

Abstract
Glutathione peroxidase (GSHPx) is a critical intracellular enzyme involved in detoxification of hydrogen peroxide (H2O2) to water. In the present study we examined the susceptibility of mice with a disruption of the glutathione peroxidase gene to the neurotoxic effects of malonate, 3-nitropropionic acid (3-NP), and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP). Glutathione peroxidase knock-out mice showed no evidence of neuropathological or behavioral abnormalities at 2–3 months of age. Intrastriatal injections of malonate resulted in a significant twofold increase in lesion volume in homozygote GSHPx knock-out mice as compared to both heterozygote GSHPx knock-out and wild-type control mice. Malonate-induced increases in conversion of salicylate to 2,3- and 2,5-dihydroxybenzoic acid, an index of hydroxyl radical generation, were greater in homozygote GSHPx knock-out mice as compared with both heterozygote GSHPx knock-out and wild-type control mice. Administration of MPTP resulted in significantly greater depletions of dopamine, 3,4-dihydroxybenzoic acid, and homovanillic acid in GSHPx knock-out mice than those seen in wild-type control mice. Striatal 3-nitrotyrosine (3-NT) concentrations after MPTP were significantly increased in GSHPx knock-out mice as compared with wild-type control mice. Systemic 3-NP administration resulted in significantly greater striatal damage and increases in 3-NT in GSHPx knock-out mice as compared to wild-type control mice. The present results indicate that a knock-out of GSHPx may be adequately compensated under nonstressed conditions, but that after administration of mitochondrial toxins GSHPx plays an important role in detoxifying increases in oxygen radicals.
Keywords: MPTP, 3-nitropropionic acid, malonate, oxidative damage, free radicals, glutathione, Parkinson's, Huntington's
The formation of hydrogen peroxide and related oxygen radicals is suspected to be involved in the mechanism of nerve cell death and in neurodegenerative diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's disease (Coyle and Puttfarcken, 1993; Beal, 1995). There is substantial evidence that the brain, which consumes large amounts of oxygen, is particularly vulnerable to oxidative damage. The relative roles of endogenous and exogenous antioxidants in protecting the brain against oxidative stress are still being clarified. The major antioxidant defenses consist of antioxidant scavengers such as glutathione, vitamin C, vitamin E, and antioxidant enzymes.
The antioxidant enzymes in the brain include Cu,Zn- and manganese superoxide dismutase, which catalyze the conversion of O2• to H2O2(Fridovich, 1989). H2O2 is then converted to H2O by either catalase or selenoglutathione peroxidases. Catalase is thought to be relatively low in the brain and is localized to peroxisomes (Gaunt and De Duve, 1976; Halliwell, 1992). The selenoglutathione peroxidases include the “classic” enzyme selenoglutathione peroxidase-I (GSHPx; GSH:H2O2 oxidoreductase, EC 1.11.19) and a more recently characterized phospholipid hydroperoxide glutathione peroxidase (Fisher et al., 1999). Among the brain glutathione peroxidases, only GSHPx is known to reduce H2O2, indicating that GSHPx may be a major protective enzyme against the action of H2O2 in the brain (Jain et al., 1991). Recent evidence showed that GSHPx also plays a major role in detoxifying peroxynitrite (ONOO−) (Sies et al., 1997). GSHPx is present both in the cytosol and in mitochondria (Vitorica et al., 1984), which are a major intracellular source of free radicals (Boveris and Chance, 1973).
Malonate and 3-nitropropionic acid (3-NP) are inhibitors of succinate dehydrogenase, which model Huntington's disease (Beal et al., 1993a,b). 1-Methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) has been extensively used to replicate the dopaminergic neuronal loss occurring in Parkinson's disease (Bloem et al., 1990). Its active metabolite 1-methyl-4-phenylpyridinium (MPP+) selectively inhibits mitochondrial complex I activity (Tipton and Singer, 1993). These neurotoxins produce impaired energy metabolism and oxidative stress, which plays a direct role in neuronal injury. We and others observed an increased formation of oxygen-derived free radicals when neurons were challenged with malonate, 3-NP, MPTP, or MPP+ (Hasegawa et al., 1990; Chiueh et al., 1992; Schulz et al., 1995a; Sriram et al., 1997; Huang and Lee, 1998).
In the present study, we investigated the importance of the glutathione system in protecting the brain from mitochondrial toxins. Our hypothesis was that an impairment of GSHPx activity, which may be compensated when occurring in isolation, may lead to irreversible cell loss when combined with increased free radical generation caused by mitochondrial toxins. Specifically, we sought to determine if GSHPx knock-out mice would be more sensitive to malonate, 3-NP, or MPTP toxicity than control mice.
MATERIALS AND METHODS
Experimental animals. Our experiments were approved by the local Animal Care Committee and were conducted in strict accordance with the National Institutes of Health guidelines for the care and use of experimental animals. All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise indicated. Mice (2- to 3-months-old) were in a B6C3F1 background. The wild-type controls were obtained from Taconic (Germantown, NY), whereas those deficient in cellular GSHPx were provided by Dr. Julie Andersen, University of Southern California (Los Angeles, CA) and bred locally. We bred the GSHPx homozygote mice with the BGC3F1 mice to produce heterozygote GSHPx knock-out mice for use as a further control for genetic background effects. The GSHPx knock-out mice were generated as previously described (Ho et al., 1997) by insertion of a neomycin resistance gene cassette into the EcoRI site located in exon 2 of the GSHPx mouse gene. This introduces a BamHI site into exon 2, which gives a 4.3 kb band on Southern blot analysis instead of the 11 kb band found in the normal controls. A herpes thymidine kinase gene cassette was placed at a second EcoRI site in the 3′ untranslated region for positive–negative selection with G418/gangcyclovir in embryonic stem cells. These mice show an 85% reduction in cortex GSHPx activity from 0.155 ± 0.021 to 0.024 ± 0.017, p < 0.001 as previously described (Lawrence and Burk, 1976).
Intrastriatal microinjections. Control (n = 10), heterozygote GSHPx knock-out mice (n = 10), and homozygous and GSHPx knock-out (n = 12) mice were anesthetized with methoxyflurane and malonate (1.4 μmol in 0.7 μl, pH 7.4) that was stereotaxically injected into the left striatum (anterior, 0.5 mm; lateral, 2 mm from bregma; ventral, 3.5 mm from dura). The injections were performed over 2 min using a 10 μl 26 gauge blunt-tipped Hamilton syringe. The needle was left in place for 5 min before being slowly withdrawn. Seven days after striatal injection animals were killed, and the brains were rapidly removed, placed in cold saline, and sectioned coronally at 1 mm intervals. Slices were stained in 2% 2,3,5-triphenyltetrazolium chloride monohydrate solution at room temperature in the dark for 30 min, and post-fixed in phosphate-buffered 4% paraformaldehyde (PFA) (Bederson et al., 1986). The lesioned area (noted by pale staining) was measured on the posterior surface of each section using Neurolucida (Microbrightfield, Colchester, VT) image analysis software. We previously showed that these measurements exhibit no significant differences from those obtained with Nissl staining (Schulz et al., 1995a). Lesion volumes (mean ± SEM) were calculated by multiplying the lesion area by the slice thickness.
Salicylate assay and 3-nitrotyrosine measurement. The salicylate hydroxyl radical trapping method was used for measuring levels of •OH radicals in striatal tissue after injection of malonate in control (n = 13), GSHPx heterozygote knock-out (n = 13), and homozygous GSHPx knock-out (n = 11) mice (Floyd et al., 1984). Salicylate (200 mg/kg, 5 ml/kg, i.p.) was administered 30 min before striatal malonate injection. Sixty minutes after malonate injection, the animals were killed, and the left and right striata were rapidly dissected from a 2-mm-thick slice on a chilled glass plate and immediately frozen at −70°C. To examine the effects of 3-NP on 3-NT levels, control (n = 8) and GSHPx knock-out (n = 8) mice received six doses of 50 mg/kg intraperitoneally at 12 hr intervals. Mice were killed 1 hr after the last dose. The striata were rapidly dissected and placed in chilled 0.1 m perchloric acid. The samples were thawed in 0.25 ml of chilled 0.1 m perchloric acid, sonicated, and centrifuged twice. Salicylate and its metabolites 2,3- and 2,5-dihydroxybenzoic acid (DHBA), tyrosine, and 3-NT were quantified in the supernatant by HPLC with 16-electrode electrochemical detection (Beal et al., 1990). Data (mean ± SEM) were expressed as the ratio of 2,3- and 2,5-DHBA to salicylate and of 3-NT to tyrosine to normalize for varying brain concentrations of salicylate and tyrosine.
Dopamine measurement. MPTP (15 mg/kg, 5 ml/kg, i.p.) was administered four times at 2 hr intervals to control (n = 10) and GSHPx knock-out (n = 10) mice. An additional set of animals of each type was also treated with 0.1 m PBS (5 ml/kg, i.p.) at the times of MPTP injections. The animals were killed at 1 week, and both striata were rapidly dissected on a chilled glass plate and frozen at −70°C. The samples were subsequently thawed in 0.25 ml of chilled 0.1m perchloric acid and sonicated. Aliquots were taken for protein quantification using a fluorometric assay (Beal et al., 1990). Other aliquots were centrifuged, and dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA) were measured in supernatants by HPLC and electrochemical detection. Concentrations of dopamine and metabolites were expressed as nanograms per milligram of protein (mean ± SEM).
MPP+ levels. To determine whether MPTP uptake or metabolism was altered, MPTP 20 mg/kg was administered intraperitoneally twice, 2 hr apart, and mice were killed 2 hr after the last dose (n = 8/group). Striatal tissue from this experiment was also used for 3-NT determinations. MPP+ levels were quantified by HPLC with UV detection at 295 nm. Samples were sonicated in 0.1m perchloric acid, and an aliquot of supernatant was injected onto a Brownlee aquapore X03–224 cation exchange column (Rainin, Woburn, MA). Samples were eluted isocratically with 90% 0.1m acetic acid and 75 mmtriethylamine HCl, pH 2.3, adjusted with formic acid and 10% acetonitrile.
Histological study. 3-NP (50 mg/kg, 5 ml/kg, i.p.) was administered eight times at 12 hr intervals to control (n = 8) and GSHPx knock-out (n = 9) mice. An additional set of animals of each type was also treated with 0.1 m PBS (5 ml/kg, i.p.) at the times of 3-NP injections. Twelve hours after the last injection, the animals were deeply anesthetized with pentobarbital and perfused with ice-cold 0.9% saline followed by 4% paraformaldehyde. Brains were post-fixed for 1 hr, rinsed in 0.1 m PBS, and then cryoprotected in a graded series of 10% and 20% glycerol/2% DMSO solution. Frozen brains were sectioned at 50 μm using a sledge microtome and Nissl-stained as previously described (Beal et al., 1989). Bilateral striatal lesion volumes were computed in serial sections through the rostrocaudal extent of each brain by videomicroscopic capture of brain sections and subsequent volume analysis using Neurolucida (Microbrightfield) image analysis software.
Statistical analysis. Results are expressed as the mean ± SEM. Statistical comparisons were made using Student'st test (unpaired) or one-way ANOVA followed by Fisher's PLSD post hoc tests.
RESULTS
The lesion volumes after intrastriatal injection of malonate in wild-type controls and GSHPx knock-out mice are shown in Figure1. Lesion volumes after malonate injections were significantly larger in homozygous GSHPx compared to both heterozygous GSHPx (p < 0.01) and wild types (p < 0.001). There was no significant difference between heterozygous GSHPx knock-out and wild-type mice. Injection of vehicle resulted in negligible lesions in both controls and GSHPx knock-out mice (0.24 ± 0.04 vs 0.34 ± 0.07 mm3). After administration of salicylate, intrastriatal injection of malonate resulted in a significant increase in 2,3 DHBA compared to the unlesioned side only in homozygous GSHPx knock-out mice (p < 0.001) (Fig.2). The level of 2,3 DHBA in the lesioned side of homozygous GSHPx knock-out was significantly higher than in the lesioned side in both heterozygous GSHPx knock-out and wild types (p < 0.001). A significant increase in 2,5 DHBA was seen in the lesioned striata in all groups, but the increase in homozygous GSHPx knock-out was significantly larger than the increase in both heterozygous GSHPx knock-out and the wild-type mice (p < 0.0001). There was no significant difference between heterozygous GSHPx knock-out and wild-type mice.
Fig. 1.

Malonate induced striatal lesion volumes in wild-type controls, heterozygote, and homozygote GSHPx knock-out mice. **p < 0.01, as compared with controls; #p < 0.05, as compared with heterozygote GSHPx knock-out mice.
Fig. 2.

Malonate induced increases in the conversion of salicylate to 2,3 and 2,5-DHBA in wild-type controls, heterozygote, and homozygote GSHPx knock-out mice. *p < 0.001, as compared with the uninjected striatum; #p < 0.001, as compared with heterozygote GSHPx knock-out and wild-type controls.
The effects of administration of MPTP in wild-type control and GSHPx knock-out mice are seen in Figure 3. We used a relatively low dose of MPTP, 4 × 15 mg/kg, which produced a small significant dopamine depletion of 15% in wild-type controls. In contrast, the same dose of MPTP produced a significant 61% depletion of dopamine in GSHPx knock-out mice that was significantly,p < 0.001, greater than that seen in controls. Depletions of DOPAC and HVA in controls did not reach significance, but they were highly significant in GSHPx knock-out mice and were significantly (p < 0.001) greater than those seen in wild-type controls. The increased sensitivity to MPTP was not caused by an alteration in uptake or metabolism of MPTP to MPP+ because striatal MPP+ levels did not significantly differ at 2 hr after MPTP administration (MPP+8.4 ± 1.3 ng/mg protein in controls and 9.5 ± 1.0 ng/mg protein in GSHPx knock-out mice). The effects of MPTP on striatal 3-NT levels are shown in Figure 4. MPTP administration in the GSHPx knock-out mice resulted in a significant increase in 3-NT levels as compared with wild-type controls (p < 0.01). We previously found that saline-injected controls had 3-NT levels of 1–2 3-NT/1000 tyrosines (Schulz et al., 1995b), consistent with the findings in Figure 6.
Fig. 3.

Effects of MPTP administered at 15 mg/kg X4 on dopamine, DOPAC, and HVA in wild-type control and GSHPx knock-out mice. *p < 0.05, ***p < 0.001, as compared to PBS-treated animals; ##p < 0.01, ###p < 0.001, as compared to wild-type treated with MPTP.
Fig. 4.

Effects of MPTP 20 mg/kg X2 on striatal 3-NT levels 2 hr after MPTP administration in wild-type control and GSHPx knock-out mice. **p < 0.01, as compared with wild-type controls.
Fig. 6.

Effects of 3-NP on 3-NT levels in wild-type control and GSHPx knock-out mice. **p < 0.01, ***p < 0.001, as compared with PBS; #p < 0.05, as compared with wild-type control.
Systemic administration of 3-NP resulted in bilateral striatal lesions in both wild-type controls and GSHPx knock-out mice (Fig.5). The areas of neuronal loss and increased gliosis within the caudate putamen were significantly (almost fourfold) greater in the GSHPx knock-out mice (Fig. 5). Striatal lesion volumes were 3.72 ± 0.26 mm3 in controls and 14.12 ± 0.92 mm3 in GSHPx knock-out mice, p < 0.01. The effects of 3-NP on 3-NT levels are shown in Figure 6. 3-Nitrotyrosine levels increased after 3-NP administration in both controls and GSHPx knock-out mice, but the increases were significantly (p < 0.05) greater in the GSHPx knock-out mice than those observed in the controls.
Fig. 5.
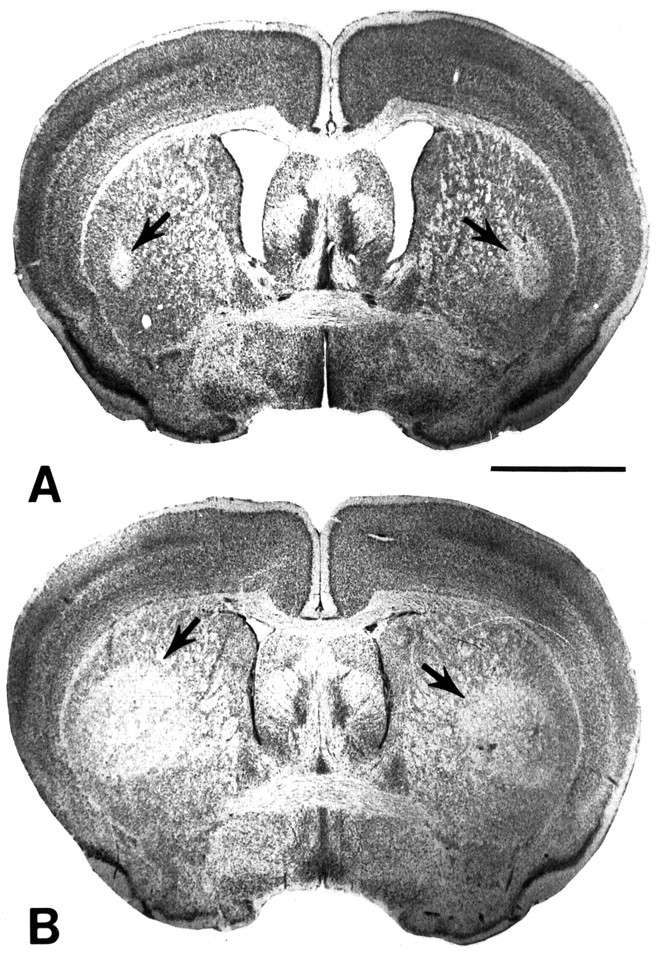
Photomicrographs of 3-NP lesions in Nissl-stained whole-brain sections through the striatum of wild-type (A) and glutathione peroxidase knock-out (B) mice. Bilateral striatal lesions are present in both A and B and are represented by staining pallor in the lateral aspect (arrows). The lesions are significantly larger in the glutathione peroxidase knock-out mouse. Scale bar, 2 mm.
DISCUSSION
The glutathione (GSH) system plays a major role in controlling cellular redox states and is a primary defense mechanism for H2O2 and peroxide removal in brain. Immunocytochemical studies showed localization of GSHPx to both brain astrocytes and neurons (Damier et al., 1993; Olanow, 1993;Trepanier et al., 1996). In cultured cerebellar astrocytes, cytosolic GSH and GSHPx were 57 and 245% higher than those found in granule cells (Huang and Philbert, 1995). Other studies also showed increased GSH in astrocytes as compared to neurons (Slivka et al., 1987; Raps et al., 1989). The ratio of mitochondrial to cytosolic GSH and mitochondrial GSHPx however is higher in cerebellar granule cells than astrocytes, suggesting that the GSHPx system may be particularly important in neuronal mitochondria. Depletion of GSH leads to mitochondrial damage and reductions in mitochondrial enzymes in brain (Jain et al., 1991; Martinez et al., 1995), and it causes calcium-mediated cell death in PC12 cells (Jurma et al., 1997). Further evidence implicating GSH in normal brain function are the observations that glutathione depletion in vivo results in dystrophic axons in dopaminergic neurons and enhances the neurotoxicity of ischemia, 6-hydroxydopamine, MPP+, and MPTP (Pileblad et al., 1989; Mizui et al., 1992; Andersen et al., 1996;Wullner et al., 1996; Nakamura et al., 1997).
The fact that mice with a knock-out of GSHPx show no neuronal degeneration up to 3 months of age is, therefore, somewhat surprising. It is, however, consistent with a previous report that mice deficient in cellular GSHPx develop normally, are fertile, and show no increase in lung toxicity to hyperoxia (Ho et al., 1997). Histological examination at 4 and 15 months of age was normal in all tissues, including the brain, and protein carbonyls and lipid peroxidation products were unaltered from controls (Ho et al., 1997). These observations suggest an alternative means of removing H2O2 under baseline physiological conditions. Although catalase activity (EC 1.11.16) was reported to be low in the brain, it is widely distributed throughout the brain (Gaunt and De Duve, 1976; Brannan et al., 1981). Both catalase and GSHPx are found in cultured astrocytes (Copin et al., 1992; Huang and Philbert, 1995; Desagher et al., 1996). H2O2 easily crosses cell membranes and therefore could leave the cell to damage neighboring cells or be detoxified by them (Halliwell, 1992). It was recently suggested that catalase was the main hydrogen peroxidase activity in astrocytes and that it protected neighboring neurons (Desagher et al., 1996). In other studies of cultured astrocytes, both GSHPx and catalase were shown to be complementary in detoxification of H2O2 (Dringen and Hamprecht, 1997). Inhibitors of either enzyme only marginally reduced the rate of disappearance of H2O2 from the incubation media, however inhibition of both enzymes strongly reduced H2O2 clearance. It therefore appears that both H2O2 detoxifying systems can increase H2O2 clearance sufficiently under physiological conditions to prevent toxicity. This is not the case with other free radical scavengers such as manganese superoxide dismutase, in which a deficiency leads to premature death with both cardiac and CNS damage (Li et al., 1995; Lebovitz et al., 1996).
We, however, wondered whether GSHPx may play a more critical role under conditions in which neuronal metabolism is stressed by mitochondrial toxins. Both malonate and 3-NP are succinate dehydrogenase inhibitors that produce striatal lesions in vivo after either local striatal or systemic administration, respectively. Studies using13C magnetic resonance spectroscopy showed that 3-NP preferentially inhibits oxidative metabolism in GABAergic neurons in vivo, whereas astrocyte metabolism was spared (Hassel and Sonnewald, 1995). The neurotoxicity of these compounds is associated with increases in OH• generation as assessed by the salicylate-trapping method, as well as with increases in 3-NT, a marker of peroxynitrite (Schulz et al., 1995c). Similarly, MPTP neurotoxicity is associated with increases in OH• generation and 3-NT (Schulz et al., 1995a). In the present study we therefore examined whether GSHPx knock-out mice would show increased susceptibility to these toxins.
The intrastriatal administration of malonate resulted in a significant twofold increase in lesion volume in GSHPx knock-out mice, as compared with both heterozygote GSHPx knock-out and wild-type control mice. Furthermore, the administration of malonate resulted in increased OH• generation, as assessed using the salicylate-trapping method in homozygous GSHPx knock-out mice, as compared with both heterozygote GSHPx knock-out and wild-type control mice. The heterozygote GSHPx knock-out mice were produced by crossing the homozygous GSHPx knock-out mice with the background strain, which should control for any genetic variation between the GHSPx knock-out mice and the original background strain. 3-Nitropropionic acid lesions were also significantly greater in GSHPx knock-out mice. Lastly, MPTP neurotoxicity, as assessed by levels of dopamine, DOPAC, and HVA, was markedly exacerbated in the GSHPx knock-out mice.
GSHPx may therefore play an important role in initially compensating for increased generation of oxidants in these illnesses. GSHPx can detoxify reactive oxygen species by catalyzing the conversion of H2O2 to H2O, but it also acts to reduce lysophospholipid hydroperoxides (Marinho et al., 1997; Fisher et al., 1999). Its role in detoxification of peroxynitrite (Sies et al., 1997) may be particularly crucial, because we and others found that inhibitors of neuronal nitric oxide synthase block malonate, 3-NP, and MPTP neurotoxicity (Schulz et al., 1995a; Hantraye et al., 1996; Przedborski et al., 1996). In the present study we found that striatal 3-NT concentrations were significantly increased after MPTP administration in GSHPx knock-out mice as compared with controls. We also found that increases in striatal 3-NT after systemic administration of 3-NP were significantly greater in GSHPx knock-out mice as compared with controls. This evidence therefore indicates that GSHPx plays an important role in the detoxification of peroxynitrite in vivo.
These results therefore indicate that although other free radical scavenging mechanisms are able to compensate for a loss of GSHPx under physiological conditions, they are inadequate in response to a metabolic stress. This has important implications for the pathogenesis of Huntington's disease (HD) and Parkinson's disease (PD). In both of these neurodegenerative diseases there is strong evidence implicating deficient energy production and increased free radical production (Beal, 1997). In HD there are increases in cerebral lactate in vivo, as assessed by magnetic resonance spectroscopy (Jenkins et al., 1993), decreases in mitochondrial complex II-III activity in postmortem tissue, and increased oxidative damage to DNA (Gu et al., 1996; Browne et al., 1997). In PD, several authors found reduced mitochondrial complex I activity in the substantia nigra and in platelets and evidence of increased oxidative damage (for review, seeBeal, 1995). GSH is significantly depleted in the substantia nigra of PD patients, as well as in incidental Lewy body disease, which may be a presymptomatic stage of PD (Dexter et al., 1994). GSHPx activity is also reduced in the substantia nigra of PD patients (Ambani et al., 1975; Kish et al., 1985).
In PD it is possible that a latent genetic defect in free radical scavenging enzymes or in mitochondrial electron enzymes may be compensated under physiological conditions, but may increase susceptibility to environmental toxins. Environmental factors could also contribute to some of the variance in age of onset of HD (Gusella et al., 1997). The present results are therefore consistent with the possibility that genetic defects may interact with environmental toxins in the pathogenesis of neurodegenerative diseases.
Footnotes
This work was supported by National Institutes of Health Grants NS16367, NS10828, NS31579, and AG11337 (M.F.B.), NS37102 and NS35255 (R.J.F.), the Veterans Administration (R.J.F.), and the American Parkinson's Disease Association (R.J.F.). The secretarial assistance of Sharon Melanson is gratefully acknowledged.
Correspondence should be addressed to Dr. M. Flint Beal, Neurology Service/WRN 408, Massachusetts General Hospital, 32 Fruit Street, Boston, MA 02114. E-mail: beal@helix.mgh.harvard.edu.
REFERENCES
- 1.Ambani LM, Van Woert MH, Murphy S. Brain peroxidase and catalase in Parkinson disease. Arch Neurol. 1975;32:114–118. doi: 10.1001/archneur.1975.00490440064010. [DOI] [PubMed] [Google Scholar]
- 2.Andersen JK, Mo. JQ, Lee FY, Harnish P, Hamill RW, McNeill TH. Effect of buthionine sulfoximine, a synthesis inhibitor of the antioxidant glutathione, on the murine nigrostriatal neurons. J Neurochem. 1996;67:2164–2171. doi: 10.1046/j.1471-4159.1996.67052164.x. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF. Aging, energy and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995;38:357–366. doi: 10.1002/ana.410380304. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF. Oxidative damage in neurodegenerative diseases. The Neuroscientist. 1997;3:21–27. [Google Scholar]
- 5.Beal MF, Kowall NW, Swartz KJ, Ferrante RJ, Martin JB. Differential sparing of somatostatin-neuropeptide Y and cholinergic neurons following striatal excitotoxin lesions. Synapse. 1989;3:38–47. doi: 10.1002/syn.890030106. [DOI] [PubMed] [Google Scholar]
- 6.Beal MF, Matson WR, Swartz KJ, Gamache PH, Bird ED. Kynurenine pathway measurements in Huntington's disease striatum: evidence for reduced formation of kynurenic acid. J Neurochem. 1990;55:1327–1339. doi: 10.1111/j.1471-4159.1990.tb03143.x. [DOI] [PubMed] [Google Scholar]
- 7.Beal MF, Brouillet E, Jenkins B, Henshaw R, Rosen B, Hyman BT. Age-dependent striatal excitotoxic lesions produced by the endogenous mitochondrial inhibitor malonate. J Neurochem. 1993a;61:1147–1150. doi: 10.1111/j.1471-4159.1993.tb03633.x. [DOI] [PubMed] [Google Scholar]
- 8.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman B. Neurochemical and histologic characterization of excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993b;13:4181–4191. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bederson JB, Pitts LH, Germano SM, Nishimura MC, Davis RL, Bartkowski HM. Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke. 1986;17:1304–1308. doi: 10.1161/01.str.17.6.1304. [DOI] [PubMed] [Google Scholar]
- 10.Bloem BR, Irwin I, Buruma OJS, Haan J, Roos RAC, Tetrud JW, Langston JW. The MPTP model: versatile contributions to the treatment of idiopathic Parkinson's disease. J Neurol Sci. 1990;97:273–293. doi: 10.1016/0022-510x(90)90225-c. [DOI] [PubMed] [Google Scholar]
- 11.Boveris A, Chance B. The mitochondrial generation of hydrogen peroxide. Biochem J. 1973;134:707–716. doi: 10.1042/bj1340707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brannan TS, Maker HS, Raes IP. Regional distribution of catalase in the adult rat brain. J Neurochem. 1981;36:307–309. doi: 10.1111/j.1471-4159.1981.tb02411.x. [DOI] [PubMed] [Google Scholar]
- 13.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MMK, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 14.Chiueh CC, Krishna G, Tulsi P, Obata T, Lang K, Huang S-J, Murphy DL. Intracranial microdialysis of salicylic acid to detect hydroxyl radical generation through dopamine autooxidation in the caudate nucleus: effects of MPP+. Free Radic Biol Med. 1992;13:581–583. doi: 10.1016/0891-5849(92)90151-6. [DOI] [PubMed] [Google Scholar]
- 15.Copin JC, Ledig M, Tholey G. Free radical scavenging systems of rat astroglial cells in primary culture: effect of anoxia and drug treatment. Neurochem Res. 1992;17:677–682. doi: 10.1007/BF00968004. [DOI] [PubMed] [Google Scholar]
- 16.Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–700. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- 17.Damier P, Hirsch EC, Zhang P, Agid Y, Javoy-Agid F. Glutathione peroxidase, glial cells and Parkinson's disease. Neuroscience. 1993;52:1–6. doi: 10.1016/0306-4522(93)90175-f. [DOI] [PubMed] [Google Scholar]
- 18.Desagher S, Glowinski J, Premont J. Astrocytes protect neurons from hydrogen peroxide toxicity. J Neurosci. 1996;16:2553–2562. doi: 10.1523/JNEUROSCI.16-08-02553.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dexter DT, Sian J, Rose S, Hindmarsch JG, Mann VM, Cooper JM, Wells FR, Daniel SE, Lees AJ, Schapira AHV, Jenner P, Marsden CD. Indices of oxidative stress and mitochondrial function in individuals with incidental Lewy body disease. Ann Neurol. 1994;35:38–44. doi: 10.1002/ana.410350107. [DOI] [PubMed] [Google Scholar]
- 20.Dringen R, Hamprecht B. Involvement of glutathione peroxidase and catalase in the disposal of exogenous hydrogen peroxide by cultured astroglial cells. Brain Res. 1997;759:67–75. doi: 10.1016/s0006-8993(97)00233-3. [DOI] [PubMed] [Google Scholar]
- 21.Fisher AB, Dodia C, Manevich Y, Chen JW, Feinstein SI. Phospholipid hydroperoxides are substrates for non-selenium glutathione peroxidase. J Biol Chem. 1999;274:21326–21334. doi: 10.1074/jbc.274.30.21326. [DOI] [PubMed] [Google Scholar]
- 22.Floyd RA, Watson JJ, Wong PK. Sensitive assay of hydroxyl radical formation utilizing high pressure liquid chromatography with electrochemical detection of phenol and salicylate hydroxylation products. J Biochem Biophys Methods. 1984;10:221–235. doi: 10.1016/0165-022x(84)90042-3. [DOI] [PubMed] [Google Scholar]
- 23.Fridovich I. Superoxide dismutases. An adaptation to a paramagnetic gas. J Biol Chem. 1989;264:7761–7764. [PubMed] [Google Scholar]
- 24.Gaunt GL, De Duve C. Subcellular distribution of d-amino acid oxidase and catalase in rat brain. J Neurochem. 1976;26:749–759. doi: 10.1111/j.1471-4159.1976.tb04448.x. [DOI] [PubMed] [Google Scholar]
- 25.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AHV. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 26.Gusella JF, Persichetti F, MacDonald ME. The genetic defect causing Huntington's disease: repeated in other contexts? Mol Med. 1997;3:238–246. [PMC free article] [PubMed] [Google Scholar]
- 27.Halliwell B. Reactive oxygen species and the central nervous system. J Neurochem. 1992;59:1609–1623. doi: 10.1111/j.1471-4159.1992.tb10990.x. [DOI] [PubMed] [Google Scholar]
- 28.Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, Beal MF. Inhibition of neuronal nitric oxide synthase prevents MPTP-induced parkinsonism in baboons. Nat Med. 1996;2:1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- 29.Hasegawa E, Takeshige K, Oishi T, Murai Y, Minakami S. 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem Biophys Res Commun. 1990;170:1049–1055. doi: 10.1016/0006-291x(90)90498-c. [DOI] [PubMed] [Google Scholar]
- 30.Hassel B, Sonnewald U. Selective inhibition of the tricarboxylic acid cycle of GABAergic neurons with 3-nitropropionic acid in vivo. J Neurochem. 1995;65:1184–1191. doi: 10.1046/j.1471-4159.1995.65031184.x. [DOI] [PubMed] [Google Scholar]
- 31.Ho YS, Magnenat JL, Bronson RT, Cao J, Gargano M, Sugawara M, Frank CD. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem. 1997;272:16644–16651. doi: 10.1074/jbc.272.26.16644. [DOI] [PubMed] [Google Scholar]
- 32.Huang H-C, Lee EHY. MPTP produces differential oxidative stress and antioxidative responses in the nigrostriatal and mesolimbic dopaminergic pathways. Free Radic Biol Med. 1998;24:76–84. doi: 10.1016/s0891-5849(97)00206-2. [DOI] [PubMed] [Google Scholar]
- 33.Huang J, Philbert MA. Distribution of glutathione and glutathione-related enzyme systems in mitochondria and cytosol of cultured cerebellar astrocytes and granule cells. Brain Res. 1995;680:16–22. doi: 10.1016/0006-8993(95)00209-9. [DOI] [PubMed] [Google Scholar]
- 34.Jain A, Martensson J, Stole E, Auld PAM, Meister A. Glutathione deficiency leads to mitochondrial damage in brain. Proc Natl Acad Sci USA. 1991;88:1913–1917. doi: 10.1073/pnas.88.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jenkins B, Koroshetz W, Beal MF, Rosen B. Evidence for an energy metabolism defect in Huntington's disease using localized proton spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 36.Jurma OP, Hom DG, Andersen JK. Decreased glutathione results in calcium-mediated cell death in PC12. Free Radic Biol Med. 1997;23:1055–1066. doi: 10.1016/s0891-5849(97)00134-2. [DOI] [PubMed] [Google Scholar]
- 37.Kish SJ, Morito C, Hornykiewicz O. Glutathione peroxidase activity in Parkinson's disease brain. Neurosci Lett. 1985;58:343–346. doi: 10.1016/0304-3940(85)90078-3. [DOI] [PubMed] [Google Scholar]
- 38.Lawrence RA, Burk RF. Glutathione peroxidase activity in selenium-deficient rat liver. Biochem Biophys Res Commun. 1976;71:952–958. doi: 10.1016/0006-291x(76)90747-6. [DOI] [PubMed] [Google Scholar]
- 39.Lebovitz RM, Zhang H, Vogel H, Cartwright J, J, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Huang T-T, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 41.Marinho HS, Antunes F, Pinto RE. Role of glutathione peroxidase and phospholipid hydroperoxide glutathione peroxidase in the reduction of lysophospholipid hydroperoxides. Free Radic Biol Med. 1997;22:871–883. doi: 10.1016/s0891-5849(96)00468-6. [DOI] [PubMed] [Google Scholar]
- 42.Martinez M, Ferrandiz ML, Diez A, Miquel J. Depletion of cytosolic GSH decreases the ATP levels and viability of synaptosomes from aged mice but not from young mice. Mech Ageing Dev. 1995;84:77–81. doi: 10.1016/0047-6374(95)01644-f. [DOI] [PubMed] [Google Scholar]
- 43.Mizui T, Kinouchi H, Chan PH. Depletion of brain glutathione by buthionine sulfoximine enhances cerebral ischemic injury in rats. Am J Physiol. 1992;262:H313–H317. doi: 10.1152/ajpheart.1992.262.2.H313. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura K, Wang W, Kang UJ. The role of glutathione in dopaminergic neuronal survival. J Neurochem. 1997;69:1850–1858. doi: 10.1046/j.1471-4159.1997.69051850.x. [DOI] [PubMed] [Google Scholar]
- 45.Olanow CW. A radical hypothesis for neuronal degeneration. Trends Neurosci. 1993;16:439–443. doi: 10.1016/0166-2236(93)90070-3. [DOI] [PubMed] [Google Scholar]
- 46.Pileblad E, Magnuson T, Fornstedt B. Reduction of brain glutathione by [SCAP]L[R]-buthionine sulfoximine potentiates the dopamine-depletion action of 6-hydroxydopamine in rat striatum. J Neurochem. 1989;52:978–980. doi: 10.1111/j.1471-4159.1989.tb02550.x. [DOI] [PubMed] [Google Scholar]
- 47.Przedborski S, Jackon-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci USA. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raps SP, Lai JCK, Hertz L, Cooper AJL. Glutathione is present in high concentrations in cultured astrocytes but not in cultured neurons. Brain Res. 1989;493:398–401. doi: 10.1016/0006-8993(89)91178-5. [DOI] [PubMed] [Google Scholar]
- 49.Schulz JB, Henshaw DR, Matthews RT, Beal MF. Coenzyme Q10 and nicotinamide and a free radical spin trap protect against MPTP neurotoxicity. Exp Neurol. 1995a;132:279–283. doi: 10.1016/0014-4886(95)90033-0. [DOI] [PubMed] [Google Scholar]
- 50.Schulz JB, Henshaw DR, Siwek D, Jenkins BG, Ferrante RJ, Cipolloni PB, Kowall NW, Rosen BR, Beal MF. Involvement of free radicals in excitotoxicity in vivo. J Neurochem. 1995b;64:2239–2247. doi: 10.1046/j.1471-4159.1995.64052239.x. [DOI] [PubMed] [Google Scholar]
- 51.Schulz JB, Matthews RT, Jenkins BG, Ferrante RJ, Siwek D, Henshaw DR, Cipolloni PB, Mecocci P, Kowall NW, Rosen BR, Beal MF. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995c;15:8419–8429. doi: 10.1523/JNEUROSCI.15-12-08419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sies H, Sharov VS, Klotz L-O, Briviba K. Glutathione peroxidase protects against peroxynitrite-mediated oxidations. J Biol Chem. 1997;272:27812–27817. doi: 10.1074/jbc.272.44.27812. [DOI] [PubMed] [Google Scholar]
- 53.Slivka A, Mytilineou C, Cohen G. Histochemical evaluation of glutathione in brain. Brain Res. 1987;409:275–284. doi: 10.1016/0006-8993(87)90712-8. [DOI] [PubMed] [Google Scholar]
- 54.Sriram K, Pai KS, Boyd MR, Ravindranath V. Evidence for generation of oxidative stress in brain by MPTP: in vitro and in vivo studies in mice. Brain Res. 1997;749:44–52. doi: 10.1016/s0006-8993(96)01271-1. [DOI] [PubMed] [Google Scholar]
- 55.Tipton KF, Singer TP. Advances in our understanding of the mechanisms of the neurotoxicity of MPTP and related compounds. J Neurochem. 1993;61:1191–1206. doi: 10.1111/j.1471-4159.1993.tb13610.x. [DOI] [PubMed] [Google Scholar]
- 56.Trepanier G, Furling D, Puymirat J, Mirault M-E. Immunocytochemical localization of seleno-glutathione peroxidase in the adult mouse brain. Neuroscience. 1996;75:231–243. doi: 10.1016/0306-4522(96)00222-9. [DOI] [PubMed] [Google Scholar]
- 57.Vitorica J, Machado A, Satrustegui J. Age-dependent variations in peroxide-utilizing enzymes from rat brain mitochondria and cytoplasm. J Neurochem. 1984;42:351–357. doi: 10.1111/j.1471-4159.1984.tb02685.x. [DOI] [PubMed] [Google Scholar]
- 58.Wullner U, Loschmann P-A, Schulz JB, Schmid A, Dringen R, Eblen F, Turski L, Klockgether T. Glutathione depletion potentiates MPTP and MPP+ toxicity in nigral dopaminergic neurones. NeuroReport. 1996;7:921–923. doi: 10.1097/00001756-199603220-00018. [DOI] [PubMed] [Google Scholar]