

ABSTRACT
DNA demethylases function in conjunction with DNA methyltransferases to modulate genomic DNA methylation levels in plants. The Arabidopsis genome contains four DNA demethylase genes, DEMETER (DME), REPRESSOR OF SILENCING 1 (ROS1) also known as DEMETER-LIKE 1 (DML1), DML2, and DML3. While ROS1, DML2, and DML3 were shown to function in disease response in somatic tissues, DME has been thought to function only in reproductive tissues to maintain the maternal-specific expression pattern of a subset of imprinted genes. Here we used promoter:β-glucuronidase (GUS) fusion constructs to show that DME is constitutively expressed throughout the plant, and that ROS1, DML2, and DML3 have tissue-specific expression patterns. Loss-of-function mutations in DME cause seed abortion and therefore viable DME mutants are not available for gene function analysis. We knocked down DME expression in a triple ros1 dml2 dml3 (rdd) mutant background using green tissue-specific expression of a hairpin RNA transgene (RNAi), generating a viable ‘quadruple’ demethylase mutant line. We show that this rdd DME RNAi line has enhanced disease susceptibility to Fusarium oxysporum infection compared to the rdd triple mutant. Furthermore, several defence-related genes, previously shown to be repressed in rdd, were further repressed in the rdd DME RNAi plants. DNA methylation analysis of two of these genes revealed increased differential promoter DNA methylation in rdd DME RNAi plants compared to WT, beyond the difference observed in the parental rdd plants. These results indicate that DME contributes to DNA demethylase activity and disease response in somatic tissues.
KEYWORDS: DEMETER, DEMETER-LIKE, ROS1, rdd, DNA-demethylation, Fusarium, Arabidopsis thaliana, Epigenetics
Introduction
DNA cytosine methylation is a major epigenetic mark in eukaryotes. In plants, the DNA methylation level in the genome is controlled by de novo DNA methylation, maintenance DNA methylation and DNA demethylation. De novo methylation is mediated by RNA-directed DNA methylation (RdDM), which can occur at all cytosine contexts, namely CG, CHG (‘H’ stands for A, C, T) and CHH sites. Methylation at symmetric CG and CHG sites can be maintained during DNA replication by METHYLTRANSFERASE 1 (MET1) and CHROMOMETHYLASE 3 (CMT3), respectively. CHH methylation at highly repetitive regions of the genome can be maintained by CMT2, but CHH methylation near genes cannot be maintained during DNA replication and depends on continuous RdDM. DNA demethylation can occur passively during DNA replication when maintenance methylation is incomplete, or through the activity of the DNA glycosylase family of DNA demethylases. These DNA demethylases remove 5-methylcytosine and replace it with unmethylated cytosine through a base-excision-repair mechanism [1].
The Arabidopsis genome contains four DNA demethylase genes, DEMETER (DME), REPRESSOR OF SILENCING 1 (ROS1) also known as DEMETER-LIKE 1 (DML1), DML2, and DML3 [1]. DME has been shown to play a major role in seed development by controlling the expression of specific imprinted maternal alleles, and mutation of the maternal copy of the DME gene leads to seed abortion [2,3]. ROS1, DML2, and DML3 are thought to account for all demethylase activities in somatic tissues. ROS1 is the dominant demethylase of the three and plays a role in maintaining the activity of transgenes and transposable elements in Arabidopsis by decreasing DNA methylation [1]. Both localized and genome-wide DNA methylation analyses have shown that DML2 and DML3 function redundantly with ROS1 to maintain low-level cytosine methylation at some loci [4,5]. DML2 and DML3 have also been shown to play a role in modulating the DNA methylation level of some heavily methylated loci in Arabidopsis [6]. In addition to maintaining genome methylation levels, the three DNA demethylase genes have recently been implicated in plant disease resistance in Arabidopsis. The single mutant ros1, and the triple mutant ros1 dml2 dml3 (rdd), have been shown to display increased susceptibility to the bacterial pathogen Pseudomonas syringae [7] and the fungal pathogen Fusarium oxysporum [8], suggesting that these DNA demethylases play a role in plant defence responses by regulating plant defence-related genes.
The expression patterns of the four DNA demethylase genes have been investigated. Choi et al. [3] reported that DME is expressed specifically in the central cells of female gametophytes, but is undetectable in leaf tissues using RT-PCR. In contrast, Ortega-Gallisteo et al. [6] detected the constitutive expression of DME as well as ROS1, DML2 and DML3 across various plant tissues using RT-PCR. Further, they used promoter:β-glucuronidase (GUS) constructs to examine the expression pattern of DML2 and DML3, and showed that both genes are strongly expressed in all tissues of Arabidopsis. Gong et al. [9] also used promoter:GUS reporter constructs to investigate the expression pattern of ROS1, and showed strong expression in all tissues of young Arabidopsis seedlings. DME, ROS1 and DML2 expression patterns are also represented in the Arabidopsis Gene Expression Visualization database (AtGenExpress [10]), which shows constitutive expression of DME but more variable expression levels of ROS1 and DML2 across different plant tissues (Supplementary Figure S1A). No DML3 expression data were recorded as DML3 was not represented on the Affymetrix ATH-1 array. However, DML3 RNA sequencing data are available in the Transcriptome Variation Analysis database (TraVA [11]), which shows the localized expression of DML3 in anthers only. In support of this, our previous microarray expression analysis [8] and mRNA sequencing (unpublished) of 3-week-old whole Col-0 Arabidopsis seedlings detected the low-level expression of DML3 along with the relatively high-level expression of DME, ROS1 and DML2 (Supplementary Figure S1B, S1C). This contradicts the reported constitutive expression pattern of DML3 [6]. Thus, the expression patterns of the four demethylase genes require further examination, but it seems clear that DME has a widespread expression in both reproductive and vegetative tissues of Arabidopsis.
The observed DME expression pattern raised the possibility that DME not only functions in reproductive tissue but also contributes to DNA demethylase activity and related biological function in the whole plant. However, this has not been investigated, possibly because a loss-of-function mutation in DME results in seed abortion, and therefore double, triple, or quadruple demethylase mutants containing a DME mutation are not available for genomic and biological function studies. Indeed, genome-wide methylation and gene expression studies utilized either single DNA demethylase mutants or the triple demethylase mutant rdd. Both DNA methylation and gene expression changes in rdd are limited, with a relatively small number of loci affected by the mutations [4,12,13]. The functional analysis of the DNA demethylases in disease resistance has also been based on the use of the single ros1 and triple rdd mutants [5,7,8].
The present study was aimed at examining the biological function of DME in somatic tissues. Using promoter:GUS fusion constructs, we confirmed the constitutive expression pattern of DME and revealed tissue-differential or tissue-specific expression patterns of ROS1, DML2 and DML3. We knocked down the expression of DME in the rdd mutant background using green tissue-specific RNAi to generate a viable rdd DME RNAi line. We used this line to demonstrate that DME plays a role in Fusarium resistance, defence gene expression and DNA demethylation in vegetative Arabidopsis tissues.
Results
DME, ROS1, DML2 and DML3 promoters show distinct expression patterns
The structures of the promoter:GUS fusion constructs are illustrated in Figure 1. The promoter fragments were amplified from the genomic region upstream of the translation start site ATG, with the size of 2.90 and 3.56 kb (DME; −2321 to +584 and −2972 to +584 relative to the transcription start site or TSS), 3.31 kb (ROS1; −2777 to +535), 2.89 kb (DML2; −2891 to −1), and 3.82 kb (DML3; −3858 to −43), respectively. These promoter fragments contain longer upstream sequences than the previously used ones for DME (−2282 to 1922 bp of the first exon) [3], ROS1 (−1565 to −25 from first ATG or −1030 to +510 relative to TSS) [9], DML2 (−2269 to +15 from first ATG or TSS) and DML3 (−1300 to +13 from first ATG or −1145 to +171 relative to TSS) [6] promoters, respectively. Two versions of the DME promoter were tested; one included the full-length upstream transposable element (TE) sequence (DME+TEp) in case the TE plays a role in DME regulation, and the second (DMEp) contained part of the TE but excluding the upstream section that overlaps with the transcription start site of the adjacent gene. The ROS1, DML2 and DML3 promoter fragments also contained TE sequence that exists in the upstream genomic sequences of the respective genes (Figure 1). To avoid the potential enhancer effects by strong constitutive promoters such as the 35S promoter [14], the promoter:GUS expression cassettes were cloned into the plant expression vector pBI101, where the selective marker gene NPTII is driven by the relatively weak nopaline synthase promoter (Nos-P), which is located distal to the inserted endogenous gene promoters (Figure 1).
Figure 1.
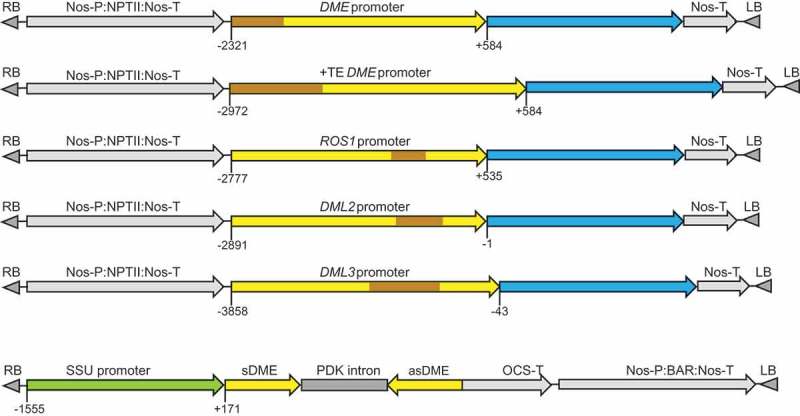
Schematic diagrams of the promoter:GUS fusion constructs and the DME hairpin RNA (hpDME) construct. Transposable element (TE) sequences are indicated in brown; blue regions indicate the GUS coding sequence. Numbers indicate the nucleotide position relative to the transcription start site (TSS).
Transgenic lines of the Col-0 ecotype were obtained, self-fertilized until the fourth generation, and then analysed for GUS expression patterns. As shown in Figure 2 and Supplementary Figure S2, the DME promoter lines showed strong GUS staining in all tissues analysed, including leaves, roots, flowers and siliques. No visible difference in GUS staining could be observed between the two versions of the DME promoter constructs, DME+TEp:GUS and DMEp:GUS. In addition, quantitative expression analysis of four independent lines for each of the DME constructs, using a fluorometric MUG assay, detected no significant difference in GUS expression levels between the two (Supplementary Figure S3). This indicated that the additional upstream TE sequence in DME+TEp:GUS is not required for DME expression. The GUS expression pattern of the two DME promoter constructs confirmed that DME is transcribed throughout the reproductive and vegetative tissues of Arabidopsis, which is consistent with the RT-PCR result of Ortega-Gallisteo et al. [6].
Figure 2.

Representative expression patterns of the promoter:GUS fusion transgenes in Arabidopsis Col-0 ecotype. Arrow indicates DML3p:GUS expression in anthers of young flowers.
The ROS1p:GUS promoter construct showed a much lower expression than the DMEp:GUS promoter constructs and displayed distinct expression patterns in different tissues. The expression level was relatively high in young leaves, particularly in the midrib areas, and in young flowers and siliques, but weaker in roots and older leaves and siliques (Figure 2 and Supplementary Figure S2). This was consistent with the ROS1 expression pattern recorded in the AtGenExpress and TraVa databases [10,11], which showed relatively high levels in the shoot apex and flower tissues but low levels in root, stem and leaves (Supplementary Figure S1A).
The DML2p:GUS construct showed weak expression in leaves and roots, with clear GUS staining mainly in the leaf midrib areas. However, it was strongly expressed in the flowers and siliques (Figure 2 and Supplementary Figure S2). This result indicated that DML2 is predominantly expressed in reproductive tissues.
The DML3p:GUS construct displayed the most tissue-specific expression pattern among the demethylase gene promoter constructs. The DML3p:GUS lines showed no detectable GUS staining in young seedlings, siliques or most flower tissues (Figure 2 and Supplementary Figure S2). However, clear GUS staining was consistently detected in young (but not old) anther tissues after an extended period of staining (up to three days), which matched the data recorded in the TraVa database [11]. Thus, DML3 is likely to be an anther-specific demethylase gene.
RNA interference knockdown of DME in rdd increases plant susceptibility to Fusarium oxysporum infection
In order to examine if DME contributes to DNA demethylase function in somatic tissues, a hairpin RNA construct (hpDME) was designed to silence DME specifically in green tissues. We used the Arabidopsis rubisco small subunit gene promoter (SSU) to drive the expression of hpDME (Figure 1). The 548 bp sequence of the DME cDNA in the hpDME construct had two short (20 and 21 nt) stretches of sequence identity with DML2 but showed no high nucleotide sequence similarities to ROS1 and DML3. This construct was transformed into rdd plants to achieve DME knockdown in the triple demethylase mutant background. We also introduced the construct into Col-0 to obtain DME knockdown lines in the wild-type background. Transgenic lines containing the hpDME construct showed no phenotypic difference to the untransformed rdd or the wild-type Col-0 plants, and were fully fertile (data not shown). Two single-copy transgenic lines in the rdd background, rdd-HP5 and rdd-HP6, were self-fertilized to generate homozygous progeny populations and used for disease response, gene expression and DNA methylation analyses. Two hpDME lines of the Col-0 background, Col-HP4 and Col-HP7, were also included. Northern blot analysis of the target DME mRNA showed that rdd-HP5 plants had little hpDME expression, hence only a slight reduction in DME mRNA level was observed (Figure 3 and Supplementary Figure S4). rdd-HP6 plants, on the other hand, showed a high level of hpDME expression and concomitant strong downregulation of DME mRNA. Similarly, Col-HP4 showed clear hpDME expression and DME downregulation, but Col-HP7 had little hpDME expression or DME downregulation (Supplementary Figure S4). It was notable that lower-molecular-weight DME-specific hybridizing signals were also detected on the northern blots, but the nature of these bands remained unclear.
Figure 3.
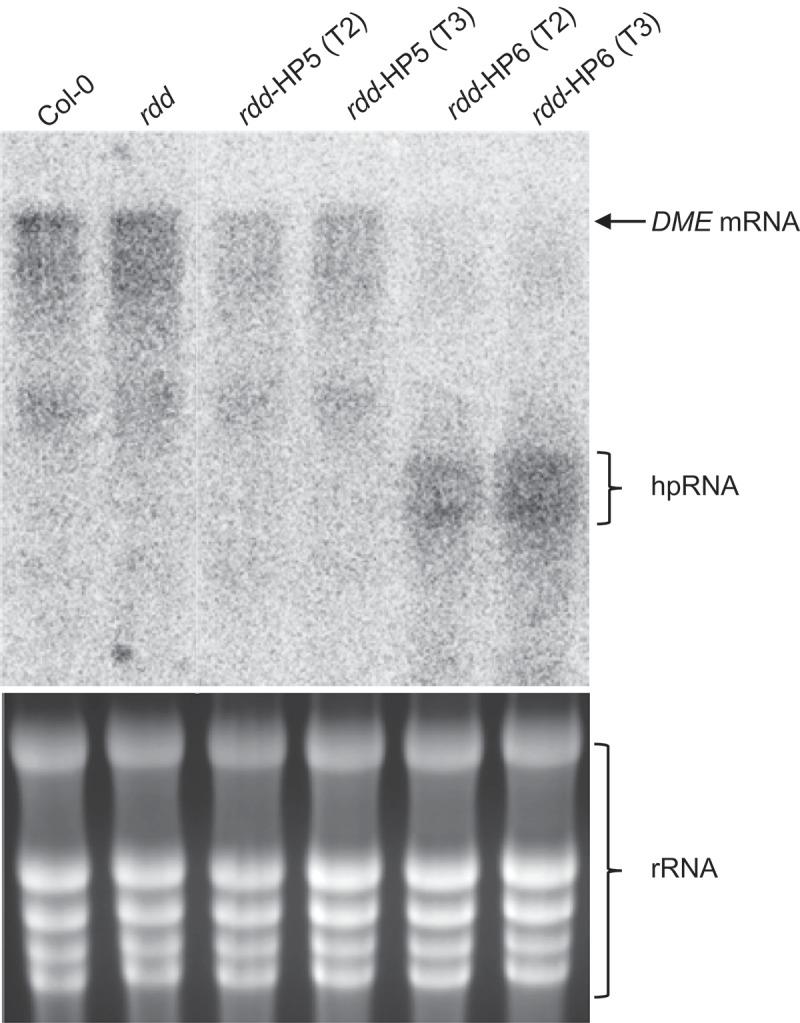
Northern blot analysis to detect DME mRNA level in Col-0, rdd and rdd DME RNAi transformants (rdd-HP5 and rdd-HP6). Top panel: Northern blot analysis probed with antisense DME RNA, which allows detection of both the DME mRNA and the hpDME-derived transcript. Bottom panel: Ethidium bromide-stained RNA agarose gel to show even loading of RNA.
Our previous study showed that rdd plants have increased susceptibility to the fungal pathogen Fusarium oxysporum compared to Col-0 [5,8], suggesting that DNA demethylases play a positive role in Fusarium resistance. To examine if DME contributes to disease resistance, the homozygous T3 and T4 generations of rdd-HP5 and rdd-HP6 transgenic lines were assayed for disease susceptibility to Fusarium, along with Col-0 and rdd plants as controls. As shown in Figure 4, the rdd-HP6 plants, with strong downregulation of DME, showed a significant increase in the chlorotic disease phenotype compared with the parental rdd line, whereas rdd-HP5 plants, with only slight downregulation of DME, displayed a disease response similar to rdd. Soil infections yielded comparable results for these lines (Supplementary Figure S5). The Col-HP4 or Col-HP7 lines showed no detectable increase in Fusarium disease phenotypes compared to the Col-0 plant (Supplementary Figure S6), suggesting that knockdown of DME expression alone is insufficient to affect plant response to Fusarium infection.
Figure 4.
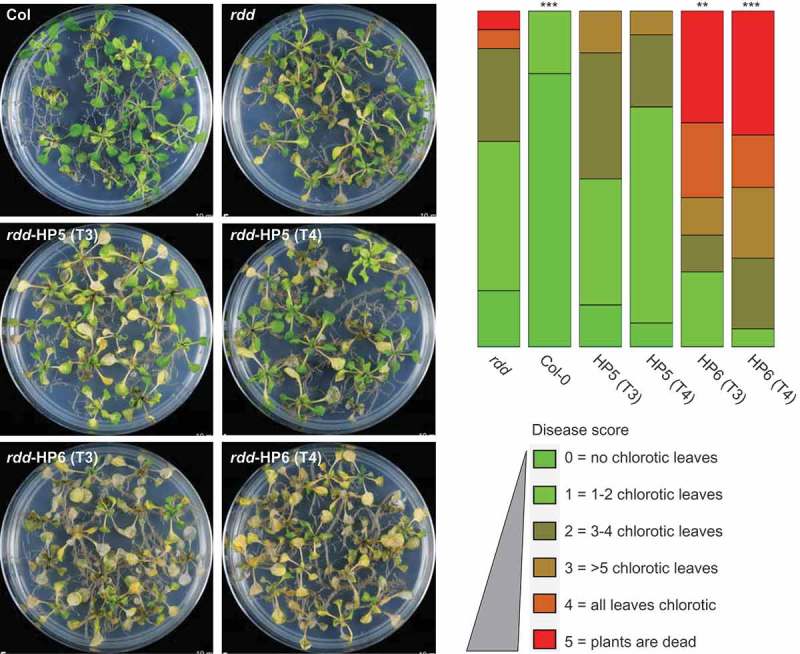
Knockdown of DME expression in rdd increases susceptibility to Fusarium oxysporum infection. Left panel: plants infected with Fusarium for 8 days at 23°C on sucrose-free MS medium. Right panel: histogram showing disease score based on the number of chlorotic leaves in each plant. Disease scoring was carried out in biological triplicates using 20–30 plants per replicate. The diagram was drawn using the software at https://github.com/wangqinhu/aid. Significance testing was performed using the Wilcox test: **p < 0.01, ***p < 0.001.
DME contributes to gene expression regulation and DNA demethylation of promoter TEs of defence-related genes
Our previous study has identified a subset of plant defence-related genes that are regulated by DNA demethylases in Arabidopsis: they contain transposable element (TE) sequences in their promoters and show differential DNA methylation in the promoter TEs and repressed gene expression in rdd compared to Col-0 [5,8]. Among the three DNA demethylase genes, ROS1 plays a dominant role, but DML2 and DML3 also function redundantly to regulate these genes [5]. To investigate if DME also contributes to defence gene regulation, we selected a number of these rdd-downregulated defence-related genes and compared their expression in Col-0, rdd, and hpDME lines in the rdd and Col-0 backgrounds.
As shown in Figure 5, these defence-related genes were downregulated in the rdd mutant compared to Col-0 plants, consistent with our previous findings [5,8]. RNAi-mediated knockdown of DME in Col-0 did not affect the expression of these genes (except for AT1G05700 which appeared to show slight downregulation in the Col-0 DME RNAi lines), which was consistent with the Fusarium infection result (Figure S6). This suggested that downregulation of DME alone is insufficient, or the level of DME downregulation in this line is not high enough, to affect the expression of these genes or the disease responses. However, RNAi-mediated downregulation of DME in the rdd background clearly affected the expression of all six defence-related genes analysed, which showed increased repression compared to the parental rdd plants (Figure 5). Consistent with the greater DME knockdown in line rdd-HP6 compared to rdd-HP5, all analysed defence-related genes showed stronger downregulation in the rdd-HP6 line. This result indicates that DME contributes to the regulation of these defence-related genes.
Figure 5.
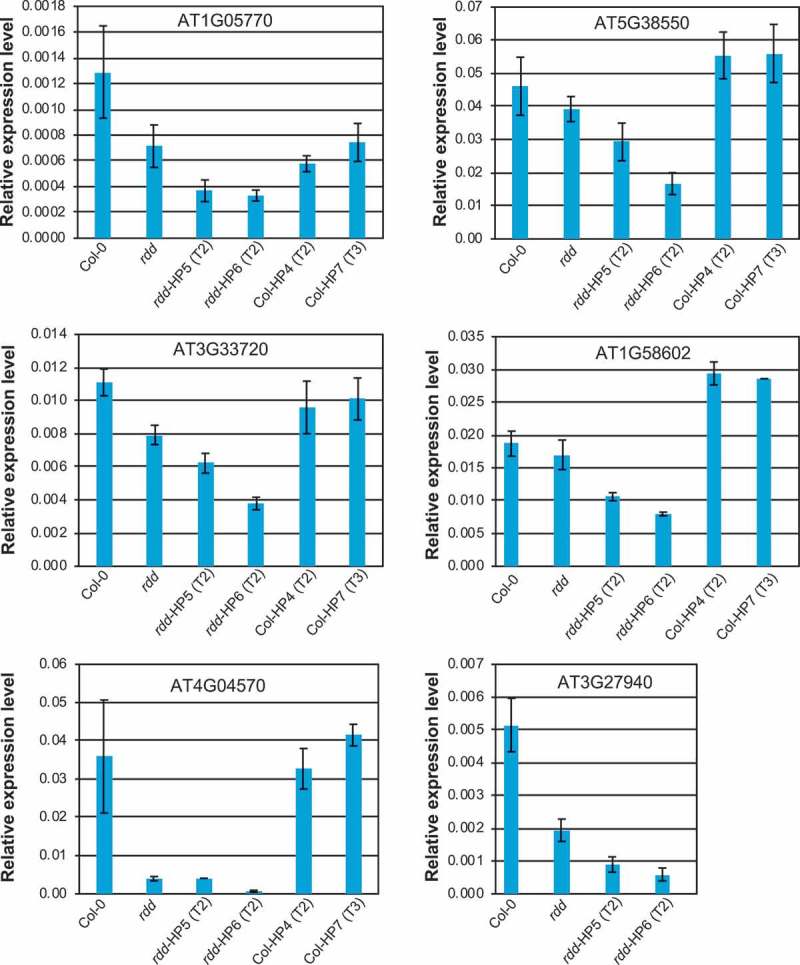
The expression levels of rdd-downregulated defence-related genes is further repressed by DME knockdown. Whole 3-week-old plants of around 20 individual siblings for each plant line were analysed as a pool. Columns indicate the average of three technical replicates with error bars showing standard deviation. The Actin 2 gene was used as internal reference for RT-qPCR.
Our previous studies have shown that the repressed expression of the defence-related genes in rdd were associated with differential DNA methylation at distinct loci coinciding with short TE sequences in the gene promoters, suggesting that ROS1, DML2, and DML3 target the localized TE sequences to modulate DNA methylation levels and consequently gene expression [5,8]. To investigate if DME contributes to this demethylase function, we used bisulfite sequencing to determine the DNA methylation levels in the differentially methylated distinct promoter TE sequences of two rdd-downregulated defence-related genes that showed stronger repression in the rdd DME RNAi lines, AT3G27940 and AT4G33720. DNA samples of Col-0, rdd, rdd-HP5 (T3 generation), and rdd-HP6 (T3 and T4 generations) were isolated from 3 week old seedlings with no reproductive tissues and used for bisulfite conversion, as in the previous studies [5,8]. Efficient bisulfite conversion of the DNA samples was confirmed by amplifying the chloroplast psaA gene sequence, known to be unmethylated, followed by digesting the PCR product with the MseI enzyme (Supplementary Figure S7). Consistent with our previous results, the rdd plants showed localized reduction of CHH methylation compared to Col-0 in the promoter TE sequences of both genes (Figure 6 and Supplementary Figure S8). The rdd-HP6 line, with strong downregulation of DME, showed a further reduction of CHH methylation levels in both gene promoter TE sequences compared to rdd. The rdd-HP5 line, with slight downregulation of DME, did not show a further reduction of CHH methylation in AT3G27940 (Figure 6) but displayed reduced CHH methylation in AT4G33720 compared to rdd (Supplementary Figure S8). The small number of CG sites (3 for AT3G27940 and 5 for AT4G33720) did not reveal a clear change in methylation level between the rdd DME RNAi lines and rdd, particularly as these cytosines already showed almost complete methylation in rdd (data not shown [8]). Thus, the bisulfite sequencing analysis of the two promoter TE regions indicated that DME contributes to DNA demethylase function in the vegetative plant tissues.
Figure 6.
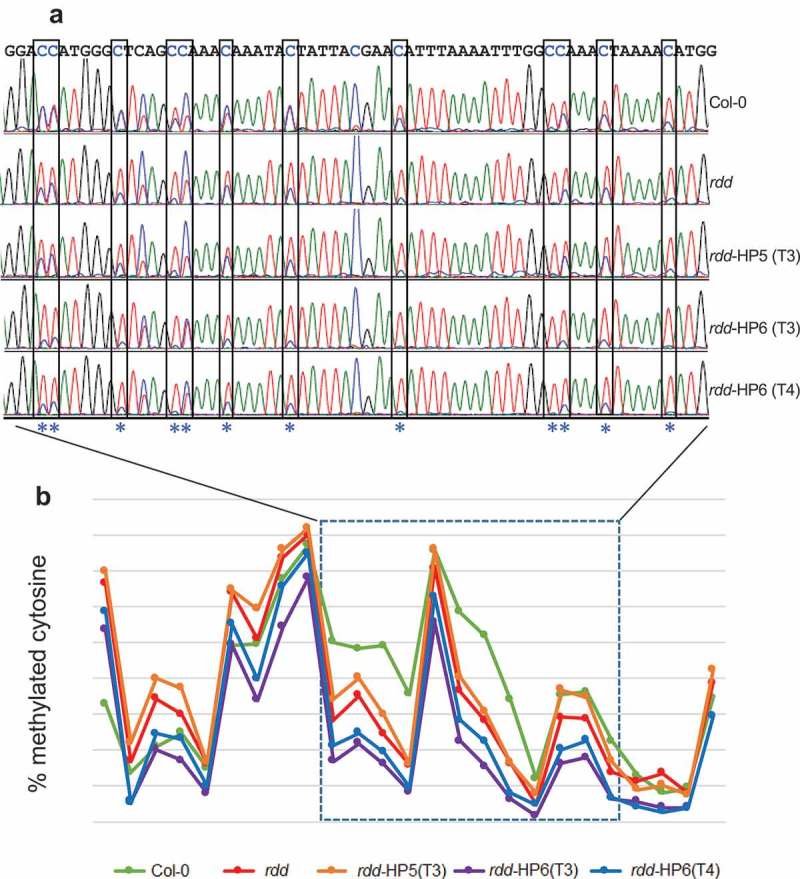
DME RNAi causes a further reduction in CHH methylation at the rdd-controlled promoter TE region of AT3G27940. a: Representative bisulfite sequencing trace file of the region showing CHH methylation changes. The CHH sites are boxed and indicated with asterisks. The ratio between blue and overlapping red peaks indicates the level of cytosine methylation. b: Cytosine methylation level of the whole bisulfite sequenced region. Each point represents one cytosine residue in the CHH context. The section for which the trace file image is shown is indicated.
Discussion
DME was initially thought to be expressed specifically in the central cell of female gametophytes, but subsequent gene expression studies by us and others suggested that DME is expressed constitutively in Arabidopsis. This prompted us to investigate if this DNA demethylase gene might function together with ROS1, DML2 and DML3 in somatic tissues and contribute to DNA demethylation activity and plant defence response.
We first examined the expression pattern of DME as well as ROS1, DML2, and DML3 using promoter:GUS fusion constructs. Our results confirmed the constitutive expression pattern of DME; the DMEp:GUS and DME+TEp:GUS constructs gave high levels of GUS expression across all tissues analysed. This result is consistent with the expression patterns recorded in AtGenExpress and TraVA [10,11].
However, the tissue-differential and tissue-specific expression patterns of ROS1p:GUS, DML2p:GUS and DML3p:GUS observed in this study, particularly of DML2p:GUS and DML3p:GUS, are different to the strong constitutive expression patterns observed by Gong et al. [9] and Ortega-Gallisteo et al. [6], who also used promoter:GUS fusion constructs to investigate gene expression. One possible explanation lies in the difference in construct configurations between the previous reports and the current study. In the ROS1, DML2 and DML3 promoter:GUS fusion constructs used by Gong et al. [9] and Ortega-Gallisteo et al. [6], the promoter:GUS expression cassettes were inserted adjacent to a cauliflower mosaic virus 35S promoter that was used to drive the expression of the selectable marker gene in the plant transformation vectors pCAMBIA1380 and pCAMBIA1381. Consequently, the ROS1, DML2 and DML3 promoters were placed in close proximity to the 35S promoter, a configuration that is known to cause constitutive expression of otherwise tissue-specific endogenous gene promoters, presumably due to the strong enhancer activity of the 35S promoter (e.g., [14]). In this study, we aimed to avoid this enhancer effect by using a plant transformation vector without a strong constitutive promoter and by placing the DNA demethylase gene promoters distal to the promoter driving the selective marker gene. Therefore, the expression patterns of ROS1, DML2 and DML3 promoter:GUS fusion constructs observed in this study are more likely to represent the endogenous gene expression patterns than those reported by Gong et al. and Ortega-Gallisteo et al. [6,9].
While the tissue-differential expression pattern of the ROS1p:GUS construct is consistent with the ROS1 expression data recorded in the AtGenExpress and TraVA databases, it was surprising that the expression level of ROS1p:GUS (as assessed by GUS staining) was weaker than that of DMEp:GUS and DME+TEp:GUS (Figure 2). This was also in contrast to the expression profile obtained from our microarray and mRNAseq analyses (Figure S1). A recent study showed that the activity of the ROS1 promoter depends on DNA methylation at a target sequence near the transcription start site [15]. It is possible that the level of DNA methylation in the transgenic ROS1 promoter did not reach the level of the endogenous promoter resulting in comparatively lower levels of GUS expression. This is worth investigating in future studies.
Consistent with widespread DME expression in somatic tissues, our results showed that RNAi-mediated knockdown of DME expression in the rdd mutant background increased the disease susceptibility to Fusarium infection. Furthermore, a number of the defence-related genes shown previously to be repressed in the rdd mutant [8], showed even stronger repression in the DME RNAi plants. This suggested that DME functions together with ROS1, DML2 and DML3 to regulate the expression of these defence-related genes mediating Fusarium resistance in Arabidopsis. These defence-related genes contain TE sequences in their promoters, and these were shown to be the target for DNA demethylases in our previous studies [5,8]. Thus, while genome-wide DNA methylation and gene expression analysis of the DME RNAi lines is required to fully evaluate the function of DME in somatic tissues, our results confirmed that DME contributes to the function of DNA demethylases in the regulation of defence-related genes and disease response in vegetative plant tissues.
In general, CHH methylation was reduced in rdd, CG methylation increased, while CHG methylation showed no clear trend of change. Our analysis here revealed further enhanced differential CHH methylation in the DME RNAi plants compared to their parental rdd plants. The function of CHH methylation in gene regulation is not fully understood. While DNA methylation at the promoters tends to repress gene expression, our previous studies showed that the reduced CHH methylation at promoter TEs in the DNA demethylase mutants often correlates with repressed gene expression [5,8]. In addition, increased expression of an rdd-regulated gene under Fusarium oxysporum infection showed increased CHH methylation at the promoter TE sequence [5]. These previous observations suggest that CHH methylation can be associated with increased gene expression. This phenomenon has also been described previously in maize, where CHH methylation at the upstream region of genes correlates positively with gene expression levels [16]. How CHH methylation positively affects gene expression remains unclear. A previous study has shown that transcriptional activation of an upstream TE can result in cryptic transcriptional initiation affecting the expression of the downstream gene [17]. It is possible that CHH methylation at such a TE sequence may play a role in silencing the TE to repress TE-initiated cryptic transcription, thereby maintaining active expression of downstream genes.
RNAi knockdown of DME in the wild-type Col-0 background did not cause increased disease response to Fusarium oxysporum infection or clear changes in defence gene expression. This result suggested that DME contributes to, but is not the dominant player, in the DNA demethylation of somatic tissues in Arabidopsis. This is consistent with ROS1 being the dominant DNA demethylase in these tissues [5]. However, it cannot be ruled out that a more dominant role of DME exists in the whole plant due to the constitutive expression pattern, but the level of DME downregulation in the Col-hpDME transgenic line was not high enough to show a stronger disease response or gene expression change. It is also possible that the genes chosen here for expression and DNA methylation analysis were not the most suitable reporters for analysing DME function, and other specific defence genes, which are not strongly downregulated in the rdd mutant, may exist as direct targets of DME.
We chose to analyse six defense-related genes previously shown to be strongly downregulated in rdd, namely AT1G05700 (LRR kinase), AT5G38550 (Mannose-binding lectin superfamily protein), AT3G33720 (Cystein-rich secretory protein, Antigen 5, PR1-related), AT1G58602 (LRR and NB-ARC domain containing disease resistance protein), AT4G04570 (Cystein-rich receptor-like kinase) and AT3G27940 (LOB-domain containing protein 26). LRR kinases, as well as Mannose-binding lectin proteins, are known receptors that are important in recognizing pathogen-associated molecular patterns and modulate gene expression in response to fungal infections. Cystein-rich receptor-like kinases, as well as pathogenesis-related (PR) proteins, are also known to be important in plant defence against pathogens. As such, each of these gene products is directly involved in the defence of Arabidopsis against fungal pathogens, which is consistent with our findings outlined here as well as previously. Hence, efficient and timely regulation of these genes could be paramount in ensuring plant survival under Fusarium oxysporum infection. Understanding the effect of DNA methylation and active DNA demethylation of short promoter TE sequences containing transcription regulatory elements holds the key to understanding the dynamics of gene transcription activation in response to fungal infection.
Our understanding of DNA demethylase gene function in Arabidopsis somatic tissues has so far been based mainly on genome-wide DNA methylation and gene expression analyses of the rdd triple mutant, which detected a relatively small number of genomic loci with differential DNA methylation and gene expression [4,12,13]. It will be interesting to examine the DNA methylation and gene expression profiles in the rdd DME RNAi lines, which could reveal more widespread changes in DNA methylation and gene expression than those observed in rdd. We anticipate that more functional roles of DNA demethylases in plants might emerge in the future.
Materials & methods
Preparation of the promoter:GUS fusion and DME hairpin constructs
Promoter:GUS fusion constructs
To create the promoter:GUS reporter gene fusion constructs, DNA sequences upstream of the ATG of each individual demethylase gene were amplified using LongAmp Taq (NEB, #M0323S), cloned into pGEM-T Easy (Promega, #A1360) and sequenced. Primer sequences used for amplifying the promoter fragments are listed in Supplementary Table 1. The promoters were then removed from pGEM-T Easy using SmaI digestion and inserted into the binary vector pBI101 at the SmaI site in front of the pre-existing GUS coding sequence. The final vectors were confirmed by sequencing.
DME hairpin construct
Using the primers DME-forward and DME-reverse (Supplementary Table 1) a 548 bp fragment of the DME coding region was amplified, cloned into the pGEM-T Easy vector and sequenced. The DME fragment was excised by restriction digestion with XbaI-ClaI and cloned in an antisense orientation in the XbaI-ClaI multi-cloning site of the pKannibal vector [18]. The DME fragment in the same pGEM-T Easy plasmid was excised by restriction digestion with XhoI-KpnI and cloned in a sense orientation in the corresponding sites of the pKannibal/DME antisense plasmid. The DME sense and antisense sequences are spaced by the PDK intron already present in the pKannibal vector. The 1.6 kb Rubisco small subunit promoter (SSU promoter) was excised from the pBC vector (Promega) using SacI-SalI digestion, and cloned into the SacI-XhoI site upstream of the DME inverted-repeat in pKannibal, replacing the 35S promoter. The SSU-DME hpRNA expression cassette was released by restriction digestion with NotI and cloned into the NotI site of the binary vector pBART. The SSU promoter was positioned adjacent to the T-DNA Right Border sequence.
Plant transformation and analysis of transgenic plants
All plant expression constructs were transferred to the Agrobacterium tumefaciens strain GV3101 using tri-parental mating in the presence of E. coli strain RK2013 [19] or by electroporation. Plants were transformed by floral dipping using Agrobacterium as described previously [20] and transgenic seeds harvested. Transgenic progeny seeds from floral-dipped plants were screened by sterilizing seeds using bleach/HCl solution [21] and germinated on MS media containing kanamycin (50 mg/L) and timentin (150 mg/L). Positive transgenic plants were self-fertilized to generate T2, T3, or T4 populations for further analysis. The presence of the constructs was confirmed by kanamycin resistance, PCR and northern blot analyses.
GUS expression analysis
Histochemical analysis of GUS expression was performed by incubating whole plants with 2 mM X-gluc (5-Bromo-4-chloro-3-indolyl-β-D-glucuronide) (Sigma, #B5285) at 37°C or room temperature followed by destaining with ethanol [22] and imaging using the Leica MZFLIII fluorescence dissector microscope. Quantitative analysis of GUS expression was carried out by MUG (4-methylumbelliferyl-β-D-glucuronide) (Sigma, #M9130) fluorometric assay as described previously [23] using pooled samples containing around 20 sibling plants (T4 generation) for each line.
Northern blot analysis and RT-qPCR
RNA was isolated from 3-week-old whole plant tissue (with no reproductive tissues) of approximately 20 individual siblings for each plant line using Trizol Reagent (Invitrogen, #15596026) according to manufacturer’s instructions. Northern blot analysis was performed essentially as described previously using the same hybridization buffer [24]: 10 µg of RNA was separated on a 1.3% formaldehyde agarose gel and the RNA fragments transferred to Hybond-N membrane (GE Healthcare Amersham, #RPN203N) by capillary transfer in 10 x SSC buffer overnight. After UV cross-linking the membrane was pre-hybridized at 55°C for 2–3 h, and then hybridized overnight with antisense RNA probe transcribed from the same pGEM-T Easy vector used to make the hpDME construct. The membrane was washed at 65°C with the phosphate-based buffers described in the Promega protocol used in [24] followed by 15-min RNase A treatment at room temperature in 2 x SSC buffer. The filter was exposed to a Phosphor Imager screen for visualization of hybridization bands.
For RT-qPCR analysis, 4 µg of total RNA was treated with RNase-free DNase I (Ambion, #AM2222) at 37°C for 20 min, and purified by phenol-chloroform and chloroform extraction and sodium-acetate-ethanol precipitation. The RNA was then reverse transcribed into cDNA using Superscript III (Invitrogen, #18,080,093) and 50 pmol of oligo-dT22 primer (22 thymines) in a 40 µl reaction according to manufacturer’s instructions. The reaction was diluted to 300 µl with water and 5 µl was used for each qPCR reaction. qPCR reactions were performed in technical triplicates on a Corbett 2000 Rotor-Gene real-time PCR machine (Corbett Research), using Fast SYBR Green Master Mix (Applied Biosystems, #4,385,610) according to the manufacturer’s instruction. The Arabidopsis Actin2 gene was used as the internal reference gene and relative expression determined using the comparative quantification method [25]. Primer sequences are listed in Supplementary Table 1.
DNA bisulfite sequencing
Approximately 2 µg of DNA was bisulfite converted using the EpiTect Plus DNA Bisulfite Kit (Qiagen, #59,124) following the manufacturer's instruction, yielding 50 µl of converted DNA solution. To check the efficiency of bisulphite conversion, PCR amplification was first performed on a 157-bp sequence of the chloroplast-encoded psaA gene. All bisulfite PCR reactions were performed using the following PCR cycles: 12 min at 94°C followed by 10 cycles of 1 min at 94°C, 2:30 min at 50°C, 1:30 min at 72°C, and 30 cycles with 1 min at 94°C, 1:30 min at 55°C, 1:30 min at 72°C, with a final extension of 10 min at 72°C. Primer sequences are listed in Supplementary Table 1. The PCR product of the psaA gene was digested with MseI restriction enzyme and separated on a 4% NuSieve agarose gel; full digestion at the two MseI sites created by C to T conversion indicates complete bisulfite conversion of the DNA (Supplementary Figure 7) [26].
Plant growth and Fusarium infection assays
Arabidopsis seeds were sown on MS agar plates [27] supplemented with 3% sucrose and kept for 3 days at 4°C, followed by germination and growth at 22°C in a 16 h/8 h light/dark cycle. Plantlets were transferred to fresh MS plates after one week.
Fusarium oxysporum f.sp. conglutinans strain 5176 (obtained from Dr Roger Shivas, Queensland Department of Primary Industries and Fisheries, Australia) was grown in Potato-Dextrose Broth (Sigma, #P6685) at 28°C and spores collected by centrifugation. A Fusarium spore solution of 2 × 106 spores/ml was prepared, and 3-week-old plants were infected by root dipping as described previously [8]. Infected plants were transferred either to MS agar without sucrose or to soil and the disease symptoms scored at the indicated time post-infection.
Supplementary material
Supplemental data for this article can be accessed here.
Acknowledgments
We thank Carl Davies for photography.
Author contribution
Conceived and designed the study: MBW, US, TM; Performed experiments: US, JL, NS, MBW; Analysed the data: US, JL, NS, CZ, MBW; Wrote the manuscript: MBW, US, NS, TM, ED.
Compliance
The work described herein was carried out in compliance with the Code of Conduct and the required Health and Safety Regulations.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet. 2009;43:143–166. PubMed PMID: 19659441; PubMed Central PMCID: PMC3137514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bauer MJ, Fischer RL.. Genome demethylation and imprinting in the endosperm. Curr Opin Plant Biol. 2011. April;14(2):162–167. . PubMed PMID: 21435940; PubMed Central PMCID: PMC3082360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Choi Y, Gehring M, Johnson L, et al. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in arabidopsis. Cell. 2002. July 12;110(1):33–42. PubMed PMID: 12150995. [DOI] [PubMed] [Google Scholar]
- [4].Penterman J, Zilberman D, Huh JH, et al. DNA demethylation in the Arabidopsis genome. Proc Natl Acad Sci U S A. 2007. April 17;104(16):6752–6757. PubMed PMID: 17409185; PubMed Central PMCID: PMC1847597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Schumann U, Lee J, Kazan K, et al. DNA-demethylase regulated genes show methylation-independent spatiotemporal expression patterns. Front Plant Sci. 2017;8:1449 PubMed PMID: 28894455; PubMed Central PMCID: PMCPMC5581395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ortega-Galisteo AP, Morales-Ruiz T, Ariza RR, et al. Arabidopsis DEMETER-LIKE proteins DML2 and DML3 are required for appropriate distribution of DNA methylation marks. Plant Mol Biol. 2008. August;67(6):671–681. PubMed PMID: 18493721. [DOI] [PubMed] [Google Scholar]
- [7].Yu A, Lepere G, Jay F, et al. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc Natl Acad Sci U S A. 2013. February 5;110(6):2389–2394. PubMed PMID: 23335630; PubMed Central PMCID: PMC3568381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Le TN, Schumann U, Smith NA, et al. DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in Arabidopsis. Genome Biol. 2014;15(9):458 PubMed PMID: 25228471; PubMed Central PMCID: PMC4189188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gong Z, Morales-Ruiz T, Ariza RR, et al. ROS1, a repressor of transcriptional gene silencing in Arabidopsis, encodes a DNA glycosylase/lyase. Cell. 2002. December 13;111(6):803–814. PubMed PMID: 12526807. [DOI] [PubMed] [Google Scholar]
- [10].Schmid M, Davison TS, Henz SR, et al. A gene expression map of Arabidopsis thaliana development. Nat Genet. 2005. May;37(5):501–506. PubMed PMID: 15806101. [DOI] [PubMed] [Google Scholar]
- [11].Klepikova AV, Kasianov AS, Gerasimov ES, et al. A high resolution map of the Arabidopsis thaliana developmental transcriptome based on RNA-seq profiling. Plant J. 2016. December;88(6):1058–1070. PubMed PMID: 27549386. [DOI] [PubMed] [Google Scholar]
- [12].Lister R, O’Malley RC, Tonti-Filippini J, et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008. May 2;133(3):523–536. PubMed PMID: 18423832; PubMed Central PMCID: PMC2723732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Stroud H, Greenberg MV, Feng S, et al. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell. 2013. January 17;152(1–2):352–364. PubMed PMID: 23313553; PubMed Central PMCID: PMC3597350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zheng X, Deng W, Luo K, et al. The cauliflower mosaic virus (CaMV) 35S promoter sequence alters the level and patterns of activity of adjacent tissue- and organ-specific gene promoters. Plant Cell Rep. 2007. August;26(8):1195–1203. PubMed PMID: 17340093. [DOI] [PubMed] [Google Scholar]
- [15].Lei M, Zhang H, Julian R, et al. Regulatory link between DNA methylation and active demethylation in Arabidopsis. Proc Natl Acad Sci U S A. 2015. March 17;112(11):3553–3557. PubMed PMID: 25733903; PubMed Central PMCID: PMCPMC4371987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gent JI, Ellis NA, Guo L, et al. CHH islands: de novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013. April;23(4):628–637. PubMed PMID: 23269663; PubMed Central PMCID: PMCPMC3613580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Barkan A, Martienssen RA. Inactivation of maize transposon Mu suppresses a mutant phenotype by activating an outward-reading promoter near the end of Mu1. Proc Natl Acad Sci U S A. 1991. April 15;88(8):3502–3506. . PubMed PMID: 1849660; PubMed Central PMCID: PMCPMC51476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Wesley SV, Helliwell CA, Smith NA, et al. Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J. 2001. September;27(6):581–590. PubMed PMID: 11576441. [DOI] [PubMed] [Google Scholar]
- [19].Ditta G, Stanfield S, Corbin D, et al. Broad host range DNA cloning system for gram-negative bacteria: construction of a gene bank of rhizobium meliloti. Proc Natl Acad Sci U S A. 1980. December;77(12):7347–7351. PubMed PMID: 7012838; PubMed Central PMCID: PMCPMC350500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. Plant J. 1998. December;16(6):735–743. PubMed PMID: 10069079. [DOI] [PubMed] [Google Scholar]
- [21].Wu Q, Smith NA, Zhang D, et al. Root-specific expression of a Jacalin Lectin family protein gene requires a transposable element sequence in the promoter. Genes (Basel) (Basel) 2018. November 13;9(11). DOI: 10.3390/genes9110550 PubMed PMID: 30428604; PubMed Central PMCID: PMCPMC6266147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Jefferson RA, Kavanagh TA, Bevan MW. GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. Embo J. 1987. December 20;6(13):3901–3907. PubMed PMID: 3327686; PubMed Central PMCID: PMCPMC553867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Chen S, Helliwell CA, Wu LM, et al. A novel T-DNA vector design for selection of transgenic lines with simple transgene integration and stable transgene expression. Funct Plant Biol. 2005;32(8):671–681. PubMed PMID: WOS:000230931100001; English. [DOI] [PubMed] [Google Scholar]
- [24].Wang MB, Wesley SV, Finnegan EJ, et al. Replicating satellite RNA induces sequence-specific DNA methylation and truncated transcripts in plants. RNA. 2001. January;7(1):16–28. PubMed PMID: 11214177; PubMed Central PMCID: PMCPMC1370065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6): 1101–1108. PubMed PMID: 18546601. [DOI] [PubMed] [Google Scholar]
- [26].Finn TE, Wang L, Smolilo D, et al. Transgene expression and transgene-induced silencing in diploid and autotetraploid Arabidopsis. Genetics. 2011. February;187(2):409–423. PubMed PMID: 21078688; PubMed Central PMCID: PMC3030486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Murashige T, Skoog F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plantarum 1962;15(3):473–497. PubMed PMID: WOS:A19621781C00014; English. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.