

Abstract
Background & Aims
The ubiquitin ligase F-box and WD repeat domain-containing 7 (FBXW7) is recognized as a tumor suppressor in many cancer types due to its ability to promote the degradation of numerous oncogenic target proteins.
Methods
Here, we investigated the role of FBXW7 in intrahepatic cholangiocarcinoma (iCCA) using mouse models ad hoc generated, iCCA cell lines, and human iCCA specimens.
Results
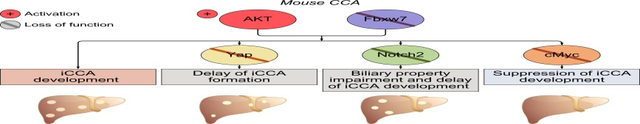
FBXW7 mRNA expression is almost ubiquitously downregulated in human iCCA specimens. To identify the molecular mechanisms whereby FBXW7 dysfunction promotes cholangiocarcinogenesis, we generated a mouse model by hydrodynamic tail vein injection of Fbxw7ΔF, a dominant negative form of Fbxw7, either alone or in association with an activated/myristylated form of AKT (myr-AKT). While forced overexpression of Fbxw7ΔF alone did not induce any appreciable abnormality in the mouse liver, co-expression with AKT triggered cholangiocarcinogenesis and mice had to be euthanized by 15 weeks post-injection. At the molecular level, a strong induction of FBXW7 canonical targets, including Yap, Notch2, and c-Myc oncoproteins, was detected. However, only c-Myc was consistently confirmed as a FBXW7 target in human CCA cell lines. Most importantly, selected ablation of c-Myc completely impaired iCCA formation in AKT/Fbxw7ΔF mice, whereas deletion of either Yap or Notch2 only delayed tumorigenesis in the same model. In human iCCA specimens, an inverse correlation between the expression levels of FBXW7 and c-Myc transcriptional activity was observed.
Conclusions
Downregulation of FBXW7 is ubiquitous in human iCCA and cooperates with AKT to induce cholangiocarcinogenesis in mice via c-Myc-dependent mechanisms. Targeting c-Myc might represent an innovative therapy against iCCA exhibiting low FBXW7 expression.
Keywords: FBXW7, cholangiocarcinogenesis, c-Myc, cholangiocarcinoma murine model, Yap, Notch2
Lay Summary
Mounting evidence implies the tumor suppressor role of FBXW7 in many cancer types, including intrahepatic cholangiocarcinoma (iCCA), by its ability to promote the degradation of numerous oncoproteins. Our present findings indicate the ubiquitous low expression of FBXW7, which is inversely correlated with c-MYC activity, in human CCA specimens. In the mouse liver, activation of Fbxw7 together with AKT overexpression induces rapid iCCA formation, which is prevented by c-Myc suppression. Thus, targeting c-MYC might be an effective treatment for human iCCA with low FBXW7 levels.
Graphical Abstract
Introduction
Cholangiocarcinoma (CCA) is the second most common primary liver cancer [1, 2]. Depending on the anatomical site, CCA is classified into intrahepatic (iCCA) and extrahepatic cholangiocarcinoma (eCCA) [3]. iCCA incidence has been rising over the last decade, while that of eCCA slightly decreased [4]. For iCCA detected at early stage, curative surgical resection is the optimal treatment strategy [5, 6]. However, less than one third of patients achieves negative tumor margins, and recurrence rate is high [5, 7]. Furthermore, in patients not meeting the narrow criteria for surgical treatment, therapeutic options are limited [8]. Therefore, substantial efforts should be devoted to unravelling the molecular mechanisms of iCCA development and progression. This would lead to novel and more effective therapeutic strategies against this pernicious disease.
The E3 ubiquitin ligase F-box and WD repeat-domain containing protein 7 (FBXW7) is a bona fide tumor suppressor gene [9, 10]. Being a substrate recognition component of the Skp1-Cul1-F-box protein-type complex, FBXW7 is responsible for the binding to and the degradation of several oncogenes, including c-MYC [11, 12], YAP [13], NOTCH1 [14, 15], mTOR [16], CCNE1 [11], and CCND1 [12]. Among them, YAP, NOTCH, and c-MYC proteins play a pivotal role in cholangiocarcinogenesis.
In human CCA, ~35% specimens were found to harbor FBXW7 mutations, resulting in failure of substrate recognition [17]. In addition, FBXW7 gene is downregulated in human CCA cells when compared with intrahepatic biliary epithelial cells [18]. Also, FBXW7 mRNA expression negatively correlates with tumor stage, metastasis, and differentiation in human iCCA specimens [18, 19]. Furthermore, low FBXW7 levels are associated with poorer prognosis and shorter survival in CCA and hepatocellular carcinoma (HCC) [19, 20] patients.
Inspired by these previous data, here we analyzed a collection of human iCCA and confirmed the reduction of FBXW7 gene expression in this tumor type, which occurred in the absence of FBXW7 mutations. Moreover, to define the molecular mechanisms whereby FBXW7 disruption contributes to cholangiocarcinogenesis, we generated a new mouse model by hydrodynamic tail vein injection (HTVi) of myr-AKT (an activated/myristylated form of AKT) and Fbxw7ΔF (AKT/Fbxw7ΔF), a dominant negative form of Fbxw7. In this iCCA model, a robust induction of Yap, Notch2, and c-Myc oncoproteins occurred along tumorigenesis. Subsequent in vitro and in vivo studies identified c-Myc as the prominent FBXW7 target responsible for cholangiocarcinogenesis in experimental and human iCCA.
Results
Low mutation rate but reduced mRNA expression of FBXW7 in human intrahepatic cholangiocarcinoma
First, we determined the FBXW7 mutation frequency in a collection of human iCCA (n=120) and corresponding non-tumorous livers. Unexpectedly and different from previous findings [17], FBXW7 was mutated only in one iCCA (0.8%). The mutation occurred in FBXW7 exon 8 and consisted of the previously reported R465H change [21, 22] (Figure 1A). As FBXW7 gene downregulation can result from promoter hypermethylation and/or loss of heterozygosity (LOH) at the FBXW7 locus [23, 24], the promoter and locus status of FBXW7 were determined. While no FBXW7 promoter hypermethylation was detected (data not shown), LOH occurred in 20 of 120 iCCA (16.7%) with wild-type FBXW7 (Figure 1B, C). Subsequently, mRNA levels of FBXW7 were investigated by real-time RT-PCR using the iCCA samples from the collection whose RNA was available (n=82). Strikingly, the vast majority of iCCA (71/82; 86.6%) exhibited lower FBXW7 mRNA levels when compared with corresponding non-tumorous livers (Figure 1D), in agreement with previous reports [19, 25] and the FBXW7 data from The Cancer Genome Atlas (TCGA, Supplementary Figure 1A). In the TCGA dataset presented on cBioportal [26, 27], containing mutation data from 35 patients, one patient (2.86%) harbored the R393Q mutation (Supplementary Figure 1B). Interestingly, this mutation and the one found in our study (R465H) have been identified as significant hotspots and indicate oncogenic events [21, 22]. Also, mRNA level of FBXW7 correlates with its copy number values (Supplementary Figure 1C). No association between FBXW7 mRNA levels and clinicopathologic features of the patients, including age, gender, etiology, presence of cirrhosis, tumor size, and tumor differentiation, was detected (data not shown). Also, since survival data from our patients’ cohort were not available, the relation between FBXW7 expression and prognosis could not be determined.
Figure 1. Downregulation of the FBXW7 gene in human intrahepatic cholangiocarcinoma (iCCA) specimens.

(A) Chromatogram showing the wild-type sequence (upper panel) and the R465H mutant form (lower panel) of FBXW7 exon 8. The red arrow indicates the mutated nucleotide. (B, C) Examples of loss of heterozygosity (LOH) at the D4S2998 (B) or D4S2934 (C) locus encompassing the FBXW7 gene. The two alleles are indicated by asterisks (*), and red arrows indicate the presence of LOH at one allele. (D) Quantitative real-time RT-PCR analysis of FBXW7 mRNA levels in normal livers (n=8), iCCA (n=82), and corresponding non-tumorous surrounding liver tissues (ST; n=82). Abbreviations: ns, not significant; *** p<0.0001 when compared to NL and ST.
Altogether, the present data indicate that downregulation of wild-type FBXW7 gene is almost ubiquitous in human iCCA.
Inactivation of Fbxw7 synergizes with activated AKT to induce hepatocyte-derived iCCA formation in mice
Subsequently, we sought to address the role of FBXW7 along cholangiocarcinogenesis in vivo. Thus, we generated a mouse model harboring a dominant negative form of Fbxw7 (Fbxw7ΔF), which was injected into the mice hydrodynamically. Inactivation of Fbxw7 alone did not result in any tumor development or appearance of liver histologic abnormalities up to 36 weeks post-injection (Supplementary Figure 2), implying that loss of Fbxw7 does not suffice to drive carcinogenesis in the mouse liver. Subsequently, to increase the malignant potential, another oncogenic insult was added to Fbxw7 inactivation, namely the induction of AKT, frequently occurring in human iCCA [28], through hydrodynamic co-injection of activated/myristylated (myr-)AKT and Fbxw7ΔF (Figure 2A). Previously, we showed that myr-AKT overexpression induces lipid-rich hepatocellular preneoplastic lesions in the mouse liver, ultimately leading to the development of HCC and, more rarely, iCCA [29] (Supplementary Figure 3). In AKT/Fbxw7ΔF livers, similar clusters of clear-cell, lipid-rich preneoplastic hepatocytes with enlarged nuclei, were detected three weeks post-injection. These hepatocellular lesions, morphologically indistinguishable from those of myr-AKT mice, were progressively replaced by iCCA lesions, starting from 6 weeks post-injection (Figure 2A; Supplementary Figure 4). Ten weeks post-injection, large iCCAs were detected in AKT/Fbxw7ΔF livers. Most iCCA exhibited large areas of necrosis in the tumor center (Supplementary Figure 4), suggesting vigorous proliferation, which was confirmed by Ki67 staining (Figure 2A). By 12–15 weeks post-injection, livers were almost completely occupied by colliding iCCA and mice were moribund (Figure 2A; Supplementary Figure 4). Liver weight and liver/body ratio gradually increased along cholangiocarcinogenesis (Supplementary Figure 5). Since AKT and Fbxw7ΔF were both HA-tagged, immunohistochemistry showed positive HA-tag immunoreactivity in both cytoplasm (for AKT) and nucleus (for Fbxw7ΔF) of tumor cells (Figure 2B).
Figure 2. Co-expression of AKT and a dominant negative form of Fbxw7 (Fbxw7ΔF) leads to intrahepatic cholangiocarcinoma development in mice.

(A) Timeline of tumor development in AKT/Fbxw7ΔF mice (upper panel); macroscopy of AKT/Fbxw7ΔF livers (second panel); histopathologic features of the lesions (third panel); Ck19 immunostaining (fourth panel). (B) HA-tag immunohistochemistry showing cytoplasmic and nuclear staining for AKT and Fbxw7ΔF. The lesions are highly proliferative, as indicated by Ki67 nuclear immunoreactivity. (C) Western blotting of pathways activated in AKT/Fbxw7ΔF cholangiocellular lesions. Gapdh was used as a loading control. (D) mRNA levels of canonical Yap (Ctgf, Cyr61; upper panel) and Notch (Hey1, Nrarp; lower panel) target genes. (E) mRNA levels of c-Myc targets (Cdk4, Ccnd2, Skp2, Tert, Ldha, Ldhc, and Odc1). Scale bar: 200μm for 10×; 100μm for 20×. Abbreviations: H&E, hematoxylin and eosin staining; NL, normal liver; T, tumor; ns, not significant. * p <0.05; ** p <0.01; *** p <0.001.
To determine whether the observed iCCA lesions derive from hepatocytes or cholangiocytes, lineage tracing technology was applied. By breeding AlbCreERT2 mice with R26REYFP mice, AlbCreERT2;R26REYFP heterozygotes were generated. Subsequently, tamoxifen was injected into AlbCreERT2;R26REYFP mice 2 weeks before hydrodynamic injection to trigger specific EYFP expression in hepatocytes (Supplementary Figure 6A). Noticeably, immunofluorescence (IF) showed that iCCAs induced by AKT/Fbxw7ΔF co-expression originated from hepatocytes, as they exhibited positive staining for both EYFP and HA-tag (Supplementary Figure 6B).
Activation of multiple oncogenic pathways in AKT/Fbxw7ΔF lesions
Next, we investigated the signaling pathways activated in AKT/Fbxw7ΔF mouse lesions. Specifically, we focused on the oncogenic cascades that are physiologically inhibited by Fbxw7, including Hippo/Yap, Notch, and c-Myc [11, 13, 14]. Western blotting and immunohistochemistry showed the activation/induction of Yap, Notch2, and c-Myc proteins (Figure 2C; Supplementary Figures 7 and 8). Yap was expressed at high level (Figure 2C) and translocated in the nucleus (a sign of its activation; Supplementary Figure 7) of tumor cells. Consequently, Yap downstream targets, namely Ctgf and Cyr61, were significantly upregulated in the iCCA lesions when compared to the non-tumorous counterparts (Figure 2D). Among the other Hippo pathway members, the Yap negative regulators Lats½ were downregulated (Supplementary Figure 8). Similarly, levels of Notch2 and its ligand Jag1 were remarkably increased in AKT/Fbxw7ΔF neoplastic lesions (Figure 2C). Accordingly, the canonical downstream targets of Notch2 (Hey1 and Nrarp) were induced at transcriptional level in AKT/Fbxw7ΔF iCCAs (Figure 2D). As for the downstream targets of c-Myc, genes involved in cell cycle and survival, such as Cdk4, Ccnd2, Skp2 and Tert, as well as energy metabolism, including Ldha, Ldhc, and Odc1, were uniformly upregulated in tumors (Figure 2E). Importantly, elevated c-Myc expression was specific for AKT/Fbxw7ΔF iCCA lesions, as it was undetectable in un-injected normal liver, AKT- or Fbxw7ΔF-only injected liver tissues (Supplementary Figure 9).
Since AKT/Fbxw7ΔF induced tumors derive from hepatocytes, we further confirmed our findings using the AML-12 normal hepatocyte cell line [30]. After transfection of myr-AKT, Fbxw7ΔF alone or together for 48h, the protein levels of Yap, Notch2, and c-Myc were elevated in Fbxw7ΔF and AKT/Fbxw7ΔF transfected cells (Supplementary Figure 10A). Interestingly, Sox9, a biliary epithelium biomarker, was also induced in Fbxw7ΔF and AKT/Fbxw7ΔF transfected AML-12 cells. AKT/Fbxw7ΔF co-expression also triggered c-Myc upregulation in RBE, KMCH, and SNU1196 human CCA cell lines (Supplementary Figure 10B).
Depletion of Yap delays without abolishing AKT/Fbxw7ΔF tumorigenesis
YAP activation occurs in over 90% of human CCA specimens [31] as well as in the AKT/Fbxw7ΔF mouse model (this study). In addition, YAP was hypothesized as a target protein of FBXW7 in HCC [13] and pancreatic cancer [32]. We therefore investigated the role of Yap in AKT/Fbxw7ΔF driven cholangiocarcinogenesis. Thus, hydrodynamic injection of AKT/Fbxw7ΔF/pCMV (as control) and AKT/Fbxw7ΔF/Cre plasmids was employed in Yapflox/flox mice (Figure 3A). Ablation of the endogenous Yap significantly delayed tumor development in AKT/Fbxw7ΔF mice (Figure 3B and 3C). By 7.7 weeks post-injection, while the control group mice developed iCCAs, no frankly malignant lesions occurred in AKT/Fbxw7ΔF/Cre livers, whereas small clusters of infiltrating lymphocytes (Figure 3C) and scattered clusters of clear-cell, lipid-rich hepatocytes were detected (Supplementary Figure 11). Twenty weeks post-injection, several tumors were observed in AKT/Fbxw7ΔF/Cre livers, with a diameter of 1mm-5mm. They consisted of clear-cell hepatocellular lesions, containing small areas of cholangiocellular differentiation inside, and they were of mixed-type morphology at this time point (Supplementary Figure 11). Noticeably, by 34 weeks post-injection, most of the hepatocellular large tumors were replaced by pure cholangiocellular lesions (Supplementary Figure 11). Both CCA and HCC lesions were highly proliferative (Figure 3C and Supplementary Figure 12). To determine whether these tumor cells were escapers (namely tumor cells retaining Yap expression), we performed immunohistochemistry with the anti-Yap antibody. Hepatocytes with no immuno-positivity for Yap expression were detectable as early as 7.7 weeks post-injection in AKT/Fbxw7ΔF/Cre livers. Importantly, all AKT/Fbxw7ΔF/Cre tumor cells were negative for Yap immunoreactivity, indicating effective Yap deletion (Figure 3C and Supplementary Figure 12). Robust Yap immunolabeling was instead detected in tumor stromal cells, which displayed Vimentin immunoreactivity (Supplementary Figure 12), and in surrounding non-tumorous hepatocytes. The observation was corroborated by Western blotting showing low Yap expression (Figure 3D), and by qRT-PCR demonstrating decreased levels of Ctgf and Cry61 genes in AKT/Fbxw7ΔF/Cre tumors (Figure 3E). Noticeably, phosphorylated/activated (p-)AKT was remarkably decreased following Yap ablation, together with profound downregulation of Jag1, a Yap target [33]. Interestingly, Notch2 levels were only mildly reduced, while c-Myc expression was decreased, in AKT/Fbxw7ΔF/Cre samples.
Figure 3. Deletion of Yap delays, without suppressing, cholangiocarcinogenesis in AKT/Fbxw7ΔF mice.

(A) Study design. Yapflox/flox conditional knockout mice were subjected to HTVi of either AKT/Fbxw7ΔF/pCMV (control, n=9) or AKT/Fbxw7ΔF/Cre (n=10) plasmids. (B) Survival curve showing the delay of intrahepatic cholangiocarcinoma development following deletion of Yap. (C) Delay of tumor development in AKT/Fbxw7ΔF mice, as revealed by macroscopic examination of the livers (upper panel), histopathology of the lesions, Ck19 immunoreactivity (as a marker of biliary tumors) and Yap immunoreactivity. (D) Western blotting of AKT/Fbxw7ΔF mouse livers from control and Yap deleted (Cre) mice. Gapdh was used as a loading control. Scale bar: 200μm for 10×; 100μm for 20×. Abbreviations: H&E, hematoxylin and eosin staining; NL, normal liver; SB, sleeping beauty transposase.
Altogether, these data indicate that Yap ablation significantly retards, but does not suppress, AKT/Fbxw7ΔF induced tumorigenesis in mice.
Notch2 activation is partially required for AKT/Fbxw7ΔF induced cholangiocarcinogenesis
We previously discovered that Notch2, instead of Notch1, is the main contributor for hepatocyte-derived iCCA formation [34]. As the present data indicate elevated expression of Notch2 and its ligand Jag1 in AKT/Fbxw7ΔF iCCA lesions, we investigated whether Notch2 signaling is required for AKT/Fbxw7ΔF induced cholangiocarcinogenesis. Thus, we hydrodynamically injected AKT/Fbxw7ΔF/pCMV and AKT/Fbxw7ΔF/Cre plasmids in Notch2flox/flox conditional knockout mice (Figure 4A). Although deletion of Notch2 delayed the fatal tumor development by 4–5 weeks (Figure 4B), both cohorts, namely mice injected with AKT/Fbxw7ΔF/pCMV and AKT/Fbxw7ΔF/Cre, developed large tumors with positive Ck19 staining (Figure 4B and 4C). Similar to that described in the AKT/Yap iCCA model [34], ablation of Notch2 significantly disrupted the biliary properties of tumor lesions, as indicated by predominance of large clear-cell hepatocellular tumors over cholangiocellular lesions and lower Ck19 and higher Hnf-4α immunoreactivity (Figures 4C and 4D; Supplementary Figure 13). Interestingly, a trend of replacement of HCC lesions with cholangiocellular lesions was observed in later stages of tumor development also in this model (Supplementary Figure 13). Western blotting confirmed the efficient deletion of Notch2, which was paralleled by decreased Jag1 and Sox9 expression (Figure 4E). Yap, the oncogenic effector of the Hippo pathway, was downregulated following Notch2 ablation (Figure 4E), confirming the positive signal loop between Yap and Notch pathways [33]. Intriguingly, c-Myc expression was not affected by Notch2 ablation.
Figure 4. Deletion of Notch2 retards, without impairing, cholangiocarcinogenesis in AKT/Fbxw7ΔF mice.

(A) Study design. Notch2lox/flox conditional knockout mice were subjected to HTVi of either AKT/Fbxw7ΔF/pCMV (control, n=8) or AKT/Fbxw7ΔF/Cre (n=10) plasmids. (B) Delay of tumor development in AKT/Fbxw7ΔF mice, as revealed by macroscopic examination of the livers and histopathology of the lesions, is accompanied by reduction of the Ck19 biliary marker and increase of the Hnf-4α hepatocellular marker. (C) Survival curve showing the delay of intrahepatic cholangiocarcinoma development following deletion of Notch2. (D) Analysis of Ck19-positive areas in control (pCMV) and Notch2-depleted (Cre) livers. (E) Western blotting of AKT/Fbxw7ΔF livers from control and Notch2 deleted mice. Scale bar: 200μm. Abbreviations: H&E, hematoxylin and eosin staining; NL, normal liver. *** p <0.001.
Overall, the present data imply a relevant but dispensable role of Notch2 in AKT/Fbxw7ΔF dependent cholangiocarcinogenesis.
c-Myc is necessary for cholangiocarcinogenesis in AKT/Fbxw7ΔF mice
Subsequently, we investigated the role of c-Myc, another important target of FBXW7 [11], in AKT/Fbxw7ΔF mice. Thus, MadMyc, a dominant negative form of c-Myc [35], and the AKT/Fbxw7ΔF plasmids, were simultaneously hydrodynamically delivered into the mouse liver and tumor development was monitored. Hydrodynamic injection of AKT/Fbxw7ΔF/pT3 construct was used as control (Figure 5A). Strikingly, inhibition of c-Myc completely suppressed iCCA formation in AKT/Fbxw7ΔF mice (Figures 5B and 5C). Indeed, mice were healthy with no gross liver tumors by 31 weeks post-injection. Consistently, histopathological evaluation revealed the absence of premalignant and malignant lesions in AKT/Fbxw7ΔF/MadMyc livers (Figure 5C). Importantly, sporadic HA-positive cells were detected in AKT/Fbxw7ΔF/MadMyc livers (Supplementary Figure 14A), and Western blotting revealed the ectopically expressed HA-tagged AKT, Fbxw7ΔF and MadMyc, leading to the inhibition of c-Myc expression (Supplementary Figure 14B). In striking contrast, numerous highly proliferative iCCAs emerged in the liver parenchyma of AKT/Fbxw7ΔF/pT3 mice, with strong immunoreactivity for the biliary marker Ck19 and the proliferation marker Ki67 (Figure 5C). All AKT/Fbxw7ΔF/pT3 mice had to be euthanized by 10–12 weeks post-injection (Figure 5B).
Figure 5. Inactivation of c-Myc completely abolishes cholangiocarcinogenesis in AKT/Fbxw7ΔF mice.

(A) Study design. FVB/N mice were subjected to HTVi of either AKT/Fbxw7ΔF/pT3 (control, n=6) or AKT/Fbxw7ΔF/MadMyc (n=8) plasmids. MadMyc is a dominant negative form of c-Myc. (B) Survival curve of AKT/Fbxw7ΔF/pT3 and AKT/Fbxw7ΔF/MadMyc mice. (C) Inhibition of tumor development in AKT/Fbxw7ΔF/MadMyc mice, as revealed by macroscopic examination of the livers and histopathology of the lesions, is accompanied by immunoreactivity for the Ck19 biliary marker limited to normal biliary epithelial cells (indicated by arrows) and low/absent immunoreactivity for the proliferation marker Ki67. Scale bar: 100μm. Abbreviation: H&E, hematoxylin and eosin staining.
c-MYC is a pivotal player downstream of FBXW7 in human iCCA
Next, we investigated the potential targets of FBXW7 in human iCCA. We transfected RBE, KMCH, and SNU1196 CCA cell lines with lentivirus particles encoding EGFP (as control), FBXW7 (wild-type), and FBXW7ΔF, and the expression of known canonical FBXW7 targets was analyzed. Protein levels of CCNA, CCND1, CCNE, GSK3β, HSF1, MCL-1, mTOR, NOTCH1, NOTCH2, PCNA, RICTOR and YAP did not show any consistent change following FBXW7 modulation (Figure 6). Additional proteins involved in tumor development, including p-AKT, p-ERK, p21, etc. were also analyzed, and their levels were not consistently affected by the same approach in the three cell lines (Supplementary Figure 15). In striking contrast, c-MYC was consistently downregulated following FBXW7 wild-type overexpression and strongly induced by FBXW7 inhibition (via FBXW7ΔF transfection) in the three cell lines (Figure 6). Accordingly, transcriptional activity of c-MYC exhibited the same pattern of modulation following transient transfection of control, FBXW7, and FBXW7ΔF plasmids in RBE cells and KMCH cells (Supplementary Figure 16).
Figure 6.

Western blot analysis showing c-MYC as a consistent target of FBXW7 in cholangiocarcinoma (CCA) cell lines. GAPDH was used as a loading control.
Finally, we investigated the relationship between FBXW7 and c-MYC in the collection of human iCCA specimens (n=82). No significant correlation was detected between FBXW7 and c-MYC mRNA levels (Figure 7A), whereas a strong, inverse correlation was found between FBXW7 mRNA levels and those of c-MYC transcriptional activity (Figure 7B). Subsequently, we determined the protein levels of FBXW7 and c-MYC in the same sample collection using immunohistochemistry (Figure 8). Upregulation of FBXW7 and c-MYC in iCCA when compared with non-tumorous surrounding livers occurred in 14 and 44 of 82 specimens (17.1% and 53.6%, respectively). FBXW7 and c-MYC were concomitantly upregulated only in 4 of 82 samples (4.9%). The remaining 10 iCCA specimens showing elevated levels of FBXW7 displayed c-MYC downregulation. On the other hand, 39 of 44 (88.6%) iCCA specimens showing high levels of c-MYC exhibited absent/weak immunoreactivity for FBXW7.
Figure 7. Expression levels of the FBXW7 gene are inversely correlated with c-MYC activity in human intrahepatic cholangiocarcinoma (iCCA).

(A) Absence of significant correlation between FBXW7 and c-MYC mRNA levels, as assessed by linear regression analysis. (B) A significant, negative correlation was found between mRNA levels of FBXW7 and c-MYC activity using the same statistical approach.
Figure 8. Immunohistochemical patterns of FBXW7 and c-Myc in human intrahepatic cholangiocarcinoma specimens.

Three representative cases (iCCA 1–3) are shown. Specifically, iCCA 1 (upper panel) recapitulates the most frequent pattern observed in the human iCCA collection, consisting of tumors with low levels of FBXW7 and strong immunoreactivity for c-MYC. iCCA 2 (middle panel) shows instead the example of an iCCA with high immunoreactivity for FBXW7 and weak/punctate staining of c-MYC protein. Finally, iCCA 3 (lower panel), consisting of a tumor displaying concomitant, elevated immunolabeling for both FBXW7 and c-MYC. CK19 staining was used as a biliary differentiation marker of the tumors. Scale bar: 100μm. Abbreviation: H&E, hematoxylin and eosin staining.
Altogether, these findings support a central role played by c-MYC both in mouse and human cholangiocarcinogenesis driven by FBXW7 inactivation.
Discussion
Here, we demonstrated that inactivation of Fbxw7 synergizes with activated AKT to induce hepatocyte-derived iCCA formation in mice. By conducting loss of function experiments using Yapflox/flox and Notch2flox/flox conditional knockout mice, we discovered that inhibition of each of the two oncogenic pathways delays without suppressing AKT/Fbxw7ΔF-induced iCCA development. In particular, histopathological analysis revealed that hepatocellular lesions appear first in all models. Subsequently, cholangiocellular lesions emerge, co-existing with the hepatocellular ones. Along tumor progression, the cholangiocellular lesions replace either completely (in the AKT/Fbxw7ΔF model) or partly (in AKT/Fbxw7ΔF depleted of Yap or Notch2) the hepatocellular lesions. These findings suggest the need of additional alterations for cholangiocellular lesions to develop in these models and, in the same time, the growth advantage of cholangiocellular over hepatocellular lesions once these alterations are acquired.
By employing a comprehensive analysis of in vitro and in vivo approaches as well as validation in human iCCA specimens, we identified c-Myc as a crucial mediator of cholangiocarcinogenesis following downregulation of FBXW7. Although Yap and Notch are considered to be canonical Fbxw7 targets [13, 14], our in vitro studies demonstrates that neither YAP nor NOTCH2 levels (nor related signaling cascades) are consistently influenced by FBXW7-mediated degradation in human iCCA cells. Similarly, several other FBXW7 targets, including mTOR, CCND1, and CCNE, were heterogeneously modulated following FBXW7 manipulation. The discrepancy between our findings and the literature requires further elucidation. Presumably, the recognition substrates for FBXW7 differ depending on the context and the tissue type as well as the genetic background/alterations present. This might also explain the reason why, besides c-MYC, most FBXW7 target proteins were heterogeneously affected by FBXW7 modulation in distinct CCA cell lines. Similarly, we found that c-MYC activation was AKT-independent both in vitro and in vivo models of iCCA, against the assumption that c-Myc is an AKT target [36]. Once again, tissue- and context-dependent mechanisms might be responsible for these unexpected results.
c-MYC is a known substrate for FBXW7 [11, 37, 38]. Alterations of FBXW7 leading to c-MYC accumulation have been detected in T-cell acute lymphoblastic leukemia (T-ALL) [38], melanoma [15], skin carcinogenesis [39], prostate cancer [40], and HCC [41]. In CCA, one group reported that overexpression of FBXW7 in QBC-939 and MZ-CHA1 CCA cell lines decreased the expression of c-MYC [11]. The study also showed that FBXW7 overexpression inhibits xenograft CCA tumor growth in nude mice [11]. In our study, c-MYC levels were profoundly downregulated by FBXW7 overexpression in three human CCA cell lines. Conversely, when transfecting a dominant negative form of FBXW7, c-MYC expression was strongly upregulated in the same cells. Based on our analysis of various genes induced by c-Myc in mouse iCCA tissues, c-Myc most likely regulates multiple processes during cholangiocarcinogenesis, including tumor cell proliferation, survival and metabolism. Most importantly, we found that impairing c-Myc activity triggers inhibition of cholangiocarcinogenesis induced by inactivation of FBXW7 along with AKT activation in vivo. The results were validated in vitro using AML-12 normal mouse hepatocytes (Supplementary Figure 10A), indicating that c-Myc is required for iCCA initiation. To investigate the role of c-Myc in iCCA progression, we infected the three CCA cell lines with tetracycline (Tet) inducible MadMyc lentivirus. Upon Doxycycline treatment, MadMyc expression was induced and inhibited CCA cell proliferation and colony formation (Supplementary Figure 17). Thus, c-MYC is a pivotal player in iCCA initiation and progression.
Another important finding of our study is that, although FBXW7 was almost ubiquitously downregulated in iCCA specimens, mutations in its coding region as well as LOH at the gene locus were rare, and no hypermethylation at the FBXW7 locus was detected. Thus, other molecular mechanisms are involved in the downregulation of FBXW7 in human iCCA. Previous studies have suggested that long non-coding RNAs and microRNAs as well as various oncogenes, such as Pin1, EZH2, CNS6, and Usp28 can downregulate FBXW7 in cancer [42, 43]. Clearly, additional studies, which are beyond the scope of the present work, are required to identify the pivotal molecular mechanisms responsible for FBXW7 downregulation in human iCCAs. Nonetheless, additional events might limit FBXW7 activity in cholangiocellular tumors independent of FBXW7 transcription. Indeed, we recently identified alternative splicing forms of FBXW7 in 27/82 (32.9%) iCCA, which were instead not detected in corresponding non-tumorous livers (Supplementary Figure 18). The presence of these splicing forms has been shown to result in very low translational efficiency of the FBXW7 protein in prostate, bladder, and kidney tumors [44]. Although this issue should be more comprehensively addressed, the present findings imply the existence of several, distinct mechanisms at play in human iCCA to suppress FBXW7 expression and/or activity.
Finally, our data might possess relevant therapeutic implications. As loss of FBXW7 is extremely frequent in iCCA, targeting c-MYC might be highly detrimental for the growth of many of these tumors. Unfortunately, c-MYC is not easily druggable and no effective drugs against c-MYC are commercially available. Nonetheless, bromodomain and extra-terminal (BET) inhibitors recently provided encouraging results for the treatment of c-MYC driven tumors [45, 46]. Thus, BET inhibitors (and similar small inhibitors) should be considered as potentially important drugs for innovative therapies against human iCCA.
Materials and Methods
Constructs and reagents
Constructs applied include PT3-EF1α, pT3-EF1α-HA-myr-AKT (human), pT3-EF5α-HA-Fbxw7ΔF (human), PT3-EF1α-MadMyc (human), pCMV, pCMV-Cre and pCMV-sleeping beauty (SB) transposase.
Animals
Mice were housed, fed, and monitored according to protocols approved by the Committee for Animal Research at the University of California, San Francisco (San Francisco, CA).
Human tissue samples
DNAs from 8 normal livers, 120 frozen iCCAs and corresponding non-tumorous surrounding livers were used for sequencing analysis of the FBXW7 gene as reported [47]. Among them, 82 iCCA specimens were available as frozen specimens and used for loss of heterozygosity (LOH) and promoter methylation analyses, as described [48, 49]. The iCCA samples were collected at the Medical Universities of Greifswald (Greifswald, Germany) and Sassari (Sassari, Italy). Clinicopathological features of iCCA samples are reported in Supplementary Table 1. Institutional Review Board approval was obtained at the local Ethical Committee of the Medical Universities of Greifswald and Sassari. Informed consent was obtained from all individuals.
Statistical analysis
The Prism 7.0 software (GraphPad, San Diego, CA) was used to analyze the data, presented as Means ± SD. Comparisons between two groups were performed with two-tailed unpaired t test when dataset achieve Gaussian distribution or non-parametric test when sample size was small. Welch correction was applied when necessary. P values < 0.05 were considered statistically significant.
Please refer to Supplementary Materials and Methods for detailed information.
Supplementary Material
Highlights.
Downregulation of the FBXW7 tumor suppressor gene was identified as a universal feature of human intrahepatic cholangiocarcinoma (iCCA).
Hydrodynamic transfection of inactivated Fbxw7 synergized with an activated form of AKT to induce rapid iCCA in mice.
Cholangiocarcinogenesis was prevented by c-Myc suppression, while being delayed by either Yap or Notch 2 depletion in these mice.
Inhibition of c-MYC might represent an innovative therapeutic strategy for the treatment of human iCCA with low FBXW7.
Acknowledgments
Financial support: This study is supported by NIH grants R01CA190606 to XC, P30DK026743 for UCSF Liver Center; grant from the Deutsche Forschungsgemeinschaft DFG (grant number RI2695/1–1) to SR, China Scholarship Council State Scholarship Fund No. 201606240132 to HW and Grants from National Natural Science Foundation of China (grant number 61006403) to YZ. Additional support was provided by the Helen Diller Family Comprehensive Cancer Center Director’s Fund.
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- [2].Sia D, Villanueva A, Friedman SL, Llovet JM. Liver Cancer Cell of Origin, Molecular Class, and Effects on Patient Prognosis. Gastroenterology 2017;152:745–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lee SY, Cherqui D. Operative management of cholangiocarcinoma. Semin Liver Dis 2013;33:248–261. [DOI] [PubMed] [Google Scholar]
- [4].Khan SA, Emadossadaty S, Ladep NG, Thomas HC, Elliott P, Taylor-Robinson SD, et al. Rising trends in cholangiocarcinoma: is the ICD classification system misleading us? J Hepatol 2012;56:848–854. [DOI] [PubMed] [Google Scholar]
- [5].Yamamoto M, Takasaki K, Otsubo T, Katsuragawa H, Katagiri S. Recurrence after surgical resection of intrahepatic cholangiocarcinoma. J Hepatobiliary Pancreat Surg 2001;8:154–157. [DOI] [PubMed] [Google Scholar]
- [6].Rizvi S, Gores GJ. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 2013;145:1215–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Hyder O, Hatzaras I, Sotiropoulos GC, Paul A, Alexandrescu S, Marques H, et al. Recurrence after operative management of intrahepatic cholangiocarcinoma. Surgery 2013;153:811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Mertens JC, Rizvi S, Gores GJ. Targeting cholangiocarcinoma. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2018;1864:1454–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cao J, Ge MH, Ling ZQ. Fbxw7 Tumor Suppressor: A Vital Regulator Contributes to Human Tumorigenesis. Medicine (Baltimore) 2016;95:e2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Mol Cancer 2018;17:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Li M, Ouyang L, Zheng Z, Xiang D, Ti A, Li L, et al. E3 ubiquitin ligase FBW7alpha inhibits cholangiocarcinoma cell proliferation by downregulating c-Myc and cyclin E. Oncol Rep 2017;37:1627–1636. [DOI] [PubMed] [Google Scholar]
- [12].Kanatsu-Shinohara M, Onoyama I, Nakayama KI, Shinohara T. Skp1-Cullin-F-box (SCF)-type ubiquitin ligase FBXW7 negatively regulates spermatogonial stem cell self-renewal. Proceedings of the National Academy of Sciences of the United States of America 2014;111:8826–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tu K, Yang W, Li C, Zheng X, Lu Z, Guo C, et al. Fbxw7 is an independent prognostic marker and induces apoptosis and growth arrest by regulating YAP abundance in hepatocellular carcinoma. Mol Cancer 2014;13:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Malyukova A, Dohda T, von der Lehr N, Akhoondi S, Corcoran M, Heyman M, et al. The tumor suppressor gene hCDC4 is frequently mutated in human T-cell acute lymphoblastic leukemia with functional consequences for Notch signaling. Cancer Res 2007;67:5611–5616. [DOI] [PubMed] [Google Scholar]
- [15].Aydin IT, Melamed RD, Adams SJ, Castillo-Martin M, Demir A, Bryk D, et al. FBXW7 mutations in melanoma and a new therapeutic paradigm. J Natl Cancer Inst 2014;106:dju107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, et al. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008;321:1499–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, et al. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res 2007;67:9006–9012. [DOI] [PubMed] [Google Scholar]
- [18].Yang H, Lu X, Liu Z, Chen L, Xu Y, Wang Y, et al. FBXW7 suppresses epithelial-mesenchymal transition, stemness and metastatic potential of cholangiocarcinoma cells. Oncotarget 2015;6:6310–6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Enkhbold C, Utsunomiya T, Morine Y, Imura S, Ikemoto T, Arakawa Y, et al. Loss of FBXW7 expression is associated with poor prognosis in intrahepatic cholangiocarcinoma. Hepatol Res 2014;44:E346–352. [DOI] [PubMed] [Google Scholar]
- [20].Imura S, Tovuu LO, Utsunomiya T, Morine Y, Ikemoto T, Arakawa Y, et al. Role of Fbxw7 expression in hepatocellular carcinoma and adjacent non-tumor liver tissue. J Gastroenterol Hepatol 2014;29:1822–1829. [DOI] [PubMed] [Google Scholar]
- [21].Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nature biotechnology 2016;34:155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chang MT, Bhattarai TS, Schram AM, Bielski CM, Donoghue MTA, Jonsson P, et al. Accelerating Discovery of Functional Mutant Alleles in Cancer. Cancer discovery 2018;8:174–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gu Z, Mitsui H, Inomata K, Honda M, Endo C, Sakurada A, et al. The methylation status of FBXW7 beta-form correlates with histological subtype in human thymoma. Biochemical and biophysical research communications 2008;377:685–688. [DOI] [PubMed] [Google Scholar]
- [24].Cassia R, Moreno-Bueno G, Rodriguez-Perales S, Hardisson D, Cigudosa JC, Palacios J. Cyclin E gene (CCNE) amplification and hCDC4 mutations in endometrial carcinoma. The Journal of pathology 2003;201:589–595. [DOI] [PubMed] [Google Scholar]
- [25].Ishii N, Araki K, Yokobori T, Watanabe A, Tsukagoshi M, Kubo N, et al. Poor prognosis in cholangiocarcinoma patients with low FBXW7 expression is improved by chemotherapy. Oncol Lett 2017;13:3653–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schmitz KJ, Lang H, Wohlschlaeger J, Sotiropoulos GC, Reis H, Schmid KW, et al. AKT and ERK½ signaling in intrahepatic cholangiocarcinoma. World J Gastroenterol 2007;13:6470–6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology 2011;140:1071–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wu JC, Merlino G, Fausto N. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proceedings of the National Academy of Sciences of the United States of America 1994;91:674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pei T, Li Y, Wang J, Wang H, Liang Y, Shi H, et al. YAP is a critical oncogene in human cholangiocarcinoma. Oncotarget 2015;6:17206–17220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang Q, Zhang Y, Parsels JD, Lohse I, Lawrence TS, Pasca di Magliano M, et al. Fbxw7 Deletion Accelerates Kras(G12D)-Driven Pancreatic Tumorigenesis via Yap Accumulation. Neoplasia 2016;18:666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kim W, Khan SK, Gvozdenovic-Jeremic J, Kim Y, Dahlman J, Kim H, et al. Hippo signaling interactions with Wnt/beta-catenin and Notch signaling repress liver tumorigenesis. J Clin Invest 2017;127:137–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Wang J, Dong M, Xu Z, Song X, Zhang S, Qiao Y, et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018;37:3229–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xin B, Yamamoto M, Fujii K, Ooshio T, Chen X, Okada Y, et al. Critical role of Myc activation in mouse hepatocarcinogenesis induced by the activation of AKT and RAS pathways. Oncogene 2017;36:5087–5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proceedings of the National Academy of Sciences of the United States of America 2008;105:6584–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 2004;23:2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].King B, Trimarchi T, Reavie L, Xu L, Mullenders J, Ntziachristos P, et al. The ubiquitin ligase FBXW7 modulates leukemia-initiating cell activity by regulating MYC stability. Cell 2013;153:1552–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ishikawa Y, Hosogane M, Okuyama R, Aoyama S, Onoyama I, Nakayama KI, et al. Opposing functions of Fbxw7 in keratinocyte growth, differentiation and skin tumorigenesis mediated through negative regulation of c-Myc and Notch. Oncogene 2013;32:1921–1932. [DOI] [PubMed] [Google Scholar]
- [40].Xi Z, Yao M, Li Y, Xie C, Holst J, Liu T, et al. Guttiferone K impedes cell cycle re-entry of quiescent prostate cancer cells via stabilization of FBXW7 and subsequent c-MYC degradation. Cell Death Dis 2016;7:e2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tu K, Zheng X, Zhou Z, Li C, Zhang J, Gao J, et al. Recombinant human adenovirus-p53 injection induced apoptosis in hepatocellular carcinoma cell lines mediated by p53-Fbxw7 pathway, which controls c-Myc and cyclin E. PLoS One 2013;8:e68574. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [42].Yeh CH, Bellon M, Nicot C. FBXW7: a critical tumor suppressor of human cancers. Molecular Cancer 2018;17:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Goeppert B, Ernst C, Baer C, Roessler S, Renner M, Mehrabi A, et al. Cadherin-6 is a putative tumor suppressor and target of epigenetically dysregulated miR-429 in cholangiocarcinoma. Epigenetics 2016;11:780–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Liu Y, Ren S, Castellanos-Martin A, Perez-Losada J, Kwon YW, Huang Y, et al. Multiple novel alternative splicing forms of FBXW7α have a translational modulatory function and show specific alteration in human cancer. Plos One 2012;7:e49453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jung M, Gelato KA, Fernández-Montalván A, Siegel S, Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics 2015;7:487–501. [DOI] [PubMed] [Google Scholar]
- [46].Li X, Zhang XA, Xie W, Li X, Huang S. MYC-mediated synthetic lethality for treatment of hematological malignancies. Current Cancer Drug Targets 2015;15−. [DOI] [PubMed] [Google Scholar]
- [47].Lee JW, Soung YH, Kim HJ, Park WS, Nam SW, Kim SH, et al. Mutational analysis of the hCDC4 gene in gastric carcinomas. European Journal of Cancer 2006;42:2369–2373. [DOI] [PubMed] [Google Scholar]
- [48].Spruck CH, Heimo S, Olle S, Müller HM, Michael H, Elisabeth MH, et al. hCDC4 gene mutations in endometrial cancer. Cancer Research 2002;62:4535. [PubMed] [Google Scholar]
- [49].Zhaodi G, Kenichi I, Hidetoshi M, Akira H. Promoter hypermethylation is not the major mechanism for inactivation of the FBXW7 beta-form in human gliomas. Genes & Genetic Systems 2008;83:347–352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.