

Lysine succinylation is a prevalent protein modification that regulates multiple critical cellular processes. Here, we performed a quantitative succinylome analysis in the model soil bacterium Streptomyces coelicolor after characterization of a specific desuccinylase ScCobB2. Comparison of the ΔScCobB2 to the wild-type succinylome identified a total of 673 unique succinylated sites, and among which, 144 protein sites are statistically hypersuccinylated in ΔScCobB2 cells. Analyses of these hypersuccinylated proteins suggested they are enriched in two major pathways, protein biosynthesis and carbon metabolism. We propose that ScCobB2 has critical regulatory roles in S. coelicolor cellular physiology.
Keywords: Posttranslational Modification, Acetylation*, Metabolites, Quantification, Bacteria, Enzyme Modification, Evolution, Microbiology, Desuccinylase, Lysine Succinylation, ScCobB2, Succinylome
Graphical Abstract

Highlights
Identification of the first evolutionary divergent sirtuin ScCobB2 in bacteria.
Implementing a global quantitative succinylome between ΔScCobB2 and WT cells.
ScCobB2 regulates S. coelicolor protein biosynthesis and carbon metabolism pathways.
The divergent sirtuin enzymes are prevalent in other groups of Actinobacteria.
Abstract
As a recently discovered protein posttranslational modification in eukaryotes, lysine succinylation has attracted increasing interest due to its ability to regulate several critical cellular processes, including catabolism, β-oxidation, and ketogenesis. Nevertheless, understanding of the regulatory mechanisms is still at an early stage due to the lack of identified specific desuccinylases in microorganisms. Here, in the model soil bacterium Streptomyces coelicolor, we biochemically characterized a sirtuin-like protein ScCobB2 as a divergent desuccinylase. Based on it, we were able to identify a total of 673 unique succinylated sites, of which 470 sites in 317 proteins were quantified by comparing the ΔScCobB2 to the wild-type succinylome via LC-MS/MS analysis. Further analyses of the quantitative succinylome revealed that at least 114 proteins representing two major pathways, protein biosynthesis and carbon metabolism, are obviously hypersuccinylated in ΔScCobB2 cells. We experimentally examined the regulatory roles of ScCobB2 on 13 hypersuccinylated proteins, including glyceraldehyde-3-phosphate dehydrogenase, aconitate hydratase, and several ribosomal proteins, the results of which suggested a high confidence in our quantitative data. This work provided the first discovery of a specific desuccinylase in bacteria and demonstrated it has pivotal regulatory roles in multiple biological processes of S. coelicolor, laying the foundation for future research of succinylation regulation in other microorganisms.
Protein posttranslational modifications (PTMs)1 play critical roles in the regulation of diverse cellular processes by altering protein properties, such as structure, stability, complex formation, or enzyme activity (1). Among the 20 amino acids in proteins, lysine, having a primary amine moiety, is one of the most frequent targets of covalent modifications, e.g. ubiquitination (2), acetylation (3–5), propionylation (6, 7), malonylation (8), crotonylation (9), and glutarylation (10), and through these modifications, the regulation of the lysine-containing proteins is broadened considerably (11, 12).
Acetylation at lysine is the most well-studied PTM in both prokaryotes and eukaryotes (3, 4). However, lysine succinylation (hereinafter referred to as succinylation) is a newly discovered PTM (13, 14), which transfers a succinyl rather than an acetyl moiety to the ε-amino group of lysine residue (Fig. 1A). Previous reports indicated that succinylation is a spontaneous process (15), while recent evidence has suggested that succinyltransferase should involve in the histone modification (16). To date, lysine succinylomes have been profiled in several eukaryotes (17, 18). Substantial enzymes enriched in the pathways of tricarboxylic acid cycle (TCA) cycle, fatty acid metabolism, and β-oxidation were identified (19, 20). The succinylation on two cellular respiration-related enzymes, pyruvate dehydrogenase and succinate dehydrogenase, and one rate-limiting ketogenic enzyme 3-hydroxy-3-methylglutaryl-CoA synthase 2 were experimentally characterized (19, 20), and all of their activities are negatively regulated by the specific desuccinylase Sirt5 (8, 21). In addition, isocitrate dehydrogenase 2 (IDH) in TCA cycle was demonstrated to be desuccinylated by Sirt5 to maintain cellular NADPH homeostasis and enhance cellular antioxidant defenses in mice (22).
Fig. 1.

Bioinformatics analyses identify two different sirtuin proteins in S. coelicolor. (A) The chemical structure of succinylated lysine. Substrates and reactions of succinylation or desuccinylation of lysine are shown correspondingly. While desuccinylation can be catalyzed by desuccinylases, the specific succinyltransferase responsible for succinylation has not been identified yet. Spontaneous succinylation occurs by succinyl-CoA treatment as reported (15). O-SADPR: 2′-O-succinyl-ADP-ribose. (B) Similarity comparison of ScCobB1 or ScCobB2 to the mammalian sirtuins and the E. coli deacylase CobB. Schematic representation of the sirtuin proteins shows a conserved silent information regulator 2 domain. (C) Phylogenetic analysis of sirtuin proteins using MEGA software. S. coelicolor ScCobB2 showed an evolutionarily closer relationship to the desuccinylase Sirt5 rather than the deacetylases Sirt1–3 of mammalian cells. The sequences of human sirtuins were obtained from NCBI at http://www.ncbi.nlm.nih.gov/.
Although succinylation has been identified in multiple prokaryotic species (23–26), its regulatory role upon their cell physiology remains elusive due to the lack of identified specific desuccinylases in these organisms. In Escherichia coli, only one sirtuin functioning as a bifunctional enzyme named CobB has been characterized (14). The CobB was proven to be not only a deacetylase but also a functional desuccinylase. Meanwhile, the global succinylome of Mycobacterium tuberculosis has been profiled, and, as in E. coli, a CobB-like bifunctional protein (Rv1151c) was identified (24, 25). However, by analyzing the sirtuin enzymes in different microorganisms, we found that most bacteria possess more than one sirtuin protein in their genomes (Fig. S1). Even though there are less than seven sirtuins as that in human cells, this observation raised the question whether these sirtuin proteins in bacteria have evolved specific functions, as that in eukaryotes.
Streptomyces coelicolor is a model soil bacterium belonging to the phylum Actinobacteria, a group of interest due to their complicated cell development and potent abilities in natural product biosynthesis. Using bioinformatics analyses, we found two predicted sirtuins in the S. coelicolor genome, one of which has been proven to be a functional deacetylase in vitro (27). Here, we identified the other sirtuin (SCO6464) as a specific succinylation regulatory enzyme both in vitro and in vivo. Based on this knowledge, a quantitative proteomic approach was employed to characterize and compare the lysine succinylomes between wild-type and SCO6464 knockout cells. A total of 673 succinylated sites in 427 proteins with diverse biological functions were identified, among which 114 proteins were shown to be hypersuccinylated in SCO6464 knockout cells. Further analysis suggested that these hypersuccinylated proteins were enriched in multiple biological pathways, such as protein biosynthesis and carbon metabolism, providing plenty of decent succinylation targets to be investigated.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Media, and Materials
The wild-type S. coelicolor M145 and its derivatives ΔSccobB1 and ΔSccobB2 were cultured in tryptic soy broth (Oxoid, UK) liquid medium or on mannitol soya flour (Oxoid, UK) solid medium at 30 °C. E. coli DH5α and E. coli BL21 (DE3) [or E. coli BL21 (DE3) ΔcobB] were used as the host strains to manipulate plasmids and express proteins, respectively. Both of them were cultured in Luria-Bertani (Oxoid, UK) medium at 37 °C. The methylation-deficient E. coli strain ET12567 was used to propagate DNA into S. coelicolor. All strains and plasmids used in this study are listed in Table S9, and all primers used are listed in Table S10. The pan anti-succinyllysine antibody used in this study is the same as that described in (28), while the anti-His-tag antibody was purchased from Abmart, Shanghai, China. The peptides modified by succinylation (K609Suc: KTRSGKSucIMRRI) or acetylation (K609Ac: KTRSGKAcIMRRI) were synthesized by BankPeptide, Inc. (Hefei, China) with > 98% purity.
Construction of ΔSccobB1 and ΔSccobB2 in S. coelicolor
The null mutants ΔSccobB1 and ΔSccobB2 were constructed, respectively, with PCR targeting (29) by in-frame replacement of the coding region of the SccobB1 or SccobB2 gene with a resistance gene in the chromosome. In brief, the target gene was replaced within a cosmid first, using the PCR-amplified apramycin cassette by the λ-Red recombination system. The resulting mutant cosmid was then propagated into S. coelicolor cells to induce a double exchange in the chromosome by homologous recombination. Correct S. coelicolor clones were selected with apramycin (50 μg ml−1) and verified by PCR.
Determination of Cellular Succinylation Levels in Wild-Type, ΔSccobB1, and ΔSccobB2 S. coelicolor
S. coelicolor cells of wild-type, ΔSccobB1, and ΔSccobB2 were grown in tryptic soy broth medium supplemented with 50 mm glucose and harvested at log phase after 2 days of growth. The cell cultures were lysed with their debris removed after centrifugation. Approximately 20-μg protein samples were separated by 12% SDS-PAGE and visualized via Coomassie blue staining. For the Western blotting assay, similar amounts of proteins (20 μg) were transferred onto nitrocellulose membranes (GE Healthcare), and standard procedures (28) were followed for the detection of their succinylation levels. Briefly, 100 mm Tris-HCl (pH 7.5) with 0.5% (v/v) Tween-20 (Sangon Biotech, China) and 1% peptone (AMRESCO) was used for blocking buffer, and 100 mm Tris-HCl (pH 7.5) with 0.05% (v/v) Tween-20 and 0.1% peptone was used for primary and secondary antibody buffers. The pan anti-succinyllysine antibody (28) with 1:1000 dilution in the primary antibody buffer was employed overnight at 4 °C. Nitrocellulose membranes were treated with an ECL Western blotting kit (GE Healthcare) and visualized using an ImageQuant LAS 4000 mini (GE Healthcare) equipped with ImageQuant TL software. Unless otherwise indicated, Coomassie blue staining was used for the loading control.
Cloning, Expression, and Purification of ScCobB1, ScCobB2, CobB, and the Target Proteins
The gene coding sequences for ScCobB1, ScCobB2, and the target proteins (ACO, IMP dehydrogenase, Aldh, GapA, Lpd, GroS, SucC, AtpA, RpL13, RpL2, RpL9, DnaK, and Tuf) were amplified from S. coelicolor genomic DNA with corresponding primers (see Table S10). The gene coding sequences for CobB and Acs were amplified from the E. coli MG1655 genome. DNA fragments were digested by EcoRI and XhoI and ligated into pET28b to construct the expression plasmids. The plasmids expressing ScCobB1, ScCobB2, and CobB were transformed into E. coli BL21 (DE3). Other plasmids expressing Acs and the target proteins were transformed into the E. coli BL21 (DE3) ΔcobB strain. The correct monoclones were grown overnight in 5 ml Luria-Bertani medium containing kanamycin (50 μg ml−1, Sangon Biotech, China) and then subcultured in 500 ml Luria-Bertani liquid medium. When the OD600 reached 0.6, cells were transferred to 16 °C, and the corresponding proteins were induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside (Sigma-Aldrich, Germany) overnight.
Cells were harvested by centrifugation at 6000 rpm for 10 min at 4 °C and resuspended in buffer containing 25 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm DTT, 10% (v/v) glycerol, and 25 mm imidazole. After passing through an EmulsiFlex-C5 cell disruptor (AVESTIN, Inc., Ottawa, Canada), cellular debris was removed by centrifuging at 13,000 g for 30 min at 4 °C. The collected supernatants were loaded onto a 5-ml nickel resin column (GE Healthcare) and purified with the ÄKTA™ FPLC System (GE Healthcare). After washing with 10 volumes of wash buffer (25 mm Tris-HCl, 150 mm NaCl, and 20 mm imidazole, pH 8.0), proteins were obtained by elution from the system (elution buffer: 25 mm Tris-HCl, 150 mm NaCl, and 500 mm imidazole, pH 8.0), resulting in >95% purity. All proteins were dialyzed with a buffer containing 25 mm Tris-HCl (pH 8.0), 150 mm NaCl, 1 mm DTT, and 10% (v/v) glycerol and then aliquoted and kept frozen at −80 °C till use.
In vitro Desuccinylation Assay
In vitro desuccinylation was completed in a 50-μl reaction mixture containing 25 mm MOPS buffer (pH 7.4, Sangon Biotech, China), 50 mm NaCl, 1 mm DTT, 1.0 mm MgCl2, 0.55 μm target proteins, 1.5 μm ScCobB2 (or 1.2 μm ScCobB1/CobB), and 1.0 mm NAD+ (or no NAD+ as a negative control). The reaction mixtures were mixed and centrifuged gently and then incubated at 37 °C for 2 h. The desuccinylated proteins were analyzed by Western blotting assay.
High-Performance Liquid Chromatography (HPLC) Determination of Peptide Desuccinylation
The succinylated peptide containing Acs Lys-609 (K609Suc: KTRSGKSucIMRRI) was synthesized by BankPeptide, Inc. (Hefei, China) and then desuccinylated using an in vitro system. The in vitro peptide deacetylation system contained 25 mm MOPS buffer (pH 7.5), 50 mm NaCl, 1 mm DTT, 1.0 mm MgCl2, 1.0 μm ScCobB2 (or ScCobB1/CobB), 1.0 mm NAD+, and 200 μm AcsK609Suc peptide. The reactions were carried out at 37 °C in a 60-μl volume for 2 h, and quenched with one volume of 10% (v/v) trifluoroacetic acid (TFA, Sigma-Aldrich) for 10 min at room temperature. Then, the mixture was centrifuged at 13,000 g for 10 min to discard the sediment, and the supernatant was analyzed by HPLC using the Aeris peptide XB-C18 column (150 × 4.6 mm, 3.6 μm; Phenomenex, CA). The mobile phase consisted of solvent A (0.1% TFA in HPLC-grade H2O) and solvent B (0.1% TFA in HPLC-grade acetonitrile). Peptides were eluted in a linear gradient of 0–80% solvent B at 1 ml min−1 over 15 min. The column temperature was set at 25 °C, and the peptides were monitored with UV light at 215-nm wavelength.
Experimental Design and Statistical Rationale
To determine the regulatory targets of ScCobB2, a quantitative succinylome analysis was performed by comparing the ΔScCobB2 succinylome to the wild-type succinylome. Three biological samples for both strains with two technical replicates were prepared and analyzed. The six samples were harvested, digested, and then tandem mass tag (TMT) labeling and affinity enrichment were performed before LC-MS/MS analysis (for detail please see sections that follow). The resulting MS/MS data were processed using MaxQuant with the integrated Andromeda search engine (v.1.4.1.2). Tandem mass spectra were searched against the UniProt S. coelicolor database (organism ID, 100226; 8038 sequences). The false discovery rate thresholds for protein, peptide, and modification sites were specified at 1%. The identified succinylation proteins were enriched by Gene Ontology (GO) annotation and KEGG pathway analyses. A two-tailed Fisher's exact test was employed to test the enrichment of the protein-containing international protein index entries against all international protein index proteins. Correction for multiple hypothesis testing was carried out using standard false discovery rate control methods (set as 1%). The GO and KEGG pathways with corrected p values < 0.001 were considered statistically significant. Statistical significance for Western blotting was determined by paired t-tests where p values < 0.01 (***) and < 0.1 (*) were considered significant.
Quantitative Succinylome Analysis between S. coelicolor Wild-Type and ΔSccobB2 Cells
Protein Extraction and Trypsin Digestion
Wild-type and ΔSccobB2 cells were grown and harvested following the same procedures as described in the preceding section (“Determination of Cellular Succinylation Levels”). The collected cells were first frozen using liquid nitrogen and then transferred to 50-ml centrifuge tubes for high-intensity ultrasonication in lysis buffer (8 m urea, 2 mm EDTA, 5 mm DTT, 1% Protease Inhibitor Mixture III, 3 μm trichostatin A, and 50 mm nicotinamide). Cell debris was removed by centrifugation at 12,000 g at 4 °C for 10 min. Finally, the protein was precipitated with cold 15% trichloroacetic acid for 2 h at −20 °C. After centrifugation at 4 °C for 10 min, the supernatant was discarded. The remaining precipitate was washed with cold acetone three times. The protein was redissolved in buffer (8 m urea, 100 mm triethylammonium bicarbonate (TEAB), pH 8.0), and the protein concentration was determined with a 2-D Quant kit (GE Healthcare) according to the manufacturer's instructions.
For digestion, the protein solution was reduced with 10 mm DTT for 1 h at 37 °C and alkylated with 20 mm iodoacetamide (Sigma-Aldrich) for 45 min at room temperature in the dark. Then protein samples were diluted by adding 100 mm TEAB to urea at a concentration less than 2 m. Finally, trypsin (Promega, WI) was added at 1:50 trypsin-to-protein mass ratio for the first digestion overnight and 1:100 trypsin-to-protein mass ratio for a second 4-h digestion.
TMT Labeling and Affinity Enrichment
After trypsin digestion, peptides were desalted by a Strata X C18 SPE column (Phenomenex) and vacuum dried. Peptides were reconstituted in 0.5 m TEAB and processed according to the manufacturer's protocol with a 6-plex TMT kit (Thermo Fisher Scientific). Briefly, one unit of TMT reagent (defined as the amount of reagent required to label 1 mg of peptide) was thawed and reconstituted in 24 μl acetonitrile (Sigma-Aldrich). The peptide mixtures were then incubated for 2 h at room temperature, pooled, desalted, and dried by vacuum centrifugation.
To enrich KSuc peptides, tryptic peptides dissolved in NETN buffer (100 mm NaCl, 1 mm EDTA, 50 mm Tris-HCl, 0.5% NP-40, pH 8.0) were incubated with prewashed antibody beads (PTM Biolabs, China) at 4 °C overnight with gentle shaking. The beads were washed four times with NETN buffer and twice with ddH2O. The bound peptides were eluted from the beads with 0.1% TFA. The eluted fractions were combined and vacuum dried. The resulting peptides were cleaned with C18 ZipTips (Millipore, Germany) according to the manufacturer's instructions, followed by LC-MS/MS analysis.
LC-MS/MS Analysis
Peptides were dissolved in 0.1% formic acid (Sigma-Aldrich) then directly loaded onto a reversed-phase pre-column (Acclaim PepMap 100, C18, 75-μm inner diameter, 2-cm length, Thermo Scientific). Peptide separation was performed using a reversed-phase analytical column (Acclaim PepMap RSLC, C18, 50-μm inner diameter, 15-cm length, Thermo Scientific). The gradient was comprised of an increase from 6% to 22% solvent B (0.1% formic acid in 98% acetonitrile) for 26 min, 22% to 35% for 8 min, and climbing to 80% in 3 min then holding at 80% for the last 3 min, all at a constant flow rate of 300 nl/min on an EASY-nLC 1000 UPLC system (Thermo Fisher Scientific). The resulting peptides were analyzed by a Q ExactiveTM Plus hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific).
The peptides were subjected to a nano-spray-ionization source followed by tandem mass spectrometry (MS/MS) in Q ExactiveTM Plus (Thermo Fisher Scientific) coupled online to the UPLC. Intact peptides were detected in the Orbitrap at a resolution of 70,000. Peptides were selected for MS/MS using the normalized collision energy setting 31; ion fragments were detected in the Orbitrap at a resolution of 17,500. A data-dependent procedure that alternated between one MS scan followed by 20 MS/MS scans was applied for the top 20 precursor ions above a threshold ion count of 1E4 in the MS survey scan with 15.0 s dynamic exclusion. The electrospray voltage applied was 2.0 kV. Automatic gain control was used to prevent overfilling of the ion trap; 5E4 ions were accumulated to generate MS/MS spectra. For MS scans, the m/z scan range was 350 to 1800. Fixed first mass was set as 100 m/z.
Data Processing and Database Searches
For TMT quantification, the ratios of the TMT reporter ion intensities in MS/MS spectra from raw datasets were used to calculate fold changes among samples (TMT-126 was used for wild-type peptide labeling and TMT-128 was used for ΔScCobB2 peptide labeling in this case). For each sample, the quantification was medium-normalized at peptide level to center the distribution of quantitative values. Protein quantitation was then calculated as the median ratio of corresponding unique or razor peptides for a given protein. To assess the reliability of our quantification measurements between the biological/technical replicates, an average relative standard deviation analysis was performed to calculate the detection accuracy for all the quantified sites either in the wild-type or ΔScCobB2 samples (including three biological replicates and two technical replicates data). Similar to what was seen in previous TMT quantitation experiments (30), the relative standard deviation of the quantified sites in both wild-type and ΔScCobB2 cells are about 10% in this case (Fig. S2), suggesting a relatively high accuracy of our quantification measurements.
Tandem mass spectra were searched against the UniProt Streptomyces coelicolor database (UniProt Release 2017_01) concatenated with a reverse decoy database. Trypsin/P was specified as the cleavage enzyme allowing up to four missing cleavages, five modifications per peptide, and five charges. Mass error was set to 20 ppm for the first search, 5 ppm for the main search, and 0.02 Da for fragment ions. Carbamidomethylation on Cys was specified as a fixed modification, and oxidation on Met, succinylation on Lys, and acetylation on protein N termini were specified as variable modifications. Minimum peptide length was set at 7. All other parameters in MaxQuant were set to default values. The score cutoff of KSuc peptide was set as > 40. The site localization probability was set as >0.75.
Bioinformatics Analysis
Sequence Alignment and Phylogenetic Analysis
Homology searches were performed with the BLAST program from the National Center for Biotechnology Information. Multiple sequence alignments were conducted with the ClustalX program. Phylogenetic trees were constructed using the bootstrap method with 1000 replications to construct neighbor-joining trees by the MEGA 4.0 program.
Functional Enrichment Analysis
The GO annotation proteome was derived from the UniProt-GOA database (http://www.ebi.ac.uk/GOA/). The database of Kyoto Encyclopedia of Genes and Genomes (KEGG) was used to annotate the pathways. We annotated the proteins using the KEGG online service tool KAAS and then mapped the annotation result on the KEGG pathway database using KEGG Mapper. To identify the enriched GO/KEGG pathways, the Functional Annotation Tool of DAVID Bioinformatics Resources (31) and InterPro database (32) were used by against the background of the S. coelicolor proteome. A two-tailed Fisher's exact test was employed to test the protein enrichments. The GO/KEGG pathway/domains with a corrected p value <0.01 were considered significant.
Motif Analysis for Lysine Succinylation Substrates
Programs Motif-X (33) and IceLogo (34) were used to analyze the model of identified succinylation sequences of succinyl-21-mers (10 amino acids upstream and downstream of the succinylation site). The S. coelicolor proteome sequences were used as the background database parameter, and other parameters were set to default values. In addition, an in-house R-package script (35) was used to generate the position-specific heat map by plotting the log10 of the ratio of frequencies.
Protein-Protein Interaction Network
All identified succinylated protein were searched against the STRING database version 9.1 (36) for protein-protein interactions. Only interactions between proteins belonging to the searched dataset were selected, thereby excluding external candidates. The interaction network from STRING was visualized in Cytoscape (37). A graph of the clustering algorithm, molecular complex detection (MCODE) (38), was utilized to analyze densely connected regions. MCODE is part of the plug-in toolkit of the network analysis and was visualized by Cytoscape software as well.
RESULTS
SCO6464 Is an NAD+-Dependent Desuccinylase in S. coelicolor
Sirtuin enzymes are connected intimately with DNA repair, cell metabolism, longevity, and bacterial pathogenesis (39–41). Mammals employ seven sirtuins to coordinate different PTMs, e.g. deacetylase Sirt1–3 (42), desuccinylase Sirt5 (21), and defatty-acylase Sirt6 (43). Much fewer sirtuins have been found in prokaryotes. By homologous BLAST using the amino acids of human desuccinylase Sirt5 as the query sequence, we identified two sirtuin homologs named SCO0452 and SCO6464 that are encoded in S. coelicolor (Fig. 1B). Both of the two predicted sirtuins share high identities to E. coli CobB. We therefore designated SCO0452 and SCO6464 as ScCobB1 and ScCobB2, respectively (Fig. 1B). In contrast to ScCobB1, ScCobB2 showed higher similarity to the desuccinylase Sirt5 than to the deacetylases Sirt1–3 by phylogenetic analysis (Fig. 1C), implying the two sirtuins have evolved divergently and may have specific functions in S. coelicolor.
ScCobB1 has been reported as a deacetylase (27), while no study has been conducted for ScCobB2 so far. To evaluate the biochemical activity of ScCobB2, the protein encoded by SCO6464 was heterogeneously purified from E. coli BL21 (DE3) and incubated with E. coli acetyl-CoA synthetase (Acs), a conserved central metabolic enzyme known to be modified by both succinylation and acetylation (14, 25). The succinylation level of Acs was measured by Western blotting using anti-succinyl lysine antibody (see “Materials” in “Experimental Procedures”). As shown in Fig. 2A, treatment by ScCobB2 and the cofactor NAD+ caused about a 50% decrease in succinylation in 2 h, while the ScCobB1 and NAD+ treatment showed no obvious desuccinylation effect on Acs. As a positive control, the E. coli CobB treatment reduced half of Acs succinylation as in ScCobB2 treatment. The desuccinylation of Acs by both ScCobB2 and CobB are dependent of NAD+ (Fig. 2A). These observations indicate ScCobB2 has desuccinylase activity that differs from the known deacetylase ScCobB1 (27).
Fig. 2.
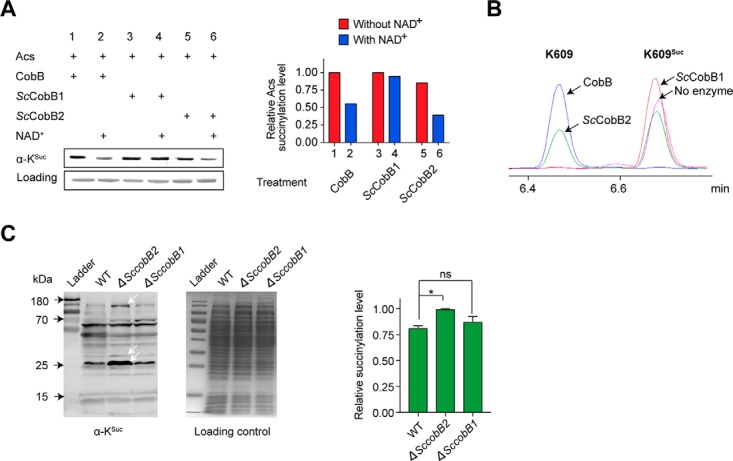
Characterization of ScCobB2 as a lysine desuccinylase both in vitro and in vivo. (A) ScCobB2 showed comparable desuccinylase activity to CobB in vitro. Using E. coli acetyl-CoA synthetase (Acs) as the substrate, Western blotting was performed to compare desuccinylase activities between ScCobB1, ScCobB2, and CobB. The Acs succinylation level was assessed using a pan-anti-succinyllysine antibody, and protein stained with Coomassie blue was set as the loading control. The succinylation level was further quantified by ImageJ and normalized against the protein loading as shown on the right panel. The value of the first sample was set as 100%. (B) The desuccinylase activity of ScCobB2 was confirmed by HPLC. The Acs peptide with succinylated Lys-609 residue was synthesized and incubated with ScCobB1, ScCobB2, or CobB at 37 °C for 1 h. Reactions were terminated by adding 1% TFA, after which the samples were analyzed by HPLC. Arrows indicate the retention time of the succinylated and nonsuccinylated peptides from different treatments. (C) The succinylation level of the whole cellular proteins increased in ΔSccobB2 in S. coelicolor. The succinylation levels of the protein lysates were assessed by Western blotting and quantified by ImageJ (on the right panel). The normalized value of ΔSccobB2 was set as 100%. The white arrows indicate the increased succinylation of ΔSccobB2 proteins compared with that of wild-type and ΔSccobB1 proteins. One representative Western-blot result of two replicates was shown.
Peptide containing the conserved K609 from Acs (25, 44) modified by succinylation was further synthesized and incubated with ScCobB1, ScCobB2, and CobB, respectively. As shown by HPLC, ScCobB2 desuccinylated about 40% of the K609Suc peptide to a non-succinylation state within 1 h, confirming that ScCobB2 functions as a desuccinylase (Fig. 2B). Consistent with the Western-blot results (Fig. 2A), robust desuccinylase activity was observed for CobB, but no desuccinylase activity was detected for ScCobB1 (Fig. 2B). We also examined whether ScCobB2 possesses deacetylase activity like the bifunctional enzyme CobB. However, both Western blotting (Fig. S3A, B) and HPLC (Fig. S3C) showed that ScCobB2 was unable to deacetylate either Acs protein or K609Ac peptide, implying that it may not be a bifunctional enzyme like CobB.
Measurement of the Lysine Succinylation Spectrum in S. coelicolor
To test if ScCobB2 has desuccinylase activity in vivo, in-frame deletion of the SccobB1 or SccobB2 gene was performed in S. coelicolor. All cells including the wild-type S. coelicolor were cultured in tryptic soy broth medium supplemented with 50 mm glucose and were harvested at the mid-log phase. Whole cellular proteins were collected and their succinylation levels were compared among ΔSccobB1, ΔSccobB2, and wild-type S. coelicolor strains. As displayed in Fig. 2C, the succinylation levels of proteins from ΔSccobB2 increased about 20%, while no significant change was detected for the proteins from ΔSccobB1, indicating that ScCobB2, rather than ScCobB1, serves as a desuccinylase in vivo.
To acquire a detailed view of succinylation regulation in S. coelicolor, quantitative succinylome analyses were conducted by comparing the succinylation levels of ΔSccobB2 to those of the wild-type strain. Both bacteria were cultured and harvested using the same conditions as in Fig. 2C. The collected cellular proteins were extracted, digested, and labeled by different TMTs. Then, all succinylated peptides from ΔSccobB2 and wild-type cells were enriched by affinity purification using pan succinyl-lysine antibody and applied to LC-MS/MS analysis (see “Materials” in “Experimental Procedures”).
By searching against the Uniprot database using the S. coelicolor proteome (organism ID, 100226), a total of 673 lysine succinylation sites in 427 proteins were identified (Table I). Comparing the ΔSccobB2 with the wild-type S. coelicolor succinylome, 470 lysine succinylation sites in 317 proteins were quantified (Fig. 3A). Among these, 144 succinylated sites in 114 proteins were up-regulated (high succinylation) while only 8 protein sites were down-regulated (low succinylation) significantly in ΔSccobB2 cells, compared with the wild-type S. coelicolor (Fig. 3B, Table I). The results confirmed ScCobB2 plays a broad regulatory role by succinylation in S. coelicolor. Detailed information for all the succinylated peptides and their matching proteins is provided in Table S1.
Table I. Summary of the quantitative succinylome in S. coelicolor.
| Identified | Quantified | ΔSccobB2/WT |
||
|---|---|---|---|---|
| Up-regulated* | Down-regulated* | |||
| Sites | 673 | 470 | 144 | 8 |
| Proteins | 427 | 317 | 114 | 8 |
WT, wild-type.
*The number was calculated statistically as significantly up-regulated or down-regulated. p value < 0.05.
Fig. 3.

Quantitative succinylome analyses between S. coelicolor wild-type and ΔSccobB2. (A) Scatter plot shows the changes of succinylated peptide intensities (i.e. the summed precursor-ion intensities of each quantifiable peptide) between S. coelicolor wild-type and ΔSccobB2 cells. (B) Histogram shows the distribution of succinylation ratios of the quantifiable sites between the wild-type and ΔSccobB2 cells. The y axis represents the number of lysine-succinylated peptides in each category. GO annotations for the up-regulated proteins in ΔSccobB2 succinylome based on biological process (C), molecular function (D), and cellular component (E) analyses.
We also checked the quality of MS data in this study. The distribution of mass errors for all the identified peptides was inspected first. As presented in Fig. S4A, the average of mass errors is nearly zero, and more than 99% of their distributions are less than 4 ppm, showing a high accuracy of our data. In addition, we calculated the distribution of peptides after trypsin digestion and found that the highest abundant group is the peptides with the lengths of eight amino acids (Fig. S4B). This result is consistent with the known property of tryptic peptides. For all the succinylated proteins, most of them were modified at only one or two sites (93%), but a few of them (7%) were modified at more than three sites (Fig. S4C), which is a typical distribution of succinylation proteins as reported before (24). All these results suggest the MS data meet the requirements for the succinylome analysis.
Quantitative Succinylome Comparison between ΔSccobB2 and the Wild-Type S. coelicolor
To better understand the quantitative succinylome, functional annotation analysis was performed for the 114 hypersuccinylated proteins in ΔSccobB2 using GO. The GO functional classification is based on three categories, i.e. biological processes, molecular function, and cellular components. Our GO functional classification demonstrated that these hypersuccinylated proteins are involved in a variety of biological processes and have diverse localizations in S. coelicolor (Fig. 3, Table S2). Based on biological process classification, most up-regulated proteins were involved in cellular processes (34.7%) and cell metabolism (34.1%) (Fig. 3A). Based on molecular function classification (Fig. 3B), the up-regulated proteins usually had DNA-binding (42.8%) or catalytic activities (41.4%). In addition, the largest number of the up-regulated proteins was remarkably enriched in the cell cytoplasm (48%) compared with other cellular components (Fig. 3C).
To determine which functional categories are preferred targeted by ScCobB2, we mapped these hypersuccinylated proteins onto KEGG pathways and evaluated their enrichment in these pathways. The results (Fig. 4A, Table S3) showed that multiple pathways featuring protein biosynthesis and carbon metabolism showed significant enrichment of succinylated proteins.
Fig. 4.

KEGG pathway analyses of the quantitative succinylome highlights the regulation of ScCobB2 in protein biosynthesis and carbon metabolism. The up-regulated succinylation proteins were enriched in protein biosynthesis and carbon metabolism pathways by KEGG pathway analyses (A). The hypersuccinylated proteins enriched in ribosome (B), pyruvate metabolism (C), TCA cycle (D), and glycolysis/gluconeogenesis (E) were mapped onto KEGG pathways and shown as in middle panels. The succinylated proteins identified in this study are highlighted in brown while the up-regulated succinylation proteins are highlighted in magenta. Typical MS/MS spectra of KSuc peptides from the enzymes in protein biosynthesis and carbon metabolism pathways were shown as in (F). Glk: glucokinase; Pgi: glucose-6-phosphate isomerase; PfkA: 6-phosphofructokinase; Fba: fructose-bisphosphate aldolase; GapA: glyceraldehyde-3-phosphate dehydrogenase; Pgk: phosphoglycerate kinase; GpmA: phosphoglycerate mutase; Eno: phosphopyruvate hydratase; Pyk: pyruvate kinase; AceE: pyruvate dehydrogenase E1 component; AceF: pyruvate dehydrogenase E2 component (dihydrolipoamide acetyltransferase); Lpd: dihydrolipoamide dehydrogenase; Adh: alcohol dehydrogenase; Aldh: aldehyde dehydrogenase; AcsA: acetyl-CoA synthetase; CS: citrate synthase; ACO: aconitate hydratase; IDH: isocitrate dehydrogenase; SucA: 2-oxoglutarate dehydrogenase E1 component; SucB: 2-oxoglutarate dehydrogenase E2 component; SucC: succinyl-CoA synthetase subunit beta; SucD: succinyl-CoA synthetase subunit alpha; FH: fumarate hydratase; MDH: malate dehydrogenase; Tuf1: elongation factor Tu-1.
Specifically, 32 out of the 56 ribosomal proteins (57%) were identified as succinylated (Fig. 4B). Among them, the succinylation level of 20 ribosomal proteins was significantly up-regulated in ΔSccobB2 succinylome, accounting for up to 63% of the total succinylated ribosomal proteins (Fig. 4B). RpoA, ET-Tu, and RpoC are the top three ribosomal complexes where most of their subunits were succinylation increased in the ΔSccobB2 cell. Hence, these results suggest the ribosome pathway is heavily regulated by ScCobB2. Moreover, the succinylation levels of 85.7% and 44.5% of enzymes in two representative metabolic pathways, i.e. pyruvate metabolism (Fig. 4C) and TCA cycle (Fig. 4D), respectively, were remarkably up-regulated with deletion of SccobB2. The pyruvate dehydrogenase complex is responsible for the conversion from pyruvate to acetyl-CoA and is composed of pyruvate dehydrogenase E1, pyruvate dehydrogenase E2, and dihydrolipoamide dehydrogenase (Lpd). In this study, the succinylation levels of these three components were increased by 1.6-, 1.6-, and 1.4-fold, respectively, in ΔSccobB2 (Fig. 4C). Similar to observations in E. coli, nearly all enzymes in the TCA cycle in the S. coelicolor succinylome were succinylated, and most of these succinylation sites were found to overlap between S. coelicolor and E. coli. However, we found that the succinylation levels of about half of TCA cycle enzymes, including α-ketoglutarate dehydrogenase components, succinyl-CoA synthetase complexes, and aconitate hydratase (ACO), were elevated in ΔSccobB2 (Fig. 4D). These results suggest both pyruvate metabolism and the TCA cycle were dominantly regulated by succinylation in S. coelicolor. Besides, it is worth emphasizing that glyceraldehyde-3-phosphate dehydrogenase (GapA), which is a rate-limit enzyme in the glycolysis pathway, was extensively succinylated at eight lysine residues, and two of them (K227, K255) were demonstrated to be regulated by ScCobB2 (Fig. 4E). It has been reported that K227 is on the conserved motif near the substrate binding pocket in GapA (45), implying that ScCobB2 may also contribute to the regulation of GapA enzyme activity in S. coelicolor. Some typical MS/MS spectra of the succinylated peptides from these enzymes are shown in Fig. 4F.
Taken together, these results emphasize that the enzymes related to protein biosynthesis and carbon metabolism in S. coelicolor were extensively modified by succinylation and highly regulated by the desuccinylase ScCobB2.
Comparison of the S. coelicolor Succinylome with Other Reported Succinylomes
Several succinylomes, including four bacterial succinylomes, E. coli (14), M. tuberculosis (24), Vibrio parahemolyticus (46), and Bacillus subtilis (23), have been released in recent years. To gain insights into the overall succinylation in S. coelicolor, we compared the S. coelicolor succinylome (containing both wild-type and ΔSccobB2 succinylomes) with the above-mentioned succinylomes (Fig. 5A). Orthologs were found in other bacterial succinylomes for most of the S. coelicolor succinylated proteins. The highest succinylation orthologous ratio was observed in S. coelicolor (66%) when comparing its succinylome with that of M. tuberculosis, which may be because M. tuberculosis is the closest relative to S. coelicolor among these bacteria. However, taking the average orthologous ratio (33%) between the S. coelicolor and M. tuberculosis genomes into consideration, the succinylation was extraordinarily enriched in the orthologs of these two organisms (Fig. 5A). The enrichment of succinylome orthologs was also observed between S. coelicolor and E. coli, and S. coelicolor and V. parahemolyticus, indicating that similar biological processes might be regulated in different species by succinylation. No orthologous enrichment was found between S. coelicolor and B. subtilis succinylomes, maybe because B. subtilis only has one-third as many succinylated proteins identified compared with other bacterial succinylomes (Fig. 5A). Strikingly, there still were 22 succinylated orthologs after comparing all the bacterial succinylomes as well as other two eukaryotic succinylomes, i.e. human (17) and rice (47) (Fig. 5B). We listed all these proteins in Fig. 5C, and most of them (16 out of 22, 72.7%) are involved in metabolic pathways, suggesting conserved succinylation regulation of cell metabolism from prokaryotes to eukaryotes. The other six orthologs include three molecular chaperones, two elongation factors, and one ribosome recycling factor, and they are all involved in protein biosynthesis, indicating that succinylation may also play a pivotal role in protein processing (17).
Fig. 5.
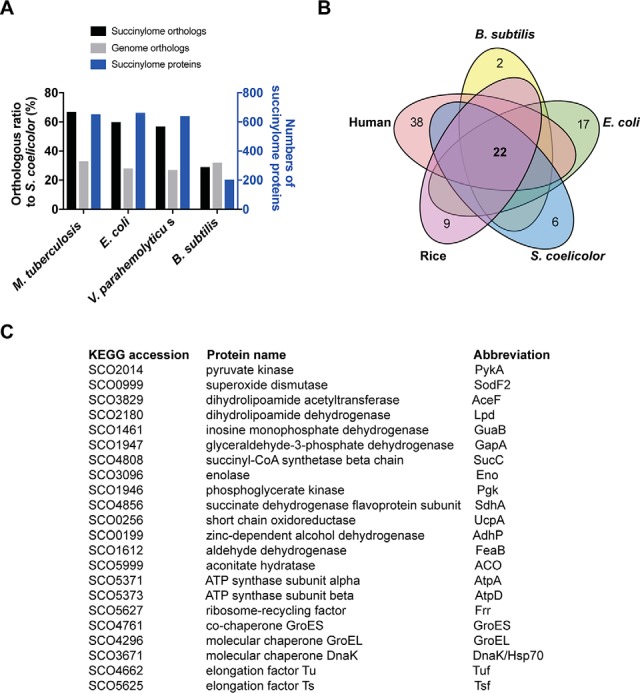
Comparative analysis of the S. coelicolor succinylome with other bacterial succinylomes. (A) Comparison of S. coelicolor succinylome or genome with that of M. tuberculosis, E. coli, V. parahemolyticus, and B. subtilis. The succinylome or genome orthologs were determined by BLASTP analysis under the conditions of a minimum of 30% identity and 70% length coverage. The corresponding orthologous ratios were calculated and shown on the left y axis, and the numbers of succinylome proteins identified in each proteomics analysis are shown on the right y axis. (B) Venn diagram outlining common succinylated proteins from S. coelicolor, B. subtilis, E. coli, rice, and humans. (C) 22 orthologs are listed and described in S. coelicolor after compared with other four succinylomes.
Similar to the observation for the up-regulated succinylome (Figs. 4A-4C), GO classification for the total succinylated proteins showed that most of them are localized in the cell plasma, are associated with protein translation and cell metabolism, and have catalytic activities (Fig. S5, Table S4). The GO enrichment analyses for the overall succinylome revealed that the top two succinylated biological processes are the protein translation and cellular metabolism (Fig. S6A). Functional enrichment analyses using the KEGG database for the overall succinylated proteins were performed as well. Similar to the above observation (Figs. 4D-4G, Table S3), the total succinylated proteins were also found to be enriched in protein biosynthesis in addition to diverse metabolic pathways (Fig. S6B, Table S5). The top three enriched categories of KEGG pathways were ribosome, aminoacyl-tRNA biosynthesis, and the TCA cycle. Therefore, all these findings implied the enriched succinylated proteins (or pathways, protein biosynthesis carbon metabolism, particularly) could be regulated by the desuccinylase ScCobB2.
Sequence Recognition Motifs of Succinylation in S. coelicolor
Datasets comprising succinylated peptides from the S. coelicolor succinylome may contribute to the determination of preferences for specific amino acid residues flanking the succinylated lysine. Here, the Motif-X program was used to search for overrepresented sequence patterns among all identified succinylated peptides. The preferred conserved consensus motifs with amino acid sequences from -10 to + 10 surrounding the succinylated lysine were extracted and analyzed (Table S6). As shown in Fig. 6A, arginine (R) and lysine (K) were frequently found upstream of KSuc, while aspartic acid (D) and proline (P) were frequently found downstream of KSuc. The most representative five motifs are **KSucP**, **RKSuc**, **KSucD**, **KKSuc**, and **R*KSuc** (* indicates a random amino acid residue). For the first four motifs, the amino acids R, K, D, and P are adjacent to the KSuc, whereas amino acid R in the last motif is at the -2 position relative to KSuc. The other two programs, i.e. IceLogo (Fig. 6B) and R program (Fig. 6C), were used to verify this observation and similar results were obtained. The analysis using IceLogo software (34) not only displayed the same overrepresented flanking sequences as in Motif-X program but also showed that alanine was the highest-frequency residue in the underrepresented flanking sequences around the KSuc (Fig. 6B). Alanine tends to be absent at the -1 and +1 positions around KSuc, suggesting it is an unpopular residue in the substrate sequences of succinylation near KSuc. In addition, a heat map generated from the R program (Fig. 6C) confirmed both overrepresented and underrepresented flanking sequences of the succinylated peptides have high confidence in this study.
Fig. 6.

Sequence properties of the succinylated peptides in the S. coelicolor succinylome. (A) Sequence Logo representation of significant motifs identified by Motif-X software. The motifs consisting of 20 residues (10 amino acids upstream and downstream of the site) surrounding the succinylated lysine are shown. (B) The position-specific under- or overrepresentation of amino acids flanking the succinylated sites was analyzed. The heat map generated from R-package script shows enrichment (red) and depletion (green) of amino acids in the specific positions. (C) IceLogo software was used to analyze the overrepresented and underrepresented flanking sequences around KSuc.
Among the succinylation motifs identified here, motif **KSucD** is the preferred substrate reported in E. coli (25), and the motifs **KSucP**, **RKSuc**, **KSucD**, and **KKSuc** have also been found as representative in mice (18). These results support the notion that the preferred sequences for lysine succinylation are conserved among different organisms, implying a similar kinetics or mechanism of succinylation.
Functional Interaction Networks of Succinylated Substrates in S. coelicolor
To determine the connections among the succinylated proteins in S. coelicolor, an interaction network was generated and visualized using Cytoscape software based on the STRING database (48). The results suggested intimate connections between succinylated proteins, and a network was constructed that consisted of more than 400 proteins, including 271 identified succinylated proteins (Table S7). Functional category analysis for these proteins retrieved several highly interconnected clusters such as ribosome biogenesis, glycolysis/gluconeogenesis, aminoacyl-tRNA biosynthesis, and oxidative phosphorylation (Fig. 7A). Consistent with our previous observation (Fig. 4 and Fig. S6), the most prominent enrichment of succinylation was in protein biosynthesis and carbon metabolism pathways. The top two identified clusters were ribosome-associated proteins (Fig. 7B) and glycolysis/gluconeogenesis enzymes (Fig. 7C). The strong physiological interactions among these succinylated protein complexes or pathway components led us to speculate that succinylation may contribute to S. coelicolor adaptation to the variable soil environments (49, 50).
Fig. 7.

The protein-protein interaction network of succinylated proteins in S. coelicolor. Interaction networks of succinylated proteins (listed in gene names) were analyzed using the MCODE plug-in toolkit in the Cytoscape software. (A) Interaction network of all succinylated proteins. The top four clusters of highly interconnected succinylated protein networks were ribosome-associated proteins, glycolysis/gluconeogenesis, aminoacyl-tRNA biosynthesis, and oxidative phosphorylation. Detailed cluster information for ribosome-associated proteins (B) and glycolysis/gluconeogenesis (C) are shown. The respective MCODE score and protein-protein interaction score are listed in Table S7.
Western Blotting Confirms ScCobB2 Regulation in Protein Biosynthesis and Cellular Metabolism
In the up-regulated succinylome of S. coelicolor (144 succinylated sites in total), 39 enzymes (45 succinylated sites) serve functions in cell metabolism and 33 proteins (42 succinylated sites) serve functions in protein translation (Table S8), accounting for 34% and 29% of the total hypersuccinylated proteins, respectively. To verify the hypothesis that succinylation primarily regulates cellular metabolism and protein biosynthesis in S. coelicolor (Fig. 8), we randomly selected 13 proteins from these 72 hypersuccinylated proteins for Western blotting test using anti-succinyllysine antibody (Table S8). After expression and purification of the 13 proteins from the E. coli ΔcobB strain, in vitro desuccinylation assays were conducted by treatment with desuccinylase ScCobB2 and cofactor NAD+. In short, 11 out of 13 samples were demonstrated to be regulated significantly by ScCobB2 after a 2-h treatment, suggesting high reliability of our comparative succinylome data (Fig. 8). Western blots showed that three ribosomal proteins, including L13 (SCO4734), L2 (SCO4705), and L9 (SCO3909) subunits, were all desuccinylated by more than 50% by ScCobB2. Enzymes involved in glycolysis (GapA), pyruvate metabolism (pyruvate dehydrogenase E1), the TCA cycle (ACO and SucC), or energy metabolism (IMP dehydrogenase) were verified as the substrates of ScCobB2 as well. Strikingly, the succinylation level of ACO was decreased to nearly zero when treated with ScCobB2 and NAD+, suggesting a very strong regulation of the TCA cycle by succinylation (Fig. 8). In summary, these results strongly support the hypothesis that ScCobB2 is a comprehensive regulatory enzyme in S. coelicolor succinylome, leaving a lot of succinylated candidates involved in multiple pathways to be investigated, either physiologically or biochemically.
Fig. 8.
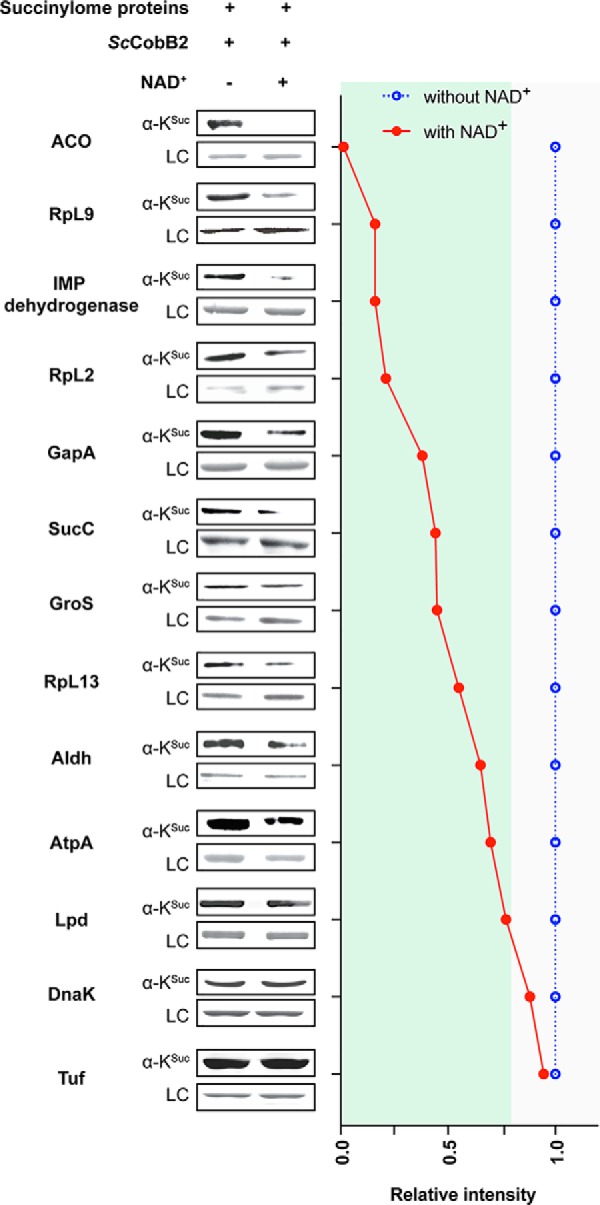
Western blotting assays confirm the high reliability of the quantitative succinylome data. Substrates including metabolic proteins, molecular chaperones, and ribosomal proteins were subjected to Western blotting with the pan-anti-succinyllysine antibody. Their response to ScCobB2 treatments were quantified and normalized against corresponding protein loading controls. We assigned the succinylation levels for each substrate without NAD+ treatments as 1.0. The relative succinylation levels for substrates by ScCobB2 and NAD+ treatments were calculated and plotted via comparing the signals from those without NAD+ treatment, respectively. Here, the relative succinylation level less than 0.8 was considered as can be desuccinylated by ScCobB2 and NAD+. ACO: aconitate hydratase; RpL9: ribosomal protein L9; GuaB: inosine monophosphate dehydrogenase; RpL2: ribosomal protein L2; GapA: glyceraldehyde-3-phosphate dehydrogenase; SucC: succinyl-CoA synthetase; GroS: 10 kDa chaperonin GroS; RpL13: ribosomal protein L13; Aldh: aldehyde dehydrogenase; AtpA: ATP synthase subunit alpha; Lpd: dihydrolipoamide dehydrogenase; DnaK: chaperone DnaK; Tuf: elongation factor Tu. For more information, please refer to Table S8.
DISCUSSION
The importance of lysine succinylation is being increasingly recognized, and thousands of succinylated proteins have been identified in both prokaryotes and eukaryotes, including E. coli (14), M. tuberculosis (24), V. parahemolyticus (46), B. subtilis (23), Saccharomyces cerevisiae (13, 51), Drosophila melanogaster (13), and human cells (13). Quantitative succinylome analysis by comparing Sirt5−/− with wild-type cells has revealed global regulation of Sirt5 in multiple metabolic pathways, including â-oxidation, ketogenesis, and cellular respiration (19, 20). However, due to the lack of characterization of specific desuccinylases, very few succinylomes have been quantified in bacteria. The sole example of comparative succinylation was documented in E. coli (14) and revealed that its succinylation levels change in response to different culture conditions. Here, we identified a specific desuccinylase ScCobB2 in the model soil bacteria S. coelicolor and performed quantitative succinylation analysis between ΔSccobB2 and wild-type cells. A total of 673 unique succinylated sites were identified, among which 470 succinylation sites in 317 proteins were quantified. The succinylation levels were up-regulated significantly in ΔSccobB2 in 114 proteins, demonstrating a critical regulatory role for succinylation in carbon metabolism and protein biosynthesis in S. coelicolor.
Although increasing numbers of succinylated proteins are being identified in different organisms, evidence suggests that the majority are not regulated by succinylases. Two independent studies have shown that only 32% and 12% of the total quantified succinylation sites, respectively, are regulated by Sirt5 desuccinylase in mitochondrial cells (19, 20). In this case in S. coelicolor, a total of 114 succinylation sites were shown to be up-regulated by ScCobB2, accounting for just one-quarter of the overall quantified sites. These data indicate there were no direct correlation between the regulation of succinylation and the modification of proteins by succinylation. Therefore, the identification of succinylated proteins that are also regulated by specific desuccinylases is important in succinylation research because only the regulatory modification is physiologically meaningful. Quantitative analysis revealed that succinylation regulates multiple pathways in S. coelicolor. The highlight of protein biosynthesis and carbon metabolism pathways by our analysis implied a close relationship between succinylation regulation and adaption to the soil environment for S. coelicolor. On the other hand, quantitative analysis is also helpful in succinylation research to hone in on the regulatory targets from thousands of succinylated proteins. In this case, we randomly selected 13 proteins from the quantitative succinylome and obtained 11 hits (85%) by Western blotting verification, suggesting high enrichment of the targets regulated by succinylation.
There are seven sirtuins (Sirt1 to Sirt7) with varied behaviors and different subcellular localizations in human cells (52). Sirt5 in eukaryotic cells is located in the mitochondrial matrix and has specific desuccinylation activity (19, 20). In contrast, only one sirtuin deacylase has been found in Enterobacter, and it shows both deacetylation and desuccinylation activities (14, 44). In Streptomyces, little is known regarding the sirtuin proteins and their activities. By using bioinformatics analysis, we identified two sirtuins, ScCobB1 and ScCobB2, in S. coelicolor. Furthermore, we demonstrated that ScCobB2 is a functional desuccinylase, which differs from the known deacetylase ScCobB1 (27). These results revealed for the first time a functional divergence of sirtuin enzymes in bacteria. We therefore collected all of the sirtuin proteins in representative bacteria and built a phylogenetic tree. Phylogenetic analysis (Fig. S1) showed that the divergent sirtuins exist not only in Streptomyces but also in other Actinobacteria, such as Mycobacterium, Corynebacterium, and Amycolatopsis. The results implied possible divergent evolution of sirtuin proteins in these subclades of bacteria, mimicking the highly evolved sirtuins in human cells. On the other hand, for the representative of Gammaproteobacteria, Alphaproteobacteria, and Firmicutes, there is only one copy of sirtuin in Shigella, Caulobacter, and Bacillus (Fig. S1), suggesting a relatively slow evolution of sirtuins as in E. coli in these microorganisms.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (53) partner repository with the dataset identifier PXD014202. Alternatively, the dataset can be accessed in MS-Viewer (54) with the search key 8gq2qf4c2e using the following URL: http://msviewer.ucsf.edu/prospector/cgi-bin/mssearch.cgi?report_title=MS-Viewer&search_key=8gq2qf4c2e&search_name=msviewer.
Supplementary Material
Acknowledgments
We thank Professor Zhong-Jun Qin at the Institute of Plant Physiology and Ecology for generously providing the cosmids for all the S. coelicolor cloning and PCR targeting. We appreciate Dr. Rui-Fang Cao and Dr. Guo-Qing Zhang at Bio-Med Big Data Center in SIBS for offering help in data processing. We also appreciate Professor Shi-Min Zhao at Fudan University for the gift of Pan-anti-succinylation antibody. We would like to thank LetPub (www.letpub.com) for providing linguistic assistance during the preparation of this manuscript.
Footnotes
* This work was supported by the National Natural Science Foundation of China (31400039 to WZ, 31430004 and 31421061 to GPZ).
 This article contains supplemental material Tables S1–S10 and Figs. S1–S6.
This article contains supplemental material Tables S1–S10 and Figs. S1–S6.
1 The abbreviations used are:
- PTM
- protein posttranslational modification
- IDH
- isocitrate dehydrogenase 2
- HPLC
- high-performance liquid chromatography
- TMT
- tandem mass tag
- GO
- Gene Ontology
- TEAB
- triethylammonium bicarbonate
- MS/MS
- tandem mass spectrometry
- KEGG
- Kyoto Encyclopedia of Genes and Genomes
- ACO
- aconitate hydratase.
REFERENCES
- 1. Hart G. W. and Ball L. E. (2013) Post-translational modifications: a major focus for the future of proteomics. Mol. Cell. Proteomics 12, 3443–3443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mattiroli F. and Sixma T. K. (2014) Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat. Struct. Mol. Biol. 21, 308–316 [DOI] [PubMed] [Google Scholar]
- 3. Kim G. W. and Yang X. J. (2011) Comprehensive lysine acetylomes emerging from bacteria to humans. Trends Biochem. Sci. 36, 211–220 [DOI] [PubMed] [Google Scholar]
- 4. Zhang J., Sprung R., Pei J., Tan X., Kim S., Zhu H., Liu C. F., Grishin N. V., and Zhao Y. (2009) Lysine acetylation is a highly abundant and evolutionarily conserved modification in Escherichia coli. Mol. Cell. Proteomics 8, 215–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hilton I. B., D'Ippolito A. M., Vockley C. M., Thakore P. I., Crawford G. E., Reddy T. E., and Gersbach C. A. (2015) Epigenome editing by a CRISPR/Cas9-based acetyltransferase activates genes from promoters and enhancers. Nature Biotechnology 33, 510–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen Y., Sprung R., Tang Y., Ball H., Sangras B., Kim S. C., Falck J. R., Peng J., Gu W., and Zhao Y. (2007) Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics 6, 812–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang K., Chen Y., Zhang Z., and Zhao Y. (2009) Identification and verification of lysine propionylation and butyrylation in yeast core histones using PTMap software. J. Proteome Res. 8, 900–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., and Yang K. (2011) The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics 10, M111.012658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tan M., Luo H., Lee S., Jin F., Yang J. S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N., Lu Z., Ye Z., Zhu Q., Wysocka J., Ye Y., Khochbin S., Ren B., and Zhao Y. (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hirschey M. D. and Zhao Y. (2015) Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol. Cell. Proteomics 14, 2308–2315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Azevedo C. and Saiardi A. (2016) Why always lysine? The ongoing tale of one of the most modified amino acids. Adv. Biol. Regul. 60, 144–150 [DOI] [PubMed] [Google Scholar]
- 12. Baslé E., Joubert N., and Pucheault M. (2010) Protein chemical modification on endogenous amino acids. Chem. Biol. 17, 213–227 [DOI] [PubMed] [Google Scholar]
- 13. Zhang Z., Tan M., Xie Z., Dai L., Chen Y., and Zhao Y. (2011) Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 7, 58–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Colak G., Xie Z., Zhu A. Y., Dai L., Lu Z., Zhang Y., Wan X., Chen Y., Cha Y. H., Lin H., Zhao Y., and Tan M. (2013) Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol. Cell. Proteomics 12, 3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wagner G. R. and Payne R. M. (2013) Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem. 288, 29036–29045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang Y., Guo Y. R., Liu K., Yin Z., Liu R., Xia Y., Tan L., Yang P., Lee J. H., Li X. J., Hawke D., Zheng Y., Qian X., Lyu J., He J., Xing D., Tao Y. J., and Lu Z. (2017) KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature 552, 273–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Weinert B. T., Schölz C., Wagner S. A., Iesmantavicius V., Su D., Daniel J. A., and Choudhary C. (2013) Lysine succinylation is a frequently occurring modification in prokaryotes and eukaryotes and extensively overlaps with acetylation. Cell Reports 4, 842–851 [DOI] [PubMed] [Google Scholar]
- 18. Cheng Y., Hou T., Ping J., Chen G., and Chen J. (2016) Quantitative succinylome analysis in the liver of non-alcoholic fatty liver disease rat model. Proteome Sci. 14, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park J., Chen Y., Tishkoff D. X., Peng C., Tan M., Dai L., Xie Z., Zhang Y., Zwaans B. M., Skinner M. E., Lombard D. B., and Zhao Y. (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Mol Cell 50, 919–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rardin M. J., He W., Nishida Y., Newman J. C., Carrico C., Danielson S. R., Guo A., Gut P., Sahu A. K., Li B., Uppala R., Fitch M., Riiff T., Zhu L., Zhou J., Mulhern D., Stevens R. D., Ilkayeva O. R., Newgard C. B., Jacobson M. P., Hellerstein M., Goetzman E. S., Gibson B. W., and Verdin E. (2013) SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metabolism 18, 920–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., Hao Q., and Lin H. (2011) Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou L., Wang F., Sun R., Chen X., Zhang M., Xu Q., Wang Y., Wang S., Xiong Y., and Guan K. L. (2016) SIRT5 promotes IDH2 desuccinylation and G6PD deglutarylation to enhance cellular antioxidant defense. EMBO Rep. 17, 811–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kosono S., Tamura M., Suzuki S., Kawamura Y., Yoshida A., Nishiyama M., and Yoshida M. (2015) Changes in the acetylome and succinylome of Bacillus subtilis in response to carbon source. PloS One 10, e0131169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie L., Liu W., Li Q., Chen S., Xu M., Huang Q., Zeng J., Zhou M., and Xie J. (2015) First succinyl-proteome profiling of extensively drug-resistant Mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. J. Proteome Res. 14, 107–119 [DOI] [PubMed] [Google Scholar]
- 25. Yang M., Wang Y., Chen Y., Cheng Z., Gu J., Deng J., Bi L., Chen C., Mo R., Wang X., and Ge R. (2015) Succinylome analysis reveals the involvement of lysine succinylation in metabolism in pathogenic Mycobacterium tuberculosis H37Rv. Mol. Cell. Proteomics 14, 796–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zheng H., He Y., Zhou X., Qian G., Lu G., Shen Y., Liu J., Li D., Li X., and Liu W. (2016) Systematic analysis of the lysine succinylome in Candida albicans. J. Proteome Res. 15, 3793–3801 [DOI] [PubMed] [Google Scholar]
- 27. Mikulik K., Felsberg J., Kudrnáčová E., Bezoušková S., Setinová D., Stodůlková E., Zídková J., and Zídek V. (2012) CobB1 deacetylase activity in Streptomyces coelicolor. Biochem. Cell Biol. 90, 179–187 [DOI] [PubMed] [Google Scholar]
- 28. Li F., He X., Ye D., Lin Y., Yu H., Yao C., Huang L., Zhang J., Wang F., Xu S., Wu X., Liu L., Yang C., Shi J., He X., Liu J., Qu Y., Guo F., Zhao J., Xu W., and Zhao S. (2015) NADP(+)-IDH mutations promote hypersuccinylation that impairs mitochondria respiration and induces apoptosis resistance. Molecular Cell 60, 661–675 [DOI] [PubMed] [Google Scholar]
- 29. Datsenko K. A., and Wanner B. L. (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhou S., Hu Y., Veillon L., Snovida S. I., Rogers J. C., Saba J., and Mechref Y. (2016) Quantitative LC-MS/MS glycomic analysis of biological samples using aminoxyTMT. Anal. Chem. 88, 7515–7522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huang D. W., Sherman B. T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M. W., Lane H. C., and Lempicki R. A. (2007) DAVID bioinformatics resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 35, W169–W175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jones P., Binns D., Chang H. Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., Pesseat S., Quinn A. F., Sangrador-Vegas A., Scheremetjew M., Yong S. Y., Lopez R., and Hunter S. (2014) InterProScan 5: Genome-scale protein function classification. Bioinformatics 30, 1236–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. MF C., and D S. (2011) Biological sequence motif discovery using Motif-X. Curr. Protoc. Bioinformatics 35, Unit 13.15–24 [DOI] [PubMed] [Google Scholar]
- 34. Colaert N., Helsens K., Martens L., Vandekerckhove J., and Gevaert K. (2009) Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 6, 786–787 [DOI] [PubMed] [Google Scholar]
- 35. Rajaram S. and Oono Y. (2010) NeatMap—Non-clustering heat map alternatives in R. BMC Bioinformatics 11, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., Jensen L. J., and von Mering C. (2011) The STRING database in 2011: Functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 39, D561–D568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Su G., Morris J. H., Demchak B., and Bader G. D. (2014) Biological network exploration with cytoscape 3. Curr. Protoc. Bioinformatics 47, 8.13.1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bader G. D., and Hogue C. W. (2003) An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choi J. E., and Mostoslavsky R. (2014) Sirtuins, metabolism, and DNA repair. Curr. Opin. Genet. Dev. 26, 24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guarente L. (2011) Sirtuins, aging, and medicine. N. Engl. J. Med. 364, 2235–2244 [DOI] [PubMed] [Google Scholar]
- 41. Ramatchandirin B., Sadasivam M., Kannan A., and Prahalathan C. (2016) Sirtuin 4 regulates lipopolysaccharide mediated Leydig cell dysfunction. J. Cell. Biochem. 117, 904–916 [DOI] [PubMed] [Google Scholar]
- 42. Hirschey M. D. (2011) Old enzymes, new tricks: Sirtuins are NAD(+)-dependent de-acylases. Cell Metabolism 14, 718–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang X., Khan S., Jiang H., Antonyak M. A., Chen X., Spiegelman N. A., Shrimp J. H., Cerione R. A., and Lin H. (2016) Identifying the functional contribution of the defatty-acylase activity of SIRT6. Nat. Chem. Biol. 12, 614–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Starai V. J., Celic I., Cole R. N., Boeke J. D., and Escalante-Semerena J. C. (2002) Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 [DOI] [PubMed] [Google Scholar]
- 45. Ventura M., Mateo F., Serratosa J., Salaet I., Carujo S., Bachs O., and Pujol M. J. (2010) Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase is regulated by acetylation. Int. J. Biochem. Cell Biol. 42, 1672–1680 [DOI] [PubMed] [Google Scholar]
- 46. Pan J., Chen R., Li C., Li W., and Ye Z. (2015) Global analysis of protein lysine succinylation profiles and their overlap with lysine acetylation in the marine bacterium Vibrio parahemolyticus. J. Proteome Res. 14, 4309–4318 [DOI] [PubMed] [Google Scholar]
- 47. He D., Wang Q., Li M., Damaris R. N., Yi X., Cheng Z., and Yang P. (2016) Global Proteome analyses of lysine acetylation and succinylation reveal the widespread involvement of both modification in metabolism in the embryo of germinating rice seed. J. Proteome Res. 15, 879–890 [DOI] [PubMed] [Google Scholar]
- 48. Szklarczyk D., Franceschini A., Wyder S., Forslund K., Heller D., Huerta-Cepas J., Simonovic M., Roth A., Santos A., Tsafou K. P., Kuhn M., Bork P., Jensen L. J., and von Mering C. (2015) STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bentley S. D., Chater K. F., Cerdeño-Tárraga A. M., Challis G. L., Thomson N. R., James K. D., Harris D. E., Quail M. A., Kieser H., Harper D., Bateman A., Brown S., Chandra G., Chen C. W., Collins M., Cronin A., Fraser A., Goble A., Hidalgo J., Hornsby T., Howarth S., Huang C. H., Kieser T., Larke L., Murphy L., Oliver K., O'Neil S., Rabbinowitsch E., Rajandream M. A., Rutherford K., Rutter S., Seeger K., Saunders D., Sharp S., Squares R., Squares S., Taylor K., Warren T., Wietzorrek A., Woodward J., Barrell B. G., Parkhill J., and Hopwood D. A. (2002) Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147 [DOI] [PubMed] [Google Scholar]
- 50. McCormick J. R. and Flärdh K. (2012) Signals and regulators that govern Streptomyces development. FEMS Microbiol. Rev. 36, 206–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xie Z., Dai J., Dai L., Tan M., Cheng Z., Wu Y., Boeke J. D., and Zhao Y. (2012) Lysine succinylation and lysine malonylation in histones. Mol. Cell. Proteomics 11, 100–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kelly G. (2010) A review of the sirtuin system, its clinical implications, and the potential role of dietary activators like resveratrol: Part 1. Altern. Med. Rev. 15, 245–263 [PubMed] [Google Scholar]
- 53. Vizcaíno J. A., Csordas A., Del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baker P. R. and Chalkley R. J. (2014) MS-viewer: A web-based spectral viewer for proteomics results. Mol. Cell. Proteomics 13, 1392–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (53) partner repository with the dataset identifier PXD014202. Alternatively, the dataset can be accessed in MS-Viewer (54) with the search key 8gq2qf4c2e using the following URL: http://msviewer.ucsf.edu/prospector/cgi-bin/mssearch.cgi?report_title=MS-Viewer&search_key=8gq2qf4c2e&search_name=msviewer.