been described, most of which are luminescence-based and rely on glucose binding to a recognition agent (e.g., boronate derivatives, concanavalin A) that may also complex with other, structurally similar monosaccharides.55 A recently reported luminescence glucose sensor made use of GOx as the recognition agent.56 The enzyme was co-immobilized in a poly(2-hydroxyethylmethacrylate-co-acrylamide) hydrogel with a palladium (II) benzoporphyrin phosphor. This species undergoes luminescence quenching by oxygen. The phosphor is interrogated at 630 nm with changes in local oxygen altering the phosphorescence lifetime (measured at ca. 810 nm).56
2.3.2. Lactate
Lactate production is monitored as a marker for failure of oxidative energy metabolism and is normally associated with hypoxia.57 Continuous lactate biosensors have been developed for blood monitoring of ICU patients, where rising lactate levels may be used to indirectly monitor tissue oxygenation or sepsis.58 Tissue lactate is a key metabolic biomarker and is often measured in conjunction with glucose to provide indicators of energy metabolism for sports medicine and neurophysiology.1,8 With the exception that lactate oxidase is used as the immobilized enzyme, lactate biosensors are otherwise very similar in construction to glucose biosensors, utilizing amperometric oxidation of hydrogen peroxide on platinum/iridium alloys8,59 or enzyme-attached redox mediators.60 Normal blood lactate levels (0.5–1.0 mM) are generally much lower than glucose levels, with concentrations above 2 mM considered elevated—partially alleviating the strict oxygen dependence of the sensor.2 Regardless, sensor response up to 10 mM is required for monitoring extremes that may be encountered during organ failure (>5 mM) or intense periods of exercise.57
2.3.3. Glutamate
Occupying nearly 90% of the cortical synapses in the human brain, glutamate is the most abundant excitatory neurotransmitter with active roles in sleep and reward-seeking behavior.61 Transient excesses of extracellular glutamate cause neuronal apoptosis, with chronic abnormal levels linked to several neurodegenerative disorders.62 Glutamate biosensors are comparable to the previous glucose/lactate biosensors in that an enzyme—glutamate oxidase (GluOx)—is used to produce hydrogen peroxide for detection, but with two distinguishing features. First, in vivo detection of glutamate is more challenging due to low basal levels in the brain (~10 µM).61,63 Oxygen also serves as the co-factor for GluOx regeneration but it is generally not rate-limiting due to the inherently lower glutamate concentrations in vivo. As such, the sensor linear dynamic range is robust, obviating the need for additional glutamate flux-limiting membranes.63 Concomitantly, the temporal resolution of the sensors is improved (<2 s). A second feature is signal interference from redox active ascorbate, which can reach ~300 µM levels. To reduce ascorbate interference, Naylor et al. co-immobilized ascorbate oxidase with GluOx on the sensor surface.61 In this configuration, ascorbate is converted into non-electroactive species (i.e., dehydroascorbate and water) and, in combination with an underlying electropolymerized selectivity layer, yielded appropriate glutamate selectivity (300:1).61
3. HOST RESPONSE TO IMPLANTED CHEMICAL SENSORS
Validating in vitro sensor analytical function is necessary as a first test, as performance shortcomings in vitro often portend sensor failure in vivo. However, even well-functioning sensors are unlikely to operate as intended in vivo due to the host response. Indeed, glucose biosensors are often championed as the most successful implantable sensors, but it is clear that poor short-term accuracy and limited useable lifetime of these sensors have severely hindered their widespread use.52 Only two subcutaneous glucose sensors were listed as approved by the United States Food and Drug Administration (FDA) in 2014: Dexcom’s G5 and Medtronic’s Enlite. The Edwards GlucoScout, a minimally-invasive ex vivo glucose sensor, is FDA-approved for up to 3 d use in blood.2,64 The GlucoScout functions by intermittently drawing up 1.2 mL blood into a bypass loop and subsequently returning the volume of blood, followed by 6 mL of heparinized calibration solution.65 In this way, the sensor is calibrated immediately after blood glucose measurement. Despite success of the GlucoScout, the sensor itself is not actually implanted nor exposed to blood continuously. To date, no intravascular chemical sensor is approved for use in humans due to poor performance and thrombosis concerns. In research settings, chemical sensors are acutely-implanted to study the brain. The vast majority of reports are limited to <1 d study durations due to unpredictable sensor response at longer implant periods.
The different biological responses to implanted chemical sensors have been thoroughly described in earlier literature for blood,66–67 subcutaneous tissue,68–70 and the central nervous system (CNS).71–72 Rather than recapitulate these prior reviews, we divert our focus to how the different aspects of the host responses vary with the in vivo location and ultimately impact the function of the implanted chemical sensors.
3.1. Blood
The ability of a sensor to operate in blood continuously, with high accuracy, and for extended periods of time remains an elusive goal in the field of chemical sensors. The foremost motivation for chemical sensing in blood is continuous monitoring of “critical care species,”—analytes that change concentration in blood relatively fast and provide important prognostic/diagnostic information relating to a patient’s overall health—including oxygen, carbon dioxide, H+/K+/Ca2+, glucose, lactate, and NO. The designs of the many such sensors have been described in a recent review by Frost and Meyerhoff.2
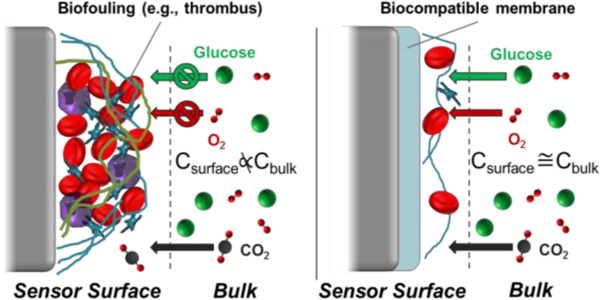
The therapeutic value of intravascular (IV) sensors is thwarted by their particularly challenging sensing environment (blood). Upon insertion into a blood vessel, a sensor will experience a highly-organized sequence of biochemical reactions (Figure 1). Virtually all materials, when implanted in blood, will attract some degree of protein adsorption dependent on the specific material surface chemistry, which will in turn mediate platelet adhesion to the surface. Glycoproteins that have been implicated in the subsequent biochemical cascades include fibrinogen (Fg), Von Willebrand factor (vWF), fibronectin (FN), and vitronectin (VN).73 It is important to note that the adsorbed protein layer may change over time to reach a steady-state (but not static) composition. For example, low affinity proteins (e.g., albumin) will rapidly accumulate on the sensor surface after initial implantation due to their large concentrations in blood. However, these proteins may be gradually displaced by other, lower abundance species with stronger adhesion affinity.2 Fibrinogen in particular has been identified as a key mediator of thrombus formation due to its large blood concentration (2–3 mg mL−1),74 propensity to adsorb onto a variety of surfaces, and ability to bind to platelet glycoprotein receptors. The consequences of initial protein adhesion to IV sensors are twofold. First, the adsorbed protein layer on the sensor serves as a passive analyte diffusion barrier. The resistance of the protein layer to molecular transport varies in time as protein is exchanged (e.g., Fg is displaced by high molecular weight kininogen), added/removed, or changing in conformation.75 Sensor signal instability or sensitivity drift are potential manifestations of protein biofouling. The second major consequence of protein adsorption is that the proteins serve as adhesion anchors for platelets and create a nidus for subsequent thrombus formation.
Figure 1.

Schematic representation of thrombus formation on the surface of a chemical sensor implanted in a blood vessel.
Platelet surface adhesion is mediated by binding interactions between Fg/vWF and the protein αIIbβ3 integrin.76 Platelet adhesion to and, in general, the thrombogenicity of a material is directly dependent on the composition and conformation of surface-adhered proteins.74 For example, adsorption of Fg is generally accompanied by protein conformational changes (i.e., decreased α-helix content) that are dependent on the underlying material’s surface chemistry.76–77 Loss of Fg secondary structure leads to a greater density of adhered platelets.76–77 Platelet adhesion triggers activation and the release of intracellular granules containing Ca2+ and a number of coagulation factors (e.g., V, VIII). This release elicits further platelet activation and aggregation. Separate from the coagulation factors secreted via platelet degranulation, the presence of a foreign surface in blood results in sequential enzymatic activation of factors XIII, XI, and IX (XIIIa, XIa, and IXa, respectively), which are serine proteases involved in the intrinsic coagulation cascade.75 Activated factor X (factor Xa) enzymatically cleaves prothrombin to thrombin, a serine protease that regulates the polymerization of fibrinogen to fibrin.73 Activation of factor X may also be stimulated via factor VIIa, which activates in response to tissue damage (i.e., the extrinsic coagulation pathway). Over time, activated/aggregated platelets amass on the sensor surface within a fibrin net. Circulating erythrocytes and leukocytes also become trapped in the fibrin gel to form a mature thrombus.
Thrombus formation on an implanted surface can be deleterious to the health of the patient, as dislodging thrombi may restrict blood flow or occlude narrower blood vessels (embolus). In the context of in vivo sensor performance, the main repercussion of thrombus formation is that the mass of entrapped cells acts as both a diffusion barrier and, for some analytes, an active transport sink. Indeed, the sensor surface concentrations of glucose and oxygen become much lower than in bulk blood due to consumption by local platelets, erythrocytes, and leukocytes. Although leukocytes (e.g., monocytes, macrophages) constitute fewer than 0.1% of circulating cells, they contribute considerably to sensor drift/inaccuracy due to excessive metabolic demands. Novak and coworkers modeled the response of amperometric glucose biosensors in blood by making use of a two compartment model of glucose transport dynamics.78 The model was based on bulk glucose diffusion through a layer of adsorbed cells using reported values for inflammatory macrophage and erythrocyte glucose metabolism (4.88×10−11 and 1.36×10−12 µmol glucose cell−1 s−1, respectively). Numerical simulations of the experimental glucose sensor signal drifts indicated that, while erythrocyte glucose metabolism did not sufficiently alter glucose flux to the sensor surface, glucose consumption by macrophages resulted in glucose depletion zones within ~100 µm of the sensor surface. Of note, the experimental signals were collected in vitro in heparinized whole blood, indicating that glucose consumption by both cell types would be more substantial under circumstances of thrombus formation.
Even for analytes that are not actively consumed as part of oxidative metabolism, in vivo sensor drift can be substantial. For example, potentiometric Na+-selective electrodes exhibit potential drift with platelet adhesion even in heparinized blood in vitro.79 It has been suggested, however, that most ion sensors are relatively insensitive to protein adhesion because their mode of operation is equilibrium-based rather than diffusion-limited.18 The local pH surrounding sensors is often decreased (relative to bulk values of 7.2–7.4) concomitant with positive drift in detected CO2 concentrations, the byproduct of cellular respiration (Figure 2).2
Figure 2.

Illustration of sensor drift associated with progressive thrombus formation on the surface of various intravascular sensors. Republished with permission from Annual Review of Analytical Chemistry 2015, 8, Frost, M.C.; Meyerhoff, M. E. “Real-time monitoring of critical care analytes in the bloodstream with chemical sensors: Progress and challenges,” pages 171–192. Copyright Annual Reviews 2015, with permission conveyed through Copyright Clearance Center.
3.2. Subcutaneous tissue
Chemical sensing in subcutaneous tissue has been pursued for diabetes management as a safer surrogate for direct glucose measurement in blood to avoid undesirable thrombosis and bacterial access to the blood stream. Unfortunately, subcutaneous chemical sensors have been essentially restricted to glucose because tissue concentrations for most analytes do not correlate well with their levels in blood.2 Despite limited applicability to glucose sensors, the host response in subcutaneous tissue has been investigated extensively. It is now well known that sensor performance is negatively impacted by the cascade of intense inflammatory events resulting from implantation. This response, collectively termed the foreign body response (FBR), includes the infiltration of the wound (sensor) site by inflammatory cells and the associated wound healing response. Indeed, the FBR may culminate either in persistent inflammation or isolation by a collagenous foreign body capsule, both of which are aberrations in the normal wound healing response (i.e., in the absence of the in vivo sensor).
The FBR is initiated upon sensor implantation and damaging of the loosely-organized, vascular tissue. Similar to the coagulation cascade, rapid accumulation of blood proteins on the sensor surface (i.e., protein biofouling) occurs within seconds of the initial insult. This process is generally believed to be irreversible.78,80 The concentration of surface-bound proteins in the thin adsorption layer may exceed their bulk (solution) concentration, with an adhered protein layer density (typical values of ~1 g protein cm−3) on some classic polymeric surfaces (e.g., polyurethane) approaching that of pure protein (~1.4 g cm−3).80 A well-known consequence of the accrued protein layer to electrochemical glucose sensors is a loss in analytical sensitivity by up to 40–80%.81 Mechanistic studies of protein adhesion on biomaterial surfaces indicate that small (<15 kDa) fragments of serum albumin (Alb) and other large proteins are the primary adsorbed biomolecules.82 The initial protein biofouling process in the subcutis is more heterogeneous than in blood. In fact, the specific identities and dynamics of these adherent proteins remain poorly understood. Nevertheless, the composition of the adsorbed protein layer is still considered to at least partially govern the pathology of the implant wound site during subsequent inflammatory phases.70
As a result of sensor implantation, platelets aggregate at damaged blood vessel sites to facilitate fibrin clot formation and deter blood loss.83 Through the release of growth factors (e.g., platelet-derived growth factor, transforming growth factor-β), platelets initiate the accompanying wound healing/inflammatory responses by recruiting circulating cells to the damaged blood vessels.83 Klueh et al. demonstrated that the microhemorrhages formed via local bleeding at the implant site resulted in inaccurate glucose biosensor performance.84 The underlying mechanism of the decreased sensor sensitivity and periods of signal dropout was attributed to glucose and oxygen consumption by erythrocytes around the sensor. Novak and coworkers disputed this result, clarifying that decreased sensor output is more likely the result of excess glucose metabolism by leukocytes (macrophages). Normal macrophage glucose metabolism (normalized per cell) is >50-fold greater than for erythrocytes.78 Importantly, Klueh et al.84 and Novak et al.78 agreed that microhemorrhages, which may form intermittently as a result of sensor micromotion,85–86 lead to the observed sensor signal dropout.
Acute inflammation occurs as the initial FBR event during the first several days of implantation, following initial biofouling and provisional matrix formation.70 Mast cells and neutrophils (phagocytic cells) infiltrate the implant site in an attempt to clear the foreign object.87 The neutrophilic response is short-lived (24–48 hours) but can be quite severe, contingent upon the degree of initial tissue trauma. In one study, Wang and coworkers implanted silicon chips in the subcutaneous tissue of Sprague-Dawley rats using different-sized needles (18, 16, and 14 gauge) and evaluated the FBR at durations between 3 and 30 days using standard tissue histological methods.88 Implantation of the silicon substrates using a larger needle size (i.e., initial trauma) lead to a more severe 3 day inflammatory response. However, the authors did not observe a significant effect of needle size on the severity of the chronic inflammatory response (>7 days).
If neutrophils are unable to remove the intruding object, the resident mast cells undergo release of cellular vesicles containing histamine and various cytokines/chemokines, including interleukins (IL-4 and IL-13),70 macrophage inflammatory protein 1α,89 and monocyte chemoattractant protein 1.89 Through this degranulation process, mast cells are largely able to orchestrate the recruitment, differentiation, and phenotypes of other inflammatory cells (i.e., monocytes/macrophages), and thus hold considerable sway over the FBR and associated sensor performance. Klueh et al. reported on the performance of glucose sensors implanted for 28 days in mast cell-sufficient and -deficient mouse models.90 Periods of sensor inaccuracy and signal dropout were frequently observed in the mast cell-sufficient model, whereas glucose sensors in the mast cell-deficient animals were more consistently able to track physiological glucose fluctuations. Histological analysis of the tissue surrounding the implanted sensors revealed a lessened inflammatory response and minimal collagen encapsulation around the implants in the deficient model at 1–4 weeks. A separate report by Egozi and coworkers indicated that the neutrophilic response in a mast cell-deficient mouse model (WBB6F1 Kitw/Kitw-v) was reduced compared to analogous WBB6F1 wild-type mice.91 Of note, mast cells are also capable of stimulating fibrocyte translocation from blood vessels to the wound site, with potential implications for subsequent collagen deposition and isolation of the sensor from the native tissue.92 Indeed, Avula et al. showed that collagen deposition was reduced at subcutaneous polyester implants in a mast cell-deficient mouse model (sash model) relative to control mice.93
Chronic inflammation and foreign body reactions characterize the host response at ≥5 days post-implantation.70 This period in the FBR strongly correlates with episodes of poor glucose sensor performance due to macrophage infiltration. Until recently, the relationships between macrophage functional polarization (phenotype), glucose metabolism, and in vivo glucose sensor performance were neglected and poorly understood. Macrophages have traditionally been classified as either pro-inflammatory (M1) or anti-inflammatory (M2, with subclasses M2a, M2b, and M2c), although it is now recognized that macrophages retain sufficient phenotypic plasticity to exist in a number of intermediate states between these two extremes.94–95 Pro-inflammatory macrophages drive the chronic inflammatory response by secretion of pro-inflammatory cytokines and chemokines (e.g., IL-1β, IL-6, IL-12, tumor necrosis factor α).96 They also produce a host of reactive oxygen (e.g., hydrogen peroxide, superoxide) and nitrogen (nitric oxide, peroxynitrite, nitrosonium) species (ROS and RNS) via respiratory bursts in the attempt to destroy the foreign object.33,97–101 The local pH can drop to acidic values (~4.0) as a result of macrophage phagocytic activity and exocytosis of acidic phagolysosomes.70,100 In this respect, GOx activity and associated sensor response are negatively impacted. In contrast, M2 macrophages are characterized by reduced phagocytic capability and glucose/oxygen metabolism,102 although it is important to note that even macrophages that fall within the M2 designation vary widely in terms of their roles in the FBR.103 With respect to in vivo sensor performance, excessive macrophage metabolic activity creates glucose and oxygen depletion zones in the direct vicinity of the implant.78,97,104–105 Klueh et al. reported that large local macrophage presence at implanted glucose biosensors lead to prolonged (>10 hours) periods of signal dropout in mouse implant models.106 The same authors produced macrophage deficient or depleted mouse models (through selective in-breeding and gene transfection methods, respectively) and demonstrated improved in vivo glucose sensor accuracy and signal stability (i.e., no signal dropout). To more directly examine macrophage metabolism in the context of implantable glucose sensors, Novak and co-workers immobilized RAW 264.7 murine macrophages in a fibrin gel surrounding commercial Medtronic glucose sensors (Figure 3A).105 A pronounced decrease in the sensor signal over the 24 hour testing period was attributed to macrophage glucose consumption (Figure 3B). The magnitude of this signal decrease was found to be dependent on macrophage phenotypic state; macrophages polarized toward a pro-inflammatory phenotype elicited an even larger decrease in the sensor signal (20% of the original glucose signal) than anti-inflammatory macrophages (~80%, similar to gels that did not contain cells). The comparatively large metabolic demands of pro-inflammatory macrophages appear to pose a significant obstacle to accurate in vivo glucose sensing. In addition to creating substantial glucose and oxygen concentration gradients surrounding the sensors, it has been speculated that the analyte levels at the sensor surface do not accurately reflect the true (bulk) analyte concentrations.107 Given that macrophages are primarily responsible for the chronic inflammatory response, macrophage phenotype may be a more reliable indicator of FBR severity than local macrophage densities. Both classically- (M1) and alternatively-activated (M2/pro-wound healing) macrophages are recognized to stimulate fibroblast activity (i.e., collagen deposition).108 In particular, expression of transforming growth factor-β by macrophages and FBGCs influences myofibroblast collagen synthesis and deposition.109 The important distinction between the two traditional subclasses of macrophages is that M1 macrophages are considered anti-angiogenic. In contrast, M2a and M2c macrophages, stimulated via IL-4/IL-13 or IL-10/glucocorticoids/secosteroids respectively, are pro-angiogenic.9,110
Figure 3.

Model of macrophage biofouling layer separating the surface of Minimed glucose sensors from the surrounding cell culture solution (A) and decrease in glucose sensor current response (I) normalized to the baseline sensor current (I0) elicited by macrophages under different stimulating conditions (B). Macrophages were either unstimulated, induced into a pro-inflammatory state using phorbol myristate or lipopolysaccharide, or polarized to an anti-inflammatory phenotype with genistein. Reprinted from Biomaterials, 2014, 35, Novak, M. T.; Yuan, F.; Reichert, W. M. “Macrophage embedded fibrin gels: An in vitro platform for assessing inflammation effects on implantable glucose sensors” pages 9563–9572, Copyright 2014, with permission from Elsevier.
Macrophages readily adhere to many foreign surfaces through interactions between β1/β2 integrins and surface-bound protein fragments (e.g., Fg fragments) and may remain at the site of an implanted material for months.111 Failure to initiate or maintain proper adhesion to foreign objects triggers frustrated phagocytosis and fusion into foreign body giant cells, which is likely an attempt to escape apoptosis after surface detachment.112 These polynuclear cells are characterized with substantial pro-inflammatory character compared to mononuclear macrophages. Giant cell formation further exacerbates local analyte depletion and increases ROS/RNS production with great propensity to damage sensor components and coatings.113–114 For example, polyetherurethanes—materials that are traditionally used as coatings for implants (e.g., in vivo sensors)—are susceptible to stress-cracking and delamination as a result of these intense foreign body reactions,113–115 both of which are widely accepted mechanisms for in vivo sensor failure.113
If unable to digest the implant, macrophages and foreign body giant cells direct the subsequent wound healing/proliferative phases and associated collagen deposition by secreting growth and angiogenic factors.83 Collagen deposition, while essential to tissue reconstruction, sequesters the implant from native tissue.116 It has been recognized since the 1990’s that the prototypical foreign body capsule poses a significant diffusion barrier to glucose.117–119 More recent work using a refined two compartment model for glucose transport dynamics showed that the presence of a dense collagen capsule does not overwhelmingly alter glucose concentrations at the sensor surface (vs. bulk concentrations).104 Rather, the foreign body capsule increases the tortuosity of the glucose diffusion path from the vasculature to the sensor surface, creating a pronounced lag (on the order of 20–30 minutes) between glucose concentrations at the sensing surface and corresponding plasma levels. Further compounding this issue, the fibrous capsule surrounding the implant is quite avascular,120 preventing efficient glucose delivery to regions inside the collagen capsule.121 Kumosa et al. observed that oxygen levels in the tissue surrounding fully subcutaneous O2 sensors also decrease over the first several weeks of implantation,22,107 which may alter glucose sensor response further due to limited O2 availability (the co-factor for GOx). Of note, the decreased oxygen levels are more likely due to inflammatory cell metabolism and poor vascularization than obstructed diffusion through the capsule. Notwithstanding, the general consequence of both capsule characteristics (avascularity and collagen density) is that in vivo glucose sensors will ultimately fail for long-term sensing applications (i.e., >7 days) due to inadequate analyte permeability and temporal lag to changing analyte concentrations.122
3.3. Neural tissue
Chemical sensing in central nervous system tissue has largely been applied to fundamental study of metabolites, ions, neurotransmitters, and biological gases. The vast majority of these reports are limited to single-day experiments due to sensor calibration challenges and sensor signal degradation due to the FBR. Unfortunately, appreciably less is known about the host response in nervous system tissue than in either blood or the subcutis due to greater tissue response heterogeneity. In addition, the majority of FBR investigations have utilized stimulating electrodes that do not depend on analyte transport. Nevertheless, useful information on the FBR has been generated in the context of electrical stimulators. The FBR in the central nervous system and subcutaneous tissue responses share many similarities, including inflammatory response timeline and many of the involved cell types.69–70,72 It is thus possible to identify key FBR events in neural tissue pertaining to in vivo sensor performance via comparison to analogous reactions in subcutaneous tissue. A key distinction between the nervous system and subcutaneous tissue responses is the existence of nervous system-resident astrocytes and microglia. Both cell types are activated upon tissue insult and play key roles in the FBR.72 Analogous to collagen capsule formation in the subcutis, the most frequently observed outcome in neural tissue is formation of a glial scar that segregates the implanted probe from surrounding neural tissue.
Implantation of the sensor into neural tissue results in a breach of the blood-brain barrier and damage to the underlying neural tissue. Excessive destruction of local vascular structures caused during device insertion will alter availability of plasma-dissolved analytes during the initial insult period due to local bleeding, blood vessel occlusion, or changes to vessel perfusion.71 Likewise, sensor implantation will also induce neuronal death and impact production of neurotransmitters, which will alter levels detected by the sensor.5 For example, Wang et al. examined dopamine production at an implanted microdialysis probe by positioning a carbon fiber ultramicroelectrode—a ~7 µm electrode that does not cause appreciable tissue damage—within ~200 µm of the microdialysis probe.14 A dopamine response was evoked via electrical stimulation and detected at the carbon fiber electrode via fast-scan cyclic voltammetry. The authors observed a ~80% decrease in the dopamine signal at the microdialysis probe when compared to uninjured tissue (i.e., no implanted microdialysis probe). The dopamine signal was decreased by only 50% (versus 80%) after sequential administration of the drugs raclopride (a dopamine D2 antagonist) and nomifensine (an inhibitor of dopamine reuptake), indicating that probe implantation induced a change in presynaptic dopamine terminal regulation. Clearly, inconsistencies in device size or implantation method will alter the initial extent of trauma and thus analyte availability.
Another consequence of tissue damage associated with sensor insertion is the recruitment of circulating inflammatory cells, which may persist around the implant for months.123 It is generally accepted that a smaller implant volume yields a milder inflammatory response.71 For instance, Karumbaiah and coworkers demonstrated that intracortical electrodes with an electrode size of 50 µm induced greater macrophage responses for acute (3 day) and chronic (12 week) implantation periods than did smaller electrodes (15 µm).124 As such, the inflammatory response can largely be avoided by reducing the initial vascular damage associated with sensor implantation. Kozai et al. reported the use of two-photon microscopy to identify brain tissue regions suitable for implantation.125 The authors demonstrated the ability to reduce inflammation by avoiding damage to well-vascularized tissue regions during implantation. Unfortunately, this task of locating optimal regions for implantation is too time-consuming for routine research use. Nesbitt et al. indicated that implants with at least one subcellular physical dimension, such as carbon-fiber ultramicroelectrodes (~7–8 µm in diameter), have a substantially reduced FBR due to the ability to avoid damage to tissue capillaries that are spaced ~60 µm apart.11 A few reports have demonstrated a negligible FBR to carbon-fiber sensors even after 4 months implantation.126–127 Even so, the number of in vivo sensor evaluations that exceed 24 hours in duration are rare, suggesting the likelihood of sensor performance degradation regardless of a favorable FBR.
The long-term FBR may still be aggravated, even for small (micron-sized) sensors, by mismatch in the Young’s modulus between the sensor and the surrounding tissue.85–86,128 Subbaroyan et al. used finite element modeling of implant-tissue strain as a function of material stiffness for typical electrode materials, including silicon (Young’s modulus ~200 GPa) and polyimide (2.8 GPa). 128 A hypothetical soft implant with a low modulus (6 MPa) was also examined for comparison to brain tissue (~6 kPa). The silicon and polyimide implants, when subjected to either tangential or radial tethering forces, were characterized with a high degree of interfacial strain. The hypothetical soft material yielded 50–90% strain relief compared to the silicon implants, but still elicited noticeable interfacial strain due to the greater material modulus relative to brain tissue. As most sensor materials (e.g., metals, polymers) have large moduli,85–86 sensor micromotion increases the propensity for local tissue irritation and intermittent bleeding.
Even with optimized sensor designs and implantation protocols, blood-brain barrier breach is unavoidable. Recruitment of circulating neutrophils and monocytes to the implant site proceeds initially (~24 h) via chemokine/cytokine release through cellular degranulation,72 akin to the initial response in subcutaneous tissue. Vasicek et al. used microdialysis probes that were implanted in the rat hippocampus to quantify several inflammatory and wound-healing cytokines that have been implicated in the subcutaneous tissue FBR.13 Although few of the cytokines were detected on the initial day of implantation, ~10–1000 pg mL−1 levels were measured for chemokine (C-C motif) ligand 2, macrophage inflammatory proteins 1α and 2, IL-1β, and IL-10 after 7 days. This increase in cytokine production is a hallmark of the chronic inflammatory response and represents a critical element of the ability of immune cells to self-direct the FBR. Skousen and coworkers utilized a cross-linked alginate hydrogel that served as a passive cytokine sink.129 Computational modeling of cytokine distributions predicted that ~400 µm thick hydrogels would reduce concentrations of macrophage chemoattractant protein-1 and tumor necrosis factor α by 25 and 40%, respectively. The alginate materials were coated onto silicon neural probes and implanted into rat brain tissue for immunohistopathological FBR evaluation. The authors observed reduced macrophage activation at alginate-coated versus bare silicon electrodes, suggesting that passive reduction of cytokine levels was a contributing factor to the reduced FBR. A reduced microglial response was also evident in tissue surrounding the alginate-coated electrodes. Importantly, microglia are one of the primary phagocytic cell types associated with chronic nervous system tissue inflammation and, once activated, carry out functions (e.g., debris removal, cytokine/chemokine secretion) that are similar to those performed by macrophages,72 including the ability to undergo frustrated phagocytosis.130 Microglia can influence neuronal survival by secreting various growth factors (e.g., brain-derived neutrophic factor, nerve growth factor),131 release of cytotoxins such as ROS/RNS species and glutamate,131 or by phagocytosing viable neurons,132 all of which directly impact neurotransmitter production and uptake in the vicinity of the sensor.
Chronic inflammation eventually coalesces into a steady-state cellular response and construction of a glial scar that isolates the implant from the surrounding neural tissue. The glial scar is largely comprised of reactive astrocytes and their secretions, such as glial fibrillary acidic protein and various proteoglycans.133 Szarowski et al. observed astrocyte infiltration of the tissue surrounding silicon microelectrodes as early as 1 day post-implantation, although their presence became progressively more concentrated at the implant surface within the first 4–6 weeks.123 The authors also noted resident microglia in the glial scar even after 12 weeks. Regarding impact to in vivo sensors, restricted ion/neurotransmitter diffusion into the glial scar has been documented as creating similar in vivo sensor performance issues as the foreign body capsule in subcutaneous tissue.134 Nevertheless, the relationship between glial scar composition and analyte diffusion remains an open area for further research and understanding.
4. BIOCOMPATIBILITY STRATEGIES
The guiding tenet in the field of implantable chemical sensors is that in vivo performance of the device should be considered with respect to key events in the appropriate host response.135–136 Consistent, long-term sensor operation in blood is entirely reliant on preventing thrombus formation.2,66,73 A similar sensor attribute is also desirable in subcutaneous and nervous tissue, but in this context avoiding/mitigating inflammation and encouraging wound healing (vs. scar tissue formation) are additional key parameters related to sensor performance.6 The biocompatibility strategies that have thus far proven effective tend to be specific to either blood or tissue and will be discussed separately. Of note, the FBR in central nervous system tissue is not as fully understood as the host responses in blood and subcutaneous tissue, and thus few strategies have been evaluated for potential biocompatibility improvements. Nevertheless, promising evidence for neural biocompatibility is highlighted where appropriate in the subsequent sections.
4.1. Blood-specific strategies
4.1.1. Hydrophilic and zwitterionic materials
Irrespective of the in vivo location, protein adhesion is the initial event experienced by all implanted sensors. Upon insertion into a blood vessel, the in vivo sensor is exposed to blood proteins that accumulate on the sensor’s surface and serve as anchors for cell attachment and ensuing thrombus formation. Strategies that prevent, or at least mitigate, protein adsorption have thus been postulated to improve the overall host response by reducing cell adhesion. Certain neutral hydrophilic polymers, namely poly(ethylene glycol) (PEG), poly(ethylene oxide) (PEO), and polyamides, have long been recognized for their resistance protein adsorption relative to other polymeric materials. 137–139 Among these materials, PEG has been the most frequently investigated due to ease of synthesis and low-fouling characteristics—however, in vivo degradation/oxidation has limited its usefulness in long-term implantable chemical sensing applications.140 The use of zwitterionic polymers (notably those prepared from monomer units of carboxybetaine, sulfobetaine, and phosphobetaine) has received greater attention recently, as these polymers have been shown to both adsorb only trace quantities of protein and inhibit thrombus formation.137–139,141
The low-fouling behavior of hydrophilic and zwitterionic surfaces is owed to layers of surface-bound water molecules that serve as an energetic barrier to protein adhesion.139 Water molecules interact with the polymer surface via hydrogen bonding or ionic interactions. The energetic properties of the adsorbed water layer are dependent on the density, orientation, and identity of the surface chemical groups. Surface-bound water molecules can be classified according to freezing point as either free water (~0 °C), bound freezing water (crystallization at <0 °C) and non-freezing water (crystallization at <−70 °C).142 Tanaka et al. investigated the thrombogenicity of copolymers prepared from different monomer ratios of 2-methoxyethylacrylate (MEMA) and 2-hydroxyethylmethacrylate (HEMA).143–144 Copolymers with low HEMA content (0–20%) exhibited reduced platelet adhesion in diluted platelet-rich plasma versus pure HEMA polymer (pHEMA; ~2×104 and 17×104 cells m−2, respectively).143 The authors observed that the lower platelet adhesion values on the 80–100% MEMA hydrogels correlated with reduced protein adhesion and greater amounts of surface-bound, freezing water versus that observed at pure pHEMA gels.143–144 Zhao and coworkers used pHEMA hydrogels to corroborate the studies by Tanaka et al.,143–144 suggesting that resistance to protein adhesion is related to the number of tightly bound water molecules on the hydrogel surface.145 However, the above studies only assessed the structure of the surface hydration layer in solutions of pure water (i.e., in the absence of proteins). Leng and coworkers used sum frequency generation vibrational spectroscopy to study surface hydration layers on zwitterionic sulfobetaine methacrylate (SBMA) and hydrophilic oligo(ethylene glycol) methacrylate (OEGMA) polymer brushes exposed to water or aqueous protein solutions.146 Vibrational spectra of the surfaces in water revealed greater proportions of strongly bound water molecules at SBMA surfaces versus OEGMA-modified interfaces. A considerable fraction of the water molecules at the OEGMA-modified surfaces were actually weakly associated. Upon exposure of the surfaces to solutions of serum albumin, lysozyme, or fibrinogen, the vibrational bands associated with the bound water molecules changed considerably at the OEGMA-modified surfaces, whereas the vibrational spectra collected on the SBMA surface appeared unchanged. Although both materials adsorbed minimal amounts of protein, the observed differences in vibrational spectra revealed a potential mechanism for reduced thrombogenicity of zwitterionic materials. Nevertheless, it is unlikely that the surface hydration layer at the SBMA surface would remain constant in more complex media (e.g., serum, blood).
Despite a still incomplete understanding of surface hydration layer dynamics, several reports have established that the non-fouling characteristics of zwitterionic surfaces in simple protein solutions actually translate to reduced platelet adhesion and thrombus formation in blood.147–149 For example, Zhang and coworkers compared protein adhesion at carboxybetaine methacrylate (pCBMA) and sulfobetaine methacrylate (pSBMA) polymer brushes prepared on gold surfaces via atom transfer radical polymerization (Figure 4).147 Both zwitterionic polymer brushes had extremely low Fg adsorption values of <0.3 ng cm−2 (from 1 mg mL−1 Fg solutions) and retained similar, low total protein adhesion (<10 ng cm−2) in blood plasma.147 Clotting times in recalcified platelet poor plasma were increased to ~25 min versus the bare gold slide that the brushes were prepared on (~10 min). Solutions of linear pSBMA and pCBMA polymers (20.9 and 22.1 kDa, respectively) were also mixed with platelet poor plasma to assess the effect of the free polymer on plasma clotting times. The pCBMA polymer elicited marked increases in clotting times (7.3 and 13 min at 1 and 10 mg mL−1 pCBMA), whereas pSBMA was similar to blood samples without additional polymer (~2.8 min). The authors noted that the unique behavior of pCBMA may be related to the anticoagulant activity of glycine betaine, a small molecule CBMA analogue.
Figure 4.

Representation of carboxybetaine methacrylate (CBMA) and sulfobetaine methacrylate (SBMA) polymer brushes prepared on gold substrates via atom transfer radical polymerization. Figure adapted with permission from Langmuir, 2006, 22, Zhang, Z.; Chao, T.; Chen, S.; Jiang, S. “Superlow fouling sulfobetaine and carboxybetaine polymers on glass slides,” pages 10072–10077. Copyright 2006 American Chemical Society.
Hydrogels consisting of pCBMA have also been coated onto IV glucose sensors to mitigate the negative effects of protein biofouling and thrombosis. Yang and coworkers chemically bonded the pCBMA gels to the surface of conventional GOx-based platinum coil glucose electrodes via reaction with trimethoxypropylsilylmethacrylate.49 The resulting sensors maintained similar sensitivity in serum as they did in phosphate buffered saline, and responded linearly to glucose concentrations of up to 20 mM. In a subsequent report, the same authors incorporated a chemical crosslinking agent (0.1% molar ratio relative to the CBMA monomer) to augment the mechanical strength of the pCBMA hydrogel.50 Sensors prepared using the improved pCBMA formulation accurately tracked glucose concentrations for up to 42 days of incubation in human blood at 4 °C. However, the stability of the sensor response at 37 °C during this period was not evaluated.
Despite the promise of zwitterionic hydrogels, particularly those prepared from polymers of CBMA, the mechanical stability of these materials in a biological environment remains concerning.150 The limited in vivo data on hemocompatibility of zwitterionic polymers in general, and whether the thromboresistivity translates to actual analytical biocompatibility benefits to, for example, the glucose sensors developed by Yang et al.,49–50 should be rigorously evaluated in the future.
4.1.2. Controlling identity and conformation of adsorbed proteins
Contrary to the strategies outlined above that aim to mitigate protein adsorption, the mass of adsorbed protein is not generally a prognostic indicator of the severity of the subsequent cellular response. Although Fg is the principal adhesive protein required for platelet adhesion, studies examining protein and platelet adhesion on polymeric surfaces have indicated little or no correlation between the amounts of adsorbed Fg and densities of adhered platelets.74,77,137,151–152 For example, Wu and coworkers interrogated the relationships between surface hydrophobicity, Fg adsorption, and platelet adhesion using custom-synthesized polyurethanes.151 Hydrophilic polyurethanes (advancing contact angle ~50°) that were synthesized using PEG diol linkers were shown to adsorb low levels of both Fg (~1.6 ng cm−2) and platelets (~2×103 cells cm−2). In the same study, more hydrophobic polyurethanes (advancing contact angle 80–110°) adsorbed greater Fg amounts (>30 ng cm−2) but varied widely in terms of platelet adhesion. One particular polyurethane formulation adsorbed 150× greater amounts of Fg than the hydrophilic polyurethanes (~250 ng cm−2), but still elicited a low platelet response similar to that observed for the PEG-modified polymers. Likewise, other reports have shown adsorbed Fg concentrations as low as 5 ng cm−2 on hydrophilic materials supporting substantial platelet adhesion.80,137,152 Some classically-utilized hydrophobic polymers (e.g., silicone rubber, polytetrafluoroethylene) adsorb greater levels of protein than more hydrophilic surfaces (PEO), yet are not facilitative of cell adhesion.153 Clearly, factors other than Fg adsorption influence the thrombogenicity of a material.
Platelet adhesion and thrombus formation is particularly sensitive to the identities of the adsorbed proteins, since adhesion is mediated by a limited number of integrins (e.g., αIIbβ3).152 Recent research has also revealed that the conformation of the adsorbed proteins may be a stronger determinant of thrombogenicity than the amount of adsorbed protein, as originally presumed. Sivaraman and Latour used adsorbed-state circular dichroism spectropolarimetry to quantify adsorption-induced Fg unfolding on alkanethiol self-assembled monolayers (SAMs) with different surface chemical functionalities (-CH3, -CF3, -NH2, -OH, and –COOH).77 For adsorption experiments carried out at Fg solution concentrations of 0.1–1.0 mg mL−1, the more hydrophobic –CH3 and –CF3 terminal SAMs elicited a greater loss in Fg α-helix content than did the –NH2, –OH, and –COOH SAMs. Decreased α-helix content on the more hydrophobic SAMs correlated consistently with greater densities of adhered platelets. At larger Fg concentrations (10 mg mL−1), the α-helix content of adsorbed Fg on all of the SAMs was similar to native Fg (due to multilayer Fg adsorption), resulting in reduced platelet densities compared to surfaces exposed to lower Fg concentrations (0.1–1 mg mL−1).
The strength of platelet adhesion is an additional factor when considering the thrombogenicity of a material. Safiullin and coworkers examined platelet/monocyte adhesion on classical blood-contacting polymers (e.g., polytetrafluoroethylene).74 Similar to the study by Sivaraman and Latour,77 maximum platelet adhesion was observed by Saffiulin et al.74 to occur at more intermediate Fg concentrations. Adhesion forces of human kidney (HEK-293) cells expressing the Fg-binding integrin receptor Mac-1 were also examined in relation to Fg adsorption using single-cell force spectroscopy.74 The force measurements revealed a maximum in the cell binding strength at an intermediate Fg concentration (~0.6 µg mL−1) with reduced binding strengths at greater Fg concentrations, indicating that further Fg adsorption actually decreases the adhesivity of the surfaces.
Although Fg has been singled out as one of the major culprits for later platelet adhesion, recent research has also implicated other proteins in this process that were originally deemed non-adhesive (i.e., serum albumin; Alb). Similar to their previous studies, Sivaraman and Latour saw adhesion-induced Alb denaturation on gold-bound thiol-based SAMs at similar concentrations to what was observed for Fg (0.1–1.0 mg mL−1).154 In vitro densities of adhered platelets increased predictably with the degree of Alb unfolding.
A mechanistic understanding of the relationships between protein adhesion and platelet aggregation has improved over the past several years, signifying a paradigm shift from mitigating the extent of protein adhesion to preventing adsorption-induced unfolding. Based on the above studies,74,76–77,154 it appears that surface-induced protein unfolding is more characteristic of the inner layer proteins, whereas the outermost proteins more closely resemble their native state. As such, surfaces that facilitate rapid multilayer protein adsorption may experience benefits of reduced platelet adhesion and thrombus formation in vivo, but this strategy has not been investigated rigorously. Preliminary studies designed to passivate surfaces by pre-adsorbing albumin155 have been unsuccessful due to eventual in vivo displacement of Alb by stronger affinity proteins (e.g., Fg, vWF)66 and the potential adhesivity of Alb itself.154 Although still unexplored, pre-adsorbing Fg multilayers may be a promising route to improved hemocompatibility.74
4.1.3. Heparin immobilization and heparin-mimicking materials
Heparin is a medium-molecular weight (~12 kDa average; range 5–40 kDa) anionic polysaccharide composed of sulfated repeating disaccharide units (e.g., 2-O-sulfo-l-iduronic acid, 6-O-sulfo-N-sulfo-glucosamine). 156–157 Heparin functions as an anticoagulant by binding to and amplifying the activity of the serine protease inhibitor Antithrombin III (AT) via a high-affinity pentasaccharide sequence.158–159 Several of the serine proteases involved in the intrinsic coagulation cascade (i.e., thrombin and factors IXa, Xa, and XIa) are inactivated by AT,160 although the basal rates of inactivation by AT are slow.161 Heparin binding accelerates the AT inactivation reactions (~2×103-fold amplification under optimal conditions),161 although the actual AT activity depends on the exact molecular weight and saccharide sequence of the heparin employed.
Systemic administration of heparin is a common approach during surgery and treatment of thrombi/emboli.66,162 However, a side effect related to heparin administration is the undesirable propensity for hemorrhage and thrombocytopenia.66,162 An approach to reducing complications associated with systemic heparin use is via surface-immobilization and controlled release methods. Both strategies have been investigated extensively in combination with synthetic (e.g., polyurethane, polyethylene terephthalate, polyvinyl chloride)163–166 and natural (e.g., silk, cellulose)20,167 polymers. Non-covalent heparin immobilization to surfaces relies on either doping heparin into polymeric films or ion pairing interactions between heparin’s anionic carboxylate or sulfate groups and appropriate cationic counterions.167–168 With non-covalent immobilization strategies, heparin gradually leaches from the material to the surrounding blood, which is useful for circumventing the issues caused by systemic heparin administration. Edagawa and coworkers modified needle-type glucose sensors with a cationic layer of polyethylenimine, which was then utilized to adsorb heparin.169 The in vivo performance of control and heparin-coated sensors was evaluated over 2 days in rabbit ear veins. Although control sensors stopped functioning after 18 hours, the heparin-coated sensors retained satisfactory accuracy (as determined via Clarke Error Grid analysis)170 over the entire 2 day implantation period, presumably due to reduced thrombosis. Unfortunately, it was unclear from this single study if the improved sensor performance was actually due to reduced thrombus formation.
An issue with both heparin adsorption and heparin release strategies is that the implanted sensor is susceptible to thrombosis once the limited supply of heparin has been exhausted. An alternative approach is covalent heparin immobilization, which provides non-depleting, surface-localized anticoagulation. The most apparent drawback associated with covalently bound heparin is the low attachment efficiency (<10%) associated with standard conjugation chemistries (i.e., carbodiimide coupling chemistries and glutaraldehyde fixation).171 A second issue involves potential conformational changes to heparin’s native structure that accompany immobilization.172 Surface conjugation may alter presentation of the Antithrombin-binding regions on heparin with implications to anticoagulant activity as well. Gore et al. reported that the chemical method employed for heparin immobilization greatly influences platelet activation and coagulation on poly(vinyl chloride) surfaces.166 Using an in vitro recirculating Chandler loop model, the authors compared covalent attachment of heparin using a single linkage at the end of the polysaccharide chain versus multipoint attachment along the side chains of the heparin molecule. Regardless of attachment chemistry, the poly(vinyl chloride) materials had similar degrees of heparin functionalization (3–7 µg cm−2). Multipoint heparin attachment led to decreased platelet counts and greater levels of prothrombin fragments 1 and 2 (F1+2) in the Chandler loop evaluation, suggestive of poor thromboresistance. In contrast, end-point attachment of heparin did not appreciably alter platelet counts and F1+2 levels compared to those in blood (i.e., prior to surface exposure), indicating a greater degree of Antithrombin binding/activation and suppression of thrombosis.
Other concerns with heparin-based strategies include heparin’s short half-life in vivo and substantial batch-to-batch variability of heparin isolates.66,173 These issues have been overcome to some extent by more efficient isolation and synthesis of ultra-low molecular weight heparins (~1.5–3.0 kDa) with better defined saccharide sequences and modifications than their larger molecular weight counterparts.174 Nevertheless, heparin immobilization does not eliminate the need for systemic heparin administration, although in many cases the doses are reduced.175–176 Issues with the quality of heparin isolates and loss of activity upon immobilization have prompted the development of sulfonated polymers designed to mimic the anticoagulant activity of native heparin.173,177–178 Heparin-mimicking polymers represent attractive alternatives to heparin-immobilization strategies due to greater anionic charge density and fewer issues regarding material stability or variability. Such materials are often produced via multilayer deposition of the sulfonated polymers with appropriate polycations with tunability of the resulting polyelectrolyte multilayer (e.g., for drug delivery). For example, Wang et al. prepared multilayers of water-soluble poly(ether sulfone) derivatives and chitosan that was functionalized with bactericidal quaternary ammonium moieties.173 The resulting films (3 alternating layers of poly(ether sulfone) and the modified chitosan) showed increased activated partial thromboplastin times, decreased thrombin production, and reduced platelet adhesion versus unmodified poly(ether sulfone) membranes. The same membranes were also capable of reducing the viability of adhered Pseudomonas Aeruginosa and Staphylococcus Aureus bacteria by ~97 and 98% after 4 hour culture, respectively. Despite the ability to impart other functionalities into heparin-mimicking polymer coatings, the ill-defined mechanism of anticoagulation remains a concern. Heparin itself complexes with and amplifies the activity of Antithrombin III via a high-affinity pentasaccharide sequence that is present in about one-third of all heparin molecules.158–159 Heparin-mimicking polymers, in contrast, do not possess the recognition sequence and appear to derive their anticoagulant activity from a high density of anionic surface charge. As such, it remains unclear if such materials can achieve the necessary level of thrombus reduction in vivo.
Regardless of the strategy, evidence to support the ability of heparin or heparin-mimicking materials to improve the analytical biocompatibility of intravascular sensors remains limited. While at least one report has verified improvements to the in vivo performance of intravascular glucose sensors,169 the required heparin amounts and release rates associated with these strategies are still unclear. As such, the achievable degrees of thrombus reduction on these materials and potential benefits to in vivo sensor performance should be systematically investigated.
4.1.4. Nitric oxide release
Nitric oxide (NO), an endogenously-produced, reactive diatomic gas, is involved in hemostasis,179 angiogenesis,180 inflammation,31–33 neurotransmission,34 and wound healing.181–182 The biosynthesis of NO occurs through the metabolism of l-arginine to l-citrulline via one of three isoforms of nitric oxide synthase (NOS): endothelial NOS (eNOS), inducible NOS (iNOS), and neuronal NOS (nNOS).29 The eNOS and nNOS isoforms are generally categorized as lower NO production enzymes (compared to iNOS), requiring elevated concentrations of Ca2+ (>70–100 nM) that facilitate binding of the enzymatic co-factor calmodulin.183 As such, both eNOS and nNOS transiently produce ñM concentrations of NO (bursts of several minutes in duration) for homeostatic processes such as blood pressure regulation and neurotransmission.29 The endothial NOS isoform is expressed by endothelial cells and platelets, whereas nNOS is expressed in the brain and skeletal muscle.184 The third isoform, iNOS, is not constitutively expressed in tissue but rather produced by immune cells (e.g., macrophages, mast cells) in response to various stimulating factors, including lipopolysaccharides, interferon-γ, and nuclear factor kappa-light-chain-enhancer of activated B cells (nuclear factor ĸB).185–186 Calmodulin binding for iNOS is less sensitive to intracellular Ca2+ levels and occurs at normal resting cell Ca2+ levels (70–100 nM).183,185 Thus, iNOS activity is largely independent of intracellular calcium and is capable of continuous, high-output (~µM) NO production, primarily as a defense against foreign pathogens.185
Nitric oxide’s role in hemostasis is primarily the result of its release from endothelial cells via eNOS at low fluxes (~6.8×10−14 µmol µm−2 s−1)187 for the purpose of discouraging platelet activation and aggregation.188–189 Inhibition of platelet activation by NO is attributed to NO binding to the heme moiety of soluble guanylate cyclase, stimulating guanosine monophosphate (cGMP) production. Increased cGMP levels result in activation of cGMP-dependent protein kinases, which in turn inactivate the G protein-coupled receptors (e.g., thromboxane A2 receptors) involved in intracellular calcium mobilization (key for factor X activation) and fibrinogen affinity for αIIbβ3 integrin.190–192 Platelets also express eNOS and influence recruitment and aggregation of other nearby platelets via NO generation. Platelets incubated with the NOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME) express greater levels of P-selectin, a glycoprotein-binding adhesion molecule involved in cellular adhesion.193–194
Low, continuous NO release from sensor surfaces in vivo was first suggested by Meyerhoff mimic the natural ability of the vascular endothelium by inhibiting platelet activation and aggregation.19 However, localized NO delivery from in vivo sensors is not straightforward due to NO’s reactive nature and short lifetime in vivo. The most successful NO-release strategies have relied on the use of NO donors (e.g., N-diazeniumdiolates or S-nitrosothiols) as dopant molecules within polymeric sensor coatings. The NO donors are purposefully selected for their ability to undergo chemical breakdown reactions via specific physiochemical triggers with ensuing NO release. N-diazeniumdiolates, formed by the base-catalyzed reaction of secondary amines with gaseous NO, degrade upon protonation of the amine coordinating the NO donor to yield two moles of NO and the regenerated parent amine. The rates of NO production in physiological buffer are predictable and dependent on solution pH, temperature, and the chemical structure of the precursor amine.195 The tunable decomposition rates render N-diazeniumdiolates the most frequently investigated class of NO donors for applications in NO release. S-nitrosothiols (RSNOs), an alternative NO donor, are readily synthesized by reaction of thiols with nitrosating reagents (e.g., acidified sodium nitrite). A number of endogenous RSNOs (e.g., S-nitrosoglutathione) serve as NO transporters that augment NO’s in vivo lifetime.196–197 Light (330–350 and 550–600 nm for primary RSNOs) and thermal irradiation both serve as RSNO decomposition triggers, causing homolytic cleavage of the S–N bond to yield NO and thiyl radicals.197 Several transition metal ions (Cu+, Ag+, and Hg2+) undergo irreversible catalytic redox reactions with RSNOs to generate NO as well.198 Due to temperature sensitivity, RSNOs are used less frequently than N-diazeniumdiolates for exogenous NO delivery.
Small molecule NO donors have been extensively utilized as dopants in sensor membranes for improving the analytical biocompatibility of a wide variety of intravascular (IV) sensors. Espadas-Torre et al. were the first to report the use of the NO donor (Z)-1-N-methyl-N-[6-(N-methylammoniohexyl)amino]-diazen-1-ium-1,2-diolate (MAHMA/NO) doped into a sensor membrane. The authors imparted NO-release capabilities to classical intravascular potentiometric H+ and K+ ion-selective electrodes using this approach.19 Both sensors were found to retain suitable potentiometric response (slope) and selectivity over Na+ upon incorporation of the NO donor. Thrombogenicity evaluation in platelet-rich plasma revealed less platelet adhesion and minimal platelet activation on the NO-releasing materials, in contrast to larger densities of activated platelets on controls (non-NO-releasing membranes). However, potential benefits of NO release to in vivo sensor performance were not evaluated in this study. Schoenfisch and coworkers reported on the in vivo analytical performance of NO-releasing IV catheter-style oxygen sensors using silicone rubber coatings incorporating the NO donor MAHMA/NO.24 Neither sensitivity nor response time was compromised with the addition of the NO donor. The analytical biocompatibility of the sensors was examined by implantation in femoral and carotid arteries of dogs for 6–24 hours without systemic anticoagulation. The NO-releasing sensors showed improved accuracy over the first 18 hours correlated with reduced in vivo platelet adhesion and thrombus formation. Importantly, the difference in accuracy between control and NO-releasing sensors was not discernable beyond 18 hours, which the authors attributed to insufficient NO fluxes being released at later time points.
The success of these initial analytical biocompatibility reports notwithstanding, NO donor leaching from the sensor membranes was identified as a concern for reasons including undesirable formation of toxic N-nitrosamine species on the parent amines.199–200 Significant effort has been devoted to reducing leaching of the NO donor constituents via the use of water-insoluble small molecule amine precursors (i.e., N,N’-dibutylhexamethylenediamine, DBHD)201 or by covalently tethering the NO donors to the polymer backbone and/or to large, macromolecular scaffolds (e.g., silica nanoparticles).202 In recent reports by the Meyerhoff research group, a more lipophilic NO donor (N-diazeniumdiolate-modified DBHD) has been utilized to limit NO donor leaching and fabricate improved NO-releasing intravascular glucose and lactate sensors.203–204 The NO-releasing glucose sensors were shown to reduce in vivo thrombus formation when implanted in the jugular veins of rabbits.204 Correspondingly, the NO-releasing sensors were better able to track rapid changes in blood glucose than analogous control sensors.203–204
A second concern associated with doping NO donors into polymeric matrices is the limited supply of NO in the resulting membranes. An innovative solution to this problem is the use of circulating S-nitrosothiols that naturally exist in blood (i.e., S-nitrosated forms of serum albumin, l-cysteine, and glutathione) to generate NO at the surface of the implanted sensor, potentially providing an unlimited supply of NO.205 In this manner, copper complexes (e.g., Cu(II)-cyclen complexes) were immobilized within polymer membranes for NO production at the blood-sensor interface via catalytic Cu2+-mediated RSNO degradation.206–207 In other work, Cha et al. immobilized an organoselenium species to polyethylenimine in order to mimic the function of glutathione peroxidase (GPx), a selenoenzyme that catalytically degrades RSNOs in the presence of free glutathione to achieve NO release.208 Unfortunately, a limitation of these methods is inconsistency in the surface NO fluxes due to varying RSNO levels in blood and tissue.
A third promising approach capable of more consistent NO production involves the electrochemical generation NO from electrodes immersed in nitrite filling solutions.209 Direct NO production from nitrite occurs at slightly positive potentials (+0.2 V vs. normal hydrogen electrode) at copper electrodes, resulting in the formation of Cu+ ions that can react with nitrite to produce NO.210 Unfortunately, the electrode is rapidly passivated at this potential via the formation of a copper oxide layer. The metallic copper surface must therefore be regenerated through the application of a second, cathodic pulse (−0.7 V). Rather than using pulse voltammetry to generate NO, Ren and coworkers reported the incorporation of Cu(II)-tri(2-pyridylmethyl)amine, a copper redox mediator, into the nitrite solution to produce NO by applying a constant potential at a noble metal electrode (e.g., gold, platinum).209 This NO-generating device was integrated into single- and dual-lumen catheters. The NO flux was tunable simply by changing the applied voltage in the range of −0.20 to −0.40 V (vs Ag|AgCl, 3 M KCl). Catheters that released NO at 1.1×10−10 mol cm−2 min−1 over a 7 hour implant study in rabbit ear veins reduced thrombus formation by ~63% versus control catheters.211 In a related report, Ren and coworkers coupled this technology to intravascular catheter-style amperometric oxygen sensors.23 The NO-releasing sensors produced >1.0×10−10 mol NO cm−2 min−1 over a 21 hour implant period in pig veins and displayed improved accuracy versus control sensors. The main drawback of this technology is the need for the separate nitrite filling solution, which places an upper limit on the size of the device and precludes compatibility with other sensor types (e.g., glucose, lactate).
Of importance, NO does not actually prevent activation of the intrinsic coagulation pathway,176 highlighting a potential limitation of all NO-release strategies. Some evidence supports simultaneous use of complementary antithrombotic methods (i.e., NO release and surface-immobilized heparin),212 but any further improvements to sensor analytical biocompatibility have yet to be proven. In addition, most of the studies discussed above have been restricted to <24 hour testing durations due to concerns regarding embolus formation and infection. It remains unclear if NO-releasing sensors can remain essentially thrombus-free for extended implant times.
4.2. Tissue-specific strategies
Classically, the FBR has been studied as a function of implant geometry and surface chemistry.68–70,101,112 A simple but useful biocompatibility principle that was established early on is that hydrophobic surfaces, such as polystyrene, generally promote more severe foreign body reactions (i.e., greater degrees of macrophage adhesion and frustrated phagocytosis) than hydrophilic surfaces (e.g., polyacrylamide).69,112 However, increased production of pro-inflammatory cytokines was routinely observed on hydrophilic surfaces and was speculated to offset the benefits of any reduced cellular response.112 As such, proper choice of implant material/composition alone has not mitigated the FBR to an extent that would benefit the analytical biocompatibility of in vivo sensors. The most successful recent tissue biocompatibility strategies are discussed below. Most proposed methods have been studied in the context of glucose sensors and aim to simultaneously reduce inflammation and guide appropriate wound healing around the implanted sensor.
4.2.1. Zwitterionic materials
In addition to the antithrombotic properties of poly(carboxy betaine)methacrylate (pCBMA) hydrogels, recent evidence suggests that antifouling zwitterionic polymeric biomaterials can markedly reduce the inflammatory response in subcutaneous tissue. In an initial study, Zhang and co-workers implanted cross-linked pCBMA and poly(hydroxyl-2-ethyl)methacrylate (pHEMA) hydrogels in a mouse FBR model.213 The zwitterionic pCBMA materials elicited reduced collagen encapsulation and greater blood vessel densities at 4 and 12 weeks post-implantation relative to pHEMA hydrogels. Macrophages surrounding the implants at 4 weeks were characterized using immunofluorescence to assess phenotypic state. In general, the macrophages surrounding the pHEMA hydrogels expressed greater levels of classical markers of inflammatory activity (iNOS, tumor necrosis factor α, IL-12), whereas the cells surrounding the pCBMA implants expressed greater levels of pro-wound healing and anti-inflammatory markers (arginase and IL-10). Of note, the favorable FBR observed for these materials appeared to be sensitive to the CBMA content of the hydrogels. Previous studies by the same group indicated that hydrogels formed from copolymers of CBMA and HEMA (~80 mol% CBMA) elicited a similar FBR relative to pHEMA hydrogels in terms of collagen and inflammatory cell densities.214 Despite reduced FBR via pCBMA materials, no reports to date have studied the utility of such biomaterials for improving the analytical biocompatibility of subcutaneous chemical sensors.
4.2.2. Porous and nanopatterned coating materials
As most surface chemical approaches do not appreciably influence the FBR, researchers have investigated the roles of porosity and surface topography on cellular behavior.215 The most successful illustrations of this approach are porous coatings that have been known to reduce the FBR to implants for more than two decades. Brauker et al. published a seminal report describing the FBR to several commercial polymers (cellulose, polytetrafluoroethylene, and acrylic copolymer) with pore sizes in the range of 0.02–15 µm.216 Vascular structures were consistently observed at large-pore (>0.8 µm) materials that enabled cellular infiltration, irrespective of the chemical composition of the membranes. Neovascularity was enhanced by a factor of 80–100 for polytetrafluoroethylene membranes with a nominal pore size of ~5 µm versus membranes with sub-cellular pore sizes (0.02 µm). Subsequent investigations by Sharkawy and coworkers began to elucidate the mechanisms through which porosity and improved FBR outcomes could impact the performance of in vivo glucose sensors.117–119 In the first of these studies, stainless steel cage implants were coated with either nonporous or porous (60 and 350 µm pores) poly(vinyl alcohol) (PVA) and implanted in rat subcutaneous tissue for tissue histology assessment after 3 and 12 weeks.117 Whereas a thick, dense, avascular collagen capsule was observed immediately proximal to the nonporous PVA implants, both porous PVA films were characterized by increased angiogenesis and decreased collagen density. Carefully removed tissue samples adjacent to the implants were used to study fluorescein diffusion through the collagen capsule (as a surrogate for glucose). The measured effective diffusion coefficient (Deff) for fluorescein through the capsule surrounding nonporous PVA implants was lower (1.11×10−6 cm2 s−1) than in subcutaneous tissue alone (2.35×10−6 cm2 s−1). In contrast, tissues surrounding the 60 and 350 µm porous PVA implants had diffusion coefficients that were more in line with native tissue (2.19×10−6 and 1.87×10−6 cm2 s−1, respectively) because of the reduced collagen density. In a follow-up study, tissue response times to changing plasma concentrations of a fluorescent tracer analyte (lissamine-rhodamine) were examined in capsular tissue surrounding the PVA implants.118 As expected, the tissue surrounding 60 µm porous PVA implants responded more quickly to changes in tracer concentrations than in capsules surrounding nonporous PVA films (~12 and 34 minutes, respectively), implicating neovascularization as an additional key parameter in tissue analyte transport.
Koschwanez and coworkers examined the effects of porosity on the FBR using commercial (Medtronic) glucose sensors.217–218 Sensors were coated with porous poly(l-lactide) (PLLA) coatings (~30 µm pore sizes) produced via a salt-leaching method. Histological evaluation of the tissue surrounding the porous implants after 2 weeks implantation in rats revealed increased blood vessel formation in the vicinity of the implant relative to the tissue adjacent to nonporous control materials (221 and 152 vessels mm−2, respectively).218 Greater total collagen was measured at the porous PLLA coatings versus controls (53 and 25%, respectively) as well, although the collagen inside the PLLA pores was less dense. The authors initiated in vivo sensor analytical performance evaluation studies as a function of coating porosity but did not observe significant differences between porous versus nonporous PLLA-coated sensors. They hypothesized that micromotion of the percutaneous sensors may have been a convoluting variable responsible for this result. Alternatively, the increased total amounts of collagen at the porous PLLA-coated sensors may have offset the beneficial effects of increased angiogenesis.
The underlying biological mechanisms and optimal porosity levels were not systematically studied by Koschwanez et al.217–218 due to pore size heterogeneity resulting from the salt-leaching process. Materials with spherical, uniform pore sizes, produced by sphere templating methods, have since been used to study the FBR.219–221 Sussman and coworkers used monodisperse ~34 and 160 µm diameter poly(methyl methacrylate) beads as templates for pHEMA hydrogel fabrication.221 After bead removal in a subsequent Soxhlet extraction step, the resulting porous pHEMA hydrogels were used to examine a potential relationship between pore size and FBR severity in mice (subcutaneous tissue). As shown in Figure 6, the authors noted the inability of cells to migrate into the nonporous pHEMA coatings, which resulted in dense collagen deposition immediately adjacent to the material-tissue interface (Figure 6A). In contrast, minimal collagen encapsulation and angiogenesis were hallmarks of the tissue reactions at pHEMA implants with pore sizes on the order of cellular dimensions (~34 µm; Figure 6B). Large-pore materials (~160 µm) invoked a more classical FBR response, with heavy collagen deposition inside the pores (Figure 6C). Other studies by Ratner and coworkers have established favorable FBR outcomes for porous materials with pore sizes on the order of cellular dimensions (~10–20 µm for most leukocytes) irrespective of the identity of the polymer.222 The prevailing hypothesis is that the inability of phagocytic cells (i.e., macrophages) to spread on the material directs them to a more reconstructive M2 phenotype.222–223 Of note, FBR-mitigating pore sizes reported by Brauker,216 Sharkawy,117–119 and Koschwanez217–218 are similar to the optimal pore sizes reported by Ratner’s group.
Figure 6.

The cellular and collagen composition of the foreign body reaction to solid and porous implants is pore-size dependent. Representative Masson’s Trichrome photomicrographs show histological responses based on pore size. Collagen is shown in blue, cellular cytoplasm in red, and cell nuclei in black. (A) non-porous implants have a dense FBC at the implant edge, (B) 34 µm porous scaffolds have highly cellular infiltrate, and (C) 160 µm porous scaffolds have a cellular infiltrate that is much richer in collagen than 34 µm scaffolds. Reprinted from Annals of Biomedical Engineering, “Porous implants modulate healing and induce shifts in local macrophage polarization in the foreign body reaction,” 42, 1508–1516 by Sussman, E. M.; Halpin, M. C.; Muster, J.; Moon, R. T.; Ratner, B. D. Copyright Annals of Biomedical Engineering 2013, with permission of Springer.
Although the porous materials above unequivocally influence the FBR, work to develop porous sensor membranes for implantable chemical sensors has been scarce, likely due to difficulties associated with thin coating deposition on the sensors and/or potential sensor-material incompatibilities (e.g., high temperatures associated with Soxhlet extraction and GOx instability). Electrospinning of polymer solutions represents an alternative approach to fabricating porous sensor membranes. The process of forming nanofibrous polymer mats by electrospinning is straightforward, requiring readily available experimental components (i.e., a syringe pump, high voltage power supply, and metal object to serve as a grounded collector).224 The electrospinning process is typically carried out by applying a high voltage (>5 kV) to a polymer solution droplet at the tip of a metal needle. Repulsive electrostatic forces within the droplet exceed attractive surface tension forces, resulting in the formation of a Taylor cone. The polymer solution stream accelerates toward electrical ground and, upon solvent loss during flight, solidifies into nanofibers that accumulate on the grounded metal collector.224–225 Although the polymer fibers formed by this method are generally polydisperse with respect to fiber diameter, both fiber size (0.01–10 µm) and porosity of the resulting fiber mats (30–95%) can be controlled by appropriate selection of solution composition (e.g., polymer concentration, polymer identity, conductivity, viscosity) and electrospinning parameters (e.g., voltage, tip-to-collector distance, collector geometry, humidity).224–225
Analogous to the porous materials produced by salt-leaching and sphere-templating methods, electrospun fiber mats are also associated with a reduced FBR.226 Optimal tissue responses have proven to be dependent on a number of material parameters of the fibers, including polymer identity and fiber/pore size. Garg and coworkers recently studied how the pore size of electrospun polydioxanone fibers impacted the phenotypes of primary mouse macrophages.227 Macrophages that were cultured on electrospun fiber mats with ~15 µm pores expressed 2–3 fold greater levels of the M2 (anti-inflammatory) marker arginase relative to the macrophages on fibers with smaller pores (~1 µm). In contrast, expression of iNOS, a classic pro-inflammatory marker, was elevated for macrophages seeded on the 1 µm pore size fiber mats versus macrophages on the larger pore size material. The authors also examined the ability of the polydioxanone fiber mats to influence the phenotypes of macrophages that were pre-polarized to either M1 or M2 states by stimulation with interferon-γ or a cocktail of IL-4/IL-13, respectively. Although the initial macrophage phenotype did impact arginase/iNOS expression, fibers with 15 µm pores consistently yielded macrophages with reduced pro-inflammatory character (i.e., greater arginase/iNOS ratios).227 Based on these results, pore sizes on the order of cellular dimensions (2–20 µm) appear to mitigate the FBR by forcing macrophages and other inflammatory cells to adopt more reconstructive phenotypes.228
Initial work indicates that the orientation of electrospun fibers also appears to be a strong determinant of the FBR. Cao et al. reported that aligned electrospun poly(caprolactone) fibers (~300–500 nm diameter) facilitated greater in vivo cell migration into the fibers compared to randomly oriented fibers in a rat subcutaneous FBR model. Tissue surrounding the aligned fibers consistently exhibited lower grades of inflammation at 1–4 weeks post-implantation compared to the tissue at random nanofiber scaffolds. Irrespective of fiber alignment, both types of fibers elicited a 75–90% decrease in collagen capsule thickness (compared to polymeric films) after 4 weeks implantation.229
The electrospinning process is amenable to modifying electrochemical glucose biosensors.48,230 Wang et al. coated epoxy polyurethane fiber mats onto miniaturized glucose sensors using a modified electrospinning setup in which the sensor was fixed onto a rotating mandrel that served as the grounded electrospinning collector.48 The sensors retained suitable glucose sensitivity and linear dynamic range (2–30 mM) at coating thicknesses of ~40–50 µm. Although the authors demonstrated the ability to successfully modify glucose sensors with electrospun fibers, future work should assess any potential analytical biocompatibility benefits using functional in vivo sensors.
4.2.3. Release of tyrosine kinase inhibitors
Tyrosine kinases are a class of enzymes involved in phosphorylation and signal transduction for many biochemical cascades. For instance, the tyrosine kinase KIT (also identified as CD117) serves as a binding receptor for stem cell factor (SCF), an important mast cell growth factor and activator.231 Inhibition of the SCF-KIT pathway limits production of histamine and the pro-inflammatory cytokine IL-4 by mast cells, suggesting suppressed degranulation. 232 As outlined in Section 3, mast cells play a crucial role in determining the chronic inflammatory response to implanted sensors.233 Of note, Klueh et al. examined the roles mast cells in the FBR using a mast cell-deficient mouse model (WBB6F1 KITw/KITw-v) produced by selective mutation to the gene encoding KIT.90 The mast cell-deficient mice consequently produced a milder FBR relative to wild-type mice.
As mast-cell deficiency has been shown to improve the accuracy of in vivo glucose sensors,233 the release of mast cell inhibitors from the surface of glucose sensors may represent a promising approach for improving in vivo sensor analytical performance. Grainger and coworkers have begun to study the impact of the KIT inhibitor masitinib on tissue biocompatibility.93,234–235 Avula et al. synthesized masitinib-releasing poly(lactic-co-glycolic acid) (PLGA) microspheres using an oil-in-water/solvent evaporation method.235 The PLGA microspheres were doped into poly(ethylene glycol)/poly(ethylene oxide) membranes and shown to release ~11.2 µg masitinib at near-constant rates for up to 30 days.93,234–235 Such polymers reduced collagen deposition at the subcutaneous implants by 75–90% in wild-type mice. Of note, the masitinib release did not reduce collagen encapsulation in mast cell-deficient mice, demonstrating masitinib selectivity toward mast cells.93,235 In a separate study, Avula et al. fabricated electrochemical glucose sensors using the masitinib-releasing coatings. 234 The signal stability of the masitinib-releasing glucose sensors was enhanced versus controls over a 21 day implantation period in mice. Although these results are initially encouraging, the authors did not assess the accuracy of the sensors, which would provide a more reliable indication of analytical biocompatibility. Future work should also examine optimal rates and amounts of masitinib release in relation to sensor performance.
4.2.4. Dexamethasone release
Dexamethasone (DX), a synthetic anti-inflammatory glucocorticoid hormone, was one of the initial release agents used to potentially improve the performance of implantable glucose sensors and has since been used in neural tissue sensing applications.52 Dexamethasone is a potent agonist for the cytosol-localized glucorticoid receptor that, upon DX binding, translocates to the cell nucleus and achieves its anti-inflammatory effects in part through transactivation/transrepression of key chemokines and cytokines (e.g., IL-1, nuclear factor ĸB, activating protein-1).236–237 Although the mechanisms of DX’s anti-inflammatory action have not been fully elucidated, DX is also known to subdue production of other pro-inflammatory mediators (e.g., tumor necrosis factor α, IL-6) through other, indirect pathways.238
A significant concern associated with DX release is systemic immune suppression. Hori et al. implanted DX-loaded sponges in the subcutaneous tissue of rats and observed indicators of a compromised immune system (i.e., thymus and spleen weight loss) at daily doses of 5–50 µg DX per implant, even though the DX was administered locally.238 Of note, DX doses lower than 0.5 µg per day did not elicit noticeable immunosuppression. Several years following this report, Ward and coworkers investigated the required DX release amounts and rates for achieving localized anti-inflammatory action without causing immune suppression in Yucatan-minipigs.239 Mock sensors were coupled to an osmotic pump that delivered DX/saline to the subcutaneously-implanted sensor at pre-determined doses (0.7, 0.28, 0.168, 0.1, or 0.05 mg kg−1 over 28 days). Doses <0.1 mg kg−1 did not incite systemic immune suppression, as illustrated by serum cortisol levels that were similar to those before DX administration. In contrast, DX doses exceeding 0.1 mg kg−1 produced a noticeable decrease in cortisol concentrations. Based on histological analysis of tissues surrounding the implants, it was concluded that the lower DX doses were still sufficient to reduce granulocyte densities at 28 days post-implantation, although the macrophage response was only lessened at DX doses greater than 0.1 mg kg−1.
It is clear from the investigation by Ward et al. that achieving localized anti-inflammatory effects without causing systemic immune suppression requires slow, precisely-controlled DX release.239 Patil et al. developed DX-loaded poly(lactide-co-gylcolide) (PLGA) microspheres capable of slow hydrolysis in physiological buffer, enabling DX release.240 The PLGA microspheres were immobilized in poly(vinyl alcohol) hydrogels that were shown to release ~4 µg DX per implant over 28 days. These hydrogels were then implanted into the subcutaneous space of rats for histopathological FBR evaluation. Regardless of the implant time (1–28 days), the authors reported reduced inflammatory cell densities at the DX-releasing hydrogels relative to control gels. Despite promising initial results in rats, this DX/PLGA formulation proved ineffective at mitigating the tissue response in Göttingen minipigs because of inconsistent temporal DX-release characteristics.241 The DX-releasing microspheres exhibited a characteristic triphasic release profile with a large initial (~1 day) DX burst, a sustained 10-day lag period where no DX was released, and zero-order DX-release kinetics thereafter (for 19 days).241 A comparison of the FBR in both mice and pigs revealed that the 10-day lag phase was the source of inconsistency. Kastellorizios and coworkers designed a new microsphere synthesis via the co-precipitation of PLGA and DX, which eliminated the lag phase in the DX-release profile.242 The optimized materials both lessened inflammation and elicited a modest reduction in collagen deposition in the swine model.242
Another reported method for DX delivery relies on incorporating DX into the perfusate solution of microdialysis probes, thereby promoting DX release via retrodialysis.10 The use of microdialysis probes has been helpful in understanding the FBR with respect to DX due to the ability to monitor cytokine/chemokine levels in addition to standard tissue histology. Keeler and coworkers examined macrophage production of chemokine (C-C motif) ligand 2 (CCL2), a pro-inflammatory chemokine, in the subcutaneous tissue surrounding microdialysis probes that released a water soluble analogue of DX (dexamethasone-21-phosphate; DXP).9 Levels of CCL2 in the dialysate solutions were decreased for the DXP-releasing probes relative to controls at 4–6 days post-implantation, corresponding to the expected timeframe in which macrophages infiltrate the implant site. The macrophages in the tissue samples were analyzed via fluorescence immunohistochemistry for expression of CD68 and CD163 (general and M2c macrophage markers, respectively) in order to determine if altered macrophage polarization was responsible for the differences in CCL2 concentrations. As expected based on the chemokine analysis, ~66% of the macrophages surrounding the DX-releasing implants stained positive for the M2c marker CD163 versus 18% CD163+ cells for controls. In another study, Kozai et al. studied the tissue surrounding DXP-releasing microdialysis probes implanted in the brain cortex of mice.15 Two-photon microscopy was used to image green fluorescent protein expressed from brain microglia of transgenic mice 6 hours after probe implantation. A smaller radius of microglia activation surrounding DXP-releasing probes (93.0 µm) was observed relative to that for control probes (177.1 µm), indicating a reduced inflammatory response.
Despite the promising histological and immunohistochemical data involving DX-release strategies, few reports have demonstrated improvements in analytical biocompatibility of either microdialysis probes or in vivo sensors.12,46,243 Nesbitt and coworkers examined studied dopamine production in rat brain (striatum) tissue surrounding DXP-releasing microdialysis probes.12 A carbon microfiber electrode was positioned close to the implanted microdialysis probes and was used to measure tissue dopamine levels via fast-scan cyclic voltammetry. Dopamine levels 100 µm from the control probe surface were diminished, presumably due to an acute microglial response. Indeed, the DXP-releasing probes were characterized with both reduced gliosis and greater dopamine concentrations in the surrounding tissue. However, dopamine levels at the DXP-releasing probes were still reduced compared to uninjured tissue, indicating that other factors (e.g., neuronal death) may also contribute to the reduced dopamine signal. Klueh et al. implanted electrochemical glucose sensors into the subcutis of mice and evaluated the influence of daily intraperitoneal DX injections (1, 6, or 10 mg kg−1) on sensor performance.243 The glucose sensitivity of control sensors degraded rapidly almost immediately after implantation (<1 nA mM−1), with total loss of the glucose response of the sensor within ~24 hours. DX treatment helped maintain glucose sensitivity for up to 7 days (3–20 nA mM−1). Histological analysis of tissue samples from the implant site confirmed that a reduced inflammatory response was at least partially responsible for the improved sensor function. The promising sensor performance data notwithstanding, systemic DX administration in this manner is not a viable approach for improving device function outside of the research setting.
An additional concern that is routinely observed in tissue studies of DX-releasing materials involves inhibited angiogenesis.244–246 Proposed strategies for overcoming DX-induced ischemia have revolved around the concurrent release of endogenous angiogenic stimulators, including vascular endothelial growth factor (VEGF)244,247–248 and platelet-derived growth factor (PDGF).248 However, growth factor release strategies are not straightforward due to issues with stability and controlled delivery.249 Price and coworkers suggested the use of l-3,4-dihydroxyphenylalanine (L-DOPA) as a more reliable alternative to either VEGF or PDGF.249 Using a chlorioallantoic membrane model, the authors demonstrated that DX that was released from a hydrogel inhibited angiogenesis, but simultaneous delivery of DX and L-DOPA increased the formation of blood vessel sprouts relative to controls (i.e., hydrogels that did not contain DX or L-DOPA). While initial results appear promising, achieving optimal delivery rates of both species from sensor coatings remains an arduous task. Another tactic for offsetting poor angiogenesis involves the combination of DX release with a more passive biocompatibility approach, such as material porosity. Vallejo-Heligon and coworkers reported the development of porous (~90% porosity) DX-releasing Tecoflex 93A polyurethane coatings via a salt-leaching process.46–47 The coatings, capable of releasing 600 µg DX over 15 days, decreased inflammatory cell infiltration relative to non-DX-releasing porous coatings after 3 day implantation in rats (Figure 7).47 Medtronic glucose sensors were then modified with the porous DX-releasing coatings with subsequent evaluation of in vivo analytical performance over a 3 week implantation period in rats.46 Sensors modified with the porous DX-releasing membranes better maintained glucose sensitivities and response times to changing glucose concentrations than did the corresponding control sensors (Figure 8). Importantly, the authors demonstrated that the FBR benefits provided by porosity alone were insufficient to improve sensor performance and actually worsened the sensor response time, a potential result of increased collagen deposition. This work also verified that vascularity and collagen deposition at the DX-releasing sensors were improved relative to tissue surrounding sensors coated with nonporous PU, demonstrating that porosity was necessary to counteract the anti-angiogenic effects of DX.
Figure 7.

Photomicrographs of hematoxylin & eosin stained tissue samples surrounding bare Tygon tubing (left column) and Tygon coated with porous Tecoflex PU (center column) or porous, DX-releasing PU (right column). N=3 separate implants for each material. Reprinted from Acta Biomaterialia, 2014, 10, Vallejo-Heligon, Klitzman, B; Reichert, W. M. “Characterization of porous, dexamethasone-releasing polyurethane coatings for glucose sensors” pages 4629–4638, Copyright 2014, with permission from Elsevier.
Figure 8.

Sensitivities of Medtronic Sof-sensor glucose sensors with or without additional porous PU coatings. Only sensors that were modified with the DX-releasing porous PU coating maintained their initial sensitivity after 3 weeks implantation. Reprinted from Acta Biomaterialia, 2016, 30, Vallejo-Heligon, S. G.; Brown, N. L.; Reichert, W. M.; Klitzman, B. “Porous, Dexamethasone-loaded polyurethane coatings extend performance window of implantable glucose sensors in vivo” pages 106–115, Copyright 2015, with permission from Elsevier.
4.2.5. Nitric oxide release
In addition to roles in hemostasis, NO is an intrinsic component of both the innate immune response31–33 and tissue reconstruction.34,182 Nitric oxide’s function in the immune response has traditionally been considered from the viewpoint of its antimicrobial action. Indeed, NO and its reactive by products (e.g., N2O3, ONOO−) work in concert to eradicate bacteria.33 Evidence also suggests that NO may also regulate the recruitment of cells to the implant site during the acute inflammatory response.181 Although the mechanisms for NO’s involvement in chemotaxis are not fully understood, NO is known to alter expression of key inflammatory cell mediators and growth factors, including tumor necrosis factor α,250–251 chemokine (C-C motif) ligand 2,250 RANTES,252 IL-1β,250,253 and IL-6.251 On the other hand, the involvement of NO as an angiogenic agent during tissue reconstruction has been well-studied. Angiogenesis during the reconstructive end-stages of the FBR requires NO derived from eNOS.180 Angiogenic factors, such as VEGF and transforming growth factor β, stimulate NO production.254–255 Nitric oxide may also upregulate VEGF via a positive feedback loop.256
The anti-inflammatory and pro-angiogenic capacities of NO indicate that NO-releasing materials may prove useful for mitigating the FBR and improving the analytical biocompatibility of tissue-implanted sensors. As for other NO-release systems (see Section 4.1.4), NO’s reactivity and short lifetime in vivo necessitates the use of chemically stable NO donors. Hetrick and coworkers reported the covalent attachment of secondary amines (for N-diazeniumdiolate formation) within an alkoxysilane-based xerogel.257 Specifically, isobutyltrimethoxysilane underwent co-condensation with N-(6-aminohexyl)aminopropyltrimethoxysilane to form the silica xerogel. The secondary amines were subsequently reacted with NO to form N-diazeniumdiolate NO donors. The tissue biocompatibility of these coatings was then evaluated in the subcutaneous space of a rodent model. After 3 and 6 week implantation periods, tissue histology indicated that the NO-releasing xerogels, which released 1.35 µmol NO cm−2 over 72 hours, decreased inflammatory cell density and collagen capsule thickness/density immediately adjacent to the implants. Quantitative immunohistochemistry for CD-31 (an endothelial cell adhesion molecule) revealed greater vascularization (angiogenesis) at both 1 and 3 weeks for the NO-releasing substrates relative to controls. However, this work did not identify optimal NO-release payloads or kinetics. In subsequent work, Nichols and coworkers evaluated the severity of the FBR in a swine model as a function of NO-release kinetics.258 Silica particles modified with either N-diazeniumdiolate or S-nitrosothiol NO donors259 were employed as polyurethane dopant molecules to fabricate NO-releasing polymer coatings. The resulting polyurethane/silica composites were diverse with respect to their NO-release kinetics, releasing 2.7–9.3 µmol NO cm−2 for 1–14 day durations depending on the type of silica nanoparticle dopant. Materials capable of releasing NO for at least 2 days both decreased the inflammatory response over the first week post-implantation and lead to reduced collagen capsule thickness at 3 and 6 weeks. In contrast, more rapid NO-releasing materials (~24 hour NO-release durations) did not decrease inflammation and were observed to increase collagen density relative to non-NO-releasing control materials. Although the anti-inflammatory effects of NO were localized to the tissue immediately surrounding the implants, these results indicate the need for precise control of NO-release kinetics for achieving the optimal tissue response. Additionally, further study is required for understanding NO’s involvement with key FBR cell types and pathways.
A critical, yet frequently overlooked aspect of biocompatibility is whether reductions in the FBR actually translate to improved sensor performance. With knowledge that NO mitigates the FBR, Nichols et al. evaluated the glucose recovery of NO-releasing microdialysis probes in a rat model as a measure of subcutaneous tissue mass transfer resistance.260 An NO-saturated buffer solution (1.9 mM NO) was used as the probe perfusate to achieve approximately constant NO release (~162 pmol cm−2 s−1 for 8 hours daily) from the microdialysis probes via retrodialysis over a 2 week implantation period. Although the glucose recovery of NO-releasing and control probes remained constant during the first 6 days of implantation (~15–25% glucose recovery), only the NO-releasing probes maintained adequate recovery values over the entire 14 day implantation period (Figure 9E). After 8 days, control probe glucose recovery was approximately half that of the NO-releasing probes and further diminished to ≤5% after 10 days. Tissue histology analysis revealed that the NO-releasing probes induced lower degrees of inflammation and collagen encapsulation than the control probes (Figure 9A–D), indicating that the superior glucose recovery for the NO-releasing probes was due in part to a reduced FBR. Based on the favorable improvements to subcutaneous glucose transport, the authors suggested potential benefits of NO release to glucose sensor performance for tissue-based platforms.
Figure 9.

Photomicrographs of Masson’s trichrome (A, C) or hematoxylin & eosin (B, D) stained tissues surrounding NO-releasing (A, B) and control (C, D) microdialysis probes after 14 d implantation in Sprague-Dawley rats. The photomicrographs in (A,C) stain blue for collagen fibers and those in (B, D) stain cell nuclei purple. The graph in (E) presents the glucose recovery of NO-releasing (red, triangle) and control (black, inverted triangle) microdialysis probes as a function of implantation time. Reprinted with permission from Analytical Chemistry, 2011, 83, Nichols, S. P.; Le, N. N.; Klitzman, B.; Schoenfisch, M. H. “Increased in vivo glucose recovery via nitric oxide release,” pages 1180–1184. Copyright 2011 American Chemical Society.
To date, two separate research studies have demonstrated benefits of NO release to in vivo chemical sensors, both in the context of subcutaneous glucose sensors.42,44 Gifford et al. functionalized percutaneous, needle-type electrochemical glucose biosensors to release NO by doping N-diazeniumdiolate-modified N,N’-dibutylhexanediamine into polyurethane/polydimethylsiloxane glucose sensor membranes.42 The NO-releasing sensors and analogous controls were implanted in the subcutis of rats for performance evaluations up to 48 hours in duration. The NO-releasing sensors better maintained glucose sensitivity throughout implantation (~12% sensitivity decrease over 48 hours) than did control sensors (32% decrease). On the day of implantation, 99.7% of blood glucose determinations made by the NO-releasing sensors were reported to be clinically acceptable by Clarke Error grid analysis of the sensor data in comparison to paired reference measurements (i.e., a handheld glucometer). In contrast, fewer measurements by control sensors met the criteria for a clinically acceptable measurement (96.3%). However, the clinical accuracy of NO-releasing sensors after 48 hours was similar to controls. Histological analysis of tissues surrounding the sensors indicated reduced inflammatory response for the NO-releasing sensors at 24 hours but not at 48 hours, highlighting a potential source for decreased NO-releasing sensor performance. The authors hypothesized that the NO release, limited only to 18 hours in duration, was insufficient to improve sensor performance beyond 24 hours. Using a macromolecular NO-release scaffold,182,202 Soto and coworkers investigated in vivo glucose sensor performance as a function of NO-release kinetics.44 Silica nanoparticles, modified with either N-diazeniumdiolate or S-nitrosothiol NO donors, were doped into polyurethane membranes to fabricate sensors with distinct NO-release durations (16 hours and 3.1 days, respectively). Control and NO-releasing sensors were implanted for 10 day periods in the subcutaneous tissue of swine. Sensor performance was assessed by numerical accuracy metrics by comparing blood glucose measurements between the sensors and a reference method (glucometer). The NO-releasing sensors were associated with a lower mean absolute relative deviation (MARD; 18–26%) than control sensors (35–47%), indicative of improved accuracy (Figure 10B–C). After 3 days, the numerical accuracy (MARD) of the NO-releasing sensors became similar to controls, as expected based on the limited NO-release duration (16 hours). Sensors capable of releasing NO for longer periods (3.1 days) had nearly constant MARD values throughout the entire 10 day implantation period, a result the authors attributed to the extended NO release.
Figure 10.

Schematic of needle-type electrochemical glucose biosensor (A). The sensors were modified to store NO by doping NO-releasing silica nanoparticles into the outermost, glucose flux-limiting polyurethane layer. The graphs in (B, C) display the in vivo sensor numerical accuracy (mean absolute relative deviation) of NO-releasing (black, square) and control (red, circle) for the N-diazeniumdiolate- (B) and S-nitrosothiol-based (C) membranes. Figure adapted with permission from Analytical Chemistry, 2014, 86, Soto, R. J.; Privett, B. J.; Schoenfisch, M. H. “In vivo analytical performance of nitric oxide-releasing glucose biosensors,” pages 7141–7149. Copyright 2014 American Chemical Society.
The in vivo tissue response and sensor performance studies demonstrate the potential of NO-releasing materials to mitigate the FBR and improve the performance of implantable continuous glucose monitoring devices. To date, these studies indicate that the FBR is only affected during periods of active NO release. Given the limited NO-release durations of the functional glucose sensors used in investigations by Gifford42 and Soto44 (<3 days), it remains unclear as to whether sensors capable of more long-term NO release (5–14 days) will further enhance in vivo glucose biosensor performance and/or sensor lifetimes.
5. Conclusions
In vivo chemical sensors are central technologies in analytical chemistry that can transform disease management and understanding of physiological that cannot be studied with other, non-invasive measurements. Unfortunately, applications of in vivo sensors are often limited by poor accuracy. Such insufficient analytical biocompatibility results from the deleterious host response and is the foremost obstacle to sensor utility. Numerous strategies have been worked on to address sensor biocompatibility. Rather than relying only on a selection of optimized implant materials or geometries, the most promising approaches aim to guide the host responses away from recognizing the sensor as a foreign body. Although several such strategies have been shown to reduce thrombosis or alter the FBR, further evaluation with respect to functional sensor assessments has been largely ignored. Validating potential benefits of improved biocompatibility with sensor performance testing represents a vital step in translating the impact of chemical sensors for fundamental research investigations and disease management.
Figure 5.

Schematic representation of different NO-release strategies used for fabricating NO-releasing in vivo chemical sensors. The electrochemical NO generation figure was adapted with permission from ACS Applied Materials and Interfaces, 2014, 6, Ren, H.; Wu, J.; Xi, C.; Lehnert, N.; Major, T.; Bartlett, R. H.; Meyerhoff, M. E. “Electrochemically modulated nitric oxide (NO) releasing biomedical devices via copper(II)-tri(2-pyridylmethyl)amine mediated reduction of nitrite,” pages 3779–3783. Copyright 2014 American Chemical Society.
ACKNOWLEDGEMENTS
R.J.S. and J.B.T. gratefully acknowledge dissertation fellowships from the Royster Society of Fellows at the University of North Carolina. The authors express their gratitude to Professor R. Mark Wightman and Nathan Rodeberg for fruitful discussions during the preparation of this review.
BIOGRAPHIES
Robert J. Soto is a graduate student in the Department of Chemistry at the University of North Carolina at Chapel Hill working under the direction of Prof. Mark Schoenfisch. His graduate research is focused on chemical sensor design, biocompatibility, and methods to evaluate in vivo sensor analytical performance. He holds a B.S. degree in Chemistry and Mathematics from the University of Arizona.
Jackson R. Hall is a graduate student in the Department of Chemistry at the University of North Carolina at Chapel Hill working under the direction of Prof. Mark Schoenfisch. His graduate research is focused on the chemistry of hydrogen sulfide and related measurement strategies. He holds a B.S. degree in Chemistry from the University of Richmond.
Micah D. Brown is a graduate student in the Department of Chemistry at the University of North Carolina at Chapel Hill working under the direction of Prof. Mark Schoenfisch. His graduate research is focused on electrochemical methodologies that facilitate accurate measurement of nitric oxide in physiological milieu. He holds a B.S. degree in Chemistry from the University of Rochester.
James B. Taylor is a graduate student in the Department of Chemistry at the University of North Carolina at Chapel Hill working under the direction of Prof. Mark Schoenfisch. His graduate research is focused on the development of assays for elucidating the direct effects of nitric oxide on inflammatory cells involved in the foreign body response. He holds a B.S. degree in Chemistry from the University of Maryland, Baltimore County.
Mark H. Schoenfisch is Professor of Chemistry at the University of North Carolina at Chapel Hill. His research interests include analytical sensors for biological and point-of-care applications, biomaterials that mitigate the foreign body response to implantable devices, and nitric oxide release scaffolds as therapeutics against various diseases.
Footnotes
The authors declare no competing interests.
REFERENCES
- 1.Wilson GS.; Johnson MA. Chem Rev 2008, 108, 2462–81. [DOI] [PubMed] [Google Scholar]
- 2.Frost MC.; Meyerhoff ME. Annu Rev Anal Chem 2015, 8, 171–92. [DOI] [PubMed] [Google Scholar]
- 3.Kissinger PT.; Hart JB.; Adams RN. Brain Res 1973, 55, 209–13. [DOI] [PubMed] [Google Scholar]
- 4.Williams DF. In Definitions in Biomaterials : Proceedings of a Consensus Conference of the European Society for Biomaterials, Chester, England, March 3–5, 1986, Amsterdam; New York, 1987; Elsevier: Amsterdam; New York. [Google Scholar]
- 5.Paul DW.; Stenken JA. Analyst 2015, 140, 3709–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ratner BD. J Cardiovasc Trans Res 2011, 4, 523–527. [DOI] [PubMed] [Google Scholar]
- 7.Bucher ES.; Wightman RM. Annu Rev Anal Chem 2015, 8, 239–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gowers SAN.; Curto VF.; Seneci CA.; Wang C.; Anastasova S.; Vadgama P.; Yang G-Z.; Boutelle MG. Anal Chem 2015, 87, 7763–7770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Keeler GD.; Durdik JM.; Stenken JA. Acta Biomater 2015, 23, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keeler GD.; Durdik JM.; Stenken JA. Acta Biomater 2015, 12, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nesbitt KM.; Jaquins-Gerstl A.; Skoda EM.; Wipf P.; Michael AC. Anal Chem 2013, 85, 8173–8179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nesbitt KM.; Varner EL.; Jaquins-Gerstl A.; Michael AC. ACS Chem Neurosci 2015, 6, 163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vasicek TW.; Jackson MR.; Poseno TM.; Stenken JA. ACS Chem Neurosci 2013, 4, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Y.; Michael AC. J Neurosci Methods 2012, 208, 34–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kozai TDY.; Jaquins-Gerstl AS.; Vazquez AL.; Michael AC.; Cui XT. Biomaterials 2016, 87, 157–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho EJ.; Lee J-W.; Ellington AD. Annu Rev Anal Chem 2009, 2, 241–264. [DOI] [PubMed] [Google Scholar]
- 17.Kraft MD.; Btaiche IF.; Sacks GS.; Kudsk KA. Am J Health Syst Pharm 2005, 62, 1663–1682. [DOI] [PubMed] [Google Scholar]
- 18.Bakker E.; Qin Y. Anal Chem 2006, 78, 3965–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espadas-Torre C.; Oklejas V.; Mowery K.; Meyerhoff ME. J Am Chem Soc 1997, 119, 2321–2322. [Google Scholar]
- 20.Badr IH.; Abdel-Sattar R.; Keshk SM. Carbohydr Polym 2015, 134, 687–94. [DOI] [PubMed] [Google Scholar]
- 21.Mistlberger G.; Crespo GA.; Bakker E. Annu Rev Anal Chem 2014, 7, 483–512. [DOI] [PubMed] [Google Scholar]
- 22.Kumosa LS.; Routh TL.; Lin JT.; Lucisano JY.; Gough DA. Biomaterials 2014, 35, 8287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ren H.; Coughlin MA.; Major TC.; Aiello S.; Rojas Pena A.; Bartlett RH.; Meyerhoff ME. Anal Chem 2015, 87, 8067–8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schoenfisch MH.; Mowery KA.; Rader MV.; Baliga N.; Wahr JA.; Meyerhoff ME. Anal Chem 2000, 72, 1119–1126. [DOI] [PubMed] [Google Scholar]
- 25.Schoenfisch MH.; Zhang H.; Frost MC.; Meyerhoff ME. Anal Chem 2002, 74, 5937–41. [DOI] [PubMed] [Google Scholar]
- 26.Huttmann SE.; Windisch W.; Storre JH. Ann Am Thorac Soc 2014, 11, 645–652. [DOI] [PubMed] [Google Scholar]
- 27.Severinghaus JW.; Bradley AF. J Appl Physiol 1958, 13, 515–520. [DOI] [PubMed] [Google Scholar]
- 28.Xie X.; Bakker E. Anal Chem 2013, 85, 1332–1336. [DOI] [PubMed] [Google Scholar]
- 29.Nathan C.; Xie Q.-w. Cell 1994, 78, 915–918. [DOI] [PubMed] [Google Scholar]
- 30.Ignarro LJ.; Buga GM.; Wood KS.; Byrns RE.; Chaudhuri G. Proc Natl Acad Sci U S A 1987, 84, 9265–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coleman JW. Int Immunopharmacol 2001, 1, 1397–406. [DOI] [PubMed] [Google Scholar]
- 32.Korhonen R.; Lahti A.; Kankaanranta H.; Moilanen E. Curr Drug Targets Inflamm Allergy 2005, 4, 471–9. [DOI] [PubMed] [Google Scholar]
- 33.MacMicking J.; Xie QW.; Nathan C. Annu Rev Immunol 1997, 15, 323–50. [DOI] [PubMed] [Google Scholar]
- 34.Bult H.; Boeckxstaens GE.; Pelckmans PA.; Jordaens FH.; Maercke YMV.; Herman AG. Nature 1990, 345, 346–347. [DOI] [PubMed] [Google Scholar]
- 35.Hunter RA.; Privett BJ.; Henley WH.; Breed ER.; Liang Z.; Mittal R.; Yoseph BP.; McDunn JE.; Burd EM.; Coopersmith CM.; Ramsey JM.; Schoenfisch MH. Anal Chem 2013, 85, 6066–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bedioui F.; Villeneuve N. Electroanalysis 2003, 15, 5–18. [Google Scholar]
- 37.Privett BJ.; Shin JH.; Schoenfisch MH. Chem Soc Rev 2010, 39, 1925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hall CN.; Garthwaite J. Nitric Oxide 2009, 21, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wink DA.; Darbyshire JF.; Nims RW.; Saavedra JE.; Ford PC. Chem Res Toxicol 1993, 6, 23–7. [DOI] [PubMed] [Google Scholar]
- 40.Diabetes Control and Complications Trial Group the Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus, New England Journal of Medicine. 1993; Vol. 329, p 977–986. [DOI] [PubMed] [Google Scholar]
- 41.Heller A.; Feldman B. Chem Rev 2008, 108, 2482–2505. [DOI] [PubMed] [Google Scholar]
- 42.Gifford R.; Batchelor MM.; Lee Y.; Gokulrangan G.; Meyerhoff ME.; Wilson GS. J Biomed Mater Res A 2005, 75, 755–66. [DOI] [PubMed] [Google Scholar]
- 43.Shichiri M.; Yamasaki Y.; Kawamori R.; Hakui N.; Abe H. The Lancet 1982, 320, 1129–1131. [DOI] [PubMed] [Google Scholar]
- 44.Soto RJ.; Privett BJ.; Schoenfisch MH. Anal Chem 2014, 86, 7141–7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vaddiraju S.; Legassey A.; Qiang L.; Wang Y.; Burgess DJ.; Papadimitrakopoulos F. J Diabetes Sci Technol 2013, 7, 441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vallejo-Heligon SG.; Brown NL.; Reichert WM.; Klitzman B. Acta Biomater 2016, 30, 106–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vallejo-Heligon SG.; Klitzman B.; Reichert WM. Acta Biomater 2014, 10, 4629–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang N.; Burugapalli K.; Song W.; Halls J.; Moussy F.; Ray A.; Zheng Y. Biomaterials 2013, 34, 888–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang W.; Bai T.; Carr LR.; Keefe AJ.; Xu J.; Xue H.; Irvin CA.; Chen S.; Wang J.; Jiang S. Biomaterials 2012, 33, 7945–7951. [DOI] [PubMed] [Google Scholar]
- 50.Yang W.; Xue H.; Carr LR.; Wang J.; Jiang S. Biosens Bioelectron 2011, 26, 2454–2459. [DOI] [PubMed] [Google Scholar]
- 51.Wilson GS.; Hu Y. Chem Rev 2000, 100, 2693–2704. [DOI] [PubMed] [Google Scholar]
- 52.Nichols SP.; Koh A.; Storm WL.; Shin JH.; Schoenfisch MH. Chem Rev 2013, 113, 2528–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen X.; Matsumoto N.; Hu Y.; Wilson GS. Anal Chem 2002, 74, 368–372. [DOI] [PubMed] [Google Scholar]
- 54.Mao F.; Mano N.; Heller A. J Am Chem Soc 2003, 125, 4951–4957. [DOI] [PubMed] [Google Scholar]
- 55.Colvin AE.; Jiang H. J Biomed Mater Res A 2013, 101A, 1274–1282. [DOI] [PubMed] [Google Scholar]
- 56.Unruh RM.; Roberts JR.; Nichols SP.; Gamsey S.; Wisniewski NA.; McShane MJ. J Diabetes Sci Technol 2015, 9, 985–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phypers B.; Pierce JT. Continuing Education in Anaesthesia, Critical Care & Pain 2006, 6, 128–132. [Google Scholar]
- 58.Shapiro NI.; Howell MD.; Talmor D.; Nathanson LA.; Lisbon A.; Wolfe RE.; Weiss JW. Ann Emerg Med 2005, 45, 524–8. [DOI] [PubMed] [Google Scholar]
- 59.Pribil MM.; Laptev GU.; Karyakina EE.; Karyakin AA. Anal Chem 2014, 86, 5215–5219. [DOI] [PubMed] [Google Scholar]
- 60.Manna B.; Retna Raj C. J Mater Chem B 2016, 4, 4585–4593. [DOI] [PubMed] [Google Scholar]
- 61.Naylor E.; Aillon DV.; Gabbert S.; Harmon H.; Johnson DA.; Wilson GS.; Petillo PA. J Electroanal Chem 2011, 656, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lewerenz J.; Maher P. Front Neurosci 2015, 9, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hu Y.; Mitchell KM.; Albahadily FN.; Michaelis EK.; Wilson GS. Brain Research 1994, 659, 117–125. [DOI] [PubMed] [Google Scholar]
- 64.Foubert LA.; Lecomte PV.; Nobels FR.; Gulino AM.; De Decker KH. Diabetes Technol Ther 2014, 16, 858–66. [DOI] [PubMed] [Google Scholar]
- 65.Smith JL.; Rice MJ. J Diabetes Sci Tech 2015, 9, 782–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brisbois EJ.; Handa H.; Meyerhoff ME., Recent Advances in Hemocompatible Polymers for Biomedical Applications in Advanced Polymers in Medicine, Puoci F., Editor Springer International Publishing: 2015; pp 481–511. [Google Scholar]
- 67.Ratner BD. J Biomater Sci Polym Ed 2000, 11, 1107–19. [DOI] [PubMed] [Google Scholar]
- 68.Anderson JM. Ann Rev Mater Res 2001, 31, 81–110. [Google Scholar]
- 69.Anderson JM.; Jones JA. Biomaterials 2007, 28, 5114–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson JM.; Rodriguez A.; Chang DT. Sem Immunol 2008, 20, 86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kozai TD.; Jaquins-Gerstl AS.; Vazquez AL.; Michael AC.; Cui XT. ACS Chem Neurosci 2015, 6, 48–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Polikov VS.; Tresco PA.; Reichert WM. J Neurosci Methods 2005, 148, 1–18. [DOI] [PubMed] [Google Scholar]
- 73.Wo Y.; Brisbois EJ.; Bartlett RH.; Meyerhoff ME. Biomater Sci 2016, 4, 1161–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Safiullin R.; Christenson W.; Owaynat H.; Yermolenko IS.; Kadirov MK.; Ros R.; Ugarova TP. Biomaterials 2015, 67, 151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gorbet MB.; Sefton MV. Biomaterials 2004, 25, 5681–703. [DOI] [PubMed] [Google Scholar]
- 76.Sivaraman B.; Latour RA. Biomaterials 2011, 32, 5365–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sivaraman B.; Latour RA. Biomaterials 2010, 31, 832–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Novak MT.; Yuan F.; Reichert WM. J Diabetes Sci Tech 2013, 7, 1547–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pawlak M.; Grygolowicz-Pawlak E.; Crespo GA.; Mistlberger G.; Bakker E. Electroanalysis 2013, 25, 1840–1846. [Google Scholar]
- 80.Horbett TA., Proteins: Structure, Properties, and Adsorption to Surfaces in Biomaterials Science: An Introduction to Materials in Medicine, Ratner BD.; Hoffman AS.; Schoen FJ.; Lemons JE., Eds. Academic Press Inc: 1996; pp 133–141. [Google Scholar]
- 81.Thomé-Duret V.; Gangnerau MN.; Zhang Y.; Wilson GS.; Reach G. Diabetes & metabolism 1996, 22, 174–178. [PubMed] [Google Scholar]
- 82.Gifford R.; Kehoe JJ.; Barnes SL.; Kornilayev BA.; Alterman MA.; Wilson GS. Biomaterials 2006, 27, 2587–98. [DOI] [PubMed] [Google Scholar]
- 83.Martin P.; Leibovich SJ. Trends Cell Biol 2005, 15, 599–607. [DOI] [PubMed] [Google Scholar]
- 84.Klueh U.; Liu Z.; Feldman B.; Henning TP.; Cho B.; Ouyang T.; Kreutzer D. J Diabetes Sci Technol 2011, 5, 583–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Helton KL.; Ratner BD.; Wisniewski NA. J Diabetes Sci Technol 2011, 5, 632–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Helton KL.; Ratner BD.; Wisniewski NA. J Diabetes Sci Technol 2011, 5, 647–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wu Y.; Meyerhoff ME. Talanta 2008, 75, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang Y.; Vaddiraju S.; Gu B.; Papadimitrakopoulos F.; Burgess DJ. J Diabetes Sci Technol 2015, 9, 966–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tang L.; Jennings TA.; Eaton JW. Proc Natl Acad Sci USA 1998, 95, 8841–8846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klueh U.; Kaur M.; Qiao Y.; Kreutzer DL. Biomaterials 2010, 31, 4540–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Egozi EI.; Ferreira AM.; Burns AL.; Gamelli RL.; Dipietro LA. Wound Repair Regen 2003, 11, 46–54. [DOI] [PubMed] [Google Scholar]
- 92.Thevenot PT.; Baker DW.; Weng H.; Sun M-W.; Tang L. Biomaterials 2011, 32, 8394–8403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Avula MN.; Rao AN.; McGill LD.; Grainger DW.; Solzbacher F. Acta Biomater 2014, 10, 1856–63. [DOI] [PubMed] [Google Scholar]
- 94.Mosser DM.; Edwards JP. Nat Rev Immunol 2008, 8, 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mantovani A.; Biswas SK.; Galdiero MR.; Sica A.; Locati M. J Pathol 2013, 229, 176–85. [DOI] [PubMed] [Google Scholar]
- 96.Martinez FO. Eur J Immunol 2011, 41, 1531–1534. [DOI] [PubMed] [Google Scholar]
- 97.Shin JH.; Schoenfisch MH. Analyst 2006, 131, 609–615. [DOI] [PubMed] [Google Scholar]
- 98.Wilson GS.; Ammam M. Febs j 2007, 274, 5452–61. [DOI] [PubMed] [Google Scholar]
- 99.Wilson GS.; Gifford R. Biosens Bioelectron 2005, 20, 2388–2403. [DOI] [PubMed] [Google Scholar]
- 100.Wilson GS.; Hu Y. Chem Rev 2000, 100, 2693–704. [DOI] [PubMed] [Google Scholar]
- 101.Liu WF.; Ma M.; Bratlie KM.; Dang TT.; Langer R.; Anderson DG. Biomaterials 2011, 32, 1796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhu L.; Zhao Q.; Yang T.; Ding W.; Zhao Y. Int Rev Immunol 2015, 34, 82–100. [DOI] [PubMed] [Google Scholar]
- 103.Murray Peter J.; Allen Judith E.; Biswas Subhra K.; Fisher Edward A.; Gilroy Derek W.; Goerdt S.; Gordon S.; Hamilton John A.; Ivashkiv Lionel B.; Lawrence T.; Locati M.; Mantovani A.; Martinez Fernando O.; Mege J-L.; Mosser David M.; Natoli G.; Saeij Jeroen P.; Schultze Joachim L.; Shirey Kari A.; Sica A.; Suttles J.; Udalova I.; van Ginderachter Jo A.; Vogel Stefanie N.; Wynn Thomas A. Immunity 2014, 41, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Novak MT.; Yuan F.; Reichert WM. Anal Bioanal Chem 2010, 398, 1695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Novak MT.; Yuan F.; Reichert WM. Biomaterials 2014, 35, 9563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Klueh U.; Frailey J.; Qiao Y.; Antar O.; Kreutzer DL. Biomaterials 2014, 35, 3145–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Novak MT.; Reichert WM. J Diabetes Sci Technol 2015, 9, 993–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gratchev A.; Guillot P.; Hakiy N.; Politz O.; Orfanos CE.; Schledzewski K.; Goerdt S. Scand J Immunol 2001, 53, 386–92. [DOI] [PubMed] [Google Scholar]
- 109.Li AG.; Quinn MJ.; Siddiqui Y.; Wood MD.; Federiuk IF.; Duman HM.; Ward WK. J Biomed Mater Res A 2007, 82, 498–508. [DOI] [PubMed] [Google Scholar]
- 110.Spiller KL.; Anfang RR.; Spiller KJ.; Ng J.; Nakazawa KR.; Daulton JW.; Vunjak-Novakovic G. Biomaterials 2014, 35, 4477–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McNally AK.; Anderson JM. The American Journal of Pathology 2002, 160, 621–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brodbeck WG.; Shive MS.; Colton E.; Nakayama Y.; Matsuda T.; Anderson JM. J Biomed Mater Res 2001, 55, 661–8. [DOI] [PubMed] [Google Scholar]
- 113.Zhao Q.; Topham N.; Anderson JM.; Hiltner A.; Lodoen G.; Payet CR. J Biomed Mater Res 1991, 25, 177–183. [DOI] [PubMed] [Google Scholar]
- 114.Zhao QH.; McNally AK.; Rubin KR.; Renier M.; Wu Y.; Rose-Caprara V.; Anderson JM.; Hiltner A.; Urbanski P.; Stokes K. J Biomed Mater Res 1993, 27, 379–88. [DOI] [PubMed] [Google Scholar]
- 115.Zdrahala RJ.; Zdrahala IJ. J Biomater Appl 1999, 14, 67–90. [DOI] [PubMed] [Google Scholar]
- 116.Kenneth Ward W. J Diabetes Sci Technol 2008, 2, 768–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sharkawy AA.; Klitzman B.; Truskey GA.; Reichert WM. J Biomed Mater Res 1997, 37, 401–12. [DOI] [PubMed] [Google Scholar]
- 118.Sharkawy AA.; Klitzman B.; Truskey GA.; Reichert WM. J Biomed Mater Res 1998, 40, 598–605. [DOI] [PubMed] [Google Scholar]
- 119.Sharkawy AA.; Klitzman B.; Truskey GA.; Reichert WM. J Biomed Mater Res 1998, 40, 586–97. [DOI] [PubMed] [Google Scholar]
- 120.Sieminski AL.; Gooch KJ. Biomaterials 2000, 21, 2232–41. [DOI] [PubMed] [Google Scholar]
- 121.Worthington KS.; Wiley LA.; Mullins RF.; Tucker BA.; Nuxoll E. J Biomed Mater Res B Appl Biomater 2015. [DOI] [PMC free article] [PubMed]
- 122.Klueh U.; Dorsky DI.; Kreutzer DL. Biomaterials 2005, 26, 1155–63. [DOI] [PubMed] [Google Scholar]
- 123.Szarowski DH.; Andersen MD.; Retterer S.; Spence AJ.; Isaacson M.; Craighead HG.; Turner JN.; Shain W. Brain Res 2003, 983, 23–35. [DOI] [PubMed] [Google Scholar]
- 124.Karumbaiah L.; Saxena T.; Carlson D.; Patil K.; Patkar R.; Gaupp EA.; Betancur M.; Stanley GB.; Carin L.; Bellamkonda RV. Biomaterials 2013, 34, 8061–8074. [DOI] [PubMed] [Google Scholar]
- 125.Kozai TD.; Marzullo TC.; Hooi F.; Langhals NB.; Majewska AK.; Brown EB.; Kipke DR. J Neural Eng 2010, 7, 046011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Clark JJ.; Sandberg SG.; Wanat MJ.; Gan JO.; Horne EA.; Hart AS.; Akers CA.; Parker JG.; Willuhn I.; Martinez V.; Evans SB.; Stella N.; Phillips PE. Nat Methods 2010, 7, 126–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Clark JJ.; Sandberg SG.; Wanat MJ.; Gan JO.; Horne EA.; Hart AS.; Akers CA.; Parker JG.; Willuhn I.; Martinez V.; Evans SB.; Stella N.; Phillips PE. M. Nat Meth 2010, 7, 126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Subbaroyan J.; Martin DC.; Kipke DR. J Neural Eng 2005, 2, 103–13. [DOI] [PubMed] [Google Scholar]
- 129.Skousen JL.; Bridge MJ.; Tresco PA. Biomaterials 2015, 36, 33–43. [DOI] [PubMed] [Google Scholar]
- 130.Weldon DT.; Rogers SD.; Ghilardi JR.; Finke MP.; Cleary JP.; O’Hare E.; Esler WP.; Maggio JE.; Mantyh PW. J Neurosci 1998, 18, 2161–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Giulian D.; Vaca K.; Corpuz M. J Neurosci 1993, 13, 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Brown GC.; Neher JJ. Nat Rev Neurosci 2014, 15, 209–216. [DOI] [PubMed] [Google Scholar]
- 133.Bovolenta P.; Fernaud-Espinosa I. Prog Neurobiol 2000, 61, 113–32. [DOI] [PubMed] [Google Scholar]
- 134.Roitbak T.; Sykova E. Glia 1999, 28, 40–8. [DOI] [PubMed] [Google Scholar]
- 135.Williams DF. J Biomed Eng 1989, 11, 185–191. [DOI] [PubMed] [Google Scholar]
- 136.Williams DF. Biomaterials 2008, 29, 2941–2953. [DOI] [PubMed] [Google Scholar]
- 137.Gombotz WR.; Guanghui W.; Horbett TA.; Hoffman AS. J Biomed Mater Res 1991, 25, 1547–1562. [DOI] [PubMed] [Google Scholar]
- 138.Deible CR.; Petrosko P.; Johnson PC.; Beckman EJ.; Russell AJ.; Wagner WR. Biomaterials 1999, 20, 101–109. [DOI] [PubMed] [Google Scholar]
- 139.Chen S.; Li L.; Zhao C.; Zheng J. Polymer 2010, 51, 5283–5293. [Google Scholar]
- 140.Browning MB.; Cereceres SN.; Luong PT.; Cosgriff-Hernandez EM. J Biomed Mater Res A 2014, 102, 4244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Venault A.; Zheng Y-S.; Chinnathambi A.; Alharbi SA.; Ho H-T.; Chang Y.; Chang Y. Langmuir 2015, 31, 2861–2869. [DOI] [PubMed] [Google Scholar]
- 142.Tanaka M.; Hayashi T.; Morita S. Polym J 2013, 45, 701–710. [Google Scholar]
- 143.Tanaka M.; Mochizuki A.; Ishii N.; Motomura T.; Hatakeyama T. Biomacromolecules 2002, 3, 36–41. [DOI] [PubMed] [Google Scholar]
- 144.Tanaka M.; Mochizuki A.; Shiroya T.; Motomura T.; Shimura K.; Onishi M.; Okahata Y. Coll Surf A 2002, 203, 195–204. [Google Scholar]
- 145.Zhao C.; Zhao J.; Li X.; Wu J.; Chen S.; Chen Q.; Wang Q.; Gong X.; Li L.; Zheng J. Biomaterials 2013, 34, 4714–4724. [DOI] [PubMed] [Google Scholar]
- 146.Leng C.; Hung HC.; Sun S.; Wang D.; Li Y.; Jiang S.; Chen Z. ACS Appl Mater Interfaces 2015, 7, 16881–8. [DOI] [PubMed] [Google Scholar]
- 147.Zhang Z.; Zhang M.; Chen S.; Horbett TA.; Ratner BD.; Jiang S. Biomaterials 2008, 29, 4285–4291. [DOI] [PubMed] [Google Scholar]
- 148.Zhang Z.; Chen S.; Chang Y.; Jiang S. J Phys Chem B 2006, 110, 10799–10804. [DOI] [PubMed] [Google Scholar]
- 149.Zhang Z.; Chao T.; Chen S.; Jiang S. Langmuir 2006, 22, 10072–10077. [DOI] [PubMed] [Google Scholar]
- 150.He Y.; Tsao H-K.; Jiang S. J Phys Chem B 2012, 116, 5766–5770. [DOI] [PubMed] [Google Scholar]
- 151.Wu Y.; Simonovsky FI.; Ratner BD.; Horbett TA. J Biomed Mater Res A 2005, 74A, 722–738. [DOI] [PubMed] [Google Scholar]
- 152.Horbett TA., Adsorbed Proteins on Biomaterials in Biomaterials Science: An Introduction to Materials in Medicine, Ratner BD.; Hoffman AS.; Schoen FJ.; Lemons JE., Eds. Academic Press, Inc: 2013; pp 394–407. [Google Scholar]
- 153.Jenney CR.; Anderson JM. J Biomed Mater Res 2000, 49, 435–47. [DOI] [PubMed] [Google Scholar]
- 154.Sivaraman B.; Latour RA. Biomaterials 2010, 31, 1036–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Andrade JD.; Hlady V., Protein Adsorption and Materials Biocompatibility: A Tutorial Review and Suggested Hypotheses in Biopolymers/Non-Exclusion Hplc, Springer Berlin Heidelberg: Berlin, Heidelberg, 1986; pp 1–63. [Google Scholar]
- 156.Murugesan S.; Xie J.; Linhardt RJ. Curr Top Med Chem 2008, 8, 80–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bentolila A.; Vlodavsky I.; Haloun C.; Domb AJ. Polym Adv Technol 2000, 11, 377–387. [Google Scholar]
- 158.Hirsh J.; Anand SS.; Halperin JL.; Fuster V. Arterioscler Thromb Vasc Biol 2001, 21, 1094–1096. [DOI] [PubMed] [Google Scholar]
- 159.Lindahl U.; Thunberg L.; Bäckström G.; Riesenfeld J.; Nordling K.; Björk I. J Biol Chem 1984, 259, 12368–12376. [PubMed] [Google Scholar]
- 160.Yang L.; Manithody C.; Rezaie AR. Blood 2004, 104, 1753–1759. [DOI] [PubMed] [Google Scholar]
- 161.Björk I.; Lindahl U. Mol Cell Biochem 1982, 48, 161–182. [DOI] [PubMed] [Google Scholar]
- 162.Kelton JG.; Warkentin TE. Blood 2008, 112, 2607–16. [DOI] [PubMed] [Google Scholar]
- 163.Kara F.; Aksoy EA.; Yuksekdag Z.; Aksoy S.; Hasirci N. Appl Surf Sci 2015, 357, Part B, 1692–1702. [Google Scholar]
- 164.Song Y.-q.; Gao Y.-l.; Pan Z.-c.; Zhang Y.; Li J.-h.; Wang K.-j.; Li J.-s.; Tan H.; Fu Q. Chin J Polym Sci 2016, 34, 679–687. [Google Scholar]
- 165.Kolar M.; Mozetič M.; Stana-Kleinschek K.; Fröhlich M.; Turk B.; Vesel A. Materials 2015, 8, 1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gore S.; Andersson J.; Biran R.; Underwood C.; Riesenfeld J. J Biomed Mater Res B Appl Biomater 2014, 102, 1817–24. [DOI] [PubMed] [Google Scholar]
- 167.Seib FP.; Herklotz M.; Burke KA.; Maitz MF.; Werner C.; Kaplan DL. Biomaterials 2014, 35, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Chou C-C.; Hsin S-W.; Lin H-C.; Yeh C-H.; Wu R.; Cherng W-J. Surf Coat Technol.
- 169.Edagawa K.; Fuchiwaki Y.; Yasuzawa M. J Electrochem Soc 2014, 161, B3111–B3115. [Google Scholar]
- 170.Clarke WL. Diabetes Technol Ther 2005, 7, 776–9. [DOI] [PubMed] [Google Scholar]
- 171.Michanetzis GPA.; Katsala N.; Missirlis YF. Biomaterials 2003, 24, 677–688. [DOI] [PubMed] [Google Scholar]
- 172.Fry AK.; Schilke KF.; McGuire J.; Bird KE. J Biomed Mater Res B 2010, 94B, 187–195. [DOI] [PubMed] [Google Scholar]
- 173.Wang L.; Su B.; Cheng C.; Ma L.; Li S.; Nie S.; Zhao C. J Mater Chem B 2015, 3, 1391–1404. [DOI] [PubMed] [Google Scholar]
- 174.Xu Y.; Masuko S.; Takieddin M.; Xu H.; Liu R.; Jing J.; Mousa SA.; Linhardt RJ.; Liu J. Science 2011, 334, 498–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Conn G.; Kidane AG.; Punshon G.; Kannan RY.; Hamilton G.; Seifalian AM. Expert Rev Med Devic 2006, 3, 245–261. [DOI] [PubMed] [Google Scholar]
- 176.Major TC.; Brisbois EJ.; Jones AM.; Zanetti ME.; Annich GM.; Bartlett RH.; Handa H. Biomaterials 2014, 35, 7271–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.He C.; Shi Z-Q.; Ma L.; Cheng C.; Nie C-X.; Zhou M.; Zhao C-S. J Mater Chem B 2015, 3, 592–602. [DOI] [PubMed] [Google Scholar]
- 178.Wang L.; Li H.; Chen S.; Nie C.; Cheng C.; Zhao C. ACS Biomater Sci Eng 2015, 1, 1183–1193. [DOI] [PubMed] [Google Scholar]
- 179.Radomski MW.; Palmer RMJ.; Moncada S. The Lancet 1987, 330, 1057–1058. [DOI] [PubMed] [Google Scholar]
- 180.Cooke JP.; Losordo DW. Circulation 2002, 105, 2133–2135. [DOI] [PubMed] [Google Scholar]
- 181.Schwentker A.; Vodovotz Y.; Weller R.; Billiar TR. Nitric Oxide 2002, 7, 1–10. [DOI] [PubMed] [Google Scholar]
- 182.Carpenter AW.; Schoenfisch MH. Chem Soc Rev 2012, 41, 3742–3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nathan C. Faseb j 1992, 6, 3051–64. [PubMed] [Google Scholar]
- 184.O W Griffith a.; Stuehr DJ. Ann Rev Physiol 1995, 57, 707–734. [DOI] [PubMed] [Google Scholar]
- 185.Bogdan C. Nat Immunol 2001, 2, 907–16. [DOI] [PubMed] [Google Scholar]
- 186.Aktan F. Life Sciences 2004, 75, 639–653. [DOI] [PubMed] [Google Scholar]
- 187.Vaughn MW.; Kuo L.; Liao JC. Am J Physiol 1998, 274, H2163–76. [DOI] [PubMed] [Google Scholar]
- 188.Cooke JP.; Stamler J.; Andon N.; Davies PF.; McKinley G.; Loscalzo J. Am J Physiol 1990, 259, H804–12. [DOI] [PubMed] [Google Scholar]
- 189.Stamler J.; Mendelsohn ME.; Amarante P.; Smick D.; Andon N.; Davies PF.; Cooke JP.; Loscalzo J. Circ Res 1989, 65, 789–95. [DOI] [PubMed] [Google Scholar]
- 190.Wang G-R.; Zhu Y.; Halushka PV.; Lincoln TM.; Mendelsohn ME. Proc Natl Acad Sci 1998, 95, 4888–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Gries A.; Bode C.; Peter K.; Herr A.; Bohrer H.; Motsch J.; Martin E. Circulation 1998, 97, 1481–7. [DOI] [PubMed] [Google Scholar]
- 192.Walford G.; Loscalzo J. J Thromb Haemost 2003, 1, 2112–8. [DOI] [PubMed] [Google Scholar]
- 193.Murohara T.; Parkinson SJ.; Waldman SA.; Lefer AM. Arterioscler Thromb Vasc Biol 1995, 15, 2068–2075. [DOI] [PubMed] [Google Scholar]
- 194.Freedman JE.; Loscalzo J.; Barnard MR.; Alpert C.; Keaney JF.; Michelson AD. J Clin Invest 1997, 100, 350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Keefer LK. ACS Chem Biol 2011, 6, 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Broniowska KA.; Diers AR.; Hogg N. Biochim Biophys Acta 2013, 1830, 3173–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Riccio DA.; Nugent JL.; Schoenfisch MH. Chem Mater 2011, 23, 1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.McCarthy CW.; Guillory Ii RJ.; Goldman J.; Frost MC. ACS Appl Mater Interfaces 2016. [DOI] [PubMed]
- 199.Mowery KA.; H Schoenfisch M.; Saavedra JE.; Keefer LK.; Meyerhoff ME. Biomaterials 2000, 21, 9–21. [DOI] [PubMed] [Google Scholar]
- 200.Coneski PN.; Schoenfisch MH. Org Lett 2009, 11, 5462–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Batchelor MM.; Reoma SL.; Fleser PS.; Nuthakki VK.; Callahan RE.; Shanley CJ.; Politis JK.; Elmore J.; Merz SI.; Meyerhoff ME. J Med Chem 2003, 46, 5153–61. [DOI] [PubMed] [Google Scholar]
- 202.Riccio DA.; Schoenfisch MH. Chem Soc Rev 2012, 41, 3731–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Yan Q.; Major TC.; Bartlett RH.; Meyerhoff ME. Biosens Bioelectron 2011, 26, 4276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Wolf AK.; Qin Y.; Major TC.; Meyerhoff ME. Chin Chem Lett 2015, 26, 464–468. [Google Scholar]
- 205.Yang J.; Welby JL.; Meyerhoff ME. Langmuir 2008, 24, 10265–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Hwang S.; Meyerhoff ME. Biomaterials 2008, 29, 2443–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Liu K.; Meyerhoff ME. J Mater Chem 2012, 22, 18784–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Cha W.; Meyerhoff ME. Biomaterials 2007, 28, 19–27. [DOI] [PubMed] [Google Scholar]
- 209.Ren H.; Wu J.; Xi C.; Lehnert N.; Major T.; Bartlett RH.; Meyerhoff ME. ACS Appl Mater Interfaces 2014, 6, 3779–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Hofler L.; Koley D.; Wu J.; Xi C.; Meyerhoff ME. RSC Advances 2012, 2, 6765–6767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ren H.; Colletta A.; Koley D.; Wu J.; Xi C.; Major TC.; Bartlett RH.; Meyerhoff ME. Bioelectrochemistry 2015, 104, 10–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zhou Z.; Meyerhoff ME. Biomaterials 2005, 26, 6506–17. [DOI] [PubMed] [Google Scholar]
- 213.Zhang L.; Cao Z.; Bai T.; Carr L.; Ella-Menye J-R.; Irvin C.; Ratner BD.; Jiang S. Nat Biotech 2013, 31, 553–556. [DOI] [PubMed] [Google Scholar]
- 214.Zhang Z.; Chao T.; Liu L.; Cheng G.; Ratner BD.; Jiang S. J Biomater Sci Polym Ed 2009, 20, 1845–1859. [DOI] [PubMed] [Google Scholar]
- 215.Chen S.; Jones JA.; Xu Y.; Low HY.; Anderson JM.; Leong KW. Biomaterials 2010, 31, 3479–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Brauker JH.; Carr-Brendel VE.; Martinson LA.; Crudele J.; Johnston WD.; Johnson RC. J Biomed Mater Res 1995, 29, 1517–24. [DOI] [PubMed] [Google Scholar]
- 217.Koschwanez HE.; Reichert WM.; Klitzman B. J Biomed Mater Res A 2010, 93, 1348–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Koschwanez HE.; Yap FY.; Klitzman B.; Reichert WM. J Biomed Mater Res A 2008, 87, 792–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bota PC.; Collie AM.; Puolakkainen P.; Vernon RB.; Sage EH.; Ratner BD.; Stayton PS. J Biomed Mater Res A 2010, 95, 649–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Bryant SJ.; Cuy JL.; Hauch KD.; Ratner BD. Biomaterials 2007, 28, 2978–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Sussman EM.; Halpin MC.; Muster J.; Moon RT.; Ratner BD. Ann Biomed Eng 2014, 42, 1508–16. [DOI] [PubMed] [Google Scholar]
- 222.Bryers JD.; Giachelli CM.; Ratner BD. Biotechnol Bioeng 2012, 109, 1898–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Brown BN.; Ratner BD.; Goodman SB.; Amar S.; Badylak SF. Biomaterials 2012, 33, 3792–3802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Teo WE.; Ramakrishna S. Nanotechnology 2006, 17, R89. [DOI] [PubMed] [Google Scholar]
- 225.Li D.; Xia Y. Adv Mater 2004, 16, 1151–1170. [Google Scholar]
- 226.Vacanti NM.; Cheng H.; Hill PS.; Guerreiro JDT.; Dang TT.; Ma M.; Watson S.; Hwang NS.; Langer R.; Anderson DG. Biomacromolecules 2012, 13, 3031–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Garg K.; Pullen NA.; Oskeritzian CA.; Ryan JJ.; Bowlin GL. Biomaterials 2013, 34, 4439–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Agarwal S.; Wendorff JH.; Greiner A. Polymer 2008, 49, 5603–5621. [Google Scholar]
- 229.Cao H.; McHugh K.; Chew SY.; Anderson JM. J Biomed Mater Res A 2010, 93, 1151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Koh A.; Lu Y.; Schoenfisch MH. Anal Chem 2013, 85, 10488–10494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Dubreuil P.; Letard S.; Ciufolini M.; Gros L.; Humbert M.; Casteran N.; Borge L.; Hajem B.; Lermet A.; Sippl W.; Voisset E.; Arock M.; Auclair C.; Leventhal PS.; Mansfield CD.; Moussy A.; Hermine O. PLoS One 2009, 4, e7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Reber L.; Da Silva CA.; Frossard N. Eur J Pharmacol 2006, 533, 327–340. [DOI] [PubMed] [Google Scholar]
- 233.Zdolsek J.; Eaton JW.; Tang L. J Transl Med 2007, 5, 31–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Avula M.; Jones D.; Rao AN.; McClain D.; McGill LD.; Grainger DW.; Solzbacher F. Biosens Bioelectron 2016, 77, 149–156. [DOI] [PubMed] [Google Scholar]
- 235.Avula MN.; Rao AN.; McGill LD.; Grainger DW.; Solzbacher F. Biomaterials 2013, 34, 9737–46. [DOI] [PubMed] [Google Scholar]
- 236.Re F.; Muzio M.; De Rossi M.; Polentarutti N.; Giri JG.; Mantovani A.; Colotta F. J Exp Med 1994, 179, 739–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Auphan N.; DiDonato JA.; Rosette C.; Helmberg A.; Karin M. Science 1995, 270, 286–290. [DOI] [PubMed] [Google Scholar]
- 238.Hori Y.; Hu D-E.; Yasui K.; Smither RL.; Gresham GA.; Fan T-PD. Brit J Pharmacol 1996, 118, 1584–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ward WK.; Hansen JC.; Massoud RG.; Engle JM.; Takeno MM.; Hauch KD. J Biomed Mater Res A 2010, 94, 280–7. [DOI] [PubMed] [Google Scholar]
- 240.Patil SD.; Papadimitrakopoulos F.; Burgess DJ. Diabetes Technol Ther 2004, 6, 887–97. [DOI] [PubMed] [Google Scholar]
- 241.Kastellorizios M.; Papadimitrakopoulos F.; Burgess DJ. J Control Release 2015, 202, 101–7. [DOI] [PubMed] [Google Scholar]
- 242.Kastellorizios M.; Tipnis N.; Papadimitrakopoulos F.; Burgess DJ. Mol Pharm 2015, 12, 3332–8. [DOI] [PubMed] [Google Scholar]
- 243.Klueh U.; Kaur M.; Montrose DC.; Kreutzer DL. J Diabetes Sci Technol 2007, 1, 496–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Norton LW.; Koschwanez HE.; Wisniewski NA.; Klitzman B.; Reichert WM. J Biomed Mater Res A 2007, 81, 858–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Moura SAL.; Lima LDC.; Andrade SP.; Silva-Cunha Junior AD.; Órefice RL.; Ayres E.; Da Silva GR. J Pharm Sci 100, 2886–2895. [DOI] [PubMed] [Google Scholar]
- 246.Nakao S.; Hata Y.; Miura M.; Noda K.; Kimura YN.; Kawahara S.; Kita T.; Hisatomi T.; Nakazawa T.; Jin Y.; Dana MR.; Kuwano M.; Ono M.; Ishibashi T.; Hafezi-Moghadam A. Am J Pathol 171, 1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Patil SD.; Papadmitrakopoulos F.; Burgess DJ. J Control Release 2007, 117, 68–79. [DOI] [PubMed] [Google Scholar]
- 248.Kastellorizios M.; Papadimitrakopoulos F.; Burgess DJ. J Control Release 2015, 214, 103–11. [DOI] [PubMed] [Google Scholar]
- 249.Price CF.; Burgess DJ.; Kastellorizios M. J Control Release 2016, 235, 176–181. [DOI] [PubMed] [Google Scholar]
- 250.Wetzler C.; Kampfer H.; Pfeilschifter J.; Frank S. Biochem Biophys Res Commun 2000, 274, 689–96. [DOI] [PubMed] [Google Scholar]
- 251.Deakin AM.; Payne AN.; Whittle BJ.; Moncada S. Cytokine 1995, 7, 408–16. [DOI] [PubMed] [Google Scholar]
- 252.Frank S.; Kämpfer H.; Wetzler C.; Stallmeyer B.; Pfeilschifter J. Biochem J 2000, 347, 265–273. [PMC free article] [PubMed] [Google Scholar]
- 253.Hill JR.; Corbett JA.; Kwon G.; Marshall CA.; McDaniel ML. J Biol Chem 1996, 271, 22672–8. [DOI] [PubMed] [Google Scholar]
- 254.Inoue N.; Venema RC.; Sayegh HS.; Ohara Y.; Murphy TJ.; Harrison DG. Arterioscler Thromb Vasc Biol 1995, 15, 1255–61. [DOI] [PubMed] [Google Scholar]
- 255.van der Zee R.; Murohara T.; Luo Z.; Zollmann F.; Passeri J.; Lekutat C.; Isner JM. Circulation 1997, 95, 1030–7. [DOI] [PubMed] [Google Scholar]
- 256.Dulak J.; Jozkowicz A.; Dembinska-Kiec A.; Guevara I.; Zdzienicka A.; Zmudzinska-Grochot D.; Florek I.; Wojtowicz A.; Szuba A.; Cooke JP. Arterioscler Thromb Vasc Biol 2000, 20, 659–66. [DOI] [PubMed] [Google Scholar]
- 257.Hetrick EM.; Prichard HL.; Klitzman B.; Schoenfisch MH. Biomaterials 2007, 28, 4571–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Nichols SP.; Koh A.; Brown NL.; Rose MB.; Sun B.; Slomberg DL.; Riccio DA.; Klitzman B.; Schoenfisch MH. Biomaterials 2012, 33, 6305–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Soto RJ.; Yang L.; Schoenfisch MH. ACS Appl Mater Interfaces 2016, 8, 2220–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Nichols SP.; Le NN.; Klitzman B.; Schoenfisch MH. Anal Chem 2011, 83, 1180–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]