

Abstract
Objective
To investigate the pathogenicity of a novel homozygous BRAT1 variant in 2 siblings with nonprogressive cerebellar ataxia (NPCA) through functional studies on primary and immortalized patient cell lines.
Methods
BRAT1 protein levels and ataxia-telangiectasia mutated (ATM) kinase activity in patient-derived and control cell lines were assessed by Western blotting. The impact of the novel BRAT1 variants on mitochondrial function was also assessed, by comparing patient and control cell lines for rates of oxygen consumption and for phosphorylation (S293) of the E1⍺ subunit of pyruvate dehydrogenase (PDH).
Results
Two male siblings with NPCA, mild intellectual disability, and isolated cerebellar atrophy were found to be homozygous for a c.185T>A (p.Val62Glu) variant in BRAT1 by whole exome sequencing. Western blotting revealed markedly decreased BRAT1 protein levels in lymphocytes and/or fibroblast cells from both affected siblings compared to control cell lines. There were no differences between the patient and control cells in ATM kinase activation, following ionizing radiation. Mitochondrial studies were initially suggestive of a defect in regulation of PDH activity, but there was no evidence of increased phosphorylation of the E1⍺ subunit of the PDH complex. Measurement of oxygen consumption rates similarly failed to identify differences between patient and control cells.
Conclusions
Biallelic pathogenic variants in BRAT1 can be associated with NPCA, a phenotype considerably milder than previously reported. Surprisingly, despite the molecular role currently proposed for BRAT1 in ATM regulation, this disorder is unlikely to result from defective ATM kinase or mitochondrial dysfunction.
BRAT1 (BRCA1-associated ataxia telangiectasia mutated [ATM] activator 1) mutations were initially identified to cause rigidity and multifocal seizure syndrome, lethal neonatal (RMFSL, OMIM #614498).1 Nonlethal phenotypes were subsequently described in several patients with biallelic BRAT1 mutations, with clinical features ranging from severe developmental delays, ataxia, and seizures to “atypical” RMFS with no lethality.2–8 The underlying mechanism for this phenotypic variability is unknown. We report 2 brothers with a novel homozygous BRAT1 variant who present with a much milder phenotype than reported in the literature to date, with mild intellectual disability, nonprogressive cerebellar ataxia (NPCA), and stable cerebellar atrophy on serial brain imaging. Functional studies confirmed markedly decreased levels of BRAT1 protein in both affected patients' cells compared to controls.
Clinical report
We describe 2 brothers with NPCA and ocular motor apraxia born to a healthy, nonconsanguineous couple of Pakistani descent. The family history was negative for ataxia, developmental delays, seizures, or other neurologic disorders.
Sibling 1, currently 24 years of age, was born spontaneously at term. Pregnancy and delivery were uneventful except for clubfoot noted at birth. He rolled at 3 months, sat without support at 7–8 months, pulled to stand at 12 months, and crawled at 18 months. At the age of 15 months, parents noted gait instability with frequent falls. Limb ataxia was formally documented at 3 years of age. He spoke his first words at 4 years, was able to ambulate independently, and continued to make steady developmental gains. By 8 years, he was able to feed himself, drink from a cup, write single letters and numbers, and perform simple mathematical calculations. He completed education at the college level and is fully independent for all activities of daily living, although he uses a walker for longer distances. Neurologic examination revealed normal head circumference and mild dysarthria with monophasic scanning speech. He had mild ocular motor apraxia, saccadic smooth pursuit, and gaze-evoked nystagmus (video 1), and fundoscopy was normal. Dysmetria and dysdiadochokinesia were evident, and he had truncal titubation. Gait was ataxic and he was unable to complete tandem gait (video 2). Extensive metabolic and other laboratory investigations, including microarray and serum alpha-fetoprotein (AFP) were normal, as were EMG and nerve conduction studies. Serial brain imaging at 9 and 16 years of age revealed stable isolated cerebellar atrophy (figure 1, A–D). Molecular testing of FXN, APTX, SETX, and spinocerebellar ataxia types 1, 2, 3, 6, 7, 8 was normal. Muscle biopsy showed no overt histopathologic abnormalities, and CoQ10 levels in muscle were normal. Mitochondrial studies of patient-derived fibroblasts revealed a decreased native pyruvate dehydrogenase (PDH) level of 0.38 nmoles/min/mg protein compared with that of control (0.73) but the activated PDH (with dichloroacetate, [DCA]) was restored to the normal levels of 1.12, compared with control (1.22). The cellular lactate/pyruvate ratio of 6.1 was low in patients compared with control (12.09).
Figure 1. Pedigree and brain imaging.

(A) Family pedigree. (B) Neuroimaging findings in the affected siblings. (B.a–B.d) Serial brain imaging for sibling 1. (B.a) Sagittal T1 image at 9 years showing normal cerebrum and brainstem, with diffuse cerebellar atrophy and moderately dilated fourth ventricle with prominent CSF spaces surrounding the cerebellum. (B.b) Coronal T2 image at 9 years showing cerebellar atrophy of both hemispheres and vermis. (B.c) Sagittal T1 and (B.d) coronal T2 images for sibling 1 at the age of 16 years, shows similar findings of stable severe cerebellar atrophy. (Be–B.h) Serial brain imaging for sibling 2. (B.e) Sagittal T1 image at 3 years showing cerebellar volume loss with prominent CSF spaces and fourth ventricle; brain and brainstem are normal. (B.f) Coronal T1 at 3 years showing small vermis and cerebellar hemispheres. (B.g) Sagittal T2 and (B.h) coronal T2 images for sibling 2 at the age of 5 years showing stable cerebellar atrophy.
Sibling 1 at 23 years of age. He has full range of extra-ocular movements, but end-gaze nystagmus, saccadic smooth pursuit and difficulty initiating saccades.Download Supplementary Video 1 (7.7MB, wmv) via http://dx.doi.org/10.1212/000359_Video_1
Sibling 1 at 23 years of age. He has a wide-based ataxic gait with unsteadiness when turning, and is unable to complete tandem gait.Download Supplementary Video 2 (7.2MB, wmv) via http://dx.doi.org/10.1212/000359_Video_2
Sibling 2, currently 7 years of age, was born spontaneously at term following a normal pregnancy. He first sat at 8 months, stood with support at 12 months, but did not walk until 21 months. He spoke his first words at 12 months but did not speak in full sentences until 4.5 years of age. His hearing is normal but he wears glasses for myopia. He attends a regular grade 2 program, with an individualized educational plan, speech therapy, occupational therapy, and physical therapy support. He has no swallowing difficulties or bulbar symptoms, and is able to walk without assistance, but with a clearly ataxic gait. Neurologic examination revealed normal head circumference, and funduscopy was normal. He had dysarthria, ocular motor apraxia, saccadic smooth pursuit, gaze-evoked nystagmus, dysmetria, dysdiadochokinesia, and mild wide-based ataxic gait with unsteadiness while turning. He was unable to perform tandem gait. Metabolic investigations including serum AFP, microarray, EMG, and NCS were normal. Brain imaging at the ages of 3 and 5 years revealed stable isolated cerebellar atrophy, similar to his brother (figure 1, E–H).
Methods
Standard protocol approvals, registrations, and patient consents
This study was approved by the Research Ethics Board of The Hospital for Sick Children (#1000009004). Tissue samples were obtained with appropriate informed consent according to the declaration of Helsinki principles of medical research involving human subjects. The parents provided written informed consent to participate in this study, including publication of videos.
Whole exome sequencing and BRAT1 expression
Whole exome sequencing of genomic DNA extracted from blood lymphocytes from both siblings identified a homozygous missense c.185T>A (p.Val62Glu) variant in BRAT1. There are no homozygotes for this BRAT1 variant in large databases of genetic variation including the Genome Aggregation Database (gnomad.broadinstitute.org/; accessed July 2019). The parents were each heterozygous carriers of the variant. This mutation, which is located in the middle of the second exon encoding a putative CIDE-N domain, markedly reduced the level of BRAT1 protein in patient-derived cell lines from both affected siblings (figure 2, A and B). This reduction was not due to decreased levels of BRAT1 mRNA, which were similar in the patients' and parental cells (figure 2C), suggesting instead that it might reflect instability of the mutant BRAT1 protein. Indeed, although we could not restore normal levels of BRAT1 protein in the patient cells by incubation with the proteasome inhibitor MG132 (figure 2B), incubation with cycloheximide to prevent nascent protein synthesis revealed that the half-life of the mutant protein was much shorter than the parental BRAT1 (figure 2D). This instability could reflect incorrect folding of the mutant protein, and/or reduced binding to one or more as yet unidentified protein partners.
Figure 2. BRAT1 expression in patient cells.

(A) Quantification of BRAT1 protein in control (1BR), parent (Father), and BRAT1 patient (Sib1) hTERT fibroblasts normalized to tubulin levels using Image studio lite. (B) BRAT1 protein levels measured in BRAT1 patients' (Sib1 and Sib2) and parents' (Mother and Father) LCLs before and after incubation with the proteasome inhibitor MG132 by Western blotting. p53 was used as a positive control. (C) BRAT1 mRNA levels in parents' (Mother and Father) and patients' (Sib1 and Sib2) LCLs as measured by qPCR. (D) BRAT1 protein levels measured in BRAT1 patient (Sib1) and parent (Father) LCLs before and after incubation with the protein synthesis inhibitor CHX by Western blotting. Note that default exposure levels were adjusted. p53 was used as a positive control. CHX = cycloheximide; LCL= lymphoblastoid cell line.
ATM kinase activation studies
BRAT1 has been reported to be important for ATM phosphorylation at Ser1981 and for activation of this protein kinase by DNA damage.9 We therefore compared ATM autophosphorylation and transphosphorylation of its downstream targets (e.g., Chk2, p53, and H2AX) in patient and parental control cells lines, following ionizing radiation. Surprisingly, however, we failed to detect a defect in the BRAT1 patient cells in ATM kinase activity (figure 3).
Figure 3. ATM activation in BRAT1 patient cells.

Western blot analysis of the phosphorylation of ATM kinase and its downstream substrates in indicated BRAT1 patients (Sib1 and Sib2), parents (Mother and Father), and control (1BR) (A) LCLs, (B) primary fibroblasts and/or (C) hTERT fibroblasts before and after ionizing irradiation (IR, 5 Gy) as indicated. ATM = ataxia telangiectasia mutated.
Mitochondrial studies
Despite initial indications of a possible PDH dysfunction, analysis of the E1⍺ subunit of this protein for phosphorylation at S293 by Western blotting failed to identify a difference between patient and parent control cells, even with the addition of DCA (figure 4A). Moreover, measurement of oxygen consumption rates in fibroblasts did not reveal any differences between the patient and the parental control cells (figure 4B).
Figure 4. Analysis of PDH E1α phospho293 (S293) expression and oxygen consumption rates in parent and BRAT1 patient primary fibroblasts.
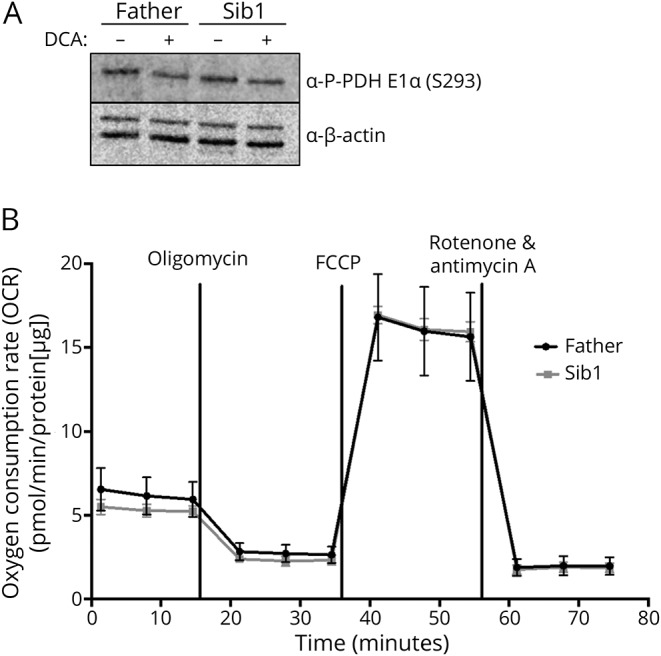
(A) Representative Western blot analysis showing no observed differences in PDH E1α S293 expression in parent (Father) and BRAT1 patient (Sib1). Protein lysates were extracted from untreated cells or fibroblasts treated with 5 mM DCA (B) OCRs of the same fibroblast cells using the Seahorse XFe96 Extracellular Flux Analyzer (Seahorse Bioscience). OCR was normalized to protein to eliminate differences in growth rate between cell lines (n = 8 wells/cell line). DCA = dichloroacetate; PDH = pyruvate dehydrogenase; OCR = oxygen consumption rates.
Data availability
Detailed methods are available as supplemental data (e-Methods, links.lww.com/NXG/A185). All data presented in this study are available on reasonable request.
Discussion
We describe 2 brothers, both homozygous for a pathogenic variant in BRAT1, with NPCA, ocular motor apraxia, and isolated cerebellar atrophy, which was nonprogressive on serial brain imaging. Seizures have not been observed in either of the affected siblings. While both siblings have mild intellectual disability, the elder brother completed education at college level and is independent for ambulation at the age of 24. Variable but consistently severe phenotypes have been associated with biallelic BRAT1 mutations ranging from RMFSL to severe neurodevelopmental disorder with cerebellar atrophy with or without seizures (NEDCAS, OMIM #618056). The reason for this variable phenotype is not well understood, but the possibility of hypomorphic BRAT1 variants, which have less disruptive effects on protein function resulting in a milder phenotype has been considered.3,4 The nonprogressive nature and considerably milder clinical course of the patients we describe are consistent with NPCA, which has never been previously associated with biallelic mutations in BRAT1.
BRAT1 has been reported to interact with the tumor-suppressor BRCA1 (breast cancer type 1) and ATM, and is believed to play a role in the activity of these proteins during cellular responses to DNA damage.9 However, the exact mechanism(s) by which mutations in BRAT1 trigger neurodegeneration and to what extent a defect in ATM function contributes to this disease are unknown. We therefore evaluated a potential effect of the novel BRAT1 variant on ATM protein kinase activity following DNA damage. Surprisingly, however, BRAT1 patient cells did not exhibit a defect in ATM protein kinase activity following DNA damage induced by exposure to ionizing radiation. This may reflect differences in the cell lines and experimental systems employed in the studies.9 Nevertheless, our data strongly suggest that defective ATM activity does not contribute to the BRAT1 disease phenotype.
One patient with biallelic mutations in BRAT1 exhibited severely reduced cytochrome c oxidase enzyme histochemical staining on muscle biopsy, suggestive of mitochondrial dysfunction.7 In another study, loss of BRAT1 expression caused decreased cell proliferation and increased glucose uptake and reactive oxygen species production, also suggestive of mitochondrial impairment.10 In the current study, analysis of fibroblasts from sibling 1 revealed decreased native PDH, again consistent with a mitochondrial defect. However, a muscle biopsy from sibling 1 was essentially normal, and DCA activation restored PDH to normal levels, suggesting that mitochondrial function was intrinsically normal. Indeed, we were unable to demonstrate a deleterious impact of the BRAT1 variant on either PDH regulation or oxygen consumption rate in the patient cells.
A major consequence of the Val62Glu homozygous variant in BRAT1 appears to be reduced BRAT1 protein levels and it is possible that this mutation also affects one or more as yet unidentified activities of this protein. We note that the Val62Glu mutation resides within a putative CIDE-N domain, a domain identified previously in apoptotic nucleases.
Finally, we highlight here that biallelic pathogenic variants in BRAT1 can be associated with NPCA, a phenotype considerably milder than previously reported, but which should be considered in the differential diagnosis of NPCA.
Acknowledgment
The authors thank the family for participating in this study. This work was selected for study by the Care4Rare Canada (Enhanced Care for Rare Genetic Diseases in Canada) Consortium Gene Discovery Steering Committee: Kym Boycott (lead; University of Ottawa), Alex MacKenzie (co-lead; University of Ottawa), Jacek Majewski (McGill University), Michael Brudno (University of Toronto), Dennis Bulman (University of Ottawa), and David Dyment (University of Ottawa). HH is funded by an ERC Advanced Investigator Award (SIDSCA; 694996) to KWC. The study was supported by the Charles University, project GA UK fellowship (project No. 1212219) to ZC.
Glossary
- AFP
alpha-fetoprotein
- ATM
ataxia telangiectasia mutated
- DCA
dichloroacetate
- NPCA
non-progressive cerebellar ataxia
- PDH
pyruvate dehydrogenase
Appendix 1. Authors
Appendix 2. Co-investigators
Study funding
This study was supported by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute, Ontario Research Fund, Genome Quebec, Children's Hospital of Eastern Ontario Foundation, and The Hospital for Sick Children. The study was funded by an ERC Advanced Investigator Award to KWC (SIDSCA; 694996). The study was supported by the Charles University, project GA UK fellowship (project No. 1212219) to ZC.
Disclosure
Disclosures available: Neurology.org/NG.
References
- 1.Puffenberger EG, Jinks RN, Sougnez C, et al. . Genetic mapping and exome sequencing identify variants associated with five novel diseases. PLoS One 2012;7:e28936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanes I, Kozenko M, Callen DJ. Lethal neonatal rigidity and multifocal seizure syndrome—A misnamed disorder? Pediatr Neurol 2015;53:535–540. [DOI] [PubMed] [Google Scholar]
- 3.Mundy SA, Krock BL, Mao R, Shen JJ. BRAT1-related disease—identification of a patient without early lethality. Am J Med Genet A 2016;170:699–702. [DOI] [PubMed] [Google Scholar]
- 4.Srivastava S, Olson HE, Cohen JS, et al. . BRAT1 mutations present with a spectrum of clinical severity. Am J Med Genet A 2016;170:2265–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernández-Jaén A, Álvarez S, So EY, et al. . Mutations in BRAT1 cause autosomal recessive progressive encephalopathy: report of a Spanish patient. Eur J Paediatr Neurol 2016;20:421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Valence S, Cochet E, Rougeot C, et al. . Exome sequencing in congenital ataxia identifies two new candidate genes and highlights a pathophysiological link between some congenital ataxias and early infantile epileptic encephalopathies. Genet Med 2019;21:553–563. [DOI] [PubMed] [Google Scholar]
- 7.Horn D, Weschke B, Knierim E, et al. . BRAT1 mutations are associated with infantile epileptic encephalopathy, mitochondrial dysfunction, and survival into childhood. Am J Med Genet A 2016;170:2274–2281. [DOI] [PubMed] [Google Scholar]
- 8.Smith NJ, Lipsett J, Dibbens LM, Heron SE. BRAT1-associated neurodegeneration: intra-familial phenotypic differences in siblings. Am J Med Genet A 2016;170:3033–3038. [DOI] [PubMed] [Google Scholar]
- 9.Aglipay JA, Martin SA, Tawara H, Lee SW, Ouchi T. ATM activation by ionizing radiation requires BRCA1-associated BAAT1. J Biol Chem 2006;281:9710–9718. [DOI] [PubMed] [Google Scholar]
- 10.So EY, Ouchi T. BRAT1 deficiency causes increased glucose metabolism and mitochondrial malfunction. BMC Cancer 2014;14:548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sibling 1 at 23 years of age. He has full range of extra-ocular movements, but end-gaze nystagmus, saccadic smooth pursuit and difficulty initiating saccades.Download Supplementary Video 1 (7.7MB, wmv) via http://dx.doi.org/10.1212/000359_Video_1
Sibling 1 at 23 years of age. He has a wide-based ataxic gait with unsteadiness when turning, and is unable to complete tandem gait.Download Supplementary Video 2 (7.2MB, wmv) via http://dx.doi.org/10.1212/000359_Video_2
Data Availability Statement
Detailed methods are available as supplemental data (e-Methods, links.lww.com/NXG/A185). All data presented in this study are available on reasonable request.