

Abstract
We have studied the mechanisms of auditory hair cell death after insults in vitro and in vivo. We show DNA fragmentation of hair cell nuclei after ototoxic drug and intense noise trauma. By using phospho-specific c-Jun-N-terminal kinase (JNK) and c-Jun antibodies in immunohistochemistry, we show that the JNK pathway, associated with stress, injury, and apoptosis, is activated in hair cells after trauma. CEP-1347, a derivative of the indolocarbazole K252a, is a small molecule that has been shown to attenuate neurodegeneration by blocking the activation of JNK (Maroney et al., 1998). Subcutaneously delivered CEP-1347 attenuated noise-induced hearing loss. The protective effect was demonstrated by functional tests, which showed less hearing threshold shift in CEP-1347-treated than in nontreated guinea pigs, and by morphometric methods showing less hair cell death in CEP-1347-treated cochleas. In organotypic cochlear cultures, CEP-1347 prevented neomycin-induced hair cell death. In addition to hair cells, CEP-1347 promoted survival of dissociated cochlear neurons. These results suggest that therapeutic intervention in the JNK signaling cascade, possibly by using CEP-1347, may offer opportunities to treat inner ear injuries.
Keywords: cochlea, noise trauma, aminoglycoside toxicity, cell death, c-Jun, therapy
Significant hearing loss compromising communication occurs in ∼10% of the population, and more than one-third of us will have substantial hearing loss by old age. In most cases, the auditory impairment results from hair cell death in the organ of Corti, the auditory organ. Because hair cells do not regenerate in the mammalian cochlea, loss of each cell, e.g., because of noise or toxic drugs, is irreversible and cumulative. If enough hair cells are lost, the end result is profound deafness. There is no medical treatment so far for this condition. The ability to prevent hair cell damage has in part been unattainable because the molecular mechanisms of noise- and ototoxic drug-induced trauma are largely unknown. Hair cells are innervated by the neurons of the cochlear ganglion. In most cases, cochlear neurons degenerate secondarily, after the loss of inner hair cells (IHCs), suggesting that hair cells should be the primary targets for therapeutic interventions aimed at preventing hearing loss.
In addition to immediate mechanical damage, oxidative stress associated with the formation of free radicals (see Discussion) and excitotoxicity (Basile et al., 1996) have been implicated in the pathogenesis of hair cell and hearing loss. Evidence on various cell lines and in vivo neuronal and non-neuronal model systems show that apoptotic death can be induced by both oxidative stress and excitotoxity (for review, see Pettmann and Henderson, 1998). In the inner ear, necrotic hair cell death, characterized by cellular swelling, has been demonstrated after acoustic trauma (Kellerhals, 1972). More recent data, obtained in the ototoxic drug-damaged inner ear, have suggested that hair cells may also die through apoptosis, based on the observations of nuclear fragmentation (Forge, 1985; Li et al., 1995; Liu et al., 1998; Nakagawa et al., 1998; Vago et al., 1998). However, the contribution of apoptotic hair cell death to the loss of hearing function is not known. In addition, the molecular mechanisms involved in commitment to hair cell death are unknown.
One of the signaling cascades that has been shown to mediate apoptotic death in response to a variety of stressful stimuli is the c-Jun-N-terminal kinase (JNK) pathway, also known as the stress-activated protein kinase (SAPK) pathway (Dérijard et al., 1994; Kyriakis et al., 1994). JNK activation by phosphorylation has been shown to be important for neuronal cell death after trophic factor withdrawal in vitro and after injury in vivo (Xia et al., 1995; Dickens et al., 1997; Yang et al., 1997). JNKs in turn phosphorylate c-Jun, a component of the transcription factor AP-1. Blockade of c-Jun activation and transcriptional activity in vitro has been shown to prevent neuronal cell death (Estus et al., 1994; Ham et al., 1995; Watson et al., 1998). Recent data from c-Jun phosphorylation-deficient mice (Behrens et al., 1999) show that c-Jun activation is essential for injury-induced neuronal death.
CEP-1347 is a small molecule derived from the indolocarbazole K252a (Kaneko et al., 1997). CEP-1347 has been recently shown to selectively inhibit the JNK signaling pathway and to protect neurons from insult-induced death in vitro and in vivo(Borasio et al., 1998; Glicksman et al., 1998; Maroney et al., 1998;Saporito et al., 1998, 1999). We have used this molecule to explore the role of the JNK pathway in insult-induced death in the cochlea.
MATERIALS AND METHODS
Cochlear cultures. The basal half of cochleas containing the basal and middle turns was dissected from postnatal day 2 Wistar rats. The cultures were maintained on Nuclepore (Pleasanton, CA) filters (pore size, 0.1 μm) placed on a metal grid in F-12 medium (Life Technologies, Gaithersburg, MD) containing 15% fetal bovine serum (Life Technologies). After a 2-hr-long stabilization period, explants were exposed to 100 μm neomycin sulfate (Sigma, St. Louis, MO) for 48 hr. CEP-1347 (500 nm) was added at the time of initiation of the cultures and every 12 hr thereafter.
Hair cell counts in cochlear cultures. Explants were fixed with 4% paraformaldehyde–0.5% glutaraldehyde in PBS, pH 7.4, for 30 min and dissected for surface preparations. They were stained with a 1:100 dilution of rhodamine–phalloidin (Molecular Probes, Eugene, OR) in PBS containing 0.25% Triton X-100 overnight at +4°C and mounted in Vectashield (Vector Laboratories, Burlingame, CA). Phalloidin is a specific marker for cellular F-actin. Outer hair cell (OHC) numbers were quantified with a Zeiss Axiovert 100/135 epifluorescent microscope connected to a Bio-Rad (Richmond, CA) MRC-1024 confocal laser scanning system. Hair cells were characterized as missing if no stereocilia and cuticular plate were observed. Numbers of OHCs were evaluated using a 40× objective lens and an ocular grid. Nine to 11 fields filled by 30 OHCs in each of the three rows (when all of them were present) were studied from each explant. Basal and middle coils were analyzed separately. Three separate experiments, each including four explants of both conditions (neomycin and neomycin plus CEP-1347) were analyzed.
Assessment of hair cell death and immunohistochemistry in cochlear cultures. Six, 12, and 24 hr after adding 100 μm neomycin to the medium, cochlear cultures were fixed with 4% paraformaldehyde in PBS for 30 min. The specimens were prepared for 5-um-thick paraffin sections. They were stained with a terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) kit (Fluorescein In Situ Cell Death Detection kit; Boehringer Mannheim, Mannheim, Germany), mounted in Vectashield containing 4′,6-diamidino-2-phenylindole (DAPI) nuclear counterstain (Vector Laboratories), and viewed under an Olympus (Tokyo, Japan) Provis microscope using epifluorescence. In addition to TUNEL staining, DNA fragmentation was verified under UV illumination using DAPI counterstain.
Adjacent sections were immunostained with a polyclonal phospho-c-Jun antibody (Ser73; 1:500; New England Biolabs, Beverly, MA) and a polyclonal phospho-JNK antibody (Thr183/Tyr185; 1:250 dilution; New England Biolabs). The phospho-JNK antibody detects the dually phosphorylated isoforms of JNK 1, 2, and 3. The specificity of the phospho-specific antibodies was verified by Western blotting using sorbitol-treated PC12 cells. Phospho-JNK antibody recognized the phosphorylated p54/p46 JNK and phospho-c-Jun antibody the phosphorylated p46 c-Jun in sorbitol-treated, but not in untreated PC12 cells (data not shown). For immunohistochemical detection, the avidin—biotin–peroxidase method (Elite ABC kit; Vector) and diaminobenzidine were used. Stainings were amplified using tyramide signal amplification (TSA-Indirect Kit, New England Nuclear, Boston, MA) according to Brady et al. (1999). No counterstaining was used in conjunction with the phospho-specific antibodies. In addition, a polyclonal calbindin antibody (1:10000 dilution; Swant, Bellinzona, Switzerland) was used as a marker for hair cells. Calbindin immunoreaction was detected using the ABC method and diaminobenzidine without tyramide signal amplification, and the sections were lightly counterstained with 1% toluidine blue. Analysis was done under Olympus Provis microscope and bright-field optics.
Noise lesioning. Adult Dunkin–Hartley female guinea pigs (weight, 300–500 gm) were exposed, four animals at a time, to octave band noise, with a center frequency of 4.0 kHz, 120 dB sound pressure level (SPL) for 6 hr as earlier described (Ylikoski et al., 1998).
Assessment of hair cell death after noise trauma. Fragmented hair cell nuclei were assessed from noise-exposed cochleas immediately and 1, 2, 4, 6, 8, and 14 d after noise exposure. Noise-exposed and nonexposed guinea pigs were decapitated under deep anesthesia, and inner ears were perilymphatically perfused with 4% paraformaldehyde in PBS and processed for 5-μm-thick paraffin-embedded sections (Ylikoski et al., 1998). TUNEL staining was done as described above. DNA fragmentation was also verified by DAPI nuclear counterstain (see above). Furthermore, trauma-induced nuclear fragmentation was verified by morphological analysis of contralateral cochleas that were perilymphatically fixed with 2.5% glutaraldehyde in 0.1 mphosphate buffer, pH 7.4, post-fixed with 1% osmium tetroxide, embedded in epoxy resin, and prepared for surface preparations (see below). Selected cochlear segments were then cut in transverse and horizontal planes to semithin (1.0 μm) sections that were stained with 1% toluidine blue. Analysis was done under an Olympus Provis microscope equipped with Nomarski optics.
Test-substance delivery. CEP-1347 was injected subcutaneously to noise-exposed guinea pigs at the dose of 1 mg/kg, starting 4 hr before noise exposure and continuing daily for 2 weeks. CEP-1347 was dissolved in 5% Solutol (BASF, Parsippany, NJ) in PBS, pH 7.4. Noise-exposed control guinea pigs received the vehicle alone.
Hearing tests. Auditory brainstem responses (ABRs) were measured 2 d before noise exposure, and 2, 6, and 14 d after exposure under anesthesia (xylazine, 10 mg/kg; ketamine, 40 mg/kg). One millisecond hanning windowed binaural stimuli with 0.5 msec rise and fall of frequencies 2, 4, 6, 8, 16, and 32 kHz were presented at a rate of 10 Hz. Preamplifier and custom data acquisition provided a gain of 80,000, with filtering of 0.3–10 kHz. Two responses summed from 1000 trials were obtained at each intensity level. Hearing threshold was determined from a set of responses at varying intensities. The threshold was defined as the last appearance of peak 3 or 4 in the ABR waveforms. Testing was performed in a sound-attenuated box, and animal temperature was kept at 37°C.
Cytocochleograms. Two weeks after noise exposure, control and CEP-1347-treated guinea pigs were decapitated under deep anesthesia, and inner ears were perilymphatically perfused with 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.4, post-fixed with 1% osmium tetroxide, embedded in epoxy resin, and processed for surface preparations as earlier described in detail (Ylikoski, 1974). Briefly, segments of the cochlear duct were dissected free from the modiolus, trimmed, and re-embedded in resin. The re-embedded cochlear segments were studied as surface preparations under an Olympus Provis microscope equipped with Nomarski optics. Hair cells were characterized as missing if both no cuticular plate and stereocilia in the appropriate location was observed. For cell countings, a 10 × 10 square eye reticular and a 40× objective lens were used. The percentage of missing hair cells was plotted as a function of the percentage length of the organ of Corti.
Dissociated neuronal cultures. Neuronal enriched cultures from E21 rat cochlear ganglia were prepared as previously described (Ylikoski et al., 1998). The cultures were maintained in F-14 medium (Life Technologies) containing 10% horse serum (Life Technologies). CEP-1347 (500 nm), neurotrophin-3 (NT-3; 2 ng/ml; Promega, Madison, WI), or nerve growth factor (NGF; 2 ng/ml; Promega) was added to the medium at the beginning and after each 12 hr. After 48 hr, cultures were fixed with 4% paraformaldehyde, and responses were assessed under a phase-contrast microscope. Surviving neurons were distinguished by a phase-bright cell body and definite neurites.
Statistics. Results are graphed as the mean ± SEM. Differences were assessed using paired Student's t test.p values of <0.05 and 0.01 were considered significant.
RESULTS
Hair cell death and activation of the JNK pathway after aminoglycoside-treatment in vitro
We first looked for hair cell death and involvement of the JNK signaling cascade in organotypic cochlear explants of neonatal rats. In paraffin sections of normal (nontreated) explants, the normal cellular architecture of the organ of Corti, one row of IHCs, and three rows of OHCs could be seen by using a calbindin antibody that labels hair cells (Fig. 1A). Cellular death was studied in paraffin sections stained by the TUNEL method, which labels fragmented DNA. Furthermore, when TUNEL-positive cells were found, DNA fragmentation was verified by using the DAPI counterstain. TUNEL-labeled hair cells were not found in normal explants (n = 5). In contrast, in explants (n = 15) exposed to 100 μmneomycin for 6, 12, 24, and 48 hr, TUNEL-positive hair cells were found (Fig. 1B,C). The majority of labeled hair cells were located in the basal cochlear turn where hair cells are most sensitive to ototoxic antibiotics. TUNEL-positive hair cell nuclei were found within the epithelium (Fig.1B,C), and, in addition, hair cells that had been extruded from the epithelium in the cultures showed nuclear fragmentation (data not shown).
Fig. 1.

Hair cell death and JNK and c-Jun phosphorylation in neomycin-exposed (100 μm) cochlear explants of neonatal rats. The specimens were embedded in paraffin and cut in transverse (midmodiolar) plane. A, One row of calbindin-immunoreactive inner hair cells and three rows of outer hair cells (arrows) are seen in nonexposed explants. B, TUNEL-stained outer hair cell nuclei (arrows) are seen in cultures exposed to neomycin for 12 hr. C, Higher magnification of an outer hair cell nucleus showing TUNEL-positive DNA fragmentation. D, E, Phospho-JNK and phospho-c-Jun immunolabeling, respectively, are found in the nuclei of hair cells (arrows) exposed to neomycin for 6 hr. F, Phospho-c-Jun-immunoreactive hair cells are not seen in cultures coincubated with neomycin and CEP-1347 (500 nm) for 6 hr. Arrows point to hair cells. Scale bar: A,B, D–F, 27 μm; C, 10 μm.
In cochlear explants (n = 10) exposed to 100 μm neomycin for 6 and 12 hr, phospho-JNK (Fig.1D) and phospho-c-Jun (Fig.1E)-immunoreactive hair cells were found in the lesioned regions, in the basal and lower middle cochlear turns. Only the nuclei of hair cells were stained by these phospho-specific antibodies. Hair cells situated in the apical, nonlesioned areas did not show phospho-JNK or phospho-c-Jun immunolabeling (data not shown), suggesting that the JNK pathway is involved in hair-cell stress responses. These findings formed the rationale to study the effects of CEP-1347 in the cochlea. CEP-1347 has been shown to attenuate neuronal apoptosis by blocking activation of JNK (Maroney et al., 1998). When cochlear explants (n = 5) were coincubated with 500 nm CEP-1347 and neomycin, both TUNEL-labeling (data not shown) and induction of JNK and c-Jun phosphorylation (Fig.1F) were prevented in hair cells. These data indicate that CEP-1347 is effective in blocking JNK activation in stressed hair cells.
CEP-1347 prevents neomycin-induced hair cell lossin vitro
To further examine the association between JNK activation and auditory hair cell death, we next examined the effect of CEP-1347 on neomycin-induced hair cell loss. Outer hair cell loss in cochlear explants of neonatal rats was assessed from surface preparations stained with rhodamine–phalloidin that strongly labels the mechanosensory region of hair cells, the cuticular plate and stereocilia (Fig.2A,B). In explants exposed to 100 μm neomycin for 48 hr (n = 12), ∼90% of OHCs in the basal turn and 25% of OHCs in the middle turn were lost (Fig. 2C). In cultures coincubated with 500 nm CEP-1347 and neomycin for 48 hr (n = 12), all OHCs in the middle turn and ∼90% of OHCs in the basal turn were preserved. Thus, CEP-1347 protected from neomycin-induced hair cell death in vitro.
Fig. 2.
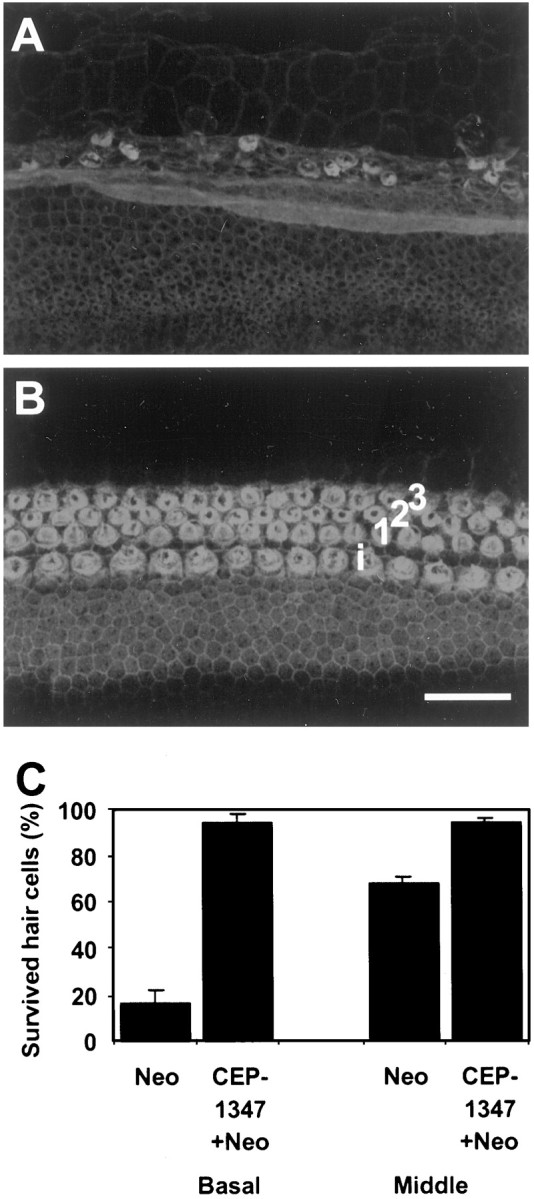
CEP-1347 (500 nm) prevents hair cell loss in cochlear explants of neonatal rats exposed to 100 μm neomycin for 48 hr. A, Neomycin causes severe hair cell degeneration in the basal turn of the cochlea, as shown in phalloidin-labeled confocal images. B, CEP-1347 prevents hair cell loss in the basal turn of the cochlea.i, Inner hair cell; 1, 2, 3, rows of outer hair cells. C, Quantification of numbers of survived outer hair cells in basal and middle turns. Histograms and bars represent mean ± SEM for three experiments, each including four explants of both the control and CEP-1347 group. Scale bar:A,B, 50 μm.
Assessment of hair cell death after noise exposure
Next, hair cell death was studied in guinea pig cochleas exposed to intense noise (120 dB SPL, 4 kHz for 6 hr). This noise regimen induced maximal damage to hair cells of the organ of Corti at the 9 mm region from the round window (second cochlear turn). Noise-exposed (n = 16) and nonexposed (n = 3) cochleas were cut in transverse (midmodiolar) plane and doublestained with the DAPI counterstain (Fig.3A) and with TUNEL method (Fig. 3B,C). In addition, resin-embedded organs of Corti of noise-exposed cochleas (n = 6) were cut in transverse (Fig. 3D) or horizontal plane (Fig. 3E,F), and stained with toluidine blue. Noise-exposed cochleas were analyzed immediately and 1, 2, 4, 6, 8, and 14 d after exposure. TUNEL-labeled hair cells were found during the first 4 d after noise exposure. Most of the labeled cells were located in the area of maximal lesion, the second cochlear turn (Fig. 3E). During the study period, the first 2 weeks after noise exposure, TUNEL labeling was not detected in the supporting cells of the organ of Corti (Fig. 3A,B) or in the cochlear neurons. In nonexposed cochleas, cells of the organ of Corti as well as neurons were not TUNEL-labeled (data not shown).
Fig. 3.

Hair cell death in the guinea pig cochlea 1 d after intense noise exposure (120 dB, 4 kHz, 6 hr). A,B, The transverse (midmodiolar) paraffin section of the organ of Corti in the second cochlear turn is doublestained with DAPI nuclear stain (A) and with TUNEL method (B). The three outer hair cells (arrow) show distorted nuclei and are TUNEL-positive. Supporting cells, seen below the hair cells, are not TUNEL-labeled.C, At high magnification, a TUNEL-labeled paraffin section shows an outer hair cell nucleus with DNA fragmentation.D, A toluidine blue-stained, resin-embedded semithin section of the organ of Corti in transverse plane shows an inner hair cell (large arrow) and three rows of OHCs (small arrows). Only OHC1 shows a fragmented nucleus (thick arrow). The section is from the area of scattered hair cell loss. De, Deiter's cells. E, A toluidine blue-stained, resin-embedded semithin section in horizontal plane and at the level of hair cell nuclei. The section is from the region of maximal trauma. Most hair cells are lost, except one outer hair cell that shows a fragmented nucleus (thick arrow).F, A section, prepared as in E, from nontraumatized region of the organ of Corti. Outer hair cells of the three rows are present, and their nuclei are not fragmented. Scale bar:A,B, 80 μm; C, 10 μm;D, 18 μm; E,F, 15 μm.
CEP-1347 attenuates noise-induced hearing lossin vivo
Data presented thus far suggest that auditory hair cells can die by an apoptotic pathway and that this pathway includes activation of JNK. We were, therefore, interested to determine whether an inhibitor of the JNK pathway could affect hearing function. To assess the effect of CEP-1347 on noise-induced hearing loss, control and CEP-1347-treated guinea pigs were exposed to 120 dB, 4 kHz for 6 hr. CEP-1347 was delivered subcutaneously, starting 2 hr before the noise exposure and continuing daily for 2 weeks. Hearing thresholds 2 d before noise exposure (baseline values) showed no significant difference between the noise-exposed control and treated group (data not shown). Noise exposure caused threshold shifts in all guinea pigs as seen 2 d after the exposure (noise-exposed control, n = 4; CEP-treated, n = 5) (Fig.4A). By day 6 after exposure, threshold shifts were significantly less in the CEP-1347 group (n = 11) than in the noise-exposed control group (n = 8, p < 0.05 at 4 and 8 kHz; p < 0.01 at 16 kHz) (Fig.4B). By 2 weeks after exposure, the difference between the two groups became even more pronounced (noise-exposed control, n = 17; CEP-treated, n = 17) (Fig. 4C). At this stage, ABR threshold shifts ranged from 28 to 45 dB SPL in noise-exposed controls, and from 12 to 22 dB SPL in CEP-1347-treated animals (p < 0.01 at all frequencies).
Fig. 4.
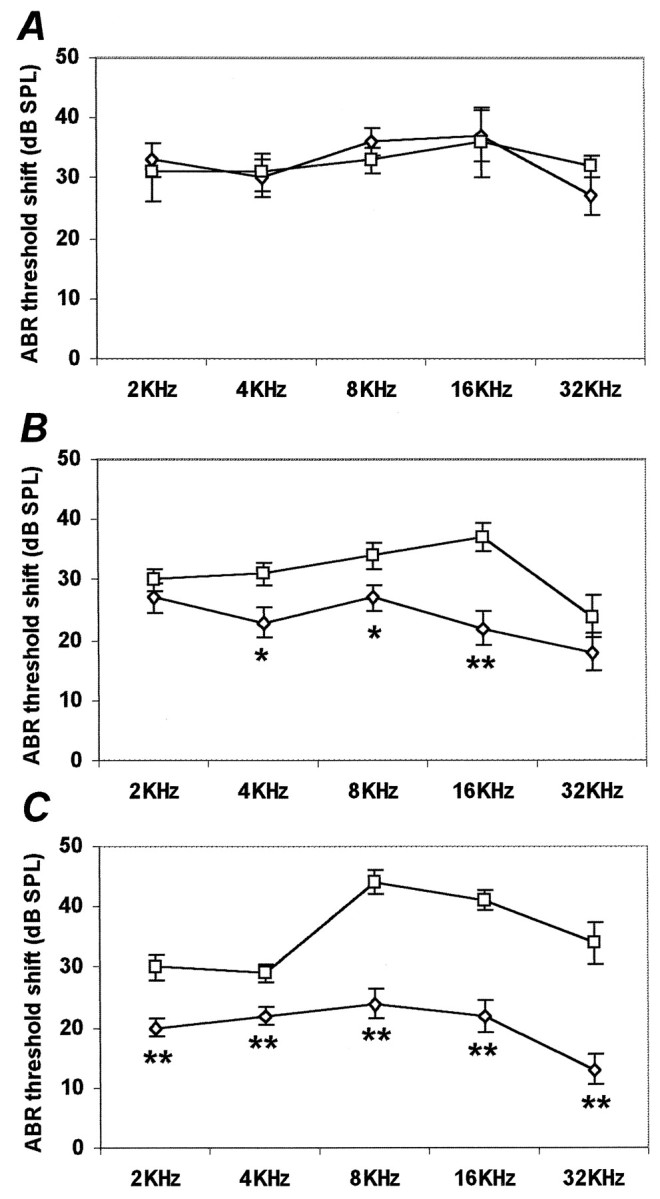
CEP-1347 attenuates intense noise-induced hearing loss in vivo. Hearing threshold shifts were assessed by auditory brainstem testing 2 (A), 6 (B), and 14 (C) d after exposing control (■; n = 4 at 2 d, 8 at 6 d, 17 at 14 d) and CEP-1347-treated (⋄;n = 5 at 2 d, 11 at 6 d, 17 at 14 d) guinea pigs to noise (120 dB, 4 kHz, 6 hr). Results show mean ± SEM. Six and 14 d after exposure, the average threshold shifts of control animals are significantly greater than that of CEP-1347-treated animals. **p < 0.01; *p < 0.05; Student's t test.
CEP-1347 attenuates noise-induced hair cell loss
At 2 weeks after noise exposure, and after final ABR recordings, the numbers of preserved and missing hair cells were assessed in resin-embedded cochlear surface preparations. Total number of OHCs per guinea pig cochlea (lost plus preserved hair cells) ranged from 6240 to 6897 (Table 1). There was a large variation in the amount of hair cell damage in noise-exposed control cochleas in which the numbers of lost hair cells ranged from 305 to 1266 (n = 13). In CEP-1347-treated cochleas (n = 13), the number of missing hair cells ranged from 106 to 291, except that one cochlea of this group showed hair cell loss that was at the range of the noise-exposed control group (Table 1). The difference between the groups is statistically significant (p < 0.01). In vehicle-treated, noise-exposed control cochleas, in addition to the missing hair cells, there were several distorted OHCs with irregular configuration and disarrayed stereocilia, but a preserved cuticular plate and nucleus in regions adjacent to the site of maximal damage. Most of the noise-exposed control cochleas showed a relatively well demarcated area of maximal damage, extending from 0.5 to 1.0 mm in the region of ∼9 mm from the round window. In addition, scattered OHC loss was found particularly along the entire upper half of the cochlea. In the region of ∼9 mm from the round window, CEP-1347-treated cochleas showed some destruction of the organ of Corti, but usually in an area extending from 0.1 to 0.2 mm only. Also scattered OHC loss was less than in noise-exposed control cochleas. The cytocochleograms (Fig.5A,B) illustrate the average percentages of missing IHCs and OHCs along the length of the organ of Corti in noise-exposed control (n = 13) and CEP-1347-treated (n = 13) cochleas. In the average cytocochleogram of the CEP-1347-treated group (Fig. 5B), the second, more apically located peak is caused by the single specimen in which the total hair cell loss corresponded to that of the control group. Altogether, these results indicate that, in our noise paradigm, CEP-1347 rescues IHCs as well as OHCs of each of the three rows of the guinea pig cochlea.
Table 1.
Number of preserved and lost IHCs and OHCs of different rows (OHC1, OHC2, OHC3) in CEP-1347-treated and control (CON) cochleas
| Cochlea # | OHCs total | Lost hair cells | OHC loss total (%) | |||
|---|---|---|---|---|---|---|
| IHC | OHC1 | OHC2 | OHC3 | |||
| CEP-0207 | 6897 | 1 | 40 | 27 | 38 | 105 (1.5) |
| CEP-1407 | 6699 | 24 | 20 | 38 | 47 | 105 (1.6) |
| CEP-0605 | 6748 | 9 | 38 | 45 | 58 | 141 (2.1) |
| CEP-2203 | 6666 | 17 | 41 | 50 | 54 | 145 (2.2) |
| CEP-2003 | 6585 | 19 | 53 | 37 | 64 | 154 (2.3) |
| CEP-1604 | 6573 | 28 | 42 | 67 | 74 | 183 (2.8) |
| CEP-2503 | 6846 | 14 | 61 | 53 | 76 | 190 (2.8) |
| CEP-0307 | 6672 | 7 | 72 | 62 | 63 | 197 (3.0) |
| CEP-0707 | 6387 | 6 | 74 | 74 | 56 | 204 (3.2) |
| CEP-0305 | 6795 | 24 | 57 | 60 | 93 | 210 (3.1) |
| CEP-2603 | 6723 | 17 | 64 | 66 | 80 | 210 (3.1) |
| CEP-1101 | 6240 | 2 | 59 | 69 | 161 | 289 (4.7) |
| CEP-0512 | 6454 | 25 | 180 | 198 | 229 | 607 (9.4) |
| CON-2007d | 6687 | 23 | 95 | 92 | 95 | 282 (4.2) |
| CON-0601 | 6423 | 2 | 65 | 149 | 114 | 328 (5.1) |
| CON-0307 | 6421 | 33 | 77 | 109 | 178 | 364 (5.7) |
| CON-1903d | 6480 | 77 | 89 | 136 | 141 | 366 (5.6) |
| CON-2007s | 6630 | 43 | 125 | 111 | 139 | 375 (5.7) |
| CON-1204 | 6897 | 56 | 102 | 139 | 184 | 425 (6.2) |
| CON-1903s | 6690 | 56 | 118 | 119 | 225 | 462 (6.9) |
| CON-2703 | 6507 | 77 | 205 | 117 | 184 | 506 (7.8) |
| CON-0405 | 6621 | 7 | 96 | 156 | 255 | 507 (7.7) |
| CON-1807 | 6621 | 7 | 96 | 156 | 255 | 507 (7.7) |
| CON-0412 | 6849 | 11 | 161 | 209 | 281 | 659 (9.5) |
| CON-0604 | 6882 | 78 | 156 | 222 | 341 | 719 (10.4) |
| CON-0407 | 6690 | 117 | 222 | 413 | 514 | 1149 (17.2) |
Guinea pigs were exposed to 120 dB, 4 kHz noise for 6 hr. Hair cell counts were done 2 weeks after the exposure.
Fig. 5.

CEP-1347 attenuates intense noise-induced hair cell loss in vivo. Quantification of hair cell numbers in vehicle-treated, noise-exposed control (A;n = 13) and CEP-1347-treated guinea pig cochleas (B;n = 13) 2 weeks after noise exposure (120 dB, 4 kHz, 6 hr). In the averaged cytocochleograms, the percentages of IHCs and OHCs are plotted for the entire length of the organ of Corti. Maximal damage is located in a region ∼50% from the round window. CEP-1347 treatment attenuates hair cell loss.
CEP-1347 promotes survival of dissociated cochlear neurons
Finally we asked whether CEP-1347 attenuates death of cochlear neurons in vitro, in addition to the hair cells. NT-3 is the most potent neurotrophic factor promoting survival of dissociated cochlear neurons of the perinatal rat, and NGF does not have any effect or its effect is very weak (Ylikoski et al., 1998). As assessed in parallel cultures, CEP-1347 was as efficacious as NT-3 in promoting survival of cochlear neurons, and NGF served as a negative control (Fig. 6).
Fig. 6.

Effects of CEP-1347 (500 nm), neurotrophin-3 (2 ng/ml), and nerve growth factor (2 ng/ml) on the survival of neonatal cochlear neurons in vitro. Numbers of neurons in cultures with added compounds are expressed as percentage of the number of neurons in control cultures. Neurons were counted after 48 hr in culture. Values represent mean ± SEM from three separate experiments.
DISCUSSION
We initiated the present study to characterize the mode of hair cell death in the traumatized cochlea. Based on morphological analysis and specific DNA labelings, fragmented hair cell nuclei were found after a 120 dB, 4 kHz noise for 6 hr. Other noise paradigms were not investigated in the present study. In addition to the present study, previous studies (Forge, 1985; Li et al., 1995; Liu et al., 1998; Nakagawa et al., 1998; Vago et al., 1998) have found DNA fragmentation in hair cells challenged with ototoxic antibiotics. Taken together, these data suggest that both mechanical and chemical agents induce hair cell death that has characteristic features of apoptosis.
Phospho-JNK and phospho-c-Jun immunoreactivity was detected in hair cells situated in the lesioned regions of neomycin-exposed explants, but not in nonlesioned areas, suggesting that activation of the JNK pathway is involved in the hair-cell stress responses. Both phosphorylated c-Jun, the transcription factor, and phosphorylated JNK, the immediate upstream kinase, were localized to the nuclei of hair cells. These immunohistochemical results are in line with previous biochemical data showing that once activated, JNKs translocate from the cytosol into the nucleus (Mizukami et al., 1997). Physiological evidence of the involvement of the JNK pathway in stressed hair cells comes from our experiments in which the indolocarbazole CEP-1347 was applied in conjunction with neomycin to organotypic cochlear cultures. CEP-1347 has been shown to block neuronal apoptosis by inhibiting the JNK signaling pathway (Maroney et al., 1998). Similarly as in neurons, CEP-1347 blocked activation of the JNK pathway in neomycin-exposed hair cells and blocked hair cell death.
JNKs are known to be activated in response to various cellular stresses such as UV irradiation, heat shock, chemotherapeutic drugs, inflammatory cytokines, osmotic imbalance, oxidative stress, and excitotoxicity (for review, see Ip and Davis, 1998). The mechanisms by which the different stressful stimuli lead to JNK activation remain largely to be determined. It has been shown that reactive oxygen species (ROS) are formed during oxidative stress and activate JNKs (for review, see Finkel, 1998). It has also been shown that JNK activation is inhibited by pretreatment of cells with antioxidants (Lo et al., 1996). ROS generation and changes in the antioxidant defense system of the cochlea have been implicated in ototoxic drug-induced hair cell damage (Hoffman et al., 1988; Garetz et al., 1994; Priuska and Schacht, 1995; Clerici et al., 1996; Hirose et al., 1997; Conlon et al., 1999). We suggest that JNK activation could be one of the intracellular pathways leading to hair cell death after ototoxic drug-induced ROS generation.
CEP-1347 protected cochlear hair cells from neomycin-induced ototoxicity in vitro. At 100 μmneomycin concentration, ∼90% of hair cells in the basal coil and 25% of hair cells in the middle coil of the cochlea were lost. Protection by CEP-1347 was prominent. It was total in the middle coil, and in the basal coil ∼90% of the hair cells were preserved. In addition to aminoglycoside antibiotics, hair cells are sensitive to the chemotherapeutic drug cisplatin. As shown in mutant fibroblastic cell lines, cisplatin causes apoptosis by activating JNKs, and fibroblasts expressing a dominant inhibitor of JNK activation are resistant to cisplatin-induced apoptosis (Zanke et al., 1996). The JNK pathway is activated in cisplatin-treated hair cells in vitro (U. Pirvola and L. Xing-Qun, unpublished observations). These data suggest that, in addition to aminoglycoside-induced hair cell death, CEP-1347 may be effective in blocking hair cell death after cisplatin treatment.
CEP-1347 protected guinea pig cochleas from damage caused by intense noise, as demonstrated by functional tests and by morphometric methods. CEP-1347-treated animals showed less ABR threshold shift and less hair cell loss than noise-exposed controls. Threshold shifts were similar in all guinea pigs 2 d after noise, indicating that the animals were similarly responsive after exposure to sound. The protective effect of CEP-1347 was evident by day 6 and even more pronounced by 2 weeks after exposure. At day 2 after lesion, most of the threshold shift is attributable to temporary threshold shift (TTS), whereas after 2 weeks the threshold shift is mainly attributable to permanent threshold shift (PTS). It is obvious that CEP-1347 cannot prevent TTS, which is thought to arise mainly from various reversible structural changes in the cochlea, such as changes in the mechanical compliance of the basilar membrane (LePage, 1989) and decreased stiffness of the hair-cell stereocilia (Saunders et al., 1986).
After exposure to intense noise, the relationship between PTS and the extent of hair cell loss is not always linear. The decreased auditory sensitivity has been explained not only by hair cell loss, but also by subtle changes within the cells of the organ of Corti (Saunders et al., 1991). Similarly as in a previous study (Fredelius et al., 1988), our noise paradigm induced threshold shift that affected a wide range of frequencies, whereas maximal hair cell loss was found in a rather restricted region. There was a linear relationship between the auditory impairment and hair cell loss at the 9 mm region from the round window, the site maximally stimulated by our noise paradigm. Fine structural changes that might contribute to the broad hearing loss were not investigated in the present study. Cytocochleograms show that, in addition to the hair cell loss in the maximal trauma area, CEP-1347 prevented scattered hair cell loss seen at lower and higher frequencies. It may be that protection of scattered hair cell loss contributed to the smaller ABR threshold shifts of the treated animals. Altogether, the present results showing reduced ABR threshold shifts in CEP-1347-treated animals indicate that CEP-1347 attenuates noise-induced hearing impairment.
Cytocochleograms prepared 2 weeks after noise exposure showed that CEP-1347 treatment rescued >70% of OHCs destined to die. A suggested cause for noise-induced hair cell death, excluding the initial death caused by pure mechanical damage, is impaired cochlear metabolism and oxidative stress. Like ototoxic drugs, noise induces ROS formation, and ROS scavengers and inhibitors have been implicated in protection against noise-induced hearing loss (Quirk et al., 1994; Hu et al., 1997; Jacono et al., 1998; Yamasoba et al., 1998). The present study shows that CEP-1347 prevents both intense noise- and aminoglycoside-induced hair cell death. In both types of traumas, ROS might activate the JNK pathway leading to hair cell apoptosis. Interestingly, in vivo CEP-1347 has been shown to attenuate loss of nigrastriatal dopaminergic neurons after exposure to a neurotoxin, 1-methyl-4-phenyl tetrahydropyridine, that causes oxidative stress and neuronal death (Saporito et al., 1999). Taken together, these data implicate the JNK pathway and suggest the therapeutic potential of CEP-1347 in non-neuronal and neuronal models of oxidative stress.
In addition to cochlear hair cells, CEP-1347 rescued developing cochlear neurons from death in vitro, indicating that the JNK signaling cascade is involved in regulation of cochlear neuron survival. In line with the present data, CEP-1347 promotes survival of several neuronal populations during development, such as dorsal root ganglion sensory neurons, sympathetic neurons, and motoneurons (Borasio et al., 1998; Glicksman et al., 1998; Maroney et al., 1998). The present results indicate that CEP-1347 is at least as efficacious as NT-3, the most potent neurotrophic factor acting on perinatal cochlear neurons (Ylikoski et al., 1998). Withdrawal of a survival factor may activate genes whose products promote cellular death (Johnson and Deckwerth, 1993), including components of the stress-activated signaling cascade. In line with this suggestion, neurotrophic factor withdrawal from neuronal cultures activates JNK (Xia et al., 1995;Dickens et al., 1997) and c-Jun (Watson et al., 1998) and leads to apoptosis. In addition to in vitro models, CEP-1347 has been shown to prevent injury-induced death of adult CNS neurons in vivo (Saporito et al., 1998, 1999). It remains to be shown whether neuronal protection can be achieved in the traumatized adult cochlea by using CEP-1347.
During recent years various agents have been identified that attenuate drug- or noise-induced hearing loss and degeneration of hair cells or neurons, in animal models either in vivo or in vitro. For many agents, such as free radical scavengers and inhibitors, the efficacy has been variable, and there has been little firm data on their molecular mechanism in the cochlea. In animal models, local delivery of neurotrophic factors into the lesioned cochlea has been shown to rescue cochlear neurons (Ernfors et al., 1996; Ylikoski et al., 1998). Neurotrophic factors have also been shown to attenuate trauma-induced hair cell loss (Keithley et al., 1998; Ruan et al., 1999), but the molecular basis for this protection is unclear and may be indirect. Clinical use of polypeptide growth factors has been limited by their pharmaceutical properties, such as low stability and difficult delivery characteristics (Yuen and Mobley, 1996). Small trophic molecules acting on intracellular signaling pathways and allowing systemic delivery represent an alternative therapeutic approach. The current study provides evidence that CEP-1347, an inhibitor of the JNK signaling pathway, may offer therapeutic potential to prevent death of both cochlear hair cells and neurons and to act against both acoustic and ototoxic trauma. This study shows that CEP-1347 rescues hair cells when the treatment is started before trauma. In clinics, the therapeutic agent should show efficacy also when the treatment is initiated after trauma. Our unpublished data show that CEP-1347 protects cochlear hair cells when dosing is started within a few days after noise exposure.
Footnotes
This work was supported by grants from the Academy of Finland, Technology Agency of Finland, and the Sigrid Jusélius Foundation. We kindly thank Maria von Numers for expert technical assistance.
Correspondence should be addressed to Ulla Pirvola, Institute of Biotechnology, University of Helsinki, P.O. Box 56, 00014 Helsinki, Finland. E-mail: ulla.pirvola@helsinki.fi.
REFERENCES
- 1.Basile AS, Huang J-M, Xie C, Webster D, Berlin C, Skolnik P. N-methyl-d-aspartate antagonists limit aminoglycoside antibiotic-induced hearing loss. Nat Med. 1996;2:1338–1343. doi: 10.1038/nm1296-1338. [DOI] [PubMed] [Google Scholar]
- 2.Behrens A, Sibilia M, Wagner E. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- 3.Borasio GD, Horstmann S, Anneser JMH, Neff NT, Glicksman MA. CEP-1347/KT7515, a JNK pathway inhibitor, supports the in vitro survival of chick embryonic neurons. NeuroReport. 1998;9:1435–1439. doi: 10.1097/00001756-199805110-00034. [DOI] [PubMed] [Google Scholar]
- 4.Brady R, Zaidi SIA, Mayer C, Katz DM. BDNF is a target-derived survival factor for arterial baroreceptor and chemoafferent primary sensory neurons. J Neurosci. 1999;19:2131–2142. doi: 10.1523/JNEUROSCI.19-06-02131.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clerici WJ, Hensley K, DiMartino DL, Butterfield DA. Direct detection of ototoxicant-induced reactive oxygen species generation in cochlear explants. Hear Res. 1996;98:116–124. doi: 10.1016/0378-5955(96)00075-5. [DOI] [PubMed] [Google Scholar]
- 6.Conlon BJ, Aran JM, Erre JP, Smith DW. Attenuation of aminoglycoside-induced cochlear damage with the metabolic antioxidant α-lipoic acid. Hear Res. 1999;128:40–44. doi: 10.1016/s0378-5955(98)00195-6. [DOI] [PubMed] [Google Scholar]
- 7.Dérijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 8.Dickens M, Rogers JS, Cavanagh J, Raitano A, Xia Z, Halpern JR, Greenberg ME, Sawyers CL, Davis RJ. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- 9.Ernfors P, Duan ML, ElShamy WM, Canlon B. Protection of auditory neurons from aminoglycoside toxicity by neurotrophin-3. Nat Med. 1996;2:463–467. doi: 10.1038/nm0496-463. [DOI] [PubMed] [Google Scholar]
- 10.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finkel T. Oxygen radicals and signaling. Curr Opin Cell Biol. 1998;10:248–253. doi: 10.1016/s0955-0674(98)80147-6. [DOI] [PubMed] [Google Scholar]
- 12.Forge A. Outer hair cell loss and supporting cell expansion following chronic gentamycin treatment. Hear Res. 1985;19:171–182. doi: 10.1016/0378-5955(85)90121-2. [DOI] [PubMed] [Google Scholar]
- 13.Fredelius L, Rask-Andersen H, Johansson B, Urquiza R, Bagger-Sjöbäck D, Wersäll J. Time sequence of degeneration pattern of the organ of Corti after acoustic overstimulation. A light microscopical and electrophysiological investigation in the guinea pig. Acta Otolaryngol (Stockh) 1988;106:81–93. doi: 10.3109/00016488809107374. [DOI] [PubMed] [Google Scholar]
- 14.Garetz SL, Rhee DJ, Schacht J. Sulfhydryl compounds and antioxidants inhibit cytotoxicity to outer hair cells of a gentamicin metabolite in vitro. Hear Res. 1994;77:75–80. doi: 10.1016/0378-5955(94)90254-2. [DOI] [PubMed] [Google Scholar]
- 15.Glicksman MA, Chiu AY, Dionne CA, Harty M, Kaneko M, Murakata C, Oppenheim RW, Prevette D, Sengelaub DR, Vaught JL, Neff NT. CEP-1347/KT7515 prevents motor neuronal programmed cell death and injury-induced dedifferentiation in vivo. J Neurobiol. 1998;35:361–370. [PubMed] [Google Scholar]
- 16.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 17.Hirose K, Hockenbery DM, Rubel EW. Reactive oxygen species in chick hair cells after gentamicin exposure in vitro. Hear Res. 1997;104:1–14. doi: 10.1016/s0378-5955(96)00169-4. [DOI] [PubMed] [Google Scholar]
- 18.Hoffman DW, Whitworth CA, Jones-King KL, Rybak LP. Potentiation of ototoxicity by glutathione depletion. Ann Otol Rhinol Laryngol. 1988;97:36–41. doi: 10.1177/000348948809700107. [DOI] [PubMed] [Google Scholar]
- 19.Hu BH, Zheng XY, McFadden SL, Kopke RD, Henderson D. R-phenylisopropyladenosine attenuates noise-induced hearing loss in the chinchilla. Hear Res. 1997;113:198–206. doi: 10.1016/s0378-5955(97)00143-3. [DOI] [PubMed] [Google Scholar]
- 20.Ip YT, Davis RG. Signal transduction by the c-Jun N-terminal kinase (JNK) –from inflammation to development. Curr Opin Cell Biol. 1998;10:205–219. doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- 21.Jacono AA, Hu B, Kopke RD, Henderson D, Van De Water T, Steinman HM. Changes in cochlear antioxidant enzyme activity after sound conditioning and noise exposure in the chinchilla. Hear Res. 1998;117:31–38. doi: 10.1016/s0378-5955(97)00214-1. [DOI] [PubMed] [Google Scholar]
- 22.Johnson EM, Deckwerth TL. Molecular mechanisms of developmental neuronal death. Annu Rev Neurosci. 1993;16:31–46. doi: 10.1146/annurev.ne.16.030193.000335. [DOI] [PubMed] [Google Scholar]
- 23.Kaneko M, Saito Y, Saito H, Matsumoto T, Matsuda Y, Vaught JL, Dionne CA, Angeles TS, Glicksman MA, Neff NT, Rotella DP, Kauer JC, Mallamo JP, Hudkins RL, Murakata C. Neurotrophic 3,9-bis[(alkylthio)methyl]-and-bis(alkoxymethyl)-K-252a derivatives. J Med Chem. 1997;40:1863–1869. doi: 10.1021/jm970031d. [DOI] [PubMed] [Google Scholar]
- 24.Keithley EM, Ma CL, Ryan AF, Louis JC, Magal E. GDNF protects the cochlea against noise damage. NeuroReport. 1998;13:2183–2187. doi: 10.1097/00001756-199807130-00007. [DOI] [PubMed] [Google Scholar]
- 25.Kellerhals B. Acoustic trauma and cochlear microcirculation. Adv Otorhinolaryngol. 1972;18:91–168. [PubMed] [Google Scholar]
- 26.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–160. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 27.LePage EL. Functional role of the olivocochlear bundle: a motor unit control system in the mammalian cochlea. Hear Res. 1989;38:177–198. doi: 10.1016/0378-5955(89)90064-6. [DOI] [PubMed] [Google Scholar]
- 28.Li L, Nevill G, Forge A. Two modes of hair cell loss from the vestibular sensory epithelia of the guinea pig inner ear. J Comp Neurol. 1995;355:405–417. doi: 10.1002/cne.903550307. [DOI] [PubMed] [Google Scholar]
- 29.Liu W, Staecker H, Stupak H, Malgrande B, Lefebvre P, Van De Water TR. Caspase inhibitors prevent cisplatin-induced apoptosis of auditory sensory cells. NeuroReport. 1998;9:2609–2614. doi: 10.1097/00001756-199808030-00034. [DOI] [PubMed] [Google Scholar]
- 30.Lo YYC, Wong JMS, Cruz TF. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem. 1996;271:15703–15707. doi: 10.1074/jbc.271.26.15703. [DOI] [PubMed] [Google Scholar]
- 31.Maroney AC, Glicksman MA, Basma AN, Walton KM, Knight E, Jr, Murphy CA, Bartlett BA, Finn JP, Angeles T, Matsuda Y, Neff NT, Dionne CA. Motoneuron apoptosis is blocked by CEP-1347 (KT 7515), a novel inhibitor of the JNK signaling pathway. J Neurosci. 1998;18:104–111. doi: 10.1523/JNEUROSCI.18-01-00104.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizukami Y, Yoshioka K, Morimoto S, Yoshida K. A novel mechanism of JNK1 activation. Nuclear translocation and activation of JNK1 during ischemia and reperfusion. J Biol Chem. 1997;272:16657–16662. doi: 10.1074/jbc.272.26.16657. [DOI] [PubMed] [Google Scholar]
- 33.Nakagawa T, Yamane H, Takayama M, Sunami K, Nakai Y. Apoptosis of guinea pig cochlear hair cells following chronic aminoglycoside treatment. Eur Arch Otorhinolaryngol. 1998;255:127–131. doi: 10.1007/s004050050027. [DOI] [PubMed] [Google Scholar]
- 34.Pettmann B, Henderson CE. Neuronal cell death. Neuron. 1998;20:633–647. doi: 10.1016/s0896-6273(00)81004-1. [DOI] [PubMed] [Google Scholar]
- 35.Priuska EM, Schacht J. Formation of free radicals by gentamicin and iron and evidence for an iron/gentamicin complex. Biochem Pharmacol. 1995;50:1749–1752. doi: 10.1016/0006-2952(95)02160-4. [DOI] [PubMed] [Google Scholar]
- 36.Quirk WS, Shivapuja BG, Schwimmer CL, Seidman MD. Lipid peroxidation inhibitor attenuates noise-induced temporary threshold shifts. Hear Res. 1994;74:217–220. doi: 10.1016/0378-5955(94)90189-9. [DOI] [PubMed] [Google Scholar]
- 37.Ruan RS, Leong SK, Mark I, Yeoh KH. Effects of BDNF anf NT-3 on hair cell survival in guinea pig cochlea damaged by kanamycin treatment. NeuroReport. 1999;10:2067–2071. doi: 10.1097/00001756-199907130-00014. [DOI] [PubMed] [Google Scholar]
- 38.Saporito MS, Brown ER, Carswell S, DiCamillo AM, Miller MS, Murakata C, Neff NT, Vaught NT, Haun FA. Preservation of cholinergic activity and prevention of neuron death by CEP-1347/KT-7515 following excitotoxic injury of the nucleus basalis magnocellularis. Neuroscience. 1998;86:461–472. doi: 10.1016/s0306-4522(98)00059-1. [DOI] [PubMed] [Google Scholar]
- 39.Saporito MS, Brown EM, Miller MS, Carswell S. CEP-1347/KT-7515, an inhibitor of c-Jun N-terminal kinase activation, attenuates the 1-methyl-4-phenyl tetrahydropyridine-mediated loss of nigrostriatal dopaminergic neurons in vivo. J Pharmacol Exp Ther. 1999;288:421–427. [PubMed] [Google Scholar]
- 40.Saunders JC, Canlon B, Flock Å. Changes in stereocilia micromechanics following overstimulation in metabolically blocked hair cells. Hear Res. 1986;24:217–225. doi: 10.1016/0378-5955(86)90020-1. [DOI] [PubMed] [Google Scholar]
- 41.Saunders JC, Cohen YE, Szymko YM. The structural and functional consequences of acoustic injury in the cochlea and peripheral auditory system: a five year update. J Acoust Soc Am. 1991;90:136–146. doi: 10.1121/1.401307. [DOI] [PubMed] [Google Scholar]
- 42.Vago P, Humbert G, Lenoir M. Amikacin intoxication induces apoptosis and cell proliferation in rat organ of Corti. NeuroReport. 1998;9:431–436. doi: 10.1097/00001756-199802160-00014. [DOI] [PubMed] [Google Scholar]
- 43.Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham H. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J Neurosci. 1998;15:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK1–p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 45.Yamasoba T, Nuttall AL, Harris C, Raphael Y, Miller JM. Role of glutathione in protection against noise-induced hearing loss. Hear Res. 1998;784:82–90. doi: 10.1016/s0006-8993(97)01156-6. [DOI] [PubMed] [Google Scholar]
- 46.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the JNK3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 47.Yuen EC, Mobley WC. Therapeutic potential of neurotrophic factors for neurological disorders. Ann Neurol. 1996;40:346–354. doi: 10.1002/ana.410400304. [DOI] [PubMed] [Google Scholar]
- 48.Ylikoski J. Guinea-pig hair cell pathology from ototoxic antibiotics. Acta Otolaryngol (Stockh) [Suppl] 1974;326:5–20. doi: 10.3109/00016487409129729. [DOI] [PubMed] [Google Scholar]
- 49.Ylikoski J, Pirvola U, Virkkala J, Suvanto P, Liang X-Q, Magal E, Altschuler R, Miller JM, Saarma M. Guinea pig auditory neurons are protected by glial cell line-derived growth factor from degeneration after noise trauma. Hear Res. 1998;124:17–26. doi: 10.1016/s0378-5955(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 50.Zanke BW, Boudreau K, Rubie E, Winnett E, Tibbles LA, Zon L, Kyriakis J, Liu F-F, Woodgett JR. The stress-activated protein kinase pathway mediates cell death following injury induced by cis-platinium, UV irradiation or heat. Curr Biol. 1996;6:606–613. doi: 10.1016/s0960-9822(02)00547-x. [DOI] [PubMed] [Google Scholar]