

Abstract
Ni-catalyzed C(sp3)-O bond activation provides a useful approach to synthesize enantioenriched products from readily available enantioenriched benzylic alcohol derivatives. The control of stereospecificity is key to the success of these transformations. To elucidate the reversed stereospecificity and chemoselectivity of Ni-catalyzed Kumada and cross-electrophile coupling reactions with benzylic ethers, a combined computational and experimental study is performed to reach a unified mechanistic understanding. Kumada coupling proceeds via a classic cross-coupling mechanism. Initial rate-determining oxidative addition occurs with stereoinversion of the benzylic stereogenic center. Subsequent transmetallation with the Grignard reagent and syn reductive elimination produces the Kumada coupling product with overall stereoinversion at the benzylic position. The cross-electrophile coupling reaction initiates with the same benzylic C-O bond cleavage and transmetallation to form a common benzylnickel intermediate. However, the presence of the tethered alkyl chloride allows a facile intramolecular SN2 attack by the benzylnickel moiety. This step circumvents the competing Kumada coupling, leading to the excellent chemoselectivity of cross-electrophile coupling. These mechanisms account for the observed stereospecificity of the Kumada and cross-electrophile couplings, providing a rationale for double inversion of the benzylic stereogenic center in cross-electrophile coupling. The improved mechanistic understanding will enable design of stereoselective transformations involving Ni-catalyzed C(sp3)-O bond activation.
Graphical Abstract
Introduction
Cross-coupling (XC) and cross-electrophile coupling (XEC) reactions that generate new C-C bonds between sp3-hybridized carbons are of great synthetic interest, particularly as medicinal chemistry shifts away from flatland.1 One of our laboratories has developed a series of such transformations employing benzylic ethers.2–6At the foundation of these projects has been the idea that the reactions would share a key elementary step: stereospecific oxidative addition of the C-O bond. This step was envisioned to control the stereochemical outcome of the reactions, and to likely be the rate-determining step of the catalytic cycles. In this manuscript, we report our collaborative computational and experimental studies to delineate the mechanistic similarities and differences between Kumada and XEC reactions of closely related benzylic ethers. This work builds on our prior collaboration to elucidate the basis for the ligand-controlled stereochemical switch in Suzuki coupling reactions of benzylic esters.7
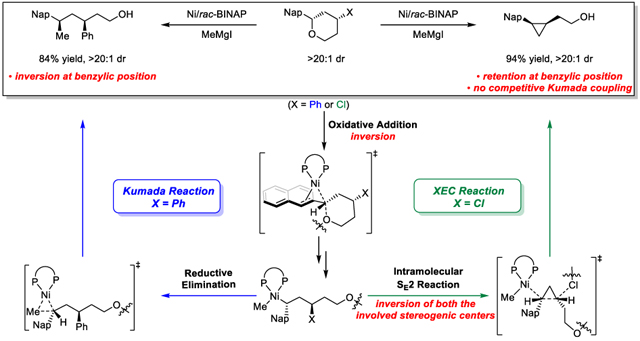
We envisioned that the reactions in Scheme 1 would serve as ideal test cases for comparison of XC and XEC reaction manifolds.3a,8 These reactions employ identical catalysts and reaction conditions as well as similar substrates, however, there are notable differences when comparing the reaction outcomes. Kumada reactions of benzylic ethers reliably proceed with inversion at the benzylic center (Scheme 1a). In contrast, XECs of closely related benzylic ethers proceed with retention at the benzylic center (Scheme 1b) and provide only XEC products, with no competitive Kumada cross-coupling observed. To reach a unified explanation for substrate-dependant chemoselectivity and stereospecificity, we undertook a combined computational and experimental mechanistic study.9 The increased understanding of the mechanistic underpinnings of selectivity in nickel-catalyzed Kumada and XEC reactions will provide a useful guide for future design of stereoselective transformations involving Ni-catalyzed C(sp3)-O bond activation.10
Scheme 1.
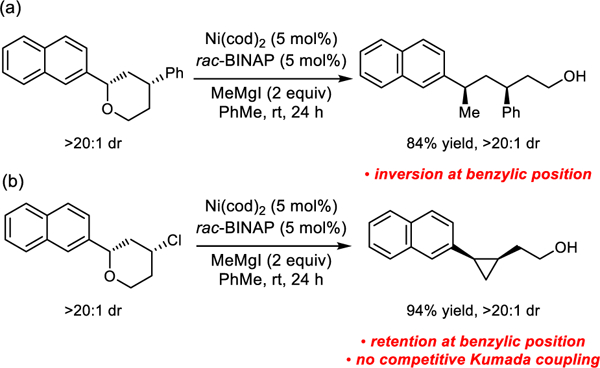
Chemoselectivity and stereospecificity of Ni-catalyzed Kumada and XEC reactions of benzylic ethers.
Computational Methods
All density functional theory (DFT) calculations were conducted with the Gaussian 09 software package.11 Geometry optimizations of all the intermediates and transition states were performed at the B3LYP12 level of theory with def2-SVP13 basis set, including solvation energy corrections and Grimme’s D3 (BJ-damping) dispersion corrections.14 Based on the optimized structures, vibrational frequencies were calculated at the same level of theory to evaluate its zero-point vibrational energy (ZPVE) and thermal corrections at 298 K. The single-point energies were computed with B3LYP functional and def2-TZVPP13,15 basis set, including solvation energy corrections and Grimme’s D3 (BJ-damping) dispersion corrections. The solvation energies were evaluated by a self-consistent reaction field (SCRF) using SMD model.16 Fragment distortion and interaction energies were calculated at the B3LYP level of theory with def2-TZVPP basis set, including Grimme’s D3 (BJ-damping) dispersion corrections without the inclusion of solvation energy corrections. The details of distortion/interaction analysis are provided in the Supporting Information. Extensive conformational searches for the intermediates and transition states have been conducted to ensure that the lowest energy conformers were located. The 3D diagrams of molecules were generated using CYLView,17 all the hydrogens were omitted in the CYLView diagrams except for the stereogenic centers.
To correct the entropy change in solution, we applied an empirical approach,18 because there is currently no widely accepted quantum mechanics-based approach to correct entropy in solution. For each component change in a reaction at 298 K and 1 atm, a correction of 4.3 kcal/mol is applied to the reaction free energy (i.e., a reaction from m- to n-components has an additional free energy correction for (n - m) × 4.3 kcal/mol). This approach has been validated through a number of computational and experimental studies. Yu and co-workers have found that the entropy corrections are overestimated by about half in several cycloaddition reactions.19 Wang and co-workers have discovered the improved description of free energy changes in a number of metal-catalyzed reactions using the same empirical approach.20
Results and Discussion
Computational study of Kumada coupling mechanism.
Two possible catalytic cycles for Ni-catalyzed Kumada coupling with benzylic ethers are shown in Scheme 2.21,22 From the active catalyst LnNi(0) species A, one of the proposed cycles begins with oxidative addition cleaving the benzylic C-O bond, generating the zwitterionic η3-nickel(II) species B with inversion at the benzylic center. Complex B undergoes transmetallation with the Grignard reagent to deliver the LnNi(benzyl)(methyl) intermediate C. Alternatively, the LnNi(0) species A can form a nickelate species F with the Grignard reagent.23 Complex F then undergoes oxidative addition to cleave the benzylic C-O bond to generate intermediate C. From C, C-C reductive elimination produces the Kumada coupling product and subsequent product liberation regenerates the active catalyst A.
Scheme 2.
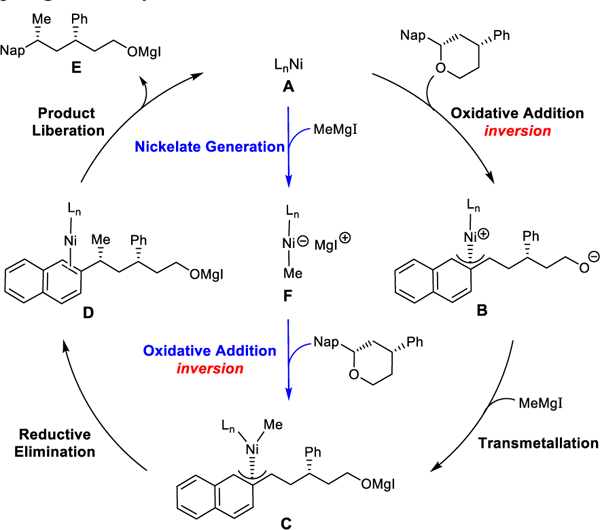
Possible catalytic cycles of Ni-catalyzed Kumada coupling of benzylic ethers.
Prior studies of nickel-catalyzed coupling reactions of allylic ethers have found that magnesium salts play a key role as Lewis acid co-catalysts.22 To evaluate magnesium coordination to the tetrahydropyrans, we examined the relative free energies of complexes of 1 with the Mg(II) species present in the reaction mixture (Scheme 3).24 MgI2 is present in the reaction mixture because of the Schlenk equilibrium25 (i.e., formation of dialkyl magnesium compounds and magnesium halide salts) and competitive Wurtz coupling that occurs during Grignard formation.26 Complexation of 1 with MgI2 and Grignard reagent MgMeI are both exergonic, with the ether-MgI2 complex 3 more favorable by 3.1 kcal/mol (−7.3 kcal/mol vs. −4.2 kcal/mol, Scheme 3). Bonding with MgI2 is stronger because of the higher Lewis acidity as compared to MgMeI. Therefore, the ether-MgI2 complex 3 was identified as the species which interacts with the nickel catalyst in the Kumada coupling.
Scheme 3.
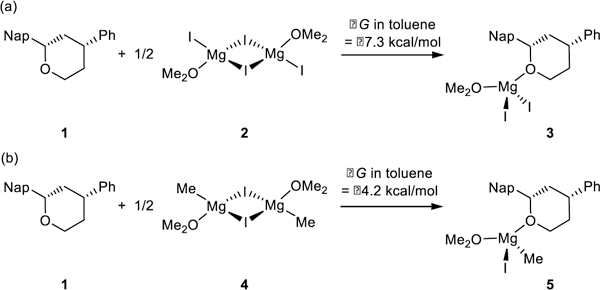
Free energy changes of complexation between benzylic ether and Mg(II) species involved in Kumada coupling.
The free energy changes of the most favorable pathway of Ni/BINAP-catalyzed Kumada coupling with benzylic ether 1 are shown in Figure 1.27 DFT-optimized structures of selected intermediates and transition states are shown in Figure 2. Based on Hartwig’s previous experimental characterization and study of the Ni(BINAP)2 complex,28 we believe this complex could exist as an off-cycle resting state and equilibrate with the active monoligated nickel catalyst. Indeed, our calculations showed that the substrate-ligand exchange of Ni(BINAP)2 (6) to form the substrate-coordinated complex 7 is 13.4 kcal/mol endergonic. Other studied nickel(0) complexes are all much less stable as compared to the catalyst resting state Ni(BINAP)2 (6); see Supporting Information for details (Figure S1). Arene complex 7 undergoes oxidative addition via TS8, via backside attack, generating the zwitterionic η3-nickel(II) species 9 with stereoinversion of the benzylic position. Alternative C-O bond cleavage transition states are all less favorable as compared to TS8. For example, we also located the similar stereoinvertive oxidative addition leading from Grignard reagent complex 5, as well as a number of possible stereoretentive C-O bond cleavage transition states, but these transition states were all significantly higher in energy than TS8 (Figure S2).
Figure 1.
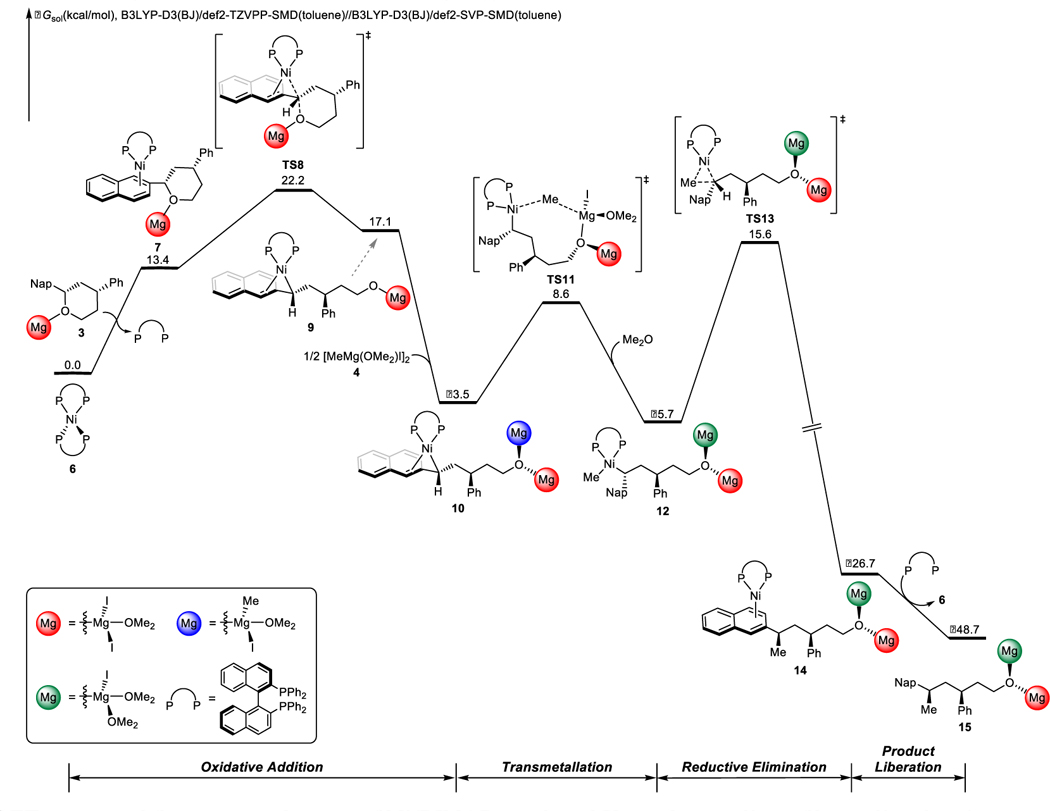
DFT-computed free energy changes of Ni/BINAP-catalyzed Kumada coupling of benzylic ether 1.
Figure 2.
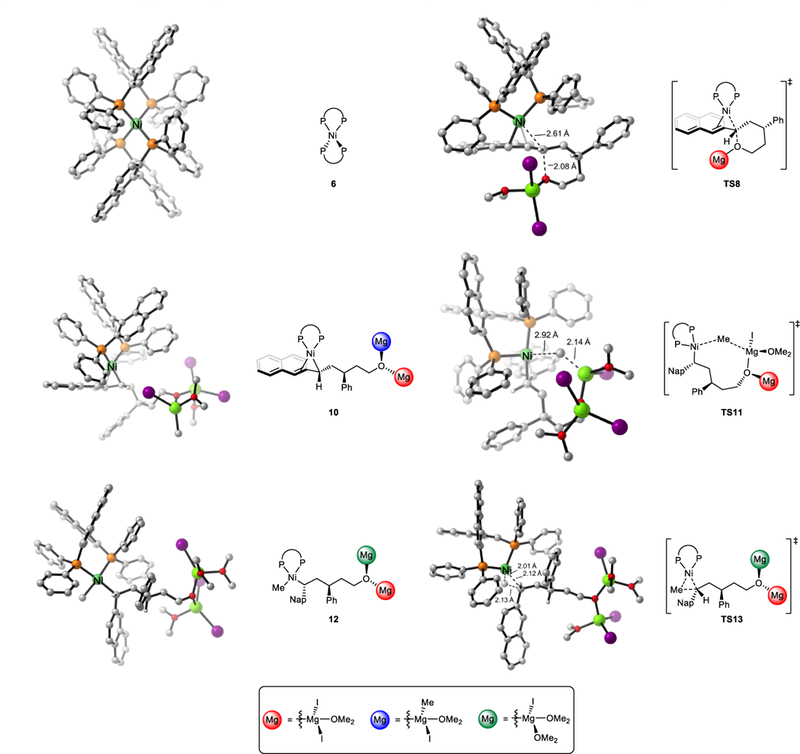
DFT-optimized structures of selected intermediates and transition states involved in Ni-catalyzed Kumada coupling.
Completion of the catalytic cycle requires, in broad strokes, transmetallation and reductive elimination. Based on our calculations, this proceeds first by coordination of Grignard reagent 4 with benzylnickel complex 9, to provide a more stable nickel(II) species 10. This complex (10) undergoes facile transmetallation via TS11 to generate the (BINAP)Ni(benzyl)(methyl) species 12. We also considered the MeMgI addition through an outer-sphere mechanism, this process is less favorable than the inner-sphere process via TS11 (Figure S3). Subsequent syn reductive elimination via TS13 generates the new C-C bond and leads to the product-coordinated complex 14. The reductive elimination can also occur with an η3-benzylnickel complex, but this alternative process is less favorable as compared to TS13 (Figure S4). Product liberation of complex 14 releases Kumada coupling product and regenerates catalyst 6. IRC analysis of key transition states were performed to verify their positions in the free energy surface (Figure S5).
Our calculations find that the alternative pathway shown in Scheme 2, involving a nickelate species (F), is infeasible. The free energy changes of the competing pathways from the Ni(0) catalyst are compared in Figure 3. The interaction between Ni(BINAP) and the Grignard reagent is quite weak, making the formation of complex 16 endergonic by 27.5 kcal/mol as compared to 6. Subsequent methyl group transfer via TS17 is 7.1 kcal/mol less favorable than the irreversible oxidative addition of the C-O bond via TS8. Therefore, methyl group transfer from the Grignard reagent to the Ni(0) complex is not competitive with oxidative addition of the benzylic ether via TS8. We also evaluated methyl group transfers involving the Grignard reagent dimer or coordination of the benzylic ether, and these processes are also not operative (Figure S6).
Figure 3.
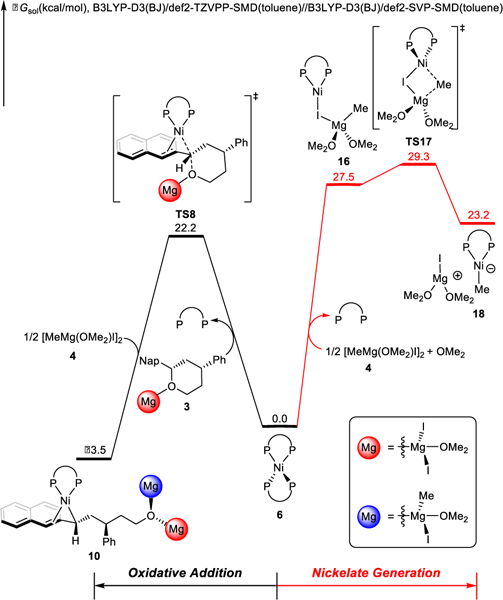
Free energy changes of the competing pathways from Ni(0) catalyst 6 in the Kumada coupling.
The rate-determining oxidative addition step explains the observed reactivity differences between naphthyl and phenyl substrates.8 The overall oxidative addition barrier of naphthyl substrate is 7.4 kcal/mol lower than that of phenyl substrate (TS8 vs. TS21, Figure 4). The coordination of naphthyl moiety is stronger than that of phenyl moiety,29 which stablizes the pre-oxidative addition intermediate (7 vs. 20), as well as the subsequent oxidative addition transition states. Our further Hammett analysis verified the oxidative addition nature of C-O bond cleavage transition state TS8. A linear correlation between the Hammett constant and the computed C-O bond cleavage rate was observed (Figure S7), suggesting that the electron-withdrawing substituent of arene would accelerate the C-O bond cleavage.
Figure 4.
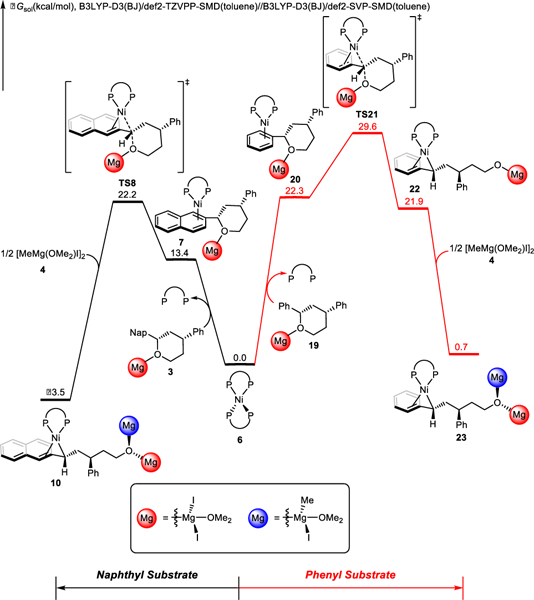
Comparison of the C-O bond cleavage between naphthyl and phenyl substrates.
Origins of stereospecificity of Kumada coupling.
Based on the free energy profile of the Ni/BINAP-catalyzed Kumada coupling with benzylic ether 1 (Figure 1), oxidative addition of the C-O bond is irreversible and determines the overall stereospecificity of the Kumada coupling. Figure 5 shows the possible competing transition states that could result in retention or inversion. The C-O bond cleavage can occur in a stereoinvertive fashion through backside attack, via transition state TS8, or a stereoretentive fashion via the concerted cyclic transition state TS24. TS8 is 37.3 kcal/mol more favorable than TS24, indicating that the C-O bond cleavage occurs with exclusive stereoinversion. These findings are consistent with prior observations that oxidative addition of allylic ethers proceeds with inversion.6b
Figure 5.
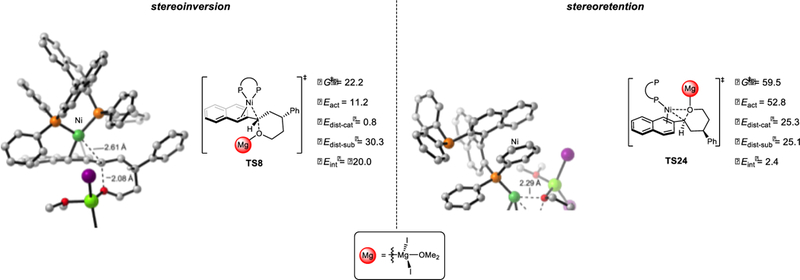
Origins of stereospecificity of C-O bond activation in the Kumada coupling. Energies are in kcal/mol.
The origins of this stereospecificity were further elucidated with distortion/interaction analysis (Figure 5).30,31 Each transition state was separated into two fragments: the Ni(BINAP) catalyst fragment and the ether-MgI2 substrate fragment. The distortion energy (ΔEdist) is the energy required for the geometric change during the C-O bond cleavage, and the interaction energy (ΔEint) reflects the strength of the interaction between the catalyst and substrate fragments in the transition state (details of distortion/interaction analysis are included in Figure S8).
Comparing the two C-O bond activation transition states, ΔEdist-cat and ΔEint are the leading causes for the stereoselectivity. The remarkable difference of ΔEdist-cat is due to the change of nickel coordination. In the stereoinvertive transition state TS8, the nickel is bisligated with two phosphine coordinations of the BINAP ligand. The stereoretentive transition state TS24 requires dissociation of one phosphine coordination to accommodate the nickel-oxygen interaction, leading to significant catalyst distortion. In addition, the interaction between catalyst and substrate is weaker in TS24: ΔEint (TS8) is −20.0 kcal/mol, while ΔEint (TS24) is 2.4 kcal/mol. Therefore, both the catalyst distortion and catalyst-substrate interaction contribute to the high stereospecificity of the Ni/BINAP-mediated oxidative addition of the C-O bond, eventually leading to the stereoinversion in the Kumada coupling.
Experimental kinetic study of the Kumada coupling.
To further validate the computed mechanistic model of the Ni/BINAP-catalyzed Kumada coupling with benzylic ether 1, experimental kinetic studies were performed. Ether 25 was chosen as the substrate for determination of the empirical rate law because the cross-coupling reaction proceeds cleanly, without side products, and at a rate that is convenient for kinetic analysis (eq 1).2a Our primary interest was to determine the order of the reaction with respect to the electrophile (25), nucleophile (MeMgl), and catalyst.
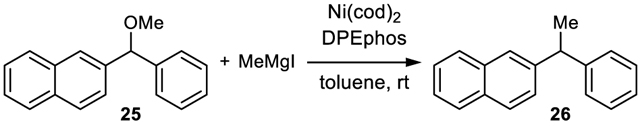 |
(1) |
Initial rate kinetic studies were performed under pseudo-first order conditions monitoring formation of product 26 by gas chromatography (Figure 6). Varying initial concentrations of substrate 25 (Figure 6a), catalyst (Figure 6b), and Grignard reagent (Figure 6c), and analysis of the initial rates demonstrated that the reaction is first order with respect to ether 25 and catalyst, and zero order with respect to the Grignard reagent.
Figure 6.
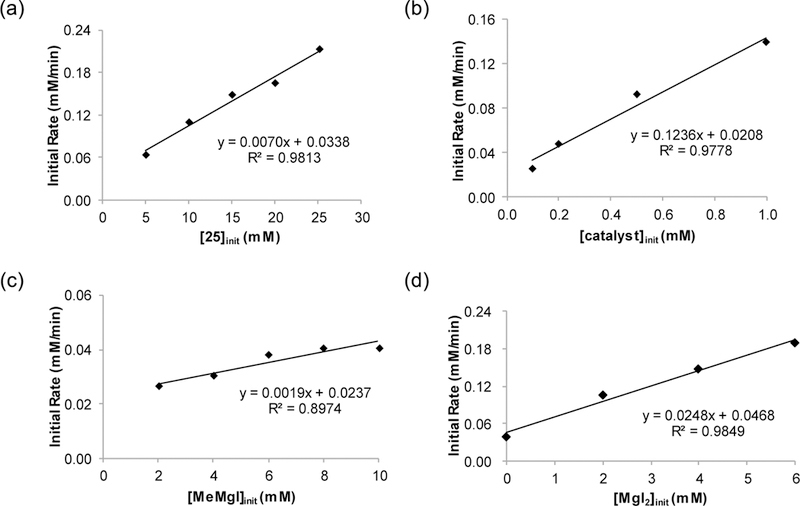
Dependence of the initial rate (mM/min) for formation of product 26 on (a) [25]init (5–25 mM) with [MeMgI] = 0.50 M, and [catalyst] = 0.1 mM in toluene; (b) [catalyst] (0.1–1.0 mM) with [25] = 10 mM, and [MeMgI] = 50 mM in toluene; (c) [MeMgI] (2–10 mM) with [25] = 50 mM and [catalyst] = 0.5 mM in toluene; (d) added [MgI2] (0–6 mM) with [25] = 50 mM, [MeMgI] = 4 mM and [catalyst] = 0.5 mM in toluene.
Despite the fact that the reaction is zero order with respect to MeMgI, we have observed discrepencies in reaction rate when different batches of methyl Grignard reagent are employed. We hypothesized that this discrepancy is caused by varying quantities of magnesium iodide salts present in the Grignard reagents.25,26 Indeed, we have observed that addition of MgI2 accelerates sluggish XC and XEC reactions.3a,8,32 These results are consistent with the important role of the magnesium salts in the calculations, serving as a Lewis acid to activate the substrate for oxidative addition, as with prior proposed mechanisms for allylic coupling reactions.22 As anticipated, Kumada coupling of substrate 25 displays first order dependance on MgI2 (Figure 6d).
The empirical rate law generated for cross-coupling of ether 25 is below (eq 2). The reaction is first order with respect to substrate 25, nickel catalyst, and Lewis acid MgI2. This rate law is consistent with rate-determining oxidative addition via TS8 (Figure 1). Therefore, the empirically determined rate law and DFT cacluations are consistent with the same mechanism.
| (2) |
We next performed an experiment inspired by Blackmond’s same excess experiments (Figure 7).33 The reaction was performed using several [MeMgI]init and monitored for formation of product 26. Plotting [MeMgl] versus time (Figure 7a) and adjusting the time axis for the 10 mM and 4 mM reactions allows for straightforward comparison of the slopes (Figure 7b). This experiment confirmed that the reaction is zero-order with respect to MeMgl. Importantly, we can also conclude that the catalyst is not degrading over the course of the reaction and that product 26 does not inhibit the reaction.
Figure 7.
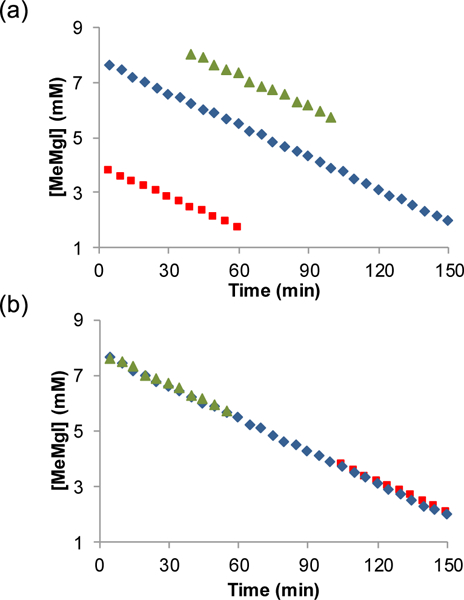
Same excess experiment. [MeMgI]init = 4 mM (■), 8 mM (♦), and 10 mM (▲). (a) Grignard concentration over time starting with different [MeMgI]init. (b) X-axis adjusted to over-lay 10 and 4 mM reactions with 8 mM reaction.
To further corroborate the computational and kinetic findings that oxidative addition is the rate-determining step of the cross-coupling reaction, we measured 13C kinetic isotope effects using the Singleton method (Scheme 4).34 Benzylic ether 27 was designed as a suitable substrate, since the 13C spectrum features disperse resonances, allowing for accurate integration. The Kumada reaction was performed on a 10 mmol scale, and the reaction was quenched when 7% of the starting material remained. Analysis of the data indicated that C3 and C4 displayed a significant (>1.01) KIE. These results are also consistent with rate-determining oxidative addition.
Scheme 4.
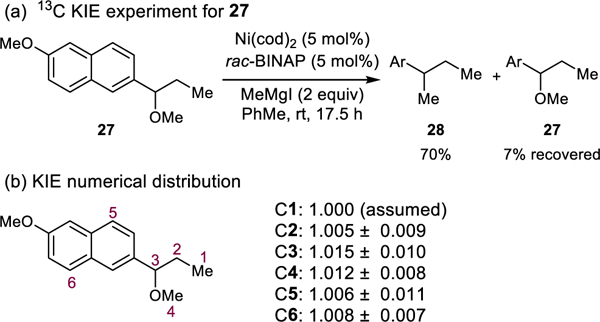
13C KIE experiment and KIE numerical distribution of Ni/BINAP-catalyzed Kumda coupling reaction.
Overall mechanism of Kumada coupling reaction.
Based on the combined computational and experimental investigations, the overall mechanism of the Kumada coupling is shown in Scheme 5. From the off-cycle resting state Ni(BINAP)2, ligand-substrate exchange generates the on-cycle arene complex 7. This species undergoes rate- and stereospecificity-determining oxidative addition, which cleaves the C-O bond of the benzylic ether and irreversibly leads to the η3-benzyl nickel(II) intermediate 9 with stereoinversion. Subsequent transmetallation and reductive elimination steps are both facile and do not alter the configuration of the benzylic stereogenic center, producing the Kumada coupling product with inversion.
Scheme 5.
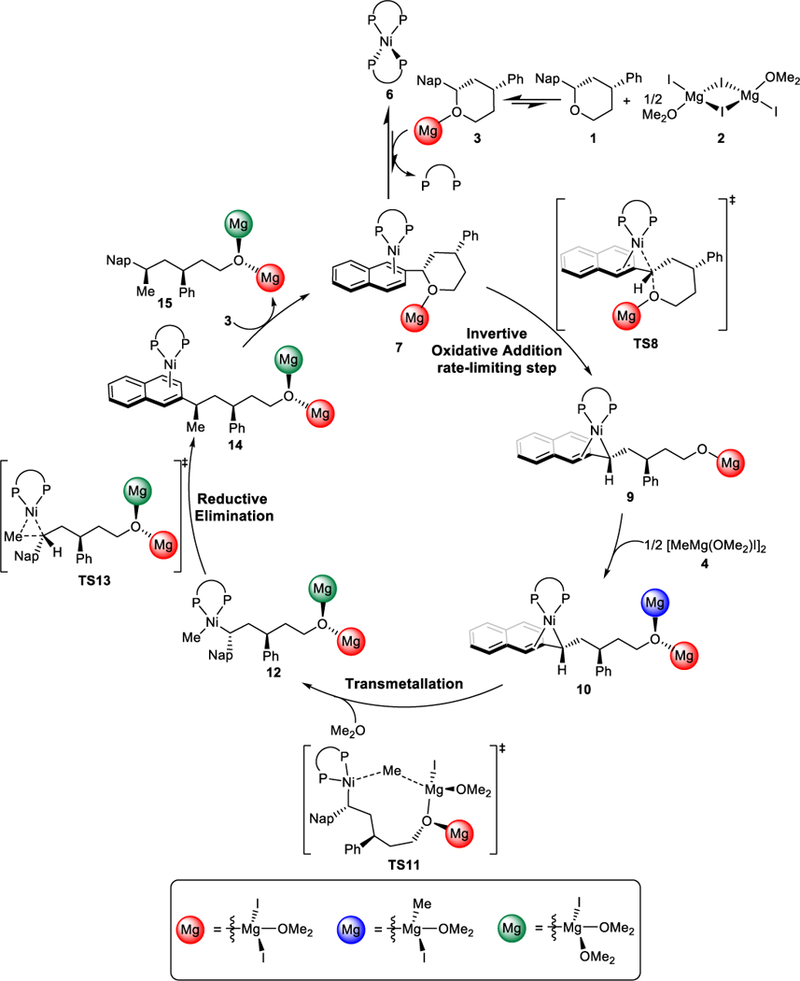
Overall mechanism of Kumada coupling.
Computational study of XEC mechanism.
We next investigated the reaction mechanism of Ni/BINAP-catalyzed XEC of 4-chlorotetrahydropyrans (Scheme 1b). A major question at the outset of these investigations was whether the reaction initates with oxidative addition of the C-O or C-Cl bond. Four proposed catalytic cycles are shown in Scheme 6; all account for the observed stereospecific transformation of both the benzylic ether and alkyl halide. Notably, these mechanisms bear little similarity to the proposed mechanisms of related XEC reactions of alkyl and aryl halides, due to the distinct substrates, reaction conditions, and stereochemical outcomes.35,36
Scheme 6.
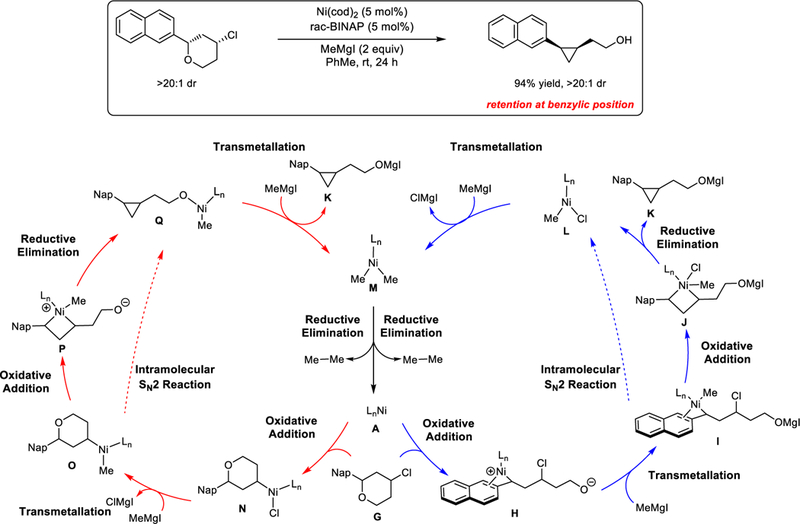
Proposed catalytic cycles of Ni-catalyzed cross-electrophile coupling of benzylic ether.
We first consider two catalytic cycles that initiate by oxidative addition of the benzylic ether (blue cycles). From the active catalyst LnNi(0) A, the benzylic C-O bond cleavage can occur to generate the zwitterionic species H. Benzylnickel complex H undergoes transmetallation with the Grignard reagent to form LnNi(benzyl)(methyl) intermediate I. Subsequent C-Cl cleavage can proceed via oxidative addition, leading to the nickel(IV) intermediate J.37 Reductive elimination would achieve cyclopropane formation. The resultant nickel(II) complex, L, can undergo a second transmetallation with the Grignard reagent to deliver LnNiMe2 species M, which undergoes reductive elimination to provide ethane and regenerate the active Ni(0) catalyst A. An alternative mechanism involves direct transformation from benzylnickel complex I to product K through an intramolecular SN2 reaction,38 in which the alkyl chloride is attacked by the nucleophilic benzylnickel fragment.39
Alternatively, catalytic cycles can be considered wherein the initial oxidative addition of nickel(0) catalyst occurs with the alkylchloride moiety (red cycles). These cycles proceed through formation of LnNi(alkyl)Cl species N. Subsequent transmetallation with the Grignard reagent leads to LnNi(alkyl)(methyl) intermediate O. From O, the C-C bond formation can occur via a sequential oxidative addition and reductive elimination (O-P-Q), or an intramolecular SN2 reaction of alkyl chloride (O-Q). Nickel alkoxide complex Q then undergoes a second transmetallation with the Grignard reagent to release the XEC product and generate LnNiMe2 (M). Reductive elimination from M regenerates the nickel(0) catalyst accompanied by the release of ethane.
To discriminate between these mechanistic possibilities, we began by evaluating the possible oxidative addition pathways that could initate the catalytic cycles. We first studied the coordination of the Lewis acidic Mg(II) species with benzylic ether 29. Both oxygen and chlorine may coordinate to the Lewis acidic Mg(II) species, and the free energy changes of the four possible complexes are shown in Scheme 7. Similar to the senario in Kumada coupling (Scheme 3), complex formation with MgI2 is more favorable than that with MeMgI (30 vs. 32; 31 vs. 33). In addition, oxygen coordination is significantly stronger than chlorine coordination (30 vs. 31; 32 vs 33), which is consistent with HSAB theory.40 Therefore, magnesium complex 30 was employed in the catalytic cycle.
Scheme 7.
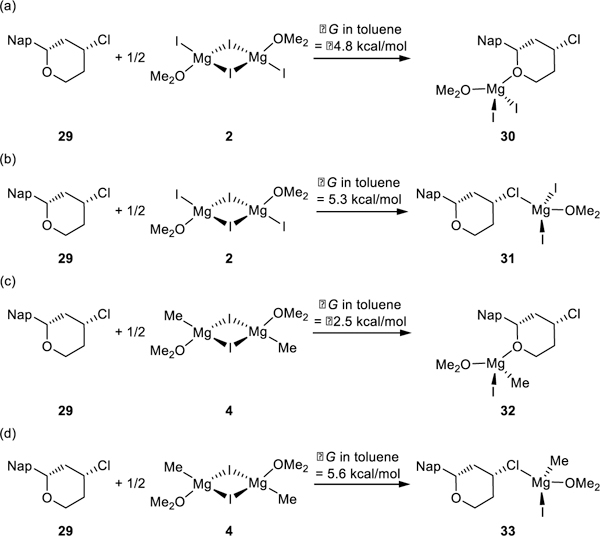
Free energy changes of complexation between substrate 29 and Mg(II) species involved in XEC.
The free energy changes of the most favorable pathway of the Ni/BINAP-catalyzed XEC with benzylic ether 29 are shown in Figures 8 and 9. DFT-optimized structures of selected intermediates and transition states are shown in Figure 10. From the catalyst resting state Ni(BINAP)2 (6), the endergonic ligand-substrate exchange delivers the (BINAP)Ni(substrate) complex 34. Arene complex 34 then undergoes oxidative addition by backside attack via TS35 to cleave the benzylic C-O bond with stereoinversion, generating the zwitterionic intermediate 36. This transition state is nearly identical to that for oxidative addition in Kumada coupling (TS8, Figure 1). Subsequent complexation with the Grignard reagent and transmetallation through TS38 generates LnNi(benzyl)(methyl) intermediate 39. From 39, coordination of the Grignard reagent assists to promote intramolecular attack of the benzylnickel moiety on the alkyl chloride via TS41. This intramolecular SN2 reaction produces cyclopropane product 42 and (BINAP)Ni(Me)Cl 43, which results in inversion at both the benzylic center and the alkyl halide.41 The very low intramolecular SN2 barrier is due to the similar geometries between the transition state TS41 and the preceding intermediate 40 (Figure S9). Hammond postulate applies to this SN2 step, varying the halide leaving group can lower the reaction barrier by making this step more exothermic (Figure S10). We also considered the alternative C-C bond formation process without the assistance of Grignard reagent, but this hypothetical process is less favorable (Figure S11).
Figure 8.
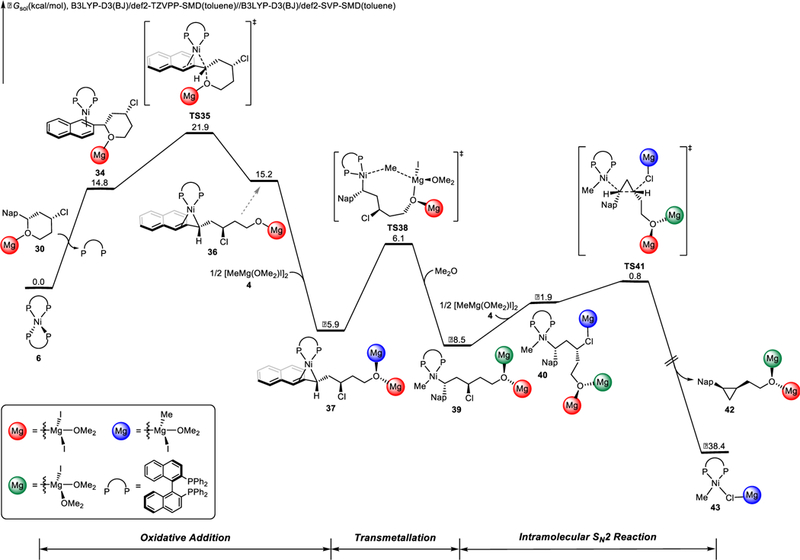
DFT-computed free energy changes of Ni/BINAP-catalyzed XEC with benzylic ether 29 (product formation).
Figure 9.
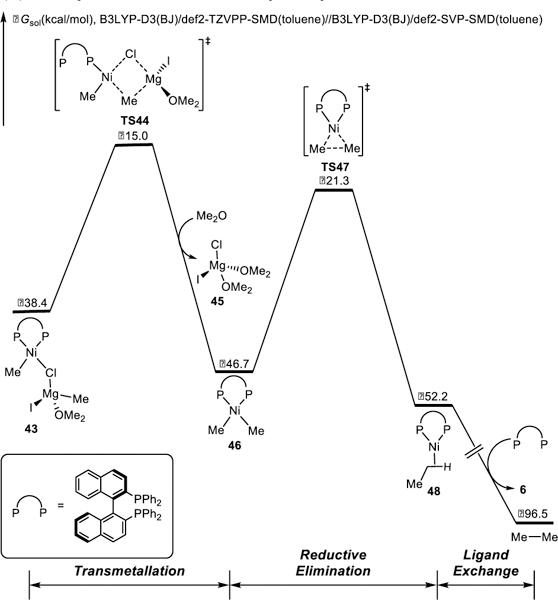
DFT-computed free energy changes of Ni/BINAP-catalyzed XEC with benzylic ether 29 (catalyst regeneration).
Figure 10.
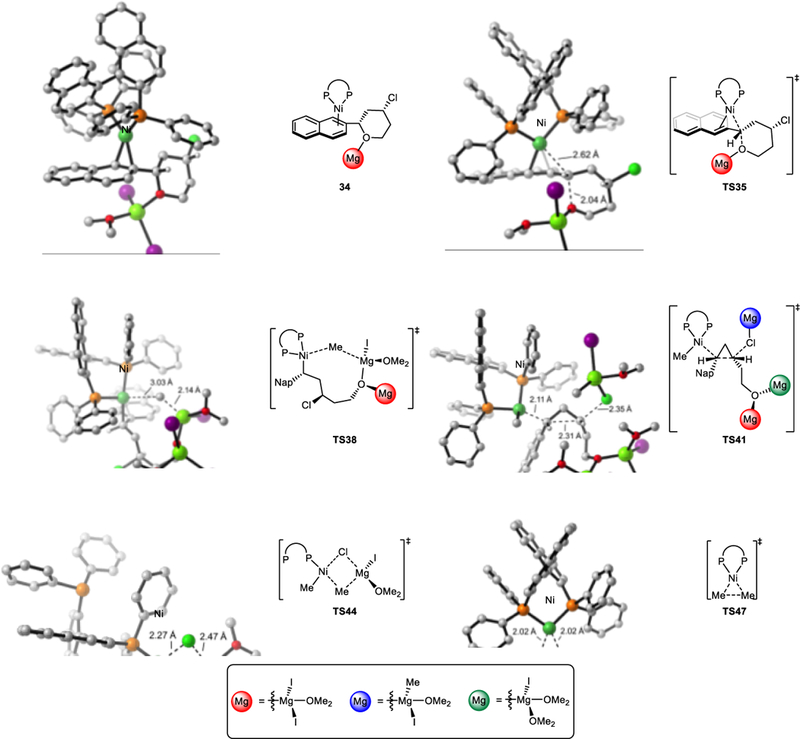
DFT-optimized structures of selected intermediates and transition states involved in Ni-catalyzed XEC.
The C-C bond formation step via TS41 can be considered as an intramolecular SN2 reaction for of chloride, or a SE2 reaction of benzylnickel moiety. We want to empharsize that the arene is not coordinated to nickel during the C-C bond formation (Figure 10). Therefore, this step is not a SE’ reaction of benzylnickel moiety. We refer this C-C bond formation as as an intramolecular SN2 reaction of the tethered alkyl chloride.
Based on the free energy profile, the oxidative addition of the C-O bond via TS35 determines the overall efficiency; subsequent cyclopropane formation via TS41 is facile. The stereospecificity of the XEC reaction is synergistically controlled by two elementary steps: oxidative addition of the benzylic ether via TS35 (which affects the benzylic stereogenic center) and the intramolecular SN2 reaction of benzylnickel intermediate via TS41 (which affects both stereogenic centers).
Completion of the catalytic cycle requires reduction of (BINAP)Ni(Me)Cl 43 to regenerate the nickel(0) catalyst. The free energy changes of the catalyst regeneration are shown in Figure 9. From 43, transmetallation with Grignard reagent occurs via TS44 to generate (BINAP)NiMe2 46. Subsequent reductive elimination via TS47 produces (BINAP)Ni(ethane) 48, which then releases ethane to regenerate the active nickel(0) catalyst for the next catalytic cycle.
The alternative catalytic cycles involve initial C−Cl bond cleavage, and the free energy changes of the competing pathways from the substrate-ligated catalyst, intermediate 34, are shown in Figure 11. Oxidative addition of the benzylic C-O bond via TS35 is 18.8 kcal/mol more favorable than oxidative addition of the C-Cl bond via TS50. We also located a number of alternative transition states for oxidative addition of the C-Cl bond; these transition states are less favorable than TS50 (Figure S12 and S13). Thus, irreversible benzylic C-O bond activation initiates the catalytic cycle. Comparing the two competing transition states, two factors contribute to the strong preference towards benzylic C-O bond activation. In TS35, naphthalene coordination stabilizes the forming nickel(II) and leads to formation of a stable π-benzylnickel complex. Naphthalene coordination in TS50 is impossible due to geometric constraints, and the product of oxidative addition is a less stable alkylnickel complex. In addition, the Lewis acidic magnesium co-catalysts favor coordination to (and thus activation of) the benzylic ether (Scheme 7). These factors combine to provide the remarkable chemoselectivity of bond activation.
Figure 11.
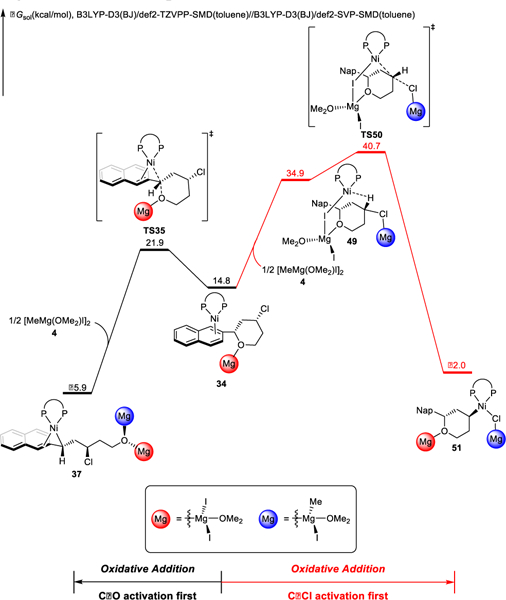
Comparison of oxidative addition of the C-O and C-Cl bonds.
An alternative pathway for C-C bond formation is sequential oxidative addition and reductive elimination via nickel(IV) intermediate J (blue cycle, Scheme 6). The free energy changes of competing pathways from intermediate 39 are shown in Figure 12. From 39, cyclization via TS41 is facile with a barrier of 9.3 kcal/mol, while oxidative addition through TS52 is significantly less favorable. Thus, the sequential oxidative addition and reductive elimination involving a nickel(IV) intermediate is not operative for this XEC reaction.
Figure 12.
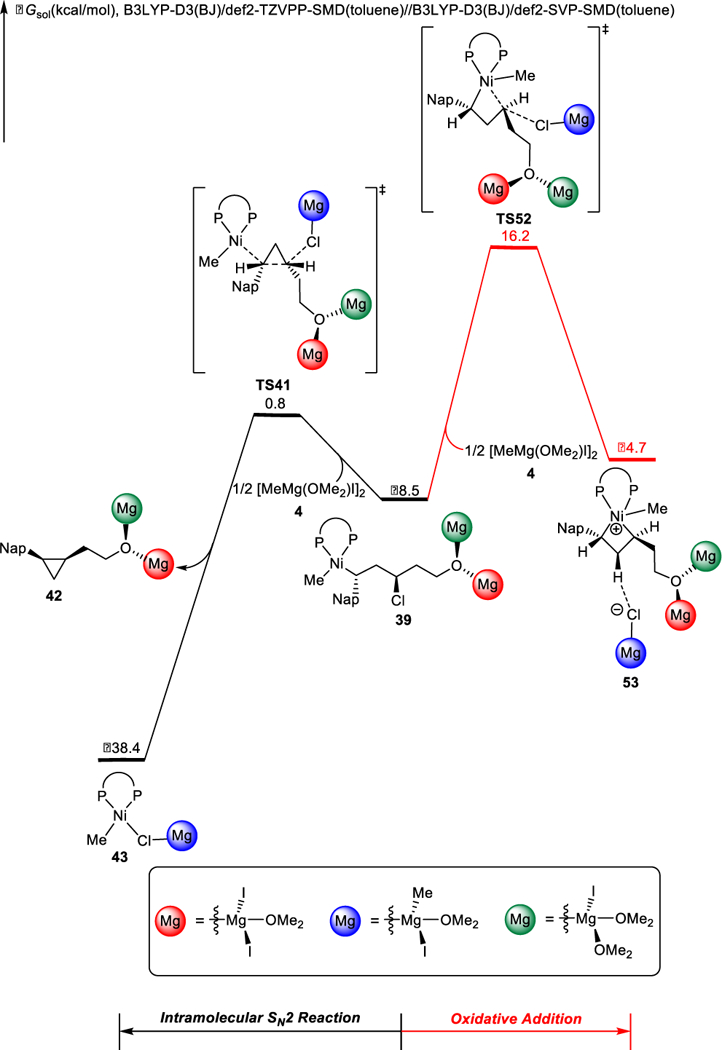
Free energy changes of the competing intramolecular SN2 reaction and oxidative addition from intermediate 39.
Chemoselectivity between XEC and Kumada coupling.
Simple Kumada coupling of substrates such as 29 was never observed as a competitive side reaction in XEC of these substrates. This observation was quite unexpected, since Kumada coupling does not require formation of a strained ring, and it was anticipated that dialkylnickel intermediates such as 39 could undergo reductive elimination. To better understand the striking chemoselectivity, we sought to compare these alternative pathways. The free energy changes of these competing pathways from alkylnickel complex 39 are shown in Figure 13. Reductive elimination transition state TS54 is 18.3 kcal/mol higher in free energy as compared to the intramolecular SN2 transition state TS41. These energy differences are consistent with the complete selectivity for XEC over XC observed experimentally (Scheme 1b). Therefore, facile cyclopropane formation drives alkylnickel complex 39 towards the desired cross-electrophile coupling, which prevents background Kumada coupling. The chemoselectivity does not depend on the stereochemistry of the alkyl chloride, both the cis- and trans-substrates exhibit the same chemoselectivity towards XEC product experimentally.3a Our computations also corroborate this trend; details are included in the Supporting Information (Figure S14).
Figure 13.
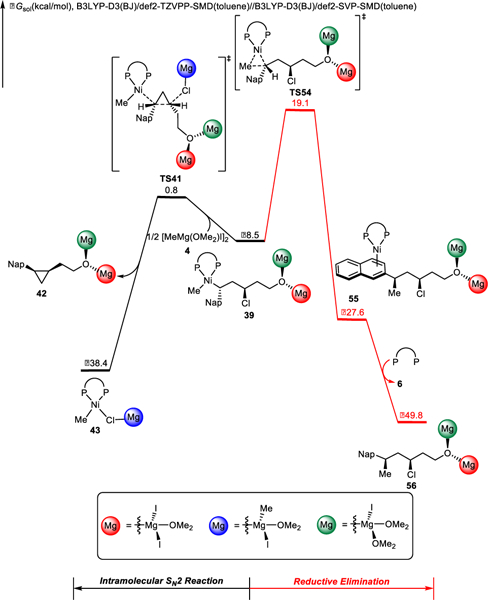
Chemoselectivity between cross-electrophile coupling and Kumada coupling of intermediate 39.
Origins of stereospecificity of XEC.
Two elementary steps, oxidative addition of the benzylic C-O bond and intramolecular SN2 attack of the benzylnickel moiety on the pendant alkyl halide, control the overall stereospecificity of the XEC reaction. The competing transition states for stereoinvertive and stereoretentive oxidative addition are shown in Figure 14a. C-O bond cleavage with inversion, via TS35, is more facile than that with retention, via TS57. Notably, the calculated transitions state for oxidative addition in the XEC reaction is almost identical to that in the Kumada coupling, consistent with the similarity of the substrates (TS8, Figure 5 versus TS35, Figure 14a).
Figure 14.
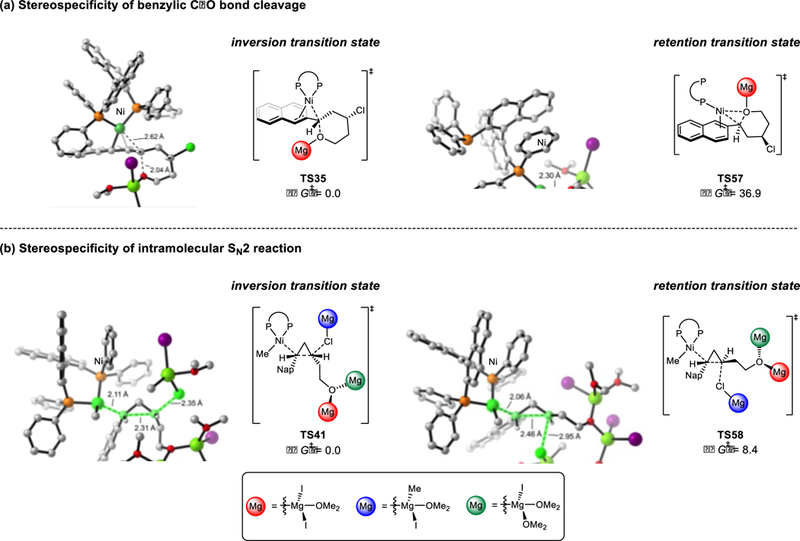
Stereospecificity of the C-O bond cleavage(a) and intramolecular SN2 reaction(b). Free energies are in kcal/mol.
Subsequent cyclopropane formation impacts the stereochemistry at both the benzylic and tertiary centers (Figure 14b). Two possible transition states are shown in Figure 14b. TS41 is the intramolecular SN2 transition state where the benzylnickel complex approaches the back side of the alkyl chloride. This attack inverts both stereogenic centers, leading to the net retention at the benzylic position and inversion at the alkyl chloride. TS58 is the competing attack of the benzylnickel complex on the front side of the alkyl chloride, which leads to stereoretention. TS41 is 8.4 kcal/mol more favorable than TS58, which indicates a strong selectivity for inversion at the alkyl chloride and is consistent with the observed high stereospecificity of the reaction. This selectivity is due to the intrinsic preference for back-side attack on alkyl halides.41 Therefore, the benzylic stereogenetic center undergoes two invertive elementary steps, oxidative addition and intramolecular SN2 attack on the alkyl chloride, resulting in net retention. In contrast, the tertiary alkyl stereogenic center only undergoes one invertive step.
Experimental analysis of XEC.
To validate the computed reaction mechanism, we measured the 13C kinetic isotope effects (KIE) using Singleton’s method.34 We anticipated that 13C KIE’s would distinguish whether the rate-determining step involves the benzylic stereocenter, the alkyl chloride, or both. Tetrahydropyran 29 was subjected to the cross-electrophile coupling reaction conditions on a 4 mmol scale. The reaction was quenched when 6% of 29 remained (Scheme 8a). NMR analysis determined that C11 was the only carbon that had a significant (>1.01) KIE (Scheme 8b). The presence of a KIE at C11 and the absence of a KIE at C13 are consistent with oxidative addition into the benzylic C-O as the rate-determining step for product formation.
Scheme 8.
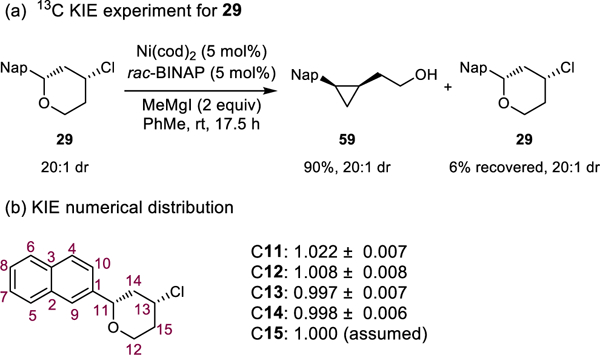
13C KIE experiment and KIE numerical distribution of Ni/BINAP-catalyzed cross-electrophile coupling.
To corroborate the computational findings that the cross-electrophile coupling reaction initiates by oxidative addition of the benzylic ether, and not at the alkyl chloride, we designed a series of competition experiments (Scheme 9). Oxidative addition into the benzylic ether of 2-naphthyltetrahydropyran 29 should be more facile than oxidative addition of 2-phenyltetrahydropyran 60 because of the lower aromatic stabilization energy of the naphthyl moiety as compared to an isolated phenyl ring (Scheme 9a).29,42 Therefore, these substrates should react with different rates if oxidative addition of the C-O bond is rate-determining, but similar rates if oxidative addition of the C-Cl bond is rate-determining. When subjected to a competition experiment, 2-naphthyltetrahydropyran 29 underwent the ring contraction to generate an 88% yield of cyclopropane 59, while 2-phenyltetrahydropyran 60 was recovered in an 89% yield without the formation of any phenylcyclopropane 61 (Scheme 9a). Similarly, allylic ether 62 should outcompete benzylic ether 29 if oxidative addition of the C-O is rate-determining (Scheme 9b). As expected, vinylcyclopropane 63 is generated in a higher yield (71%, Scheme 9b) than naphthylcyclopropane 59 (12%, Scheme 9b). These results are consistent with oxidative addition into the C-O bond as the first step of the cross-electrophile coupling reaction rather than oxidative addition into the C-Cl bond. They are also consistent with rate-determining oxidative addition of the C-O bond.
Scheme 9.
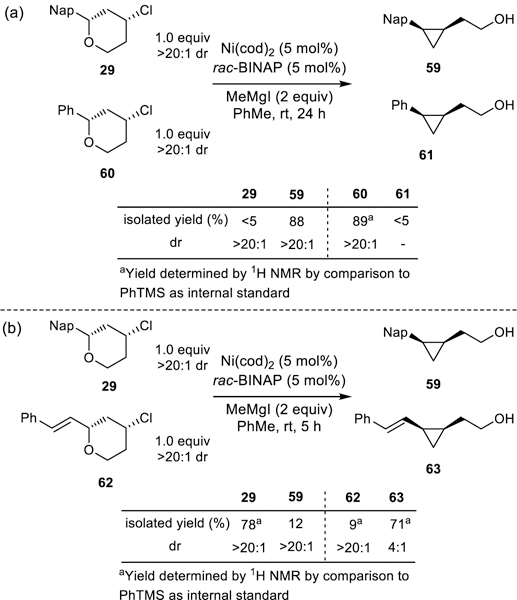
Competition experiments to distinguish rate-determining oxidative addition of C-O versus C-Cl.
Overall mechanism of XEC.
Based on the combined computational and experimental investigations, the overall mechanism of the XEC reaction is shown in Scheme 10. From the off-cycle resting state Ni(BINAP)2, ligand-substrate exchange generates the on-cycle arene complex 34. This active species undergoes oxidative addition to cleave the benzylic C-O bond with stereoinversion, leading to benzylnickel(II) intermediate 36. Subsequent transmetallation with the Grignard reagent generates the benzylnickel complex 39. The tethered alkyl chloride is then attacked by the electron-rich benzylnickel moiety in an intramolecular SN2 reaction. This C-C bond forming step generates the cyclopropane, with inversion of both stereogenic centers. Catalyst regeneration is accomplished by transmetallation with an additional equivalent of Grignard reagent and reductive elimination to form ethane.
Scheme 10.
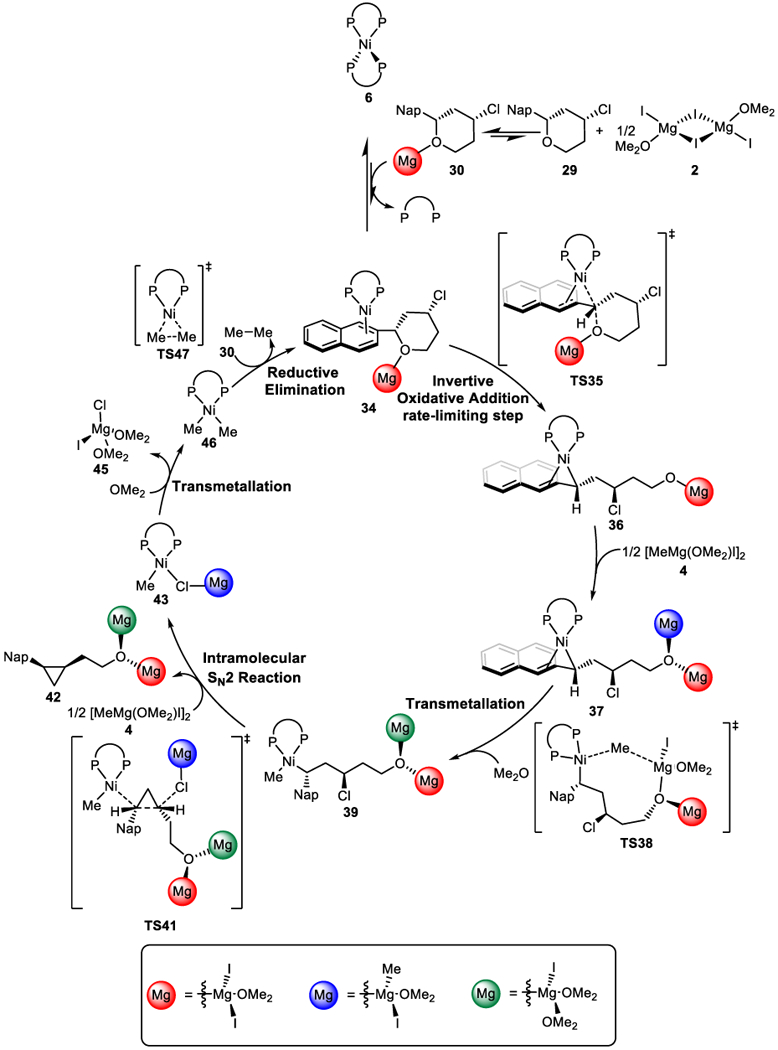
Overall mechanism of cross-electrophile coupling reaction.
Comparison between Kumada and cross-electrophile coupling reaction.
Based on above mechanistic studies, a unified mechanistic picture of Ni-catalyzed Kumada coupling and XEC reactions is shown in Scheme 11. Starting from the (BINAP)Ni(substrate) complex I, both Kumada coupling and XEC reactions proceed via stereoinvertive oxidative addition of the benzylic C-O bond. This step is facilitated by MgI2, which serves as a Lewis acid to activate the leaving group, and is rate-determining for formation of both XC and XEC products. Benzylnickel(II) intermediate II then undergoes transmetallation with the Grignard reagent to form (BINAP)Ni(benzyl)(methyl), IV. From IV, the mechanistic pathways of Kumada coupling and XEC divert. Direct C(benzyl)-C(methyl) reductive elimination produces the Kumada coupling product and regenerates the active nickel(0) catalyst, which completes the catalytic cycle of Kumada coupling. However, if the secondary alkyl chloride is present, as in an XEC reaction, facile intramolecular SN2 attack occurs to form the cyclopropane ring. Subsequent transmetallation and reductive elimination regenerates the nickel(0) catalyst for next catalytic cycle of the XEC reaction.
Scheme 11.
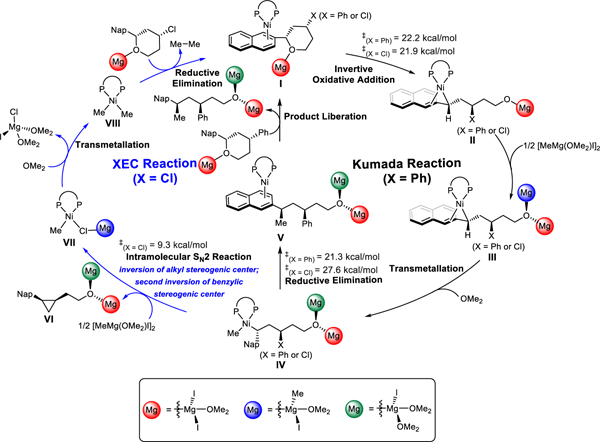
Unified mechanism for Kumada and cross-electrophile coupling reactions.
Competition experiment between Kumada coupling and XEC reactions.
To challenge our unified mechanistic understanding of the Kumada and cross-electrophile coupling reactions, we performed a competition experiment (Scheme 12). Based on the calculated free energy diagrams, both reactions share a rate-determining oxidative addition step with a similar barrier height. Therefore, in a competition reaction, a Kumada-type and cross-electrophile-type substrate should be consumed with similar rates. Indeed, subjecting a mixture of tetrahydropyrans 1 and 29 to the standard reaction conditions produced a similar amount of ring-opened Kumada product (25%) and cyclopropane XEC product (31%). These results corroborate our proposed mechanisms for the Kumada and XEC reactions, and support a similar oxidative addition event in both catalytic cycles.
Scheme 12.
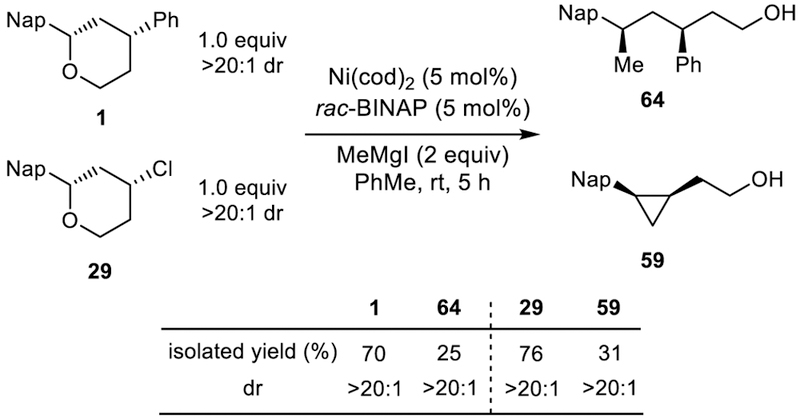
Competition experiment between Kumada and cross-electrophile coupling.
Conclusions.
In summary, the reaction mechanism and origins of chemoselectivity and stereospecificity of Ni-catalyzed Kumada and cross-electrophile coupling reactions of benzylic ethers have been defined using a combined computational and experimental approach. Kumada coupling proceeds via a classical cross-coupling mechanism. Rate- and stereospecificity-determining oxidative addition occurs to cleave the C-O bond of the benzylic ether and inverts the benzylic stereogenic center. Subsequent transmetallation with the Grignard reagent and syn reductive elimination produce the Kumada coupling product with net inversion at the benzylic stereocenter. The empirical rate law and 13C KIE’s are consistent with this proposed mechanism.
Cross-electrophile coupling initiates with the same invertive oxidative addition of the benzylic C-O bond and transmetallation step. This is supported by DFT calculations, competition experiments, and measured 13C KIE’s. At this point, the mechanisms diverge. Due to the presence of the pendant secondary alkyl chloride, the electron-rich benzylnickel moiety can attack via a facile intramolecular SN2 reaction. This efficient step produces the strained cyclopropane ring with inversion at both stereocenters. It also outcompetes the reductive elimination for Kumada coupling, leading to excellent control of chemoselectivity. Over the course of the mechanism, the benzylic position reacts twice with inversion, resulting in net retention at this position, while the alkyl chloride reacts with inversion. This mechanism is distinct from those typically proposed for XEC reactions of alkyl and aryl halides, as it does not involve alkyl radical intermediates or odd-electron nickel complexes.
These combined computational and experimental results provide a refined understanding of the reactions of ether electrophiles with nickel(0) catalysts, including the critical role of Lewis acidic co-catalysts. We characterize two-electron oxidative addition and intramolecular SN2 reactions, providing insight into the transition states that lead to stereospecific reactions. We anticipate that this improved understanding will facilitate the design of new stereospecific catalytic reactions including those that involve oxidative addition of C(sp3)-O bonds.
Supplementary Material
ACKNOWLEDGMENT
Financial support from National Natural Science Foundation of China (21702182, 21873081, X. H.), US National Institutes of Health NIGMS (R01GM100212, E. R. J.) Zhejiang University, and the Chinese “Thousand Youth Talents Plan” is gratefully acknowledged. Calculations were performed on the high performance computing system at Department of Chemistry, Zhejiang University.
Footnotes
ASSOCIATED CONTENT
Supporting Information.
Additional computational results. Coordinates and energies of DFT-computed stationary points. General information for experiments. Detailed results of experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Lovering F; Bikker J; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]; (b) Geist E; Kirschning A; Schmidt T. sp3−sp3 Coupling Reactions in the Synthesis of Natural Products and Biologically Active Molecules. Nat. Prod. Rep 2014, 31, 441–448. [DOI] [PubMed] [Google Scholar]; (c) Cherney AH; Kadunce NT; Reisman SE Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C–C Bonds. Chem. Rev 2015, 115, 9587–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jana R; Pathak TP; Sigman MS Advances in Transition Metal (Pd, Ni, Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-organometallics as Reaction Partners. Chem. Rev 2011, 111, 1417–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Choi J; Fu GC Transition Metal-Catalyzed Alkyl-Alkyl Bond Formation: An-other Dimension in Cross-Coupling Chemistry. Science 2017, 356, eaaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lucas EL; Jarvo ER Stereospecific and Stereoconvergent Cross-Couplings Between Alkyl Electrophiles. Nat. Rev. Chem 2017, 1, 0065. [Google Scholar]
- (2).(a) Taylor BLH; Swift EC; Waetzig JD; Jarvo ER Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Alkyl Ethers: Enantioselective Synthesis of Diarylethanes. J. Am. Chem. Soc 2011, 133, 389–391. [DOI] [PubMed] [Google Scholar]; (b) Harris MR; Hanna LE; Greene MA; Moore CE; Jarvo ER Retention or Inversion in Stereospecific Nickel-Catalyzed Cross-Coupling of Benzylic Carbamates with Arylboronic Esters: Control of Absolute Stereochemistry with an Achiral Catalyst. J. Am. Chem. Soc 2015, 135, 3303–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wisniewska HM; Swift EC; Jarvo ER Functional-Group-Tolerant, Nickel-Catalyzed Cross-Coupling Reaction for Enantioselective Construction of Tertiary Methyl-Bearing Stereocenters. J. Am. Chem. Soc 2013, 135, 9083–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Harris MR; Konev MO; Jarvo ER Enan-tiospecific Intramolecular Heck Reactions of Secondary Benzylic Ethers. J. Am. Chem. Soc 2014, 136, 7825–7828. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Tollefson EJ, Hanna LE; Jarvo ER Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Benzylic Ethers and Esters. Acc. Chem. Res 2015, 48, 2344–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Tollefson EJ; Erickson LW; Jarvo ER Stereospecific Intramolecular Reductive Cross-Electrophile Coupling Reactions for Cyclopropane Synthesis. J. Am. Chem. Soc 2015, 137, 9760–9763. [DOI] [PubMed] [Google Scholar]; (b) See reference 1f.
- (4).(a) For reviews and representative experimental studies of cross-coupling reactions of phenol derivatives, see: Yu D-G; Li B-J; Shi Z-J. Exploration of New C−O Electrophiles in Cross-Coupling Reactions. Acc. Chem. Res 2010, 43, 1486–1495. [DOI] [PubMed] [Google Scholar]; (b) Rosen BM; Quasdorf KW; Wilson DA; Zhang N; Resmerita A-M; Garg NK; Percec V. Nickel-Catalyzed Cross-Couplings Involving Carbon−Oxygen Bonds. Chem. Rev 2011, 111, 1346–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tobisu M; Chatani N. Cross-Couplings Using Aryl Ethers via C−O Bond Activation Enabled by Nickel Catalysts. Acc. Chem. Res 2015, 48, 1717–1726. [DOI] [PubMed] [Google Scholar]; (d) Zarate C; van Gemmeren M; Somerville RJ; Martin R. Phenol Derivatives: Modern Electrophiles in Cross-Coupling Reactions. Adv. Organomet. Chem 2016, 66, 143–222. [Google Scholar]
- (5).(a) see reference 4a.; (b) Guan B-T; Xiang S-K; Wang B-Q; Sun Z-P; Wang Y; Zhao K-Q; Shi Z-J Direct Benzylic Alkylation via Ni-Catalyzed Selective Benzylic sp3 C−O Activation. J. Am. Chem. Soc 2008, 130, 3268–3269. [DOI] [PubMed] [Google Scholar]; (c) Pound SM; Watson MP Asymmetric Synthesis via Stereospecific C–N and C–O Bond Activation of Alkyl Amine and Alcohol Derivatives. Chem. Commun 2018, 54, 12286–12301. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhou Q; Srinivas HD; Dasgupta S; Watson MP Nickel-Catalyzed Cross-Couplings of Benzylic Pivalates with Arylboroxines: Stereospecific Formation of Diarylalkanes and Triarylmethanes. J. Am. Chem. Soc 2013, 135, 3307–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Zhou Q; Cobb KM; Tan T; Watson MP Stereospecific Cross Couplings to Set Ben-zylic, All-Carbon Quaternary Stereocenters in High Enantiopurity. J. Am. Chem. Soc 2016, 138, 12057–12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Allylic ethers undergo facile and stereospecific cross-coupling employing nickel catalysts, see: Takahashi T and Kanno K. Nickel-Catalyzed Cross-Coupling Reactions In Modern Organonickel Chemistry; Tamaru Y. Ed.; Wiley-VCH: Weinheim, 2005; p47. [Google Scholar]; (b) Consiglio G; Morandini F; Piccolo O. Stereochemical Aspects of the Nickel-Catalyzed Alkylation of Allylic Alcohols. J. Am. Chem. Soc 1981, 103, 1846–1847. [Google Scholar]; (c) Kobayashi Y; Ikeda E. Nickel-Catalysed Substitution Reactions of Allylic Carbonates with Aryl-and Alkenyl-Borates. J. Chem. Soc., Chem. Commun 1994, 1789–1790. [Google Scholar]; (d) Didiuk MT; Morken JP; Hoveyda AH Phosphine-Directed Stereo- & Regioselective Ni-Catalyzed Reactions of Gri-gnard Reagents with Allylic Ethers. Tetrahedron, 1998, 54, 1117–1130. [Google Scholar]; (e) Zhou Q; Srinivas HD; Zhang S; Watson MP Accessing Both Retention and Inversion Pathways in Stereospecific, Nickel-Catalyzed Miyaura Borylations of Allylic Pivalates. J. Am. Chem. Soc 2016, 138, 11989–11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang S-Q; Taylor BLH; Ji C-L; Gao Y; Harris MR; Hanna LE; Jarvo ER; Houk KN; Hong X. Mechanism and Origins of Ligand-Controlled Stereoselectivity of Ni-Catalyzed Suzuki–Miyaura Coupling with Benzylic Esters: A Computational Study. J. Am. Chem. Soc 2017, 139, 12994–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tollefson EJ; Dawson DD; Osborne CA; Jarvo ER Stereospecific Cross-Coupling Reactions of Aryl-Substituted Tetrahydrofurans, Tetrahydropyrans, and Lactones. J. Am. Chem. Soc 2014, 136, 14951–14958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) For related computational studies on Ni-catalyzed C–O bond activation, see: Li Z; Zhang S-L; Fu Y; Guo Q-X; Liu L. Mechanism of Ni-Catalyzed Selective C−O Bond Activation in Cross-Coupling of Aryl Esters. J. Am. Chem. Soc 2009, 131, 8815–8823. [DOI] [PubMed] [Google Scholar]; (b) Hong X; Liang Y; Houk KN Mechanisms and Origins of Switchable Chemoselectivity of Ni-Catalyzed C(aryl)–O and C(acyl)–O Activation of Aryl Esters with Phosphine Ligands. J. Am. Chem. Soc 2014, 136, 2017–2025. [DOI] [PubMed] [Google Scholar]; (c) Lu Q; Yu H; Fu Y. Mechanistic Study of Chemoselectivity in Ni-Catalyzed Coupling Reactions Between Azoles and Aryl Carboxylates. J. Am. Chem. Soc 2014, 136, 8252–8260. [DOI] [PubMed] [Google Scholar]; (d) Xu H; Muto K; Yamaguchi J; Zhao C; Itami K; Musaev DG Key Mechanistic Features of Ni-Catalyzed C–H/C–O Biaryl Coupling of Azoles and Naphthalen-2-yl Pivalates. J. Am. Chem. Soc 2014, 136, 14834–14844. [DOI] [PubMed] [Google Scholar]; (e) Liu X-Q; Hsiao C-C; Kalvet I; Leiendecker M; Guo L; Schoenebeck F; Rueping M. Lewis Acid Assisted Nickel-Catalyzed Cross-Coupling of Aryl Methyl Ethers by C−O Bond-Cleaving Alkylation: Prevention of Undesired β-Hydride Elimination. Angew. Chem. Int. Ed 2016, 55, 6093–6098. [DOI] [PubMed] [Google Scholar]; (f) Yang Z-K; Wang C; Uchiyama M. DFT Studies Provide Mechanistic Insight into Nickel-Catalyzed Cross-Coupling Involving Organoaluminum-Mediated C–O Bond Cleavage. Synlett 2017, 28, 2565–2568. [Google Scholar]; (g) Schwarzer MC; Konno R; Hojo T; Ohtsuki A; Nakamura K; Yasutome A; Takahashi H; Shimasaki T; Tobisu M; Chatani N; Mori S. Combined Theoretical and Ex-perimental Studies of Nickel-Catalyzed Cross-Coupling of Methoxy-arenes with Arylboronic Esters via C–O Bond Cleavage. J. Am. Chem. Soc 2017, 139, 10347–10358. [DOI] [PubMed] [Google Scholar]; (h) Xu Z; Yang Y; Jiang J; Fu Y. Theoretical Investigation on Ni-Catalyzed C(sp3)−F Activation and Ring Contraction of Tetrahydropyrans: Exploration of an SN2 Path-way. Organometallics 2018, 37, 1114–1122. [Google Scholar]
- (10).Lucas EL; Jarvo ER Keeping Track of the Electrons. Acc. Chem. Res 2018, 51, 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zheng G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Mont-gomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Nor-mand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi R; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas O; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ; Gaussian 09, revision C.01; Gaussian Inc.: Wallingford, CT, 2016. [Google Scholar]
- (12).(a) Becke AD Density Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]; (b) Lee C; Yang W; Parr RG Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (13).Weigend F; Ahlrichs R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (14).Grimme S, Anthony J, Ehrlich S; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys 2010, 132, 154104–1–154104–19. [DOI] [PubMed] [Google Scholar]
- (15).Weigend F. Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- (16).Marenich AV; Cramer CJ; Truhlar DG Universal Solva-tion Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (17).Legault CY CYLview, version 1.0b; Université de Sherbrooke, 2009. (http://www.cylview.org). [Google Scholar]
- (18).Martin RL; Hay PJ; Pratt LR Hydrolysis of Ferric Ion in Water and Conformational Equilibrium. J. Phys. Chem. A 1998, 102, 3565–3573. [Google Scholar]
- (19).(a) Yu ZX; Houk KN Intramolecular 1,3-Dipolar Ene Reactions of Nitrile Oxides Occur by Stepwise 1,1-Cycloaddition/Retro-Ene Mechanisms. J. Am. Chem. Soc 2003, 125, 13825–13830. [DOI] [PubMed] [Google Scholar]; (b) Chen Y; Ye S; Jiao L; Liang Y; Sinha-Mahapatra DK; Hern-don JW; Yu Z-X Mechanistic Twist of the [8+2] Cycloadditions of Dienylisobenzofurans and Dimethyl Acetylenedicarboxylate: Stepwise [8+2] versus [4+2]/[1,5]-Vinyl Shift Mechanisms Revealed through a Theoretical and Experimental Study. J. Am. Chem. Soc 2007, 129, 10773–10784. [DOI] [PubMed] [Google Scholar]; (c) Liang Y; Liu S; Xia Y; Li Y; Yu Z-X Mechanism, Regioselectivity, and the Kinetics of Phosphine-Catalyzed [3+2] Cycloaddition Reactions of Allenoates and Electron-Deficient Alkenes. Chem. - Eur. J 2008, 14, 4361–4373. [DOI] [PubMed] [Google Scholar]
- (20).(a) Li H; Jiang J; Lu G; Huang F; Wang Z-X On the “Reverse Gear” Mechanism of the Reversible Dehydrogena-tion/Hydrogenation of a Nitrogen Heterocycle Catalyzed by a Cp*Ir Complex: A Computational Study. Organometalics 2011, 30, 3131–3141. [Google Scholar]; (b) Li H; Wen M; Wang Z-X Computational Mecha-nistic Study of the Hydrogenation of Carbonate to Methanol Cata-lyzed by the RuIIPNN Complex. Inorg. Chem 2012, 51, 5716–5727. [DOI] [PubMed] [Google Scholar]; (c) Wen M; Huang F; Lu G; Wang Z-X Density Functional Theory Mechanistic Study of the Reduction of CO2 to CH4 Catalyzed by an Ammonium Hydridoborate Ion Pair: CO2 Activation via Formation of a Formic Acid Entity. Inorg. Chem 2013, 52, 12098–12107. [DOI] [PubMed] [Google Scholar]; (d) Qu S; Dang Y; Song C; Wen M; Huang K−W; Wang Z−X. Catalytic Mechanisms of Direct Pyrrole Synthesis via Dehydrogenative Coupling Mediated by PNP-Ir or PNN-Ru Pincer Complexes: Crucial Role of Proton-Transfer Shuttles in the PNP-Ir System. J. Am. Chem. Soc 2014, 136, 4974–4991. [DOI] [PubMed] [Google Scholar]
- (21).Tamao K; Sumitani K; Kiso Y; Zembayashi M; Fujioka A; Kodama S; Nakajima I; Minato A; Kumada M. Nickel-Phosphine Complex-Catalyzed Grignard Coupling. I. Cross-Coupling of Alkyl, Aryl, and Alkenyl Grignard Reagents with Aryl and Alkenyl Grignard Reagents with Aryl and Alkenyl Halides: General Scope and Limitations. Bull. Chem. Soc. Jpn 1976, 49, 1958–1969. [Google Scholar]
- (22).Felkin H; Swierczevski G. Stereochemical Evidence in Favour of π-Allylnickel Intermediates in the Formation of Olefins from Allylic Alcohols and Grignard Reagents, Catalysed by Nickel Complexes. Tetrahedron Lett 1972, 13, 1433–1436. [Google Scholar]
- (23).(a) Jonas K; Pörschke KR; Krüger C; Tsay Y-H Carbani-on Complexes of Nickel(0). Angew. Chem., Int. Ed. Engl 1976, 15, 621–622. [Google Scholar]; (b) Terao J; Kambe N. Cross-Coupling Reaction of Alkyl Halides with Grignard Reagents Catalyzed by Ni, Pd, or Cu Complexes with π-Carbon Ligands(s). Acc. Chem. Res 2008, 41, 1545–1554. [DOI] [PubMed] [Google Scholar]
- (24).(a) The dimer form of Grignard reagent involves two solvent (dimethyl ether) ligands. For related mechanistic studies involving Grignard reagent, see: Ashby EC; Smith MB Concerning the Structure of the Grignard Reagent. II.1 In Diethyl Ether. Relevance of Grignard Composition to the Mechanism of Addition to Ketones. J. Am. Chem. Soc 1964, 86, 4363–4370. [Google Scholar]; (b) Jiménez-Osés G; Brockway AJ; Shaw JT; Houk KN Mechanism of Alkoxy Groups Substitution by Grignard Reagents on Aromatic Rings and Experimental Verification of Theoretical Predictions of Anomalous Reactions. J. Am. Chem. Soc 2013, 135, 6633–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Schlenk W; Schlenk W. Über die Konstitution der Grignardschen Magnesiumverbindungen. Ber. Dtsch. Chem. Ges. B 1929, 62, 920–924. [Google Scholar]
- (26).(a) Wurtz A. Sur une nouvelle classe de radicaux organiques. Annales de chimie et de physique 1855, 44, 275–312. [Google Scholar]; (b) Wurtz A. Ueber eine neue Klasse organischer Radicale. Annalen der Chemie und Pharmacie 1855, 96, 364–375. [Google Scholar]
- (27).For simplicity, DFT calculations were performed using (R)-BINAP as the ligand and (2S,4R)-1 and (2S,4R)-24. In experimental studies, it has been demonstrated that there is no match/mismatch effect between the enantiomers of BINAP and substrate. See refer-ences 3a and 8.
- (28).For a related experimental study that confirms the stability of Ni((R)-BINAP)2 species, see: Ge S-Z; Green RA; Hartwig JF Controlling First-Row Catalysts: Amination of Aryl and Heteroaryl Chlorides and Bromides with Primary Aliphatic Amines Catalyzed by a BINAP-Ligated Single-Component Ni(0) Complex. J. Am. Chem. Soc 2104, 136, 1617–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Given a comparison between π-extended systems and regular arenes, a scenario that the former binds more strongly to low-valent metal complexes in an η2-fashion than the latter is probably due to the retention of a certain degree of aromaticity. For a related study, see: Bauer DJ; Krueger C. Bonding of Aromatic Hydrocarbons to Nickel(0). Structure of Bis(tricyclohexylphosphine)(1,2-.eta.2-anthracene)nickel(0)-toluene. Inorg. Chem 1977, 16, 884–891. [Google Scholar]
- (30).(a) For early studies relates to distortion/interaction analysis, see: Kitaura K; Morokuma K. A New Energy Decomposition Scheme for Molecular Interactions within the Hartree-Fock Approximation. Int. J. Quantum Chem 1976, 10, 325–340. [Google Scholar]; (b) Ziegler T; Rauk A. A Theoretical Study of the Ethylene-Metal Bond in Complexes between Copper(1+), Silver(1+), Gold(1+), Platinum(0) or Platinum(2+) and Ethylene, Based on the Hartree-Fock-Slater Transition-State Method. Inorg. Chem 1979, 18, 1558–1565. [Google Scholar]
- (31).(a) For reviews of distortion/interaction analysis, see: van Leeuwen PWNM; Kamer PCJ; Reek JNH; Dierkes P. Ligand Bite Angle Effects in Metal-catalyzed C−C Bond Formation. Chem. Rev 2000, 100, 2741–2770. [DOI] [PubMed] [Google Scholar]; (b) Van Zeist W-J; Bickelhaupt FM The Activation Strain Model of Chemical Reactivity. Org. Biomol. Chem 2010, 8, 3118−3127. [DOI] [PubMed] [Google Scholar]; (c) Fernandez I; Bickelhaupt FM The Activation Strain Model and Molecular Orbital Theory: Understand-ing and Designing Chemical Reactions. Chem. Soc. Rev 2014, 43, 4953−4967. [DOI] [PubMed] [Google Scholar]; (d) Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed 2017, 56, 10070−10086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).(a) Greene MA; Yonova IM; Williams FJ; Jarvo ER Traceless Directing Group for Stereospecific Nickel-Catalyzed Al-kyl−Alkyl Cross-Coupling Reactions. Org. Lett 2012, 14, 4293–4296. [DOI] [PubMed] [Google Scholar]; (b) Dawson DD; Jarvo ER Stereospecific Nickel-Catalyzed Cross-Coupling Reactions of Benzylic Ethers with Isotopically-Labeled Grignard Reagents. Org. Process. Res. Dev 2015, 19, 1356– 1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Blackmond DG Reaction Progress Kinetic Analysis: A Power-ful Methodology for Mechanistic Studies of Complex Catalytic Reactions. Angew. Chem. Int. Ed 2005, 44, 4302–4320. [DOI] [PubMed] [Google Scholar]
- (34).Singleton DA; Thomas AA High-Precision Simultaneous Determination of Multiple Small Kinetic Isotope Effects at Natural Abundance. J. Am. Chem. Soc 1995, 117, 9357–9358. [Google Scholar]
- (35).(a) Biswas S; Weix DJ Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2013, 135, 16192–16197. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cherney AH, Kadunce NT; Reisman SE Catalytic Asymmetric Reductive Acyl Cross-Coupling: Synthesis of Enantioenriched Acyclic α,α-Disubstituted Ketones. J. Am. Chem. Soc 2013, 135, 7442–7445. [DOI] [PubMed] [Google Scholar]; (c) Knappke CEI; Grupe S; Gärtner D; Corpet M; Gosmini C; Jacobi von Wangelin, A. Reductive Cross-Coupling Reactions between Two Electrophiles. Chem. Eur. J 2014, 20, 6828–6842. [DOI] [PubMed] [Google Scholar]; (d) Weix DJ Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res 2015, 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) See reference 1f.
- (36).Intermolecular reactions of allylnickel complexes with alkyl bromides result in racemization of the alkyl halide and are proposed to proceed via a radical chain mechanism. See: Hegedus LS; Miller LL Reactions of n-Allylnickel Bromide Complexes with Organic Halides. Stereochemistry and Mechanism. J. Am. Chem. Soc 1975, 97, 459–460. [Google Scholar]
- (37).(a) Terao J; Watanabe H; Ikumi A; Kuniyasu H; Kambe N. Nickel-Catalyzed Cross-Coupling Reaction of Grignard Reagents with Alkyl Halides and Tosylates: Remarkable Effect of 1,3-Butadienes. J. Am. Chem. Soc 2002, 124, 4222–4223. [DOI] [PubMed] [Google Scholar]; (b) Terao J; Ikumi A; Kuniyasu H; Kambe N. Ni- or Cu-Catalyzed Cross-Coupling Reaction of Alkyl Fluorides with Grignard Reagents. J. Am. Chem. Soc 2003, 125, 5646–5647. [DOI] [PubMed] [Google Scholar]; (c) Terao J; Todo H; Watanabe H; Ikumi A; Kambe N. Nickel-Catalyzed Cross-Coupling Reaction of Alkyl Halides with Organozinc and Grignard Reagents with 1,3,8,10-Tetraenes as Additives. Angew. Chem. Int. Ed 2004, 43, 6180–6182. [DOI] [PubMed] [Google Scholar]; (d) See reference 23b.
- (38).(a) Chan TH; Fleming I. Electrophilic Substitution of Organosilicon Compounds-Applications to Organic Synthesis. Synthesis 1979, 761–786. [Google Scholar]; (b) Sakurai H. Reactions of Allylsilanes and Application to Organic Synthesis. Pure Appl. Chem 1982, 54, 1–22. [Google Scholar]
- (39).(a) Corey EJ; Semmelhack MF Organonickel Compounds as Reagents for Selective Carbon-Carbon Bond Formation Between Unlike Groups. J. Am. Chem. Soc 1967, 89, 2755–2757. [Google Scholar]; (b) Hegedus LS; Wagner SD; Waterman EL; Siirala-Hansen K. Reaction of π-Allylnickel Bromide Complexes with Ketones and Aldehydes. Synthesis of α-Methylene-γ-Butyrolactones. J. Org. Chem 1975, 40, 593–598. [Google Scholar]; (c) Johnson JR; Tully PS; Mackenzie PB; Sabat M. A Practical Reversed-Polarity Alternative to Organocuprate Conjugate Addition Chemistry. Halocarbon Coupling Reactions of Enal- and Enone-Derived Allylnickel Reagents. J. Am. Chem. Soc 1991, 113, 6172–6177. [Google Scholar]; (d) Kondo T; Ono H; Satake N; Mitsudo T; Watanabe Y. Nucleophilic and Electrophilic Allylation Reactions. Synthesis, Structure, and Ambiphilic Reactivity of (η3-Allyl)ruthenium(II) Complexes. Organometallics 1995, 14, 1945–1953. [Google Scholar]
- (40).Pearson RG Hard and Soft Acids and Bases, HSAB, Part I: Fundamental Principles. J. Chem. Educ 1968, 45, 581–587. [Google Scholar]
- (41).(a) Anh NT; Minot C. Conditions Favoring Retention of Configuration in SN2 Reactions. A Perturbational Study. J. Am. Chem. Soc 1980, 102, 103–107. [Google Scholar]; (b) Glukhovtsev MN; Pross A; Schlegel HB; Bach RD; Radom L. Gas-Phase Identity SN2 Reactions of Halide Anions and Methyl Halides with Retention of Configuration. J. Am. Chem. Soc 1996, 118, 11258–11264. [Google Scholar]
- (42).(a) see: reference 5b; (b) Tobisu M; Shimasaki T; Chatani N. Nickel-Catalyzed Cross-Coupling of Aryl Methyl Ethers with Aryl Boronic Esters. Angew. Chem, Int. Ed 2008, 47, 4866–4869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.