

Abstract
Triisopropylsilane (TIS), a hindered hydrosilane, has long been utilized as a cation scavenger for the removal of amino acid protecting groups during peptide synthesis. However, its ability to actively remove S-protecting groups by serving as a reductant has largely been mischaracterized by the peptide community. Here, we provide strong evidence that TIS can act as a reducing agent to facilitate the removal of acetamidomethyl (Acm), 4-methoxybenzyl (Mob), and tert-butyl (But) protecting groups from cysteine (Cys) residues in the presence of trifluoroacetic acid (TFA) at 37 °C. The lability of the Cys protecting groups in TFA/TIS (98/2) in this study are in the order: Cys(Mob) > Cys(Acm) > Cys(But), with Cys(Mob) being especially labile. Unexpectedly, we found that TIS promoted disulfide formation in addition to aiding in the removal of the protecting group. Our results raise the possibility of using TIS in orthogonal deprotection strategies of Cys-protecting groups following peptide synthesis as TIS can be viewed as a potential deprotection agent instead of merely a scavenger in deprotection cocktails based on our results. We also tested other common scavengers under these reaction conditions and found that thioanisole and triethylsilane were similarly effective as TIS in enhancing deprotection and catalyzing disulfide formation. Our findings reported herein show that careful consideration should be given to the type of scavenger used when it is desirable to preserve the Cys-protecting group. Additional consideration should be given to the concentration of scavenger, temperature of the reaction, and reaction time.
GRAPHICAL ABSTRACT
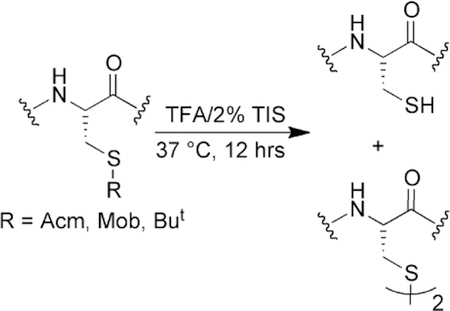
Triisopropylsilane can act as a deprotection reagent to remove S-protecing groups from cysteine. Care should be taken when using alkylsilane scavengers in peptide deprotection cocktails under conditions where it is desired to keep the protecting group on cysteine. The concentration, temperature, and length of time of the deprotection reaction with alkylsilanes are all important factors for consideration.
INTRODUCTION
Hindered hydrosilanes such as triisopropylsilane (TIS) are commonly used as cation scavengers in the deprotection of amino acid side-chains following solid-phase peptide synthesis (SPPS)[1, 2]. Protecting groups such as trityl (Trt) and tert-butyl (But) form stable cations upon acidolysis and are easily removed from N-, O-, and S-containing amino acid side-chains (for cysteine (Cys) But is not easily removed). In acid, TIS drives the equilibrium towards cleavage of the protecting group from the amino acid side-chain by donating a hydride to the resulting cation in an irreversible manner [1]. In these cases the addition of TIS to the peptide cleavage cocktail is almost an afterthought because TIS need not directly participate in breaking of a carbon-heteroatom bond.
Though TIS and other hindered hydrosilanes are exclusively thought of as protecting group scavengers in SPPS [2, 3], they are actually mild reducing agents and can reduce a carbon-heteroatom bond and can therefore directly participate in the removal of a protecting group from an amino acid side-chain. This fact has largely been misunderstood in the peptide chemistry literature. Silicon is metallic in nature, and has low electronegativity relative to hydrogen resulting in polarization of the Si-H bond [4, 5]. This polarization causes hydrogen to be hydridic, and results in a milder reducing agent than the usual aluminum-, boron-, and other metal-based hydrides [4, 5]. Thus, hydrosilanes have long been commonly employed to provide the hydride in reductions of various carbenium ion precursors and it should not be surprising that TIS can function as a deprotection reagent, such as iodine or mercuric acetate, in SPPS as we now report.
An earlier investigation into the use of various scavengers in the synthesis of a target Cys-containing peptide protected with the acetamidomethyl (Acm) group demonstrated that TIS afforded the highest yield of Cys(Acm)-protected peptide compared to other commonly used scavengers such as water, phenol, anisole, and ethanedithiol (EDT) [6]. All of the tested scavengers including TIS also yielded a mixture of other products including the fully deprotected peptide and the peptide disulfide. The use of TIS in the deprotection cocktail yielded 4% fully deprotected peptide and 20% peptide disulfide [6].
The results of Singh and coworkers were later misinterpreted in several reviews to mean that TIS helped to suppress removal of S-protecting groups [2, 3]. In contrast, we have tested TIS and other commonly used scavengers used in Fmoc peptide cleavage cocktails, and provide evidence that TIS serves as a reducing agent to actively aid in the removal of S-protecting groups including Acm, 4-methoxybenzyl (Mob), and But in the presence of trifluoracetic acid (TFA) and mild heat. In addition, we find that TIS actually helps to promote disulfide formation as initially reported by Singh and coworkers [6].
Our findings reported herein show that careful consideration should be given to the type of scavenger used when it is desirable to preserve the Cys-protecting group. Additional consideration should be given to the concentration of scavenger and reaction time. Conversely, the addition of TIS can actually facilitate the removal of the protecting group using specific reagents. The latter point is a topic of a future report.
MATERIALS AND METHODS
Materials.
Solvents for peptide synthesis were purchased from Fisher Scientific (Pittsburgh, PA). Fmoc-Cys(Acm)-OH was purchased from Advanced ChemTech. Fmoc-Cys(But)-OH and Fmoc-Cys(Mob)-OH were purchased from NovaBiochem (Burlington, MA). All other Fmoc-amino acids were purchased from RSsynthesis (Louisville, KY). 2-Chlorotrityl chloride resin SS (100–200 mesh) and 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) for solid-phase synthesis were purchased from Advanced ChemTech (Louisville, KY). Triisopropylsilane (98%), triethylsilane (99%), thioanisole (99%), and anisole (99%) were purchased from Acros Organics (Pittsburgh, PA). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was purchased from ThermoFischer Scientific. All other chemicals were purchased from either Sigma-Aldrich (Milwaukee, WI) or Fisher Scientific (Pittsburgh, PA). The HPLC system is from Shimadzu with a Symmetry® C18 −5 mm column from Waters Corp. (Milford, MA) (4.6 × 150 mm). Mass spectral analysis was performed on an Applied Biosystems QTrap 4000 hybrid triple-quadrupole/linear ion trap liquid chromatograph-mass spectrometer (SciEx, Framingham, MA).
Peptide Synthesis.
Either Fmoc-Cys(Acm)-OH, Fmoc-Cys(Mob)-OH, or Fmoc-Cys(But), were used to synthesize test peptides of sequence H-Pro-Thr-Val-Thr-Gly-Gly-Cys-Gly-OH (H-PTVTGGCG-OH). All peptides were synthesized on a 0.1 mmol scale using a glass vessel that was shaken with a model 75 Burrell wrist action shaker. For each batch, 300 mg of 2-chlorotrityl chloride resin SS (100–200 mesh, AdvancedChemtech), was swelled in dichloromethane (DCM) for 30 min. The first amino acid was directly coupled to the resin using 2% N-methylmorpholine (NMM) in DCM, shaking for 1 h. The resin was then capped using 8:1:1 DCM:methanol:NMM. Subsequent amino acids were coupled using 0.2 mmol of Fmoc-protected amino acid, 0.2 mmol of HATU coupling agent, and 2% NMM in dimethylformamide (DMF), shaking for 1 h. Preactivation of any amino acid was not performed prior to coupling. Between amino acid couplings, the Fmoc protecting group was removed via two 10 min agitations with 20% piperdine in DMF. Success of Fmoc removal steps and amino acid couplings were monitored qualitatively using a ninhydrin test [7]. Removal of the final Fmoc protecting group completed the peptide synthesis. Peptides were cleaved from the resin via a 1.5 h reaction with a cleavage cocktail consisting of TFA/TIS/H2O (96/2/2). Following cleavage and side chain deprotection, the resin was washed with TFA and DCM and the volume of the cleavage solution was reduced by evaporation with nitrogen gas. The peptide solution was then transferred by pipette into cold, anhydrous diethyl ether, where the peptides were observed to precipitate. Centrifugation at 3000 rpm on a clinical centrifuge (International Equipment Co., Boston, MA) for 10 min pelleted the peptide. Peptides were dried, then dissolved in a minimal amount of water, lyophilized, and used without further purification.
Reduction of Cys(Acm), Cys(Mob), and Cys(But) by triisopropylsilane.
Lyophilized test peptide H-PTVTGGCG-OH with either S-Acm, S-But, or S-Mob protection on the Cys residue were dissolved in neat TFA and the solution was divided into two aliquots. To one aliquot, 2% TIS was added. Both aliquots were incubated in a 37 ºC water bath for 12 h. To test TIS reduction under standard cleavage conditions, S-Acm protected peptide was dissolved in neat TFA, divided into two aliquots, 2% TIS was added to one aliquot, and both aliquots were incubated at room temperature for 2 h. For all reactions, the final reaction volume was 1.0 – 1.5 mL and contained 1.7 – 2.5 mM peptide. After incubation, peptides were precipitated with cold diethyl either, pelleted via centrifugation (as described above), and dried using nitrogen gas. For purification; dried pellets were re-dissolved in minimal amounts of neat TFA, precipitated with cold ether, pelleted via centrifugation, and dried (all as described previously). Dried peptide pellets were then dissolved in water, lyophilized, and analyzed without further purification.
Reduction of peptide disulfide by TCEP.
Cys(Acm) protected test peptide (3.45 μmol) that was previously incubated with TFA/TIS (98/2) for 12 h at 37 °C was precipitated and lyophilized as described above. The peptide was next re-dissolved in 4:1 water: acetonitrile. To this solution, 4.95 mg (17.3 μmol) of TCEP was added and allowed to react for 12 h at room temperature. HPLC analysis was then performed without further purification.
Reduction of Cys(Acm) with water, phenol, anisole, thioanisole, and triethylsilane.
Test peptide H-PTVTGGC(Acm)G-OH was dissolved in neat TFA and the solution divided into two aliquots. To one aliquot, 2% scavenger (either water, phenol, anisole, thioanisole or triethylsilane) was added. Both aliquots were incubated in a 37 °C water bath for 12 h. For all reactions, the final reaction volume was 1.0 mL with 2.0 mM peptide. After incubation, peptides were precipitated with cold diethyl ether, pelleted via centrifugation (as described above), and dried using nitrogen gas. For purification, dried pellets were re-dissolved in minimal amounts of neat TFA, precipitated with cold ether, pelleted via centrifugation, and dried (as described previously). Peptide pellets were then dissolved in a minimal amount of water, lyophilized, and analyzed without further purification.
High Pressure Liquid Chromatography.
HPLC analysis of all samples was completed using a Shimadzu with a Symmetry® C18 5 µm column from Waters (4.6 × 150 mm). Aqueous and organic phases were 0.1% TFA in distilled, deionized water (buffer A) and 0.1% TFA in HPLC‐grade acetonitrile (buffer B), respectively. Beginning with 100% buffer A, buffer B was increased by 1% up to 50% over 50 min with a 1.4 mL/min gradient elution. Buffer B was then increased by 10% up to 100% over 5 min. This method was used for analysis of each sample. Peptide elution was monitored via absorbance at both 214 and 254 nm.
RESULTS AND DISCUSSION
Previous studies by Harris and coworkers showed that it is possible to convert Cys(Acm) to Cys(5-Npys) using 2,2′-dithiobis(5-nitropyridine) (DTNP) in TFA in the presence of thioanisole [8]. This is an improvement over typical Cys(Acm) deprotection which requires use of iodine or mercuric ion under acidic conditions [9–12]. Iodine oxidation is harsh, toxic and produces many side reactions, and mercuric ion is a toxic, heavy metal [12]. Thus, using DTNP for Cys(Acm) deprotection is more gentle and produces fewer side reactions. However, the drawback to this method is that a large excess of DTNP is required for Cys(Acm) deprotection with only a 70% removal of the Acm group [8].
While working on methods to overcome the limitations of using DTNP to deprotect Cys(Acm) residues, we unexpectedly discovered that Acm was partially removed (70%) from a test peptide of sequence H-PTVTGGC(Acm)G-OH in TFA/TIS (98/2), when incubated for 12 h at 37 ºC (Figure 1B). In contrast, the same test peptide incubated in 100% TFA showed almost no conversion of Cys-Acm to Cys-SH when incubated under the same conditions (Figure 1A). We found that the deprotected peptide existed as both the Cys-SH (35%) and disulfide forms (35%). The peptide disulfide was unambiguously identified by adding 5-fold excess TCEP to the peptide reaction mixture containing 2% TIS. Under these acidic conditions, reduction of the disulfide using TCEP is slow and the reaction was incomplete, but the amount of peptide disulfide dramatically decreased with a concomitant rise in the Cys-SH form as revealed by HPLC analysis (Figure 1C).
Figure 1:
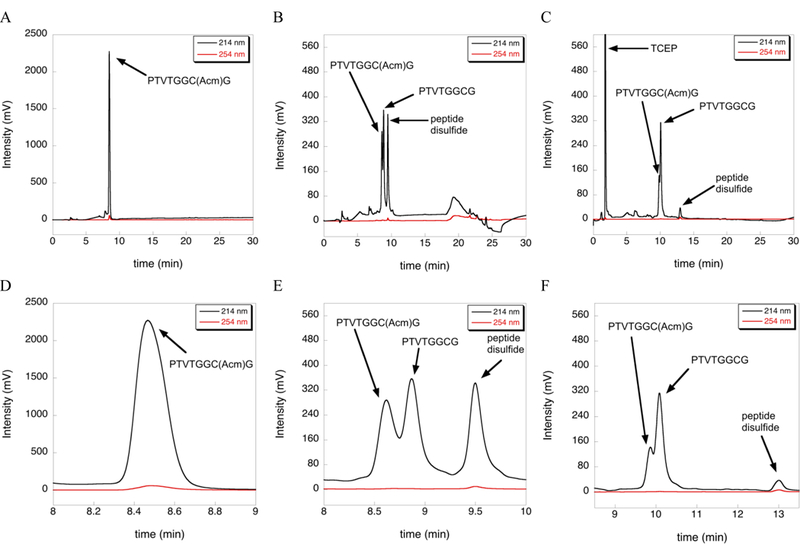
Extent of deprotection of Cys(Acm) by TIS at 37 ºC as measured by HPLC. (A) No reduction of the S-Acm bond in the Cys(Acm) test peptide was observed by HPLC after incubation in neat TFA for 12 h at 37 ºC. (B) In contrast, incubation of the test peptide with TFA/TIS (98/2) for 12 h at 37 ºC resulted in conversion of S-Acm to S-H (35%) and peptide disulfide (35%) as evidenced by the HPLC trace. (C) Addition of 5-fold excess TCEP to the reaction in (B) for 12 h resulted in nearly complete reduction of the disulfide to the reduced form. Traces D, E, and F are zoomed in views of traces A, B, and C, respectively.
Our identification of each of the peaks in Figure 1 is further supported by mass spectrometric (MS) analyses. Mass chromatograms of the reactions in Figure 1 are shown in Figure 2.
Figure 2:
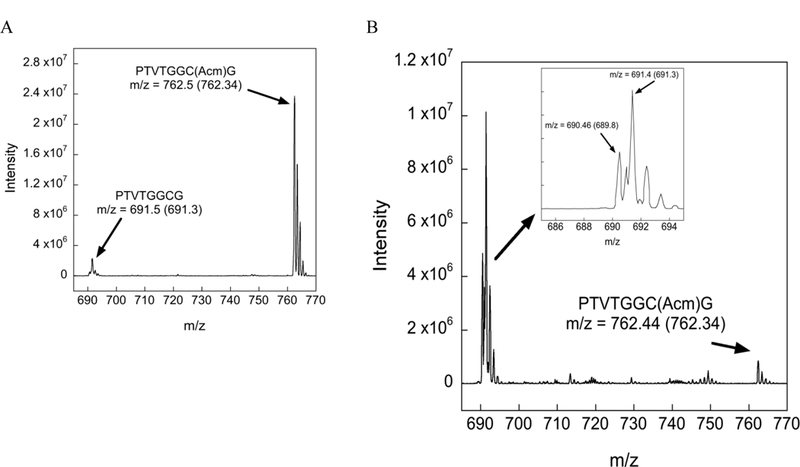
Mass chromatograms of the Cys(Acm) protected peptide under various conditions. The arrow points to the observed m/z whereas the number inside the parentheses denotes the theoretical m/z value. (A) Mass chromatogram of the Acm-test peptide after 12 h incubation in neat TFA at 37 ºC. The MS data show that nearly all of the peptide exists in the Cys(Acm) form, in agreement with HPLC analysis. (B) Mass chromatogram of the Acm-test peptide after 12 h incubation in TFA/TIS (98/2) at 37 ºC. The MS data show that inclusion of 2% TIS in the reaction mixture drives deprotection to the Cys-SH (m/z = 691.4) and disulfide (m/z = 690.46) forms in support of our HPLC analysis. Please note that the value m/z = 690.46 for the peptide disulfide correspond to the doubly charged ion (z = 2).
The deprotection reactions in Figure 1 were carried out at 37 ºC and extended reaction time. We found these conditions to be useful for new deprotection chemistry currently under development in our laboratory as mentioned above. However, use of elevated temperature allowed us to “discover” side reactions that may be absent under standard deprotection conditions. To test whether the inclusion of TIS would drive deprotection of our Cys(Acm)-containing peptide under typical deprotection conditions, we incubated the peptide in TFA/TIS (98/2) for 2 h at room temperature. HPLC analysis of this reaction shows that little or no deprotection occurred (Figure 3).
Figure 3:
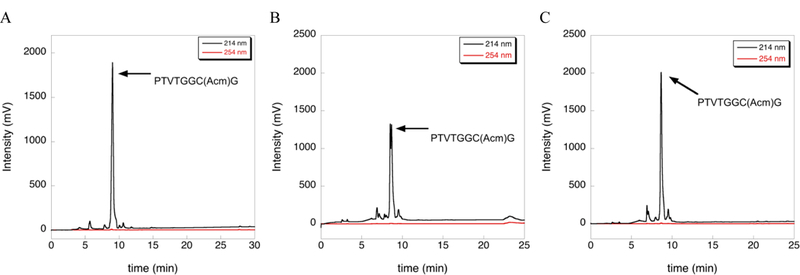
Extent of deprotection of Cys(Acm) by TIS under standard deprotection conditions as measured by HPLC. (A) HPLC chromatogram of the Acm-test peptide after cleavage from the solid support using a cleavage cocktail containing TFA/TIS/H2O (96/2/2) for 2 h at room temperature. (B) HPLC chromatogram of the Acm- test peptide after further incubation of the isolated peptide in neat TFA for 2 h at room temperature. (C) HPLC chromatogram of the Acm-test peptide after further incubation of the isolated peptide in TFA/TIS (98/2) for 2 h at room temperature. Little or no deprotection is observed as evidence by the absence of change in the HPLC chromatogram.
As demonstrated in Figures 1A and 3, the S-Acm protecting group is stable to incubation with neat TFA in agreement with earlier reports [3, 9]. However, others, both at elevated temperature and at room temperature, have questioned the stability of S-Acm. The reason for this disparity is most likely due to sequence specific effects. For example, Muttenthaler and coworkers report that S-Acm is stable to TFA at 25 ºC, but loss of ~10% of the Acm group occurs at 40 ºC of a bis-Acm protected oxytocin peptide [13]. We posit that one reason for the higher loss of the Acm group reported by Muttenthaler may be due to the peptide containing two Cys(Acm) residues whereas our peptide only contains one. Engebretsen and coworkers speculate that sequence specific effects may also play a role in the loss of the Acm group as they reported loss (11%) of both Acm groups of a fragment of human coagulation factor VII after incubation in TFA/H2O (95/5) at room temperature [14].
We next tested water, anisole, phenol, thioanisole, and triethylsilane (TES) to see if these commonly used scavengers would also aid in the removal of the Acm group using our reaction conditions (2% scavenger, 37 ºC, and extended reaction time). The results of our HPLC analyses for these scavengers are shown in Figure 4. Peak assignments in Figure 4 are based upon retention times and MS analyses of the reactions (see Figure S1 in the Supporting Information). Of the scavengers tested, water was very ineffective at aiding in the removal of the Acm group, while anisole and phenol were comparable to TIS in ability to remove the Acm group. Thioanisole under these reaction conditions removed 80–90% of the Acm group and thus was significantly better than anisol or phenol in this respect, and like TIS promoted disulfide formation. This data is consistent with previous reports which showed that anisole [15] and thioanisole [16] act to enhance the lability of S-Acm. TES was slightly better than all other scavengers tested (including TIS) in promoting the removal of the Acm group. This should not be surprising since TES is much less hindered than TIS and thus it is a better hydride donor [1]. However, one drawback of using TES is that it can reduce the indole ring of tryptophan, whereas this is much less of a problem with TIS [1]. One interesting thing to note is that like TIS, both thioanisole and TES promote disulfide bond formation whereas the other scavengers tested do not.
Figure 4:
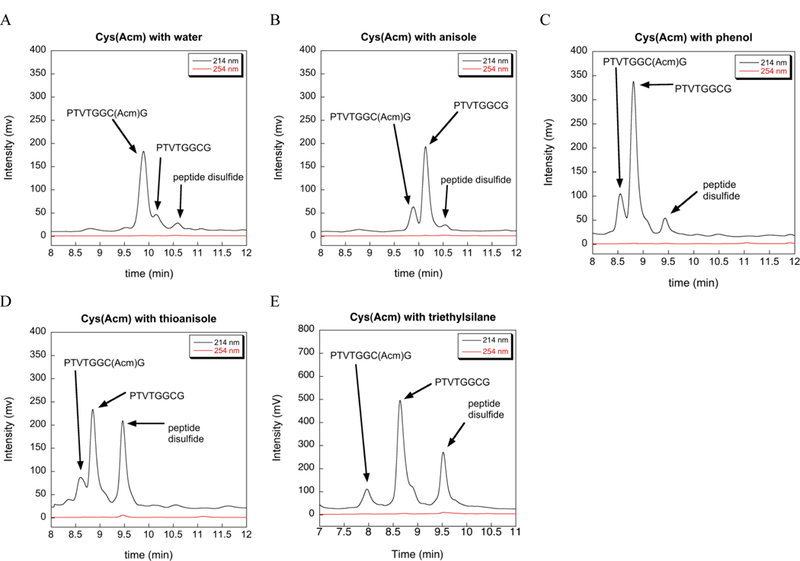
Extent of deprotection of Cys(Acm) by commonly used SPPS scavengers at 37 ºC in TFA as measured by HPLC. (A) 2% H2O. (B) 2% anisole. (C) 2% phenol. (D) 2% thioanisole. (E) 2% TES.
We next wondered about the lability of other commonly used Cys-protecting groups in the presence of TIS. We synthesized peptides H-PTVTGGC(Mob)G-OH and H-PTVTGGC(But)G-OH, isolated each peptide by ether precipitation and lyophilization, and then incubated each in TFA/TIS (98/2) at 37 ºC for 12 h as before. HPLC analyses of both of these reactions are shown in Figure 5.
Figure 5:
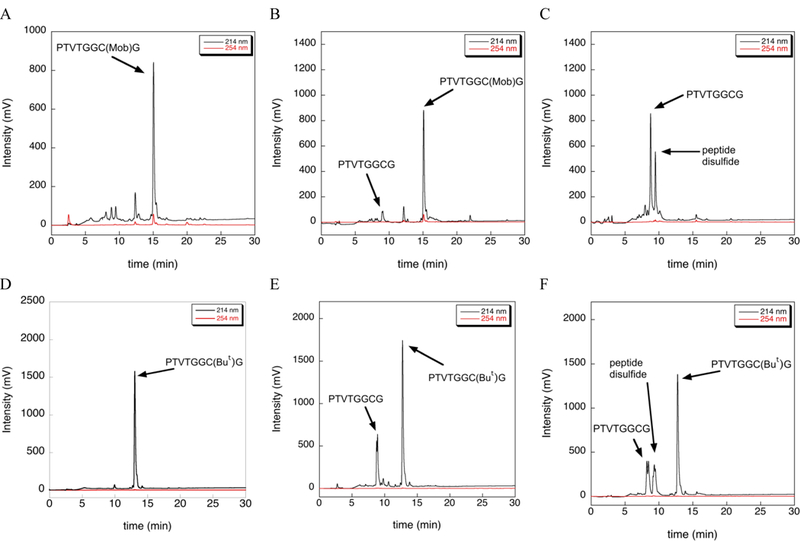
Extent of deprotection of Cys(Mob) and Cys(But) by TIS at 37 ºC as measured by HPLC. (A) HPLC chromatogram of the Cys(Mob) test peptide after cleavage from the solid support using a cleavage cocktail containing TFA/TIS/H2O (96/2/2) for 2 h at room temperature. (B) HPLC chromatogram of the Cys(Mob) test peptide after incubation in neat TFA for 12 h at 37 ºC. (C) HPLC chromatogram of the Cys(Mob) test peptide after incubation in TFA/TIS (98/2) for 12 h at 37 ºC. (D) HPLC chromatogram of the Cys(But) test peptide after cleavage from the solid support using a cleavage cocktail containing TFA/TIS/H2O (96/2/2) for 2 h at room temperature. (E) HPLC chromatogram of the Cys(But) test peptide incubation in neat TFA for 12 h at 37 ºC. (F) HPLC chromatogram of the Cys(But) test peptide after incubation in TFA/TIS (98/2) for 12 h at 37 ºC.
As is evident in Figure 5(A-C), S-Mob is mostly stable to TFA in the absence of TIS. In contrast, S-Mob is very labile in the presence of TIS at slightly elevated temperature and extended reaction time as shown by the complete conversion of Cys(Mob)-protected peptide to the Cys-SH and disulfide forms as shown by the HPLC trace in Figure 5C. We did not test shorter reaction times and it is likely that removal of Mob can be achieved using much shorter reaction times at 37 ºC.
In the case of the Cys(But)-protected peptide, ~20% of the But-protecting group is removed when incubated in neat TFA in the absence of TIS (Figure 5E), which indicates that Cys(But) is more acid labile than Cys(Mob). The addition of TIS to the reaction results in only slightly more deprotection of Cys(But), but also results in more disulfide formation (Figure 5F).
Our HPLC analyses of the products of deprotection for Cys(Mob)- and Cys(But)-containing peptides is further supported by the MS data shown in Figure 6. As shown in Figure 6B, the amount of Cys(Mob) protected peptide is almost completely diminished, while the amount of disulfide and deprotected peptide are in abundance, in agreement with the HPLC trace shown in Figure 5C. The MS data shown in Figures 6C and 6D show that the Cys(But)-containing peptide remains largely intact in the presence of 2% TIS, with a relatively small amount of peptide in the Cys-SH and disulfide forms, in agreement with the HPLC trace shown in Figure 5F.
Figure 6:
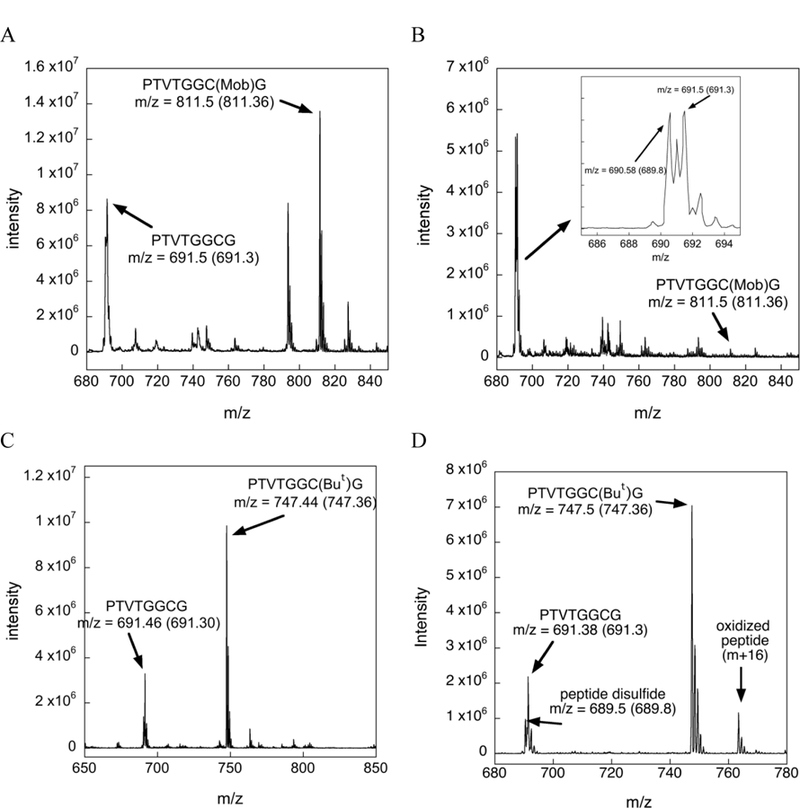
Mass chromatograms of the Cys(Mob)- and Cys(But) protected test peptides under various conditions. The arrow points to the observed m/z whereas the number inside the parentheses denotes the theoretical m/z value. (A) Mass spectrogram of the Cys(Mob) test peptide incubated in TFA for 12 h at 37 ºC. The peak at m/z = 827 corresponds to the oxidized Cys(Mob) peptide and the peak at m/z 793 corresponds to a dehydrated form of the Cys(Mob) peptide, (B) Mass spectrogram of the Cys(Mob) test peptide after incubation in TFA/TIS (98/2) for 12 h at 37 ºC. The inset shows a close up of the region between m/z 685 and m/z 695. The m/z values in the inset show the presence of the deprotected peptide and the peptide disulfide. (C) Mass spectrogram of the Cys(But) test peptide incubated in TFA for 12 h at 37 ºC. (D) Mass spectrogram of the Cys(But) test peptide incubated in TFA/TIS (98/2) for 12 h at 37 ºC.
As is evident from the deprotection reactions with Cys(Mob) and Cys(But), one effect of using TIS in the deprotection cocktail is to help catalyze disulfide formation as originally observed by Singh and coworkers [6]. While we can only speculate on the mechanism by which TIS promotes disulfide formation, it most likely involves transient bonding of silicon to sulfur. A possible mechanism by which TIS helps to deprotect Cys(Acm) and other Cys-protecting groups is shown in Figure 7.
Figure 7:
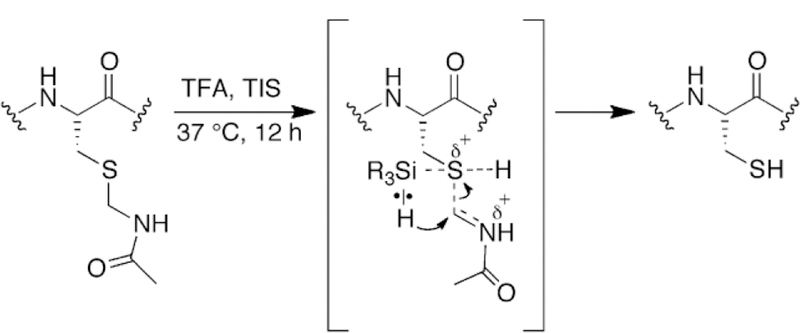
Potential mechanism of reduction of Cys(Acm) containing test peptide to Cys-SH by TIS.
The lability of the Cys protecting groups in TFA/TIS in this study are Cys(Mob) > Cys(Acm) > Cys(But). We suggest the reason for the higher lability of Cys(Mob) relative to Cys(Acm) is due to the higher stability of the resulting cation upon cleavage of the S–C bond of the protecting group. Cleavage of the S–C bond in the case of the Mob protecting group results in the formation of a 1º benzylic cation that can be stabilized by resonance from the electron donating methoxyphenyl substituent. Cleavage of the S–C bond in the case of the Acm protecting group also results in the formation of a 1º carbocation that can be stabilized by resonance from the adjacent lone pair of electrons on nitrogen, but the Acm carbocation should be less stable due to the competing resonance of the adjacent amide group.
A potential reason for the unexpected lability of Cys(Acm) compared to other alkyl protecting groups is the formation of an iminium nitrogen in the transition state as shown in Figure 7. The iminium nitrogen would result from protonation of the sulfur atom in TFA, weakening the S–C bond. As a result, a partially empty p-orbital on carbon could be stabilized by donation of the lone pair of electrons on the neighboring nitrogen, resulting in an electrophilic iminium carbon that can more easily accept a hydride from TIS in comparison to the 3º carbon of S-But for example.
If stability of the protecting group carbocation was the dominant factor in lability of the protecting group in a solution of TIS, it should be expected that But would be the most labile since it results in a 3º carbocation, but this is not observed. A likely explanation for the stability of S-But towards TIS is that steric hindrance prevents the tertiary carbon of But from accepting the hydride from TIS in agreement with our proposed mechanism in Figure 7. Thus lability of the protecting group is a combination of factors including carbocation stability and lack of steric hindrance. These factors explain the lability of Cys(Mob) towards TIS.
The role of TIS as a reductant undoubtedly explains the results of Singh and coworkers who showed that a TFA/TIS (95/5) cleavage cocktail did in fact result in moderate conversion of a Cys(Acm)-containing peptide to the Cys-SH form (4%) and 20% to the disulfide form [6] as stated in the introduction. Thus the addition of TIS results in 24% removal of the Acm protecting group under their reported conditions. Our examination of their data shows that a cocktail of TFA/anisole (95/5) resulted in an even higher percentage of Acm removal (32%). On this basis in part, Singh and coworkers concluded that TIS gave the best yields of the fully protected peptide in comparison to phenol, EDT, and anisole [6]. However their study lacked an important control since they did not report the extent of removal of the Acm group when using only neat TFA. Based on our results, we believe they would have found minimal removal of the Acm group with neat TFA.
The conclusion of Singh and coworkers is the source of subsequent incorrect statements about the role of TIS in deprotection of Cys(Acm) in TFA. For example, Houben-Weyl’s “Synthesis of Peptides and Peptidomimetics” cites the results reported by Singh, stating that:
“The type of scavenger used, e.g. anisole or thioanisole as additive to HF, or thiols added to TFA, apparently plays an important role; for example, use of TIS as a scavenger prevents partial cleavage of S-(acetamidomethyl) by TFA.”[3]
The key word in the quote above is “prevents” (bolded for our emphasis) as TIS does not prevent cleavage of the carbon-sulfur bond, but rather facilitates it.
This mischaracterization was further propagated in the review of Isidro-Llobet and coworkers on amino acid protecting groups who also state that:
“Nevertheless, it (Acm) is partially removed with HF or even TFA depending on the scavengers used. In the latter case, absence of water and use of TIS minimizes the removal.”[2].
We have again bolded a key phrase from the sentence above since our results contradict this statement. Our results are supported by the original work by Singh and coworkers who clearly showed that the presence of TIS in the deprotection cocktail resulted in the removal of Acm. Other examples of the misunderstanding regarding the abilities of TIS to act as reducing agents are present in the literature [13, 17].
Since its introduction as a scavenger for protecting group cations in 1989, the role of TIS as a reductant has been overlooked, even though the original report cites the known role of trialkylsilanes as mild reductants in TFA [1]. For example, it has been shown that TIS can reduce the indole ring of tryptophan residues to indoline [1]. Interestingly, indoles are commonly reduced via iodine oxidation, using similar techniques to those typically used to reduce the S-Acm bond [18, 19].
Our results present a cautionary tale for those attempting regioselective disulfide bond forming strategies using orthogonal pairs of Cys-protecting groups when using TIS as a scavenger. Using “standard” deprotection conditions, Cys(Mob) and Cys(Acm) will be more stable than reported here, but sequence specific effects as discussed above could result in a small percentage of deprotection that would result in low yields of the target disulfide as disulfide scrambling would inevitably occur.
If peptide disulfide bonds are stable to TIS, then our results suggest that TIS itself could be used in orthogonal deprotection strategies of Cys-protecting groups. In a hypothetical peptide with two-disulfide bonds, the first disulfide bond could be installed using a pair of Cys(Trt) residues. The Trt group could be removed in TFA with mild scavenger at room temperature, and then the peptide could be subjected to air oxidation. The second disulfide bond could then be installed using a pair of Cys(Mob) residues. The peptide could then be incubated in TFA/TIS or TFA/thioanisole at 37 ºC to remove the Mob groups, and help catalyze disulfide formation. If disulfide formation was incomplete, appropriate catalyst could be added to aid in formation of the second disulfide.
In conclusion, we hope this report provides strong evidence that TIS can reduce S-protecting group bonds under acidic conditions and draws attention to this observation to researchers in the peptide community. We will show in a subsequent report that TIS can enhance the deprotection of Cys in combination with certain deprotection reagents.
Supplementary Material
ACKNOWLEDGEMENT
This study was supported by NIH grant GM094172 to RJH.
REFERENCES
- [1].Pearson DA, Blanchette M, Baker ML, Guindon CA. Trialkylsilanes as scavengers for the trifluoroacetic acid deblocking of protecting groups in peptide synthesis. Tetrahedron Lett 1989; 30: 2739–2742. [Google Scholar]
- [2].Isidro-Llobet A, Alvarez M, Albericio F Amino acid-protecting groups. Chem Rev 2009; 109: 2455–2504. [DOI] [PubMed] [Google Scholar]
- [3].Moroder L, Musiol HJ, Schaschke N, Chen L, Hargittai B, Barany G. In Houben-Weyl, Synthesis of Peptides and Peptidomimetics; Goodman M, Felix A, Moroder L, Toniolo C. Eds.; Georg Thieme Verlag: Stuttgart, 2004; pp 384–423. [Google Scholar]
- [4].Larson GL, Halides M. Silicon-based reducing agents. Supplement to the Gelest Catalog available on-line at www gelest com. 2008.
- [5].Pesti J, Larson GL. Tetramethyldisiloxane: A practical organosilane reducing agent. Org. Process Res. Dev 2016; 20: 1164–1181. [Google Scholar]
- [6].Singh PR, Rajopadhye M, Clark SL, Williams NE. Effect of scavengers in acidolytic cleavage of Cys (Acm)-containing peptides from solid support: Isolation of an ethanedithiol disulfide adduct. Tetrahedron Lett 1996; 37: 4117–4120. [Google Scholar]
- [7].Kaiser E, Colescott R, Bossinger C, Cook P. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem 1970; 34: 595–598. [DOI] [PubMed] [Google Scholar]
- [8].Harris KM, Flemer S, Hondal RJ. Studies on deprotection of cysteine and selenocysteine side-chain protecting groups. J. Pep. Sci 2007; 13: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Veber D, John Milkowski, Sandor Varga, Robert Denkewalter, Ralph Hirschmann. Acetamidomethyl. A novel thiol protecting group for cysteine. J. Am. Chem. Soc 1972; 94: 5456–5461. [DOI] [PubMed] [Google Scholar]
- [10].Klodt J, Kuhn M, Marx UC, Martin S, Rousch P, Forssmann WG, et al. Synthesis, biological activity and isomerism of guanylate cyclase C-activating peptides guanylin and uroguanylin. Chem. Biol. Drug Des 1997; 50: 222–230. [DOI] [PubMed] [Google Scholar]
- [11].Andreu D, Albericio F, Solé NA, Munson MC, Ferrer M, Barany G. Formation of disulfide bonds in synthetic peptides and proteins. Peptide synthesis protocols: Springer; 1994. p. 91–169. [DOI] [PubMed] [Google Scholar]
- [12].Sieber P, Kamber B, Riniker B, Rittel W. Iodine oxidation of S-trityl- and S-acetamidomethyl-cysteine-peptides containing tryptophan: Conditions leading to the formation of tryptophan-2-thioethers. Helv. Chim. Acta 1980; 63: 2358–2363. [Google Scholar]
- [13].Muttenthaler M, Ramos YG, Feytens D, de Araujo AD, Alewood PF. p-Nitrobenzyl protection for cysteine and selenocysteine: A more stable alternative to the acetamidomethyl group. Pep. Sci 2010; 94: 423–432. [DOI] [PubMed] [Google Scholar]
- [14].Engebretsen M, Agner E, Sandosham J, Fischer PM. Unexpected lability of cysteine acetamidomethyl thiol protecting group. Tyrosine ring alkylation and disulfide bond formation upon acidolysis. Chem. Biol. Drug Des 1997; 49: 341–346. [DOI] [PubMed] [Google Scholar]
- [15].Van Rietschoten JPM,E & Granier C Peptides, Proceedings of the Fifth American Peptide Symposium J Wiley & Sons, New York: 1977: 522–524. [Google Scholar]
- [16].Atherton E, Sheppard RC, Ward P. Peptide synthesis. Part 7. Solid-phase synthesis of conotoxin G1. J. Chem. Soc. Perkin. Trans 1 1985: 2065–2073. [Google Scholar]
- [17].D’Hondt M, Bracke N, Taevernier L, Gevaert B, Verbeke F, Wynendaele E, et al. Related impurities in peptide medicines. J. Pharm. Biomed. Anal 2014; 101: 2–30. [DOI] [PubMed] [Google Scholar]
- [18].Robinson B Reduction of indoles and related compounds. Chem. Rev 1969; 69: 785–797. [DOI] [PubMed] [Google Scholar]
- [19].Hudson C, Robertson A. The synthesis and chemistry of DL-indoline-2-carboxylic acid. Aust. J. Chem 1967; 20: 1935–1941. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.