

Abstract
Synthesis of antibody-oligonucleotide conjugates has enabled the development of highly sensitive bioassays for specific epitopes in the laboratory and clinic. Most synthetic schemes to generate these hybrid molecules require expensive reagents, significant quantities of input antibody, and multi-step purification routes, thus limiting widespread application. Here we report a facile and robust conjugation strategy that involves “plug-and-play” antibody conjugation with succinimidyl-functionalized oligonucleotides, which are high-yielding and compatible for use directly after buffer exchange. The succinimidyl-linked oligonucleotides are synthesized with 5’-amine-modified oligonucleotides and disuccinimidyl suberate (DSS), both of which are inexpensive and commercially available. Direct incubation of the resulting stable succinimidyl-oligonucleotide conjugates with commercial antibodies yields conjugates ready for use after benchtop buffer exchange. Here we demonstrate that the resulting oligonucleotide-antibody and oligonucleotide-streptavidin conjugates retain potent and specific binding in activity-dependent proximity ligation imaging, and proximity ligation-mediated qPCR detection of endogenous proteins in native cellular contexts down to picogram levels of whole proteome. This DSS conjugation strategy should be widely applicable in the synthesis of protein-oligonucleotide conjugates.
Keywords: antibody-oligonucleotide conjugates, DSS chemistry, ADPL, PLA
Graphical Abstract
A facile and robust antibody-oligonucleotide conjugation strategy based on the homobifunctional linker disuccinimidyl suberate (DSS) is described, which involves native antibody and amine-modified oligonucleotides. Applications for ultrasensitive imaging and quantification of endogenous proteins by activity-dependent proximity ligation imaging and proximity-mediated qPCR are demonstrated.
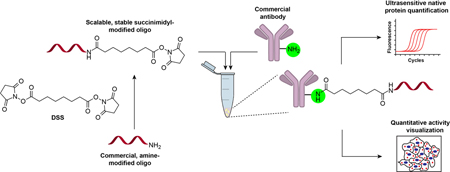
Introduction
The rapid development of nucleic acid sequencing technologies has enabled the detection and quantification of oligonucleotides in highly multiplexed formats. To introduce the benefits of nucleic acid detection to the field of proteomics, antibody-oligonucleotide conjugates, which integrate antibody target-specificity with the barcoding and amplification capability of nucleic acids, have attracted wide attention in recent years. Applications of antibody-oligonucleotide conjugates include immuno-PCR[1], proximity ligation assays (PLA)[2], proximity extension assays (PEA)[3], DNA-PAINT imaging[4], DNA-encoded antibody libraries[5], and activity-dependent proximity ligation (ADPL)[6]. All of these methods rely on the ability to synthesize antibody-oligonucleotide conjugates that retain the binding properties of the native antibody. Most synthetic schemes involve conjugation of linkers or oligonucleotides on solvent-accessible residues on the antibody surface. For example, the heterobifunctional cross-linker succinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (SMCC)[2b, 7] is used to directly modify surface lysines, which is then incubated with thiol-containing oligonucleotide followed by buffer exchange. While widely used, this approach is prone to side reaction of the linker’s maleimide moiety with reduced cysteine or proximal lysine residues on the antibody[8], producing intra- and intermolecular cross-linked species that degrade antibody activity and specificity. Even when the desired functional conjugates are obtained, the resulting species are known to have limited stability due to retro-Michael-type[9] or thiol-maleimide exchange reaction[10] decomposition. Other strategies include succinimidyl 4-hydrazinonicotinate acetone hydrazine (SANH) crosslinking to aldehyde-containing oligonucleotides[11], strain-promoted alkyne-azide cycloaddition (SPAAC) with a dibenzocyclooctyne (DBCO) linker and azide-modified oligonucleotide[12] or 4-azidobenzoyl fluoride labeling of the antibody followed by coupling to bicyclononyne (BCN)-modified oligonucleotides[13]. Despite the advantages of these new chemistries, they require the synthesis of uncommon and/or expensive oligonucleotides and linkers, relatively large amounts of input antibody, and can require tedious chromatographic purification of modified conjugates. Therefore, we sought to develop a streamlined and practical antibody modification scheme using commercially available reagents, minimal processing steps and long-term stability of isolated intermediates. Here we report such a route that involves installation of an activated ester on either terminus of a single stranded DNA of interest, which is then used to directly modify native antibodies or proteins without intervening purification steps of the modified antibody. Using this approach, we demonstrate that DSS-activation is a modular “plug-and-play” strategy to yield antibody-oligonucleotide conjugates that are useful for native protein imaging and qPCR-based protein quantification directly in cellular samples.
Results and Discussion
In our design, commercially available and affordable amino-containing single-stranded DNA (ssDNA) is reacted with an excess of the homobifunctional linker disuccinimidyl suberate (DSS, 250 equiv.)[14], and purified by high performance liquid chromatography (HPLC). The resulting activated-oligonucleotides can be stably aliquoted and stored under acidic conditions for months prior to direct introduction to antibody conjugation reactions (Fig. 1). To test this approach, a model 5’-amine modified, single-stranded 60-mer ssDNA was reacted with an excess of DSS linker at room temperature for 30 minutes, followed by ethanol precipitation and reverse phase HPLC purification. The hydrophobic nature of the succinimidyl suberate linker results in consistent chromatographic separation of the product from the starting material and side products (Fig. 2a). Modification yields above 50% were consistently observed, and the purified succinimidyl-modified ssDNA products could be lyophilized under acidic conditions (0.1% TFA), aliquoted and stably stored for several months, as confirmed by MALDI-TOF (Fig. S1). In particular, the absence of hydrolysis product confirmed the stability of the activated oligonucleotide. Using this DSS labeling scheme, a single round of synthesis and HPLC purification yielded sufficient succinimidyl-modified ssDNA for ~20 conjugations with 10 μg antibody samples that represent the lower limit of input antibody currently employed in other conjugation routes.
Fig. 1.
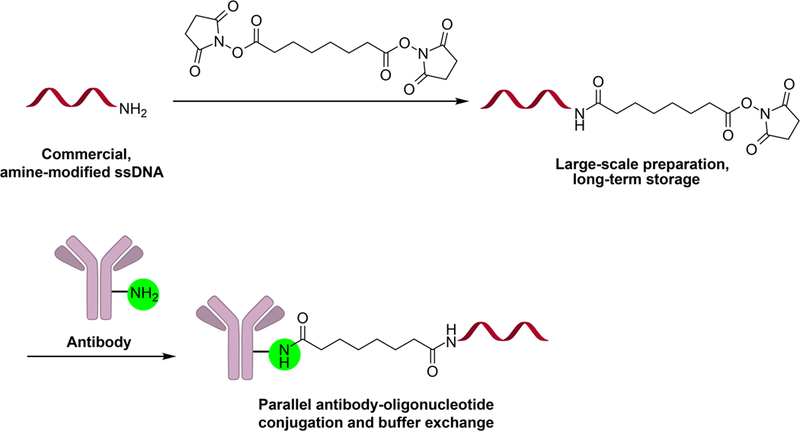
General DSS conjugation reaction schematic. Disuccinimidyl ester modification of amine-modified ssDNA, followed by parallel reaction with commercial antibodies yields stable antibody-oligonucleotide conjugates after benchtop sample buffer exchange.
Fig. 2.
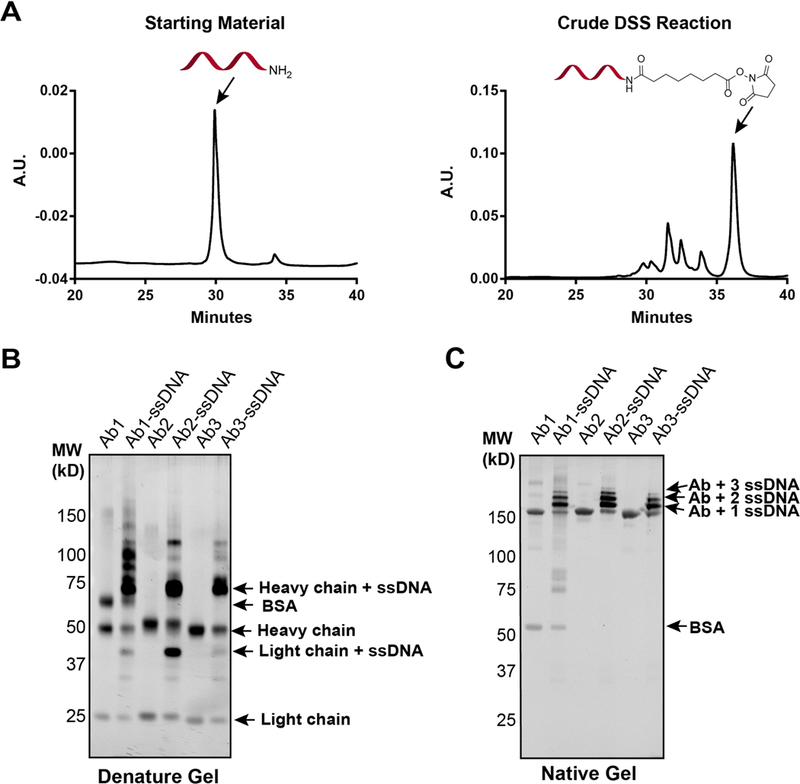
(A) HPLC chromatograms of precursor amine-modified ssDNA (left) and crude succinimidyl-modified ssDNA (right) after reaction with DSS. (B) Reducing SDS-PAGE gel analysis of three commercial antibodies before and after reaction with succinimidyl-ssDNA. Protein bands were visualized by Sybr-gold staining, revealing ssDNA-modification predominantly on heavy chains. (C) Native PAGE gel analysis of commercial antibodies before and after reaction with succinimidyl- modified ssDNA reveals high labeling efficiency to mono-, di, and tri-oligonucleotide conjugates for all three antibodies.
To test the antibody-oligonucleotide conjugation reaction, 3–4 equivalents of succinimidyl-modified ssDNA was added to ~10 μg of antibody (0.3–1 mg/mL in PBS) in 0.25 M HEPES, 0.25 M NaCl (pH 7.4) buffer. Reactions were run overnight at room temperature, quenched with 1.0 M TRIS buffer (pH 7.5), and unreacted and hydrolyzed oligonucleotide was removed by passage through a centrifugal filter. Reducing SDS-PAGE gel analysis of conjugation reactions with three commercial antibodies (anti-CTSB, anti-Myc, and anti-FAAH) confirmed the appearance of higher molecular weight conjugates primarily consistent with heavy chain and light chain monomers attached to a single oligonucleotide (Fig. 2b). The presence of bovine serum albumin (BSA), a common additive to commercial antibody stocks, did not influence conjugation reactions. Close inspection of the antibody-conjugates by non-denaturing PAGE gel electrophoresis revealed a degree of conjugation (DoC) of between one and three ssDNAs, with a single ssDNA conjugate being the most abundant species for all antibodies tested (Fig. 2c). These results were confirmed by MALDI-TOF detection of the mono-substituted, full length ssDNA-antibody conjugate (Fig. S2). Conjugation yields determined by band densitometry for the three representative commercial antibodies were 85.7, 82.8 and 79.1% for anti-CTSB, anti-Myc, and anti-FAAH, respectively. Together with functional validation of antibody activity after oligo-conjugation (Fig. S3), these data establish that DSS chemistry is a highly efficient, modular strategy for constructing antibody-oligonucleotide conjugates.
We next sought to validate the activity and potential applications of ssDNA-antibody conjugates synthesized with this modular DSS ligation strategy. We recently reported an activity-dependent proximity ligation (ADPL) platform for the spatially-resolved, quantitative imaging of active enzymes in single cells[6]. This method involves treating live cells with a biotinylated, family-wide activity-based probe targeting an enzyme family of interest, followed by cell fixation. Fixed cells are then labeled with probe-specific and protein-of-interest (POI)-specific primary antibodies, followed by secondary antibodies conjugated to barcoded, single-stranded oligonucleotides (Fig. 3A). Coincidence of the probe- and protein-directed dual antibody complexes enables specific ligation of a bridging oligonucleotide and rolling circle amplification. Finally, ADPL signal is detected by incubation with a complementary, fluorophore-labeled oligonucleotide and fluorescence microscopy[6]. We speculated that a potential liability of the dual primary-secondary antibody “sandwich” complex, which could theoretically extend up to ~40 nm from either the probe or POI[15], may result in background signal generated from complexes formed on the POI and endogenous biotinylated proteins or other non-POI targets of the probe. Based on this assumption, we sought to shorten the distance between the protein epitope and the probe recognition element (i.e. biotin) by installing the oligonucleotide barcode directly to the primary antibody-of-interest. For chemical probe recognition and labeling we sought to replace the α−biotin antibody with a streptavidin-ssDNA conjugate, which would take advantage of its smaller size and higher binding affinity to biotin. To compare the performance of the dual sandwich complex and direct ssDNA-Ab conjugates side-by-side we synthesized a barcoded ssDNA conjugate on polyclonal antibodies directed at the serine hydrolase enzyme neutral cholesterol ester hydrolase 1 (NCEH1), as well as a non-antibody protein, streptavidin, for chemical probe recognition (Fig. S3). We compared NCEH1 activity in the aggressive ovarian cancer cell line SKOV3 with the established dual antibody sandwich complex (Fig. 3B–C), as well as our direct anti-NCEH1-oligonucleotide and streptavidin-oligonucleotide conjugates. Consistent with our previous findings, the dual sandwich structure has a relatively high amount of background signal, as indicated by a probe/no-probe fluorescence ratio of 3.2-fold (Fig. 3C). In contrast, ADPL quantification of NCEH1 activity with DSS-synthesized direct oligonucleotide conjugates had significantly lower background signal in the no probe control, marked by a probe/no-probe ratio of 9.0 (Fig. 3D–F). Similar to previous results, we found omission of any component or step in the protocol resulted in significant loss of signal (Fig. S4). We note that signal with probe treatment obtained using the DSS synthesized direct oligonucleotide conjugates was lower compared to the signal obtained by dual sandwich complex, which is expected due to the lack of signal amplification caused by multivalent secondary antibodies binding to primary antibody[16].
Fig. 3.
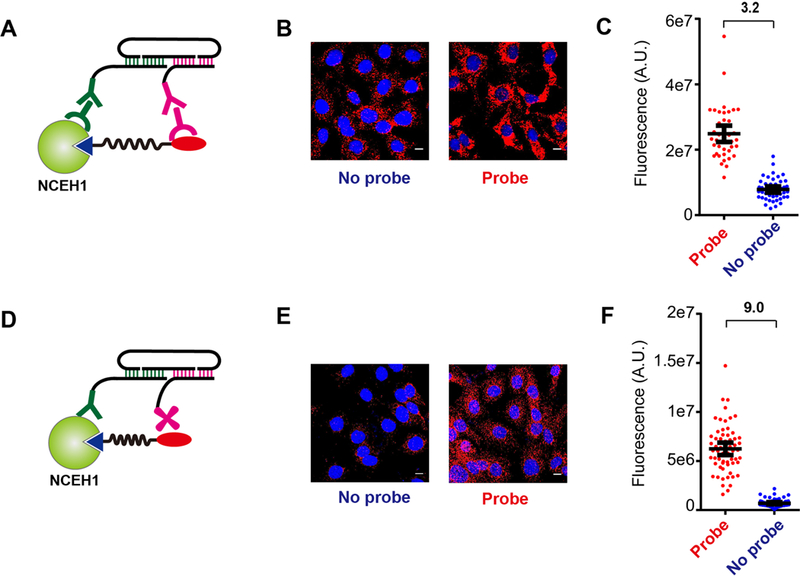
(A) Schematic depiction of the secondary-linked, “sandwich-like” activity dependent proximity ligation arrangement. (B-C) ADPL imaging (B) and quantification (C) of NCEH1 activity in SKOV3 ovarian cancer cells with or without fluorophosphonate-biotin (FP-Bio) probe treatment using secondary-linked conjugates. (D) Schematic depiction of direct ADPL detection of NCEH1 activity using antibody- and streptavidin-oligonucleotide conjugates synthesized by DSS chemistry. (E-F) ADPL imaging (E) and quantification (F) of NCEH1 activity in SKOV3 ovarian cancer cells with or without fluorophosphonate-biotin (FP-Bio) probe treatment using the direct oligonucleotide conjugates. Scale bar: 10 μm.
To determine whether protein levels alone could be quantified with direct ssDNA-antibody conjugates generated by DSS chemistry, we tested the utility of our conjugates for solution-phase PLA-qPCR quantitation of endogenous proteins in whole cell lysates[2a, 17]. This ultrasensitive quantification method requires the binding of two antibodies to distinct epitopes on the same protein molecule, thus templating proximity-based hybridization of unique barcoded oligonucleotides with splint oligonucleotide (Fig. 4A), which is unable to form at the extremely dilute concentrations in free solution. Using DSS chemistry, we separately labeled a polyclonal antibody mixture with two succinimidyl-modified ssDNAs in parallel (Fig. S5). Consistent with previous literature reports[17e, 17f], because polyclonal antibodies can bind an array of sites on the target protein the combined mixture can detect proximity of both bound oligonucleotides and template subsequent hybridization, ligation and amplification. Using these sets of barcoded ssDNA-antibody conjugates, we first performed PLA-qPCR-based detection of the prototypical human protein glyceraldehyde-3-phosphate dehydrogenase (GAPDH) with pure protein. These measurements demonstrated specific and robust qPCR signal over >4-log window of protein concentration, with up to a 4,000-fold signal dynamic range at picogram quantities of protein (Fig. 4B; Fig. S6). These results are qualitatively similar in performance to DBCO-based antibody-oligonucleotide conjugate detection of pure proteins[12], but requires fewer steps and cheaper reagents. To extend beyond pure protein and determine whether oligo-conjugates generated by DSS chemistry were compatible with ultrasensitive detection of endogenous proteins directly in complex biological samples (e.g. fluids, lysates), we performed PLA-qPCR directly in PC3 cell lysates. Similar to pure protein, we could detect endogenous GAPDH across a wide working window of total proteome concentration, with a signal dynamic range of nearly seven amplification cycles (Fig. 4C). Amplified signal was shown to be highly specific through the greater than 100-fold loss in signal upon omission of key reagents in the detection, ligation or amplification reactions, as well as through spike-in experiments with pure GAPDH (Fig. 4C–D). Combined, these data confirm that qPCR-PLA with DSS-synthesized oligo-antibody conjugates can detect endogenous proteins directly in whole cell lysate with a limit of detection (LOD) in the sub-picogram range of whole proteome (Fig. 4E). This work confirms that DSS-enabled protein-oligo conjugates can be more readily accessed compared to other conjugation methods, yet still support the ultrasensitive detection of proteins in extremely limited samples, including single cell protein levels, for both basic biology as well as diagnostic applications.
Fig. 4.
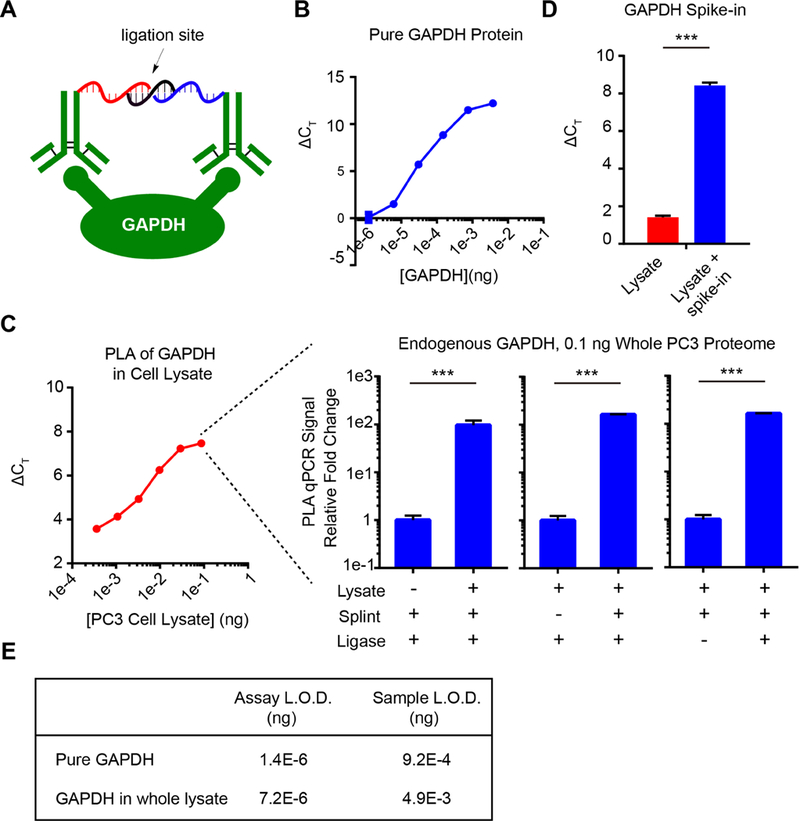
(A) Schematic depiction of dual-antibody detection of a model protein, GAPDH, for PLA-qPCR quantification. (B) PLA-qPCR detection of purified GAPDH protein across a high dynamic range. (C) PLA-qPCR detection of endogenous GAPDH protein in whole cell lysate from PC3 cells below pg level. The specificity is completely dependent on proximity ligation components: GAPDH-containing cell lysate, splint, and DNA ligase. In B and C, the X-axis displays the concentration of the protein or cell lysate, while the Y-axis is the delta Ct value normalized to ‘PBS’ blank control. (D) Purified GAPDH was spiked into the lysate and the increased PLA signal demonstrated the specificity of the detection in cell lysate. (E) Limit of detection values in the format of pure GAPDH and whole proteome amount. Sample LOD denotes the LOD of original sample input amount, whereas the assay LOD accounts for the 680-fold dilution introduced during the PLA workflow.
Conclusions
Recently, many methods have been developed for preparing covalent antibody conjugates, especially in the field of antibody-drug conjugates (ADCs)[18], which are being widely tested as cancer therapeutics. Given the high demand for convenient strategies with a low technical barrier of entry, heterobifunctional cross-linker chemistries have been widely utilized. In this report, we developed a homobifunctional DSS chemistry workflow for facile plug-and-play synthesis of antibody-oligonucleotide conjugates. Previous reports have also shown that succinimidyl-modified ssDNAs can be used in the specific cases of metal-binding or epitope-tagged proteins through the use of a template DNA strand that brings the reactive ssDNA into specific proximity on the protein[14a]. Likewise, commercial kits are available using DSS conjugation chemistry that involve pre-activation of the protein with DSS, which generates a tethered succinimidyl group directly on the antibody surface, but this approach can result in inter- and intra-protein crosslinks, lower yield and reduced activity. In contrast to these approaches, our data herein demonstrate that direct labeling with succinimidyl-modified ssDNAs can be applied generally to diverse natural proteins like antibodies and streptavidin, without requiring DNA-templated guides, while still generating specific affinity reagents that avoid unwanted background reactions. DSS conjugation has the advantages of streamlined procedures, high conjugation yield, low sample loss, and minimal perturbation to antibody function. Additionally, comparison to conjugation strategies using other heterobifunctional cross-linkers (e.g. SMCC, SANH and DBCO) with respect to oligonucleotide price, conjugation steps, purification method, and minimum antibody requirements highlights several advantages for the DSS conjugation strategy (Table 1). In terms of the oligonucleotide price, DSS chemistry only requires an amine-modified ssDNA, which is ~10-fold cheaper than thiol-modified oligonucleotides and ~100-fold cheaper than azide-modified oligonucleotides. Likewise, the disuccinimidyl linker itself is considerably cheaper than bifunctional linkers for Michael addition-like conjugations (e.g. SMCC) or click chemistry (e.g. DBCO). DSS chemistry only requires one step – DSS modification of target ssDNA – prior to direct conjugation to commercial antibodies. This one-step preparation can be performed in a parallel fashion, and the resulting succinimidyl-modified ssDNA species is stable for long-term storage and sufficient for many downstream parallel labeling reactions to generate, for example, multiplexed barcoded antibody libraries. Finally, the average labeling yield for DSS chemistry is in excess of 80%, which allows omission of FPLC or magnetic bead purification, labeling of small amounts of input antibody (~10 μg tested here), which greatly reduces the waste of precious antibodies. We anticipate DSS chemistry will serve as a useful synthetic strategy for the preparation of antibody or other protein-oligonucleotide conjugates in a range of applications.
Table 1.
Comparison of DSS to SMCC, SANH, and click chemistry
| Method | Oligo Price [a] |
Additional Reagent(s) Price |
Typical Conjugation Procedure |
Minimum Ab amount |
|---|---|---|---|---|
| DSS chemistry | amine-modified ssDNA:~$10/100nmol | DSS:$9.7/100mg [b] | 1) ssDNA-amine reaction with DSS; HPLC purification and storage | <10μg |
| 2) Parallel reaction of succinimidyl-modified ssDNA with antibody; buffer exchange | ||||
| SMCC chemistry | Thio-modified ssDNA:~$130/100nmol | sSMCC:$491.6/100mg[b] | 1) Reaction of sSMCC with antibody; desalt | ~100μg |
| 2) Reduce the ssDNA-thiol with DTT; desalt | ||||
| 3) Reaction of sSMCC-modified antibody with ssDNA-thiol | ||||
| 4) FPLC purification and concentrate for use | ||||
| SAHN chemistry | amine-modified ssDNA:~$10/100nmol | All-in-one conjugation Kit[c]:$546.0/100μgAb | 1) Reaction of ssDNA-amine with sulf-S-4FB; desalt | ~100μg |
| 2) Reaction of S-HyNic with antibody; desalt | ||||
| 3) Reaction of modified ssDNA and antibody with aniline catalyst | ||||
| 4) Magnetic-affinity, solid phase purification & elution | ||||
| Click chemistry | azide-oligo:~$1270/100 nmol | DBCO-PEG4-NHS ester:$523.2/100 mg | 1) Reaction of DBCO-PEG4-NHS ester with antibody; desalt | ~10μg |
| 2) Reacton of ssDNA-azide with DBCO-antibody; buffer exchange | ||||
representative prices from oligo vendor IDT.
prices from Thermo Fisher.
price from TriLink Biotechnologies
Experimental Section
Reagents and Materials.
All chemicals were purchased from Sigma-Aldrich unless noted otherwise and were used as received. The amine modified single strand oligonucleotides were purchased from Integrated DNA Technologies (IDT); detailed sequences are listed in Supplementary Tables 1 and 2. Commercial antibodies were purchased from Abcam (#ab58802, anti-cathepsin B), Millipore Sigma (#05–724, anti-Myc tag), Abcam (#ab54615, anti-FAAH1), respectively. Polyclonal anti-NCEH1 antibody was prepared in house by antigen immunization. Duolink PLA probes (#DUO92004 and #DUO92002) and Duolink PLA detection reagents (#DUO92007) were purchased from Sigma-Aldrich. Purified GAPDH protein (#G6091) was purchased form Sigma-Aldrich. NuPAGE Novex 4–12% Bis-Tris protein gels were purchased from Thermo Fisher Scientific (#NP0322BOX). Sybr-gold nucleic acid gel stain solution was purchased from Thermo Fisher Scientific (#S11494). Streptavidin was purchased from Leinco Technologies, Inc (#S203). IgG was purchased from Thermo Scientific (#02–6202). Absorption measurements were recorded on a Thermo Scientific Nanodrop 2000 spectrophotometer. 12-well chamber slide was purchased from Ibide (#81201). HPLC for oligonucleotide modification purification was performed using a Waters e2695 Separations Module. FPLC for streptavidin-oligonucleotide conjugate was performed using a ÄKTAexplorer with HiTrap Q HP column. Leica SP8 Laser Scanning Confocal was used to image a single focal plane to accurately detect the ADPL signal location using HyD detectors. Identical microscope acquisition parameters were set and used within experiments. Post-acquisition processing was performed using ImageJ software (NIH).
Activated NHS Ester Modified Oligonucleotides Preparation.
5’ or 3’ amine modified oligonucleotide was dissolved in water (20 nmol, 75 μL). DSS linker was dissolved in DMF (50 mM), then 75 μL of the DSS solution was added to oligonucleotide solution together with 75 μL of acetonitrile and 1 μL of triethylamine. The mixture was shaken at room temperature for 30 minutes, followed by ethanol precipitation. Briefly, 28 μL of sodium acetate (3 M, pH 5.2), 565 μL of pure ethanol and 2 μL of glycogen (20 mg/ml) were added to the mixture. After thorough vortexing, the mixture was kept in −80 °C for one hour, followed by centrifugation at 14,000 rpm for 30 minutes. The pellet was recovered by removing the supernatant, reconstituted in 0.03 M acetic acid (pH 4.5), and filtered through 0.2 μm filter. Reverse phase HPLC (phase A: 0.05 M trimethylamine/acetic acid buffer, pH 7.0; phase B: acetonitrile) was employed to purify the mixture. The HPLC gradient was 0–20% of phase B in 35 minutes. The peak of product fraction was collected and an equal volume of 0.2% trifluoroacetic acid solution was added to stabilize the activated NHS ester product. Then the modified oligonucleotide was aliquoted and lyophilized. The concentration was quantified based on the absorbance at 260 nm by Nanodrop.
Succinimidyl-modified ssDNA MALDI-TOF characterization.
50 mg/mL 3-HPA was dissolved in water and acetonitrile (1:1). 50 mg/mL ammonium citrate was dissolved in water. The 3-HPA solution was mixed with ammonium citrate solution (9:1) to act as the matrix. ~100 pmol of succinimidyl-modified ssDNA (1 μL) was spotted together with matrix (1 μL) and characterized by Bruker ultraflextreme MALDI-TOF.
Antibody-oligonucleotide Conjugate Preparation.
The antibody was dialyzed against PBS at 4 °C overnight, then concentrated using a 50 kD centrifugal filter tube. The concentration of the antibody was quantified based on absorbance at 280 nm. The typical concentration should be within 0.3–1.0 mg/ml. Lyophilized oligonucleotide (200 pmol, 3 equivalents) was dissolved in 4 μL of 1.0 M HEPES plus 1.0 M NaCl buffer (pH 7.4) and mixed with antibody (10 μg in PBS) and stirred at room temperature overnight. After quenching the reaction with 1 μL 1.0 M Tris (pH 7.5), excess oligonucleotide was removed by 50 kD centrifugal filter tube 6× for 10 minutes each. Antibody-oligonucleotide concentration was quantified by microBCA assay (Thermo Fisher Scientific, #23235) and the labeling conjugate was validated by native and denaturing PAGE gels. The gels were stained with Sybr-gold stain solution for 30 minutes at room temperature and briefly washed with water. The gel images were captured in a Chemidoc imaging system and the labeling yield was quantified by densitometry in ImageJ.
Antibody-oligonucleotide Conjugates Characterization.
The DNA-modified antibody or IgG (~1 mg/ml) was desalted using Zeba spin columns (7000 MWCO). A matrix solution was prepared by dissolving sinapinic acid (1 mg) in acetonitrile (70 μL) and water with 0.1% trifluoroacetic acid (30 μL). 1 μl of the DNA-antibody solution was deposited onto the MALDI plate and then mixed with 1 μL of MALDI matrix. The plate was allowed to dry at room temperature for ~4–5 hours. The oligonucleotides-antibody conjugates were characterized by Bruker ultraflextreme MALDI-TOF.
Streptavidin-oligonucleotide Conjugate Preparation.
DSS modified oligonucleotide (960 pmol, 0.5 equivalent) was dissolved in 20 μL 0.03M acetic acid (pH 4.5). Streptavidin (100 μg, in 30 μL PBS) was added to the oligonucleotide solution together with 15 μL of 1.0 M HEPES plus 1.0 M NaCl buffer (pH 7.4). The mixture was stirred at room temperature overnight. After quenching the reaction with 1 μL 1.0 M Tris (pH 7.5), the mixture was purified by ion exchange fast protein liquid chromatography (FPLC) in an ÄKTAexplorer with HiTrap Q HP (1 ml) column (Fig. S7 for FPLC chromatogram).
Activity-based Proximity Ligation (ADPL) with “Sandwich Probe Structure”.
SKOV3 cells were seeded in the 12-well chamber slide at 30,000 cells per well. Cells at 80–90% confluency were pulse treated with serine hydrolase family-wide inhibitor FP-Bio (2 μM) in RPMI1640 medium and incubated at 37 °C for 40 minutes. Cells were washed with PBS, fixed with 4% paraformaldehyde in PBS at room temperature for 15 minutes, washed twice with PBS for 5 minutes each at room temperature with orbital shaking, then permeabilized in 0.5% Triton X-100 in PBS at room temperature for 15 minutes, and washed twice with 0.05% Tween-20 in PBS for 5 minutes each at room temperature with orbital shaking. Prior to antibody incubation, the chamber was removed and the well boundaries delineated with the hydrophobic barrier pen (Vector laboratories, #H-4000). One-drop Duolink blocking buffer was added and the slide was incubated at 37 °C for 30 minutes in a humidified chamber. The blocking solution was removed by tapping, followed by addition of 20 μg/ml of the anti-biotin (rabbit, Abcam, #G196266) and anti-NCEH1 (mouse, 4 μg/ml of in-house polyclonal). Generally, a 20 μL solution of the two primary antibodies per well was incubated at 4 °C overnight with orbital shaking. Primary antibodies solution was removed by tapping; the slide was washed in wash buffer A (150 mM NaCl, 10 mM Tris, 0.05% Tween 20, pH 7.3) three times for 5 minutes with gentle orbital shaking. Oligo-linked secondary antibodies were then diluted 5-fold in antibody diluent buffer (Duolink anti-mouse minus and anti-rabbit plus), added to the slide and incubated at 37 °C for 1 hour with orbital shaking. The secondary antibody-probe solution was removed by tapping the slide, followed by washing in buffer A three times with gentle orbital shaking. Ligation mixture from the Duolink In Situ Detection Reagents Orange kit was diluted five-fold in water prior to addition of ligase at a 40-fold dilution. The ligation mixture was incubated at 37 °C for 30 minutes with orbital shaking, removed, and the slide was washed twice. Finally, amplification solution was diluted 5-fold in water prior to addition of polymerase at 80-fold dilution. This amplification solution was added to each well, incubated at 37 °C for 90 minutes in the dark, and removed by washing with buffer B (0.1 M NaCl, 0.2 M Tris, pH 7.3) twice for 10 minutes each, followed by a wash with 100-fold dilution of wash buffer B for 1 minute. Slides were dried at room temperature in the dark, mounted with 50 μL anti-fade mounting solution (Life technology, #P36961), covered with cover glass (Fisher, #12–545M), and sealed with nail polish.
Activity-based Proximity Ligation (ADPL) with “Direct-Conjugated Probe Structure”.
SKOV3 cells were seeded, treated with probe, fixed, permeabilized and blocked as mentioned above. Then the cells were incubated with 20 μg/ml of streptavidin-oligonucleotide 1 and 4 μg/ml of anti-NCEH1-oligonucleotide 2 at 4 °C overnight with orbital shaking. After washing, the cells were incubated with the hybridization mixture containing 250 nM bridging oligonucleotide 1, 250 nM bridging oligonucleotide 2, 0.25 mg/ml BSA, 0.25 M NaCl, 0.05% Tween 20 and 1X T4 ligation buffer (10 mM Tris-acetate, 10 mM magnesium acetate, 50 mM potassium acetate, pH 7.5). After 30 minutes, the slide was washed with 1× T4 ligation buffer for 2 minutes and incubated with the ligation mixture containing 0.1 U/μL T4 DNA ligase, 1 mM ATP, 0.25 mg/ml BSA, 0.25 M NaCl, 0.05% Tween 20 and 1X T4 ligation buffer for 30 minutes. After wash with buffer A twice, the slide was incubated with amplification buffer containing 0.25 U/μL Phi29, 0.25 mM dNTP, 0.2 mg/ml BSA, 0.05% Tween 20 and 1× RCA buffer (50 mM Tris-HCl, 10 mM magnesium chloride, 10 mM ammonium sulfate, pH 7.5) for 100 minutes. Following two washes with buffer A, the slide was incubated with detection mixture containing 10 nM detection oligonucleotide, 2× SSC buffer, 0.25 mg/ml BSA, 7.5 ng/μL Poly A, 0.05% Tween 20, and 1× DAPI for 30 minutes. The slide was washed, mounted and sealed as mentioned above.
ADPL image processing and quantification.
ImageJ was used to process all images. Lossless TIFF files were employed to quantify fluorescence intensity. To simplify the image processing workflow a Macro script to automatically process all images was created. The workflow was as follows: open all channels for each field of view; designate a color for each channel; adjust brightness/contrast for all channels (applying the same levels for all conditions within and between experiments to allow for direct comparison); merge the channels together; adjust the image unit from pixel to micrometer; add scale bars; export the processed TIFF files for quantification. For quantitative analysis single cell boundaries were identified manually using the DIC image. Then the “ROI Manager” tool in ImageJ was utilized to add all the cell outlines as a collection and overlay with the ADPL channel to measure per-cell fluorescence intensity. Typical quantitative comparisons were made using data from three or more independent fields of view per independent biological replicate condition.
PLA-qPCR Quantification in Pure GAPDH protein or Cell Lysate.
PC3 cell lysate was diluted in 3-fold aliquots. Pure GAPDH protein was diluted in 5-fold aliquots. PEG-8000 was added to a final concentration of 5% and incubated at 4 °C for 30 min, then centrifuged at 4,000 rpm for 20 minutes to remove potential assay interferences. 2 μL of each sample was added to 2 μL of probe mix resulting in a 200 pM concentration of each antibody-ssDNA probe in PBS pH 7.2, 20 μg/mL poly-A, 2 mM EDTA, 1% BSA, 0.05% goat IgG. Incubations were performed at 37 °C for 2 hours. As for the ligation reaction, 116 μL ligation solution were added containing 100 nM splint oligonucleotide, 2.5 units of Ampligase (Lucigen), 0.3 mM NAD+ (Sigma), 10 mM DTT, 20 mM Tris-HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2. Ligation proceeded at 30 °C for 15 minutes and was terminated by adding 2.5 μL of the 10-fold dilution of the USER (Uracil-Specific Excision Reagent) enzyme (NEB, #M5505S) to degrade the uracil containing connectors for another 15 minutes. 5 μL of the ligation reaction was added to a 20 μL PCR tube using 200 nM primers for 18 cycles. The product was then diluted 2-fold in 1× TE buffer prior to real-time PCR. 9 μL of the diluted pre-amplification product was added to 11 μL of qPCR mix (NEB, #M3004S) resulting in 0.9 μM of primers and 0.45 μM Taqman probe (IDT). Samples were run on a Roche LightCycler 480 with the default cycling protocol.
Supplementary Material
Acknowledegements
We thank Q. Dai, C. He, N. Li and J. Piccirilli for access to and assistance with instrumentation. We are grateful for financial support of this work from the following: NIH R00CA175399, DP2GM128199-01, and 2R01CA093577-11 (to R.E.M.), and the Duchossois Family Institute at the University of Chicago.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1] a).Sano T, Smith CL, Cantor CR, Science 1992, 258, 120–122; [DOI] [PubMed] [Google Scholar]; b) Adler M, Wacker R, Niemeyer CM, Analyst 2008, 133, 702–718. [DOI] [PubMed] [Google Scholar]
- [2] a).Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gústafsdóttir SM, Östman A, Landegren U, Nature biotechnology 2002, 20, 473; [DOI] [PubMed] [Google Scholar]; b) Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius K-J, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson L-G, Nature methods 2006, 3, 995. [DOI] [PubMed] [Google Scholar]
- [3]. Lundberg M, Eriksson A, Tran B, Assarsson E, Fredriksson S, Nucleic acids research 2011, 39, e102-e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Jungmann R, Avendaño MS, Dai M, Woehrstein JB, Agasti SS, Feiger Z, Rodal A, Yin P, Nature methods 2016, 13, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bailey RC, Kwong GA, Radu CG, Witte ON, Heath JR, Journal of the American Chemical Society 2007, 129, 1959–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Li G, Montgomery JE, Eckert MA, Chang JW, Tienda SM, Lengyel E, Moellering RE, Nature communications 2017, 8, 1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Niemeyer CM, Sano T, Smith CL, Cantor CR, Nucleic Acids Research 1994, 22, 5530–5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Chen Y, Kim MT, Zheng L, Deperalta G, Jacobson F, Bioconjugate chemistry 2016, 27, 2037–2047. [DOI] [PubMed] [Google Scholar]
- [9].Koniev O, Leriche G, Nothisen M, Remy J-S, Strub J-M, Schaeffer-Reiss C, Van Dorsselaer A, Baati R, Wagner A, Bioconjugate chemistry 2014, 25, 202–206. [DOI] [PubMed] [Google Scholar]
- [10].Ponte JF, Sun X, Yoder NC, Fishkin N, Laleau R, Coccia J, Lanieri L, Bogalhas M, Wang L, Wilhelm S, Bioconjugate chemistry 2016, 27, 1588–1598. [DOI] [PubMed] [Google Scholar]
- [11].Kozlov IA, Melnyk PC, Stromsborg KE, Chee MS, Barker DL, Zhao C, Biopolymers 2004, 73, 621–630. [DOI] [PubMed] [Google Scholar]
- [12].Gong H, Holcomb I, Ooi A, Wang X, Majonis D, Unger MA, Ramakrishnan R, Bioconjug Chem 2016, 27, 217–225. [DOI] [PubMed] [Google Scholar]
- [13].Dovgan I, Ursuegui S, Erb S, Michel C, Kolodych S, Cianferani S, Wagner A, Bioconjug Chem 2017, 28, 1452–1457. [DOI] [PubMed] [Google Scholar]
- [14] a).Rosen CB, Kodal AL, Nielsen JS, Schaffert DH, Scavenius C, Okholm AH, Voigt NV, Enghild JJ, Kjems J, Tørring T, Nature chemistry 2014, 6, 804; [DOI] [PubMed] [Google Scholar]; b) Trads JB, Tørring T, Gothelf KV, Accounts of chemical research 2017, 50, 1367–1374. [DOI] [PubMed] [Google Scholar]
- [15].Jarvius M, Paulsson J, Weibrecht I, Leuchowius K-J, Andersson A-C, Wählby C, Gullberg M, Botling J, Sjöblom T, Markova B, Molecular & cellular proteomics 2007, 6, 1500–1509. [DOI] [PubMed] [Google Scholar]
- [16].Agasti SS, Wang Y, Schueder F, Sukumar A, Jungmann R, Yin P, Chemical science 2017, 8, 3080–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17] a).Gullberg M, Gústafsdóttir SM, Schallmeiner E, Jarvius J, Bjarnegård M, Betsholtz C, Landegren U, Fredriksson S, Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 8420–8424; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fredriksson S, Dixon W, Ji H, Koong AC, Mindrinos M, Davis RW, Nature methods 2007, 4, 327; [DOI] [PubMed] [Google Scholar]; c) Schallmeiner E, Oksanen E, Ericsson O, Spångberg L, Eriksson S, Stenman U-H, Pettersson K, Landegren U, Nature methods 2007, 4, 135; [DOI] [PubMed] [Google Scholar]; d) Nong RY, Wu D, Yan J, Hammond M, Gu GJ, Kamali-Moghaddam M, Landegren U, Darmanis S, Nature protocols 2013, 8, 1234; [DOI] [PubMed] [Google Scholar]; e) Albayrak C, Jordi CA, Zechner C, Lin J, Bichsel CA, Khammash M, Tay S, Molecular cell 2016, 61, 914–924; [DOI] [PubMed] [Google Scholar]; f) Robinson PV, Tsai C.-t, de Groot AE, McKechnie JL, Bertozzi CR, Journal of the American Chemical Society 2016, 138, 10722–10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18] a).Chudasama V, Maruani A, Caddick S, Nature chemistry 2016, 8, 114; [DOI] [PubMed] [Google Scholar]; b) Beck A, Goetsch L, Dumontet C, Corvaïa N, Nature Reviews Drug Discovery 2017, 16, 315. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.