

Abstract
Cannabinoid effects on sustained conductances that control neuronal excitability have not been investigated in brain. Here, intracellular voltage-clamp recordings were performed using the rat hippocampal slice preparation to study the postsynaptic effect of cannabinoid agonists on CA1 pyramidal neurons. Superfusion of the cannabimimetics WIN55212–2 or methanandamide onto CA1 neurons elicited an inward steady-state current that reversed near the equilibrium potential for K+ and voltage-dependently activated from a threshold of approximately −70 mV. The cannabinoid receptor (CB1) antagonist SR141716 did not alter membrane properties but prevented this effect. Further investigation revealed that the inward current elicited by cannabinoids was caused by a decrease of the noninactivating voltage-dependent K+ M-current (IM). Cannabinoids had no effect in slices pretreated with the M-channel blocker linopirdine. Assessment of the IM relaxation indicated that cannabinoids decreased IM in a concentration-dependent manner, with a maximum inhibition of 45 ± 3% with WIN55212–2 (EC50 of 0.6 μm) and 41 ± 5% with methanandamide (EC50 of 1 μm). Cannabinoids did not affect the inwardly rectifying cationic h-current (Ih). The cannabinoid-induced IM decrease was prevented by SR141716 but remained unaffected by the muscarinic receptor antagonist atropine. Conversely, the cholinergic agonist carbamylcholine decreased IM in the presence of SR141716, indicating that cannabinoid and muscarinic receptor activation independently diminish IM. It is concluded that cannabinoids may postsynaptically augment the excitability of CA1 pyramidal neurons by specifically decreasing the persistent voltage-dependent IM.
Keywords: cannabinoid, brain, slice, voltage-clamp, potassium current, excitation
Cannabinoid substances have powerful psychoactive properties and alter many physiological processes, such as cognition, behavior, and nociception (Ameri, 1999). These effects are believed to be mediated via specific high-affinity binding sites present throughout the brain (Herkenham et al., 1990). A G-protein-linked receptor expressed in brain (CB1) has been cloned (Matsuda et al., 1990), and the compound SR141716 (SR1) is a selective antagonist at this receptor (Rinaldi-Carmona et al., 1994). One of the highest CB1 receptor density is found in the hippocampus, a brain structure associated with learning and memory processes, and cannabinoids appear to impair memory via activation of these receptors (Lichtman and Martin, 1996). The discovery of specific receptors led to the isolation of two endogenous ligands, the endocannabinoids anandamide (Devane et al., 1992) and 2-arachidonylglycerol (Mechoulam et al., 1995), both found in brain (Di Marzo et al., 1994; Stella et al., 1997).
Little is known on the cellular mechanisms underlying the central effects of cannabinoids, and only a few studies have been conducted at the postsynaptic level. In cultured hippocampal neurons, cannabinoid agonists increase the transient K+A-current (IA) (Deadwyler et al., 1993) and reduce currents passing through N- and P/Q type calcium channels (Twitchell et al., 1997; Shen and Thayer, 1998). Cannabinoids receptors heterologously expressed in ganglion neurons also reduce Ca2+ currents without altering the K+ A- and M-currents (Pan et al., 1996). Other studies using coexpression or transfection of CB1 receptors in non-neuronal systems showed that cannabinoids may also activate an inwardly rectifying K+ conductance (Henry and Chavkin, 1995; Mackie et al., 1995). No postsynaptic studies, however, have investigated the effect of cannabinoids on sustained (noninactivating) conductances in native brain preparations, such as the hippocampal slice.
Hippocampal neurons are under the tonic control of sustained conductances, such as IM,Ih, and leak-currents, which are active at or near resting potential and readily regulate neuronal activity (Storm, 1990). The time- and voltage-dependentIM is modulated by several neurotransmitters and plays a unique role in modulating cellular excitability, because it is the only K+current that both activates below the action potential threshold and does not inactivate (Brown and Adams, 1980; Marrion, 1997). In CA1 pyramidal neurons, IM is decreased by muscarinic agonists and serotonin (Halliwell and Adams, 1982; Colino and Halliwell, 1987) and increased by somatostatin (Moore et al., 1988). Because IM opposes membrane depolarization, substances that decrease this current augment neuronal excitability, whereas substances that increaseIM diminish neuronal excitability.
Although sustained conductances are modulated by numerous neurotransmitters, their sensitivity to cannabinoids has not been investigated in brain. Previous postsynaptic studies have been conducted with cultured neurons or non-neuronal cells. In the present study, I recorded from native neurons in a slice preparation and found that cannabinoids reduce the K+IM via activation of CB1 receptors, thus postsynaptically augmenting neuronal excitability.
MATERIALS AND METHODS
Slice preparation. Standard intracellular recording techniques were used in rat hippocampal slices as described previously (Schweitzer et al., 1993). In brief, transverse hippocampal slices (taken from male Sprague Dawley rats of 100–170 gm) 350-μm-thick were cut on a slicer and incubated in gassed (95% O2, 5% CO2) artificial CSF (ACSF) of the following composition (in mm): NaCl 130, KCl 3.5, NaH2PO4 1.25, MgSO4 1.5, CaCl2 2.0, NaHCO3 24, and glucose 10. Slices were completely submerged and continuously superfused with warm (30–31°C) ACSF at a constant rate within the range of 1–3 ml/min. Methods of superfusion, voltage-clamp recording, drug administration, and data analysis were as described previously (Schweitzer et al., 1993). Drugs were added to the ACSF with dimethylsulfoxide (0.05–0.15% final concentration). Dimethylsulfoxide did not affect membrane properties at this concentration (Schweitzer et al., 1993). R1-methanandamide, WIN55212–2, and linopirdine (DuP 996) were purchased from Research Biochemicals (Natick, MA), tetrodotoxin was from Calbiochem (La Jolla, CA), and all other chemicals were from Sigma (St. Louis, MO). SR141716 was obtained from the National Institute of Mental Health Chemical Synthesis and Drug Supply Program.
Voltage-clamp recordings. Voltage-clamp studies were performed with an Axoclamp 2A preamplifier (Axon Instruments, Foster City, CA), using sharp glass micropipettes filled with 3m KCl (impedance range of 50–85 MΩ) to penetrate CA1 pyramidal neurons. Tetrodotoxin (1 μm) was added to the bath after impalement to block Na+-dependent action potentials and synaptic transmission. In discontinuous single-electrode voltage-clamp mode, the switching frequency between current injection and voltage sampling was 3–4 kHz. Current and voltage records were filtered at 0.3 kHz, acquired by analog-to-digital sampling and acquisition software, and measured with analysis software (Axon Instruments). Values are presented as mean ± SEM. The various problems associated with voltage-clamping of neurons with extended processes were discussed previously (Halliwell and Adams, 1982; Johnston and Brown, 1983). Such problems should be minimized when studying relative conductance changes with superfusion of drugs to equilibrium conditions.
Voltage protocols. Current-voltage (I–V) curves were generated by holding neurons at −59 ± 0.2 mV (n = 47) and applying hyperpolarizing and depolarizing voltage steps (1.5 sec duration, 7 sec apart). Neurons were not depolarized beyond −40 mV because of space-clamp considerations and the likelihood of activating large Ca2+currents. I–V curves were constructed from current values measured at the end of the voltage step (steady state), and the values obtained in control condition were subtracted from those in presence of the tested substances to obtain the net current induced. Two voltage-dependent noninactivating conductances found in CA1 neurons were separately assessed. The IMrelaxation was observed at the onset of hyperpolarizing voltage steps (1 sec duration) delivered from a holding potential (VH) of −44 ± 0.3 mV (n = 49). The Ihrelaxation was observed at the onset of hyperpolarizing voltage steps delivered from a holding potential of −59 mV (Halliwell and Adams, 1982).
RESULTS
Intracellular recordings were performed from 65 CA1 pyramidal neurons using the adult hippocampal slice preparation to investigate cannabinoid effects on sustained conductances. The average resting membrane potential (RMP) was −69 ± 0.3 mV, the input resistance determined at onset of a small hyperpolarizing current step before addition of tetrodotoxin was 74 ± 2 MΩ, and the action potential amplitude from threshold was 104 ± 1 mV. Two nondegradable cannabinoid agonists were used: the methylated analog of anandamide R1-methanandamide (mAEA), and the aminoalkyndole WIN55212–2 (WIN-2).
Cannabinoids elicit an inward steady-state current
I–V relationships were generated to study the effects of cannabinoids on steady-state membrane properties in the depolarized and hyperpolarized ranges. Superfusion of mAEA (5 μm) onto CA1 pyramidal neurons elicited an inward steady-state current in the depolarized range but showed no effect at hyperpolarized potentials (Fig.1A). Current values were back near control upon washout of the drug. The net steady-state currents were obtained by subtracting current values obtained at each condition from current values in control (Fig. 1B). The mAEA component showed voltage-dependence and had a reversal potential of −87 ± 5 mV (n = 5), close to the theoretical equilibrium potential for K+(−98 mV in these experimental conditions). The conductance decrease elicited by mAEA, GmAEA, was calculated by dividing the cannabinoid-induced current by the driving force (Fig. 1C). GmAEA was voltage-dependent with an activation threshold of approximately −75 mV and amplitude of −3.1 nS at −43 mV. The mAEA effect was dose-dependent as the amplitude of the inward current increased with the drug concentration (Fig. 2). The apparent threshold response was 0.25 μm, and the maximum effect was obtained with 5 μmmAEA.
Fig. 1.
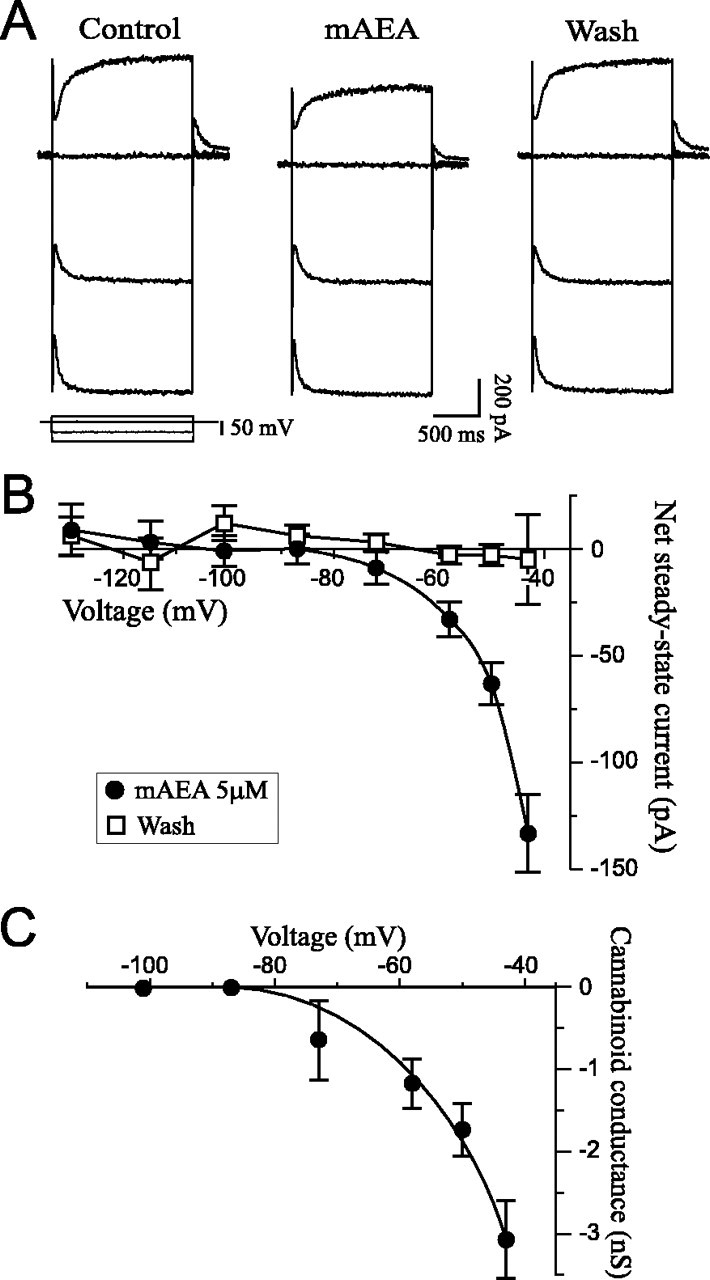
Cannabinoids elicit an inward steady-state current. A, Selected current traces obtained with anI–V protocol. This representative CA1 pyramidal neuron held at −56 mV was subjected to three different voltage steps sequentially applied and superimposed at each condition (voltage protocol at bottom left). Superfusion of 5 μm mAEA induced an inward steady-state current at depolarized potentials (170 pA at −42 mV) but had no effect in the hyperpolarized range. RMP was −69 mV. B, Net currents averaged from five neurons exposed to 5 μm mAEA. The cannabinoid elicited a voltage-dependent inward current that reversed at −87 mV, with recovery to control values on washout of the drug.C, Plot of the mAEA-induced conductance derived fromB. GmAEA was calculated asImAEA/(V − Vrev), whereImAEA is the mAEA-induced current,V is the command potential, andVrev is the reversal potential. The conductance was voltage-dependent and activated at approximately −75 mV.
Fig. 2.

The cannabinoid inward current is concentration-dependent. Averaged steady-state currents elicited with different concentrations of mAEA: 0.1 μm(n = 3), 0.25 μm(n = 4), 1 μm (n= 4), 5 μm (n = 5), and 10 μm (n = 4). The amplitude of the inward current increased with the concentration of mAEA. The threshold response was 0.25 μm, and the maximum effect was reached with 5 μm.
It was then determined whether the mAEA effect was mediated via activation of CB1 receptors by using the selective CB1 receptor antagonist SR1. Superfusion of SR1 alone (1 μm) did not elicit a measurable effect on steady-state currents throughout the potential range tested (Fig.3A,B). However, the mAEA-induced component was completely prevented by SR1, indicating that the cannabinoid effect occurred via activation of CB1 receptors. To confirm these findings, the experiments were repeated with the structurally different cannabinoid WIN-2. WIN-2 had effects similar to those of mAEA and induced a voltage-dependent inward current that reversed at −85 mV (Fig. 3C). The threshold response was 0.25 μm and the maximum inward current was obtained with 2 μm (n = 6), because superfusion of 5 μm WIN-2 did not elicit a larger effect (n = 3; data not shown). The maximum effect, however, was not as pronounced and consistent as the effect observed with mAEA, although it occurred at a lesser concentration. The effect of WIN-2 was also prevented by SR1 (Fig.3C), demonstrating involvement of CB1 receptors.
Fig. 3.
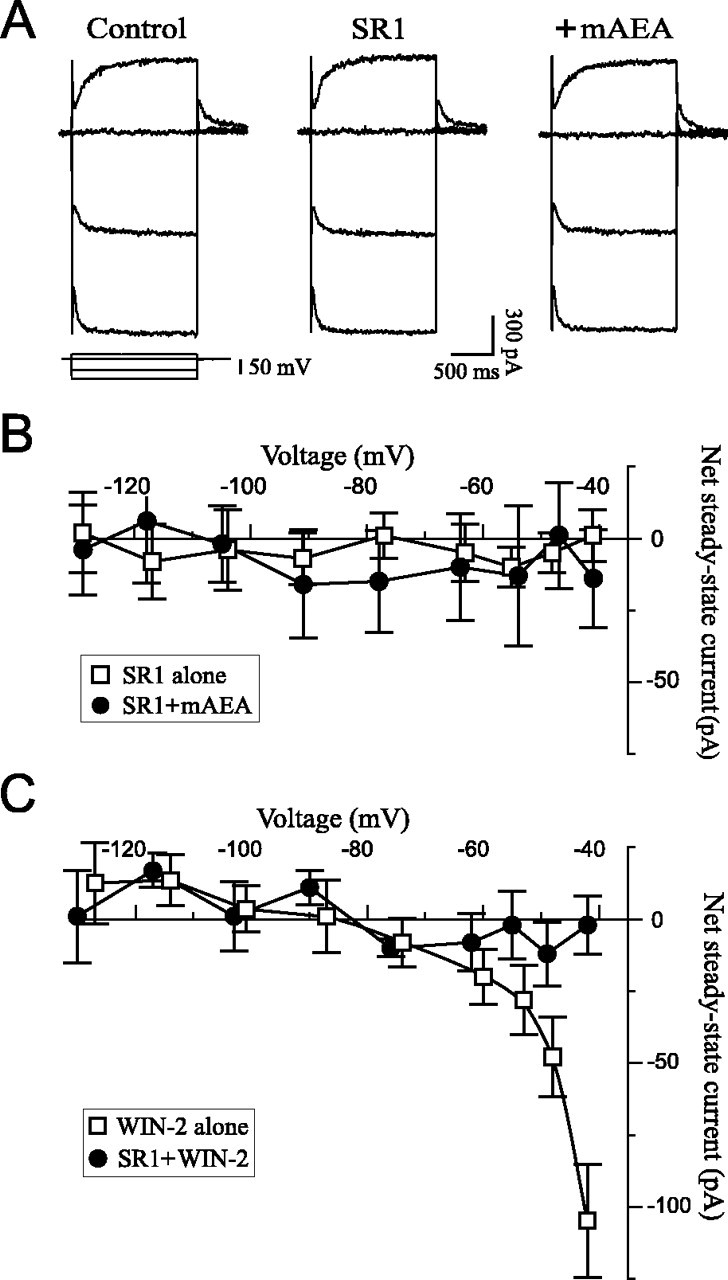
The cannabinoid inward current is elicited via activation of CB1 receptors. A, Selected current traces from a neuron exposed to the CB1 antagonist SR1 (1 μm) and mAEA (5 μm) in the presence of SR1. SR1 alone had no effect but completely prevented the mAEA response. RMP was −67 mV, andVH was −59 mV. B, Net currents averaged from seven neurons exposed to 1 μm SR1 alone and three neurons exposed to 5–10 μm mAEA in the presence of SR1. The antagonist completely prevented the mAEA effect.C, Net currents elicited by WIN-2 in the absence (2 μm; n = 6) or presence (2–5 μm; n = 5) of 1 μm SR1. WIN-2 elicited a voltage-dependent inward current that was completely prevented by SR1.
Cannabinoids decrease IM
The IM is a persistent voltage-dependent K+ outward current that activates at approximately −70 mV, thus having properties resembling the effect elicited by cannabinoids. A separate voltage protocol was used to quantify IM (see Materials and Methods) and determine whether cannabinoids decreasedIM to elicit the observed inward steady-state current at depolarized potentials. Addition of WIN-2 in the superfusate indeed reduced IMrelaxation amplitudes (Fig.4A) and concomitantly elicited an inward holding current (Fig. 4A,dotted line), consistent with closing of M-channels. All values returned toward control levels upon washout of WIN-2, although recovery was only partial. The averaged effect onIM over nine neurons is shown on Figure 4B; WIN-2 (2–5 μm) decreased IM to 55 ± 3% of control, with a recovery on washout to 85 ± 6% of control. The specific IM blocker linopirdine (Aiken et al., 1995) was used to further identifyIM as the target of the cannabinoid effect. Linopirdine elicited an inward steady-state current because ofIM inhibition (Fig. 4C) and decreased IM relaxations to 18 ± 4% of control (n = 5; data not shown). Addition of 2 μm WIN-2 in the continued presence of linopirdine did not alter steady-state currents (Fig. 4C) orIM relaxations that remained at 17 ± 4% of control, indicating that cannabinoids solely affectedIM.
Fig. 4.
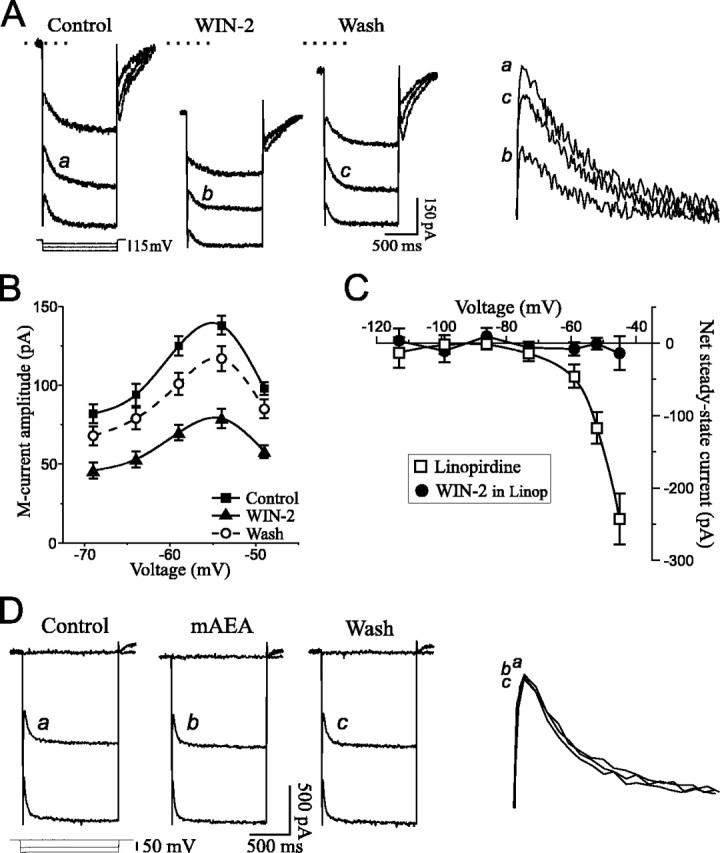
Cannabinoids decreaseIM. A, Current recordings showing IM relaxations from a neuron held at −44 mV. Hyperpolarizing voltage commands (3 steps superimposed, protocol at bottom left) were applied to deactivateIM (slow relaxation at command onset). WIN-2 elicited an IM decrease associated with an inward holding current (dotted line is control holding current). The IM relaxations identified withletters are magnified and superimposed on thefarright for comparison. RMP was −67 mV. B, Average of IMamplitude in nine cells tested with 2–5 μm WIN-2. The cannabinoid decreased IM by 44% with recovery to 85% of control upon washout. C, Net steady-state currents from five neurons exposed to the selectiveIM inhibitor linopirdine, followed by WIN-2. Linopirdine (10 μm) elicited a voltage-dependent inward current because of blockade of M-channels. Further addition of 2 μm WIN-2 had no effect, indicating that cannabinoids affected only IM. D, Recordings showing Ih relaxations observed with hyperpolarizing voltage commands to −103 and −119 mV (VH of −58 mV). Superfusion of 5 μm mAEA did not alter Ihamplitude. RMP was −68 mV.
Cannabinoids reportedly augment inwardly rectifying K+ conductances in expression systems. I investigated a possible action of cannabinoids onIh (also calledIQ), a persistent Na+–K+conductance that activates in the hyperpolarized range below −60 mV and rectifies inwardly. The Ihrelaxation amplitude was unchanged upon exposure to mAEA (Fig.4D) or WIN-2 (data not shown). On average,Ih remained at 99 ± 3% of control when neurons were exposed to 5–10 μmmAEA (n = 6) and 101 ± 2% of control when 2–5 μm WIN-2 was applied (n = 8).
The cannabinoid-induced IM decrease was concentration-dependent. Superfusion of 1 μm mAEA decreased theIM amplitude by 27% and elicited a small inward holding current (Fig.5A). A higher concentration of 5 μm mAEA elicited a stronger effect to decrease IM by 58%, concomitant with a large inward holding current (Fig. 5B). Current values returned near control levels on washout of mAEA. The dose–response relationship obtained with WIN-2 and mAEA is shown in Figure5C. WIN-2 had a maximal effect at 3 μm to decreaseIM to 55% of control, with an apparent EC50 of 0.6 μm. The maximal effect with mAEA was obtained at 6 μm to decreaseIM to 59% of control, with an apparent EC50 of 1 μm.
Fig. 5.

The cannabinoid-inducedIM decrease is concentration-dependent.A, IM recordings from a neuron exposed to 1 μm mAEA. Superfusion of mAEA decreased IM by 27% (IM relaxations magnified on the far right) and elicited a limited inward holding current. RMP was −68 mV, and VH was −47 mV.B, Superfusion of 5 μm mAEA produced a larger IM decrease (by 58% on this cell; relaxations magnified on far right) associated with a pronounced inward holding current. RMP was −71 mV, andVH was −43 mV. C, Dose–response curve of IM inhibition by WIN-2 (filled circles) or mAEA (open squares). The threshold response was below 0.2 μm, and maximal effects were obtained with 3 μm WIN-2 (EC50 of 0.6 μm;dashed line) to inhibit IM by 45% and 6 μm mAEA (EC50 of 1 μm;dotted line) to inhibitIM by 41%.
Cannabinoids decrease IM via CB1 receptors independently of muscarinic receptors
The CB1 receptor antagonist SR1 was used to determine whether the cannabinoid-induced IM decrease occurred via activation of CB1 receptors. Superfusion of 1 μm SR1 alone had no effect onIM amplitude (n = 5; data not shown). In the presence of SR1, however, a subsequent application of WIN-2 at concentrations that greatly reducedIM (1–5 μm;n = 5) was without effect (Fig.6A,B). Likewise, mAEA (5–10 μm; n = 3) did not affect IM nor elicit an inward holding current in slices pretreated with SR1 (Fig.6C), indicating that cannabinoids decreasedIM by activating CB1 receptors.
Fig. 6.

The cannabinoid-inducedIM decrease is mediated via CB1 receptors.A, IM relaxation elicited with a 10 mV hyperpolarizing step (VH of −42 mV). A first application of 1 μm WIN-2 decreasedIM by 47%. After washout of WIN-2 coincident with superfusion of 1 μm SR1, a second application of WIN-2 in the continued presence of SR1 had no effect onIM. The bottom panel shows the magnified IM relaxations. RMP was −71 mV. B, Average of IMamplitude on five neurons exposed to 1–5 μm WIN-2 in slices treated with 1 μm SR1, showing the lack of effect of the cannabinoid in presence of the CB1 antagonist. C, SR1 also prevented the IM decrease expected with superfusion of 5 μm mAEA. RMP was −67 mV, andVH was −48 mV.
A possible involvement of muscarinic receptors in the cannabinoid effect was investigated by treating the slices with the muscarinic receptor antagonist atropine. In the presence of 1 μmatropine, the nondegradable cholinergic agonist carbamylcholine (carbachol, 5 μm) did not affectIM because of blockade of muscarinic receptors. Addition of 2 μm WIN-2 in the presence of atropine, however, greatly decreased theIM relaxation (Fig.7A). On average, atropine alone did not affect IM, but addition of WIN-2 together with atropine decreasedIM to 56 ± 5% of control (n = 5) (Fig. 7B), a value similar to that observed in the absence of the muscarinic antagonist (55 ± 3% of control) (Fig. 4B). To ensure that the well known muscarinic-induced IM inhibition occurred independently of CB1 receptors, additional experiments were conducted with SR1 and carbachol. In the presence of the cannabinoid receptor antagonist, WIN-2 no longer alteredIM, but further addition of 5 μm carbachol greatly decreasedIM (Fig. 7C). On average, carbachol was more efficacious than cannabinoids and decreasedIM to 20 ± 6% of control (n = 4; 15 mV hyperpolarizing step). These results show that cannabinoid and muscarinic receptor agonists independently diminish IM.
Fig. 7.

Cannabinoid and muscarinic receptor agonists independently decrease IM. A,IM relaxation elicited with a 10 mV hyperpolarizing step (VH of −47 mV) in the presence of the muscarinic receptor antagonist atropine (1 μm). Carbachol (CCh, 5 μm) had no effect on IM because of blockade of muscarinic receptors, but addition of 2 μm WIN-2 in the continued presence of atropine decreased IM. RMP was −67 mV. B, AverageIM amplitude on five cells exposed to 1 μm atropine, followed by 2 μm WIN-2. The cannabinoid-induced IM decrease was unaffected by the muscarinic receptor antagonist. C,IM relaxation elicited with a 10 mV hyperpolarizing step (VH of −44 mV) in the presence of SR1. WIN-2 had no effect on IMbecause of blockade of CB1 receptors, but 5 μm CCh decreased IM (washout performed in atropine). RMP was −69 mV.
The cannabinoid effects on the IMrelaxation are summarized for comparison in Figure8. WIN-2 decreasedIM by 45 ± 3% when applied alone and by 44 ± 5% in the presence of atropine. SR1 alone did not affect IM (2 ± 3% increase) but prevented WIN-2 from inhibiting IM(3 ± 5% decrease). Similar to WIN-2, mAEA decreasedIM by 41 ± 5% in absence of SR1 and by 6 ± 6% when the CB1 receptor antagonist was present.
Fig. 8.

Summary chart of IMinhibition by cannabinoids. Superfusion of SR1 alone did not affectIM amplitude (2% augmentation). WIN-2 decreased IM by 45%, an effect prevented in the presence of SR1 (3% decrease) but unaltered by atropine (44% decrease). Comparable results were obtained with mAEA that decreasedIM by 41% in absence of SR1 and by 6% in presence of the CB1 antagonist.
DISCUSSION
The results showed that cannabinoids acting at CB1 receptors elicited a postsynaptic excitatory effect on CA1 pyramidal neurons by decreasing the persistent voltage-dependentIM.
Cannabinoids decrease the persistentIM
In the presence of tetrodotoxin to block neurotransmission, cannabinoids elicited an inward current that voltage-dependently increased with depolarization. The current reversed at −87 mV, indicating that K+ was the carrier, and activated at approximately −75 mV. Such properties were reminiscent ofIM, a time- and voltage-dependent persistent K+ current that activates between −80 and −70 mV, and the I–V relationship profile of the cannabinoid effect was consistent with a decrease ofIM. Although the inwardly rectifyingIh activates only at hyperpolarized potentials, the IM andIh relaxations appear similar. The results showed that neither WIN-2 nor mAEA alteredIh. Moreover, WIN-2 had no effect on neurons pretreated with the M-channel blocker linopirdine (Aiken et al., 1995), verifying that cannabinoids solely affectedIM. However, I–Vrelationships were not performed beyond −40 mV because of space-clamp considerations, and a cannabinoid action on conductances active at more depolarized potentials is possible.
The cannabinoid effect was dose-dependent. WIN-2 and mAEA had a comparable efficacy and decreased IMto a similar level, although WIN-2 appeared more potent. The EC50 values of 0.6 and 1 μm are comparable with the 1–2 μm range reported for synaptic inhibition in brain slices (Lévénès et al., 1998; Szabo et al., 1998) but much higher than the 10–20 nm range reported for Ca2+ current inhibition in hippocampal cultures (Twitchell et al., 1997; Shen and Thayer, 1998). Such discrepancy is usually attributed to limited drug penetration and inferior access to the recorded neurons in slice preparations.
Cannabinoid and muscarinic receptor activation independently decrease IM
The inward steady-state current andIM decrease elicited by mAEA and WIN-2 were both prevented in slices treated with SR1, demonstrating that cannabinoids activated CB1 receptors. A previous report showed that endocannabinoids are detected in hippocampal slices subjected to similar experimental conditions, including the presence of tetrodotoxin (Stella et al., 1997). In the present study, SR1 applied alone had no effect on the recorded currents, indicating that endocannabinoids may not tonically affect postsynaptic properties in the slice preparation.
Cholinergic agonists acting at muscarinic receptors decreaseIM. Because cannabinoids have been shown to inhibit the release of acetylcholine in hippocampus (Gifford and Ashby, 1996) and carbachol reportedly enhances the production of the endocannabinoid 2-arachidonylglycerol in rat aorta (Mechoulam et al., 1998b), experiments using receptor antagonists were conducted to investigate possible interactions. The presence of atropine did not alter the extent of IM inhibition by WIN-2. Conversely, carbachol decreasedIM in the presence of SR1, indicating that cannabinoid and muscarinic receptor agonists independently decrease IM.
Postsynaptic actions of cannabinoids
The cannabinoid modulation of persistent conductances has not been investigated in brain neurons, precluding an adequate comparison with the present effect. In cultured hippocampal neurons, cannabinoids augment the transient K+IA and may therefore modulate the excitatory synaptic input (Deadwyler et al., 1995). Although this conductance does not readily influence neuronal activity, its augmentation denotes an inhibitory action of cannabinoids. Experiments conducted in non-neuronal expression systems showed that cannabinoids increased an inwardly rectifying K+ conductance (Henry and Chavkin, 1995; Mackie et al., 1995). The augmentation of such conductance generates a small outward current to inhibit neuronal activity, in contrast to the present results that point to increased excitability. Such differences can be explained by the use of totally different preparations, native brain slices versus non-neuronal systems expressing CB1 receptors. As well, the lack of effect of cannabinoids on IM andIA in ganglion neurons transiently expressing CB1 receptors may be because of an ineffective coupling of the adequate second messenger systems (Pan et al., 1996).
The identification of the intracellular mechanisms ofIM inhibition remains under intense investigation. A rise of intracellular Ca2+ levels may play a key role in the decrease of IM by various transmitters (for review, see Marrion, 1997). Cannabinoids can increase intracellular Ca2+ levels via phospholipase C in cell lines (Sugiara et al., 1997). Cannabinoids also enhance the depolarization-induced increase of intracellular Ca2+ by a mechanism involving phospholipase C and Ca2+ release from inositol triphosphate-sensitive Ca2+stores in cerebellar neurons (Netzeband et al., 1999). Interestingly, a recent study showed that bradykinin inhibitsIM in ganglion neurons via phospholipase C and Ca2+ release from inositol triphosphate-sensitive Ca2+stores (Cruzblanca et al., 1998). Such a mechanism could be involved in the cannabinoid inhibition of IM in CA1 pyramidal neurons in which an increase in intracellular concentrations of inositol triphosphate reportedly decreaseIM (Dutar and Nicoll, 1988).
Cannabinoids and eicosanoids have opposite effects
Arachidonic acid and its metabolites, the eicosanoids, are potent signaling molecules implicated in several forms of neuromodulation (Meves, 1994; Piomelli, 1994). Although arachidonic acid is produced upon degradation of anandamide and 2-arachido-nylglycerol (Mechoulam et al., 1998a), the fatty acid and its lipoxygenase metabolites augmentIM in CA1 pyramidal neurons (Schweitzer et al., 1990), an effect opposite to those of cannabinoids. Interestingly, arachidonic acid also decreases the hippocampalIA (Keros and McBain, 1997), whereas cannabinoids increase it (Deadwyler et al., 1993). Furthermore, cannabinoids prevent hippocampal long-term potentiation (Collins et al., 1994; Stella et al., 1997), whereas arachidonic acid elicits this phenomenon (Williams et al., 1989). Thus, cannabinoids and eicosanoids act on similar targets in hippocampus but in an opposite direction.
The arachidonic acid produced upon endocannabinoid degradation has to be rapidly removed to prevent further biological effects. Indeed, very little arachidonic acid resulting from endocannabinoid hydrolysis is detected using cellular assays, because the fatty acid appears to be immediately reincorporated into membrane phospholipids (Mechoulam et al., 1998a). The IM decrease via arachidonic acid activation of protein kinase C reported in cultured cells (Schmitt and Meves, 1993) is also an unlikely mechanism, especially because a recent study indicates that stimulation of protein kinase C phosphorylates CB1 receptors and prevents cannabinoid actions (Garcia et al., 1998). Evidently, the eicosanoids do not mediate cannabinoid effects. Still, the fact that these two closely related families of lipidic mediators have opposite effects is puzzling.
Functional implications
Because IM is a persistent current active near the threshold for action potential initiation, it has a major influence on neuronal excitability and responsiveness to synaptic inputs (Marrion, 1997). The primary role ofIM is to clamp the membrane potential near rest. When depolarizing events occur,IM activates to hyperpolarize the membrane back toward resting potential and prevents excessive depolarizations. Thus, IM participates in the mechanism of spike frequency adaptation to slow the firing of action potentials (Aiken et al., 1995) and also plays a major role in the termination of bursting activity in CA1 neurons (Azouz et al., 1996). By reducing IM, cannabinoids diminish the ability of neurons to counteract depolarizations, favoring increased firing of action potentials and prolonged bursting.
Interestingly, cannabinoids reinforce bursting activity in CA1 hippocampus (Xue et al., 1993) and increase neuronal firing rate and bursting activity in the ventral tegmentum and substantia nigra pars compacta in vivo (French et al., 1997), an effect consistent with IM inhibition. An alteration ofIM could also be involved in the dual effects of cannabinoids on neurons of the solitary tract nucleus (Himmi et al., 1998). The present results indicate that, in addition to presynaptic disinhibitory effects associated with decreased GABAergic transmission (Miller and Walker, 1995; Chan and Yung, 1998; Szabo et al., 1998), cannabinoids may also directly increase neuronal activity via postsynaptic actions. It should be noted, however, that hippocampal pyramidal neurons reportedly possess few CB1 receptors (Tsou et al., 1998), and an indirect effect is always possible despite the blockade of neurotransmission by tetrodotoxin.
Recent reports have attributed the occurrence of an epileptic syndrome to mutations of the K+ channel genes KCNQ2 and KCNQ3 (Biervert et al., 1998; Charlier et al., 1998). Further work demonstrated that the combination of KCNQ2 and KCNQ3 subunits, highly expressed in hippocampus, form native M-channels (Wang et al., 1998). These data strongly implicate IM in the control of seizure. Cannabinoid research performed before the identification of specific receptors showed that Δ9 -tetrahydrocannabinol has both convulsant and anticonvulsant effects (for review, see Martin, 1986). Although the mechanisms implicated in these actions were not determined, the anticonvulsant effect could be possibly attributed to the cannabinoid inhibition of glutamate release (Ameri, 1999). On the other hand, and consistent with the alteration of M-channel expression in some form of epilepsy, the cannabinoid inhibition ofIM could play a role in the reported convulsant action.
Conclusion
The activation of CB1 receptors postsynaptically decreasesIM in CA1 pyramidal neurons. This action will diminish the ability of neurons to counteract depolarizing events and may play an important role in response to hyperexcitability and bursting in hippocampus. Cannabinoids can therefore increase neuronal excitability by altering IMbut can also decrease hippocampal activity by inhibiting neurotransmitter release and synaptic plasticity. Surprisingly, cannabinoids and eicosanoids have opposite effects on hippocampal electrophysiology.
Footnotes
This work was funded by National Institute on Drug Abuse (NIDA) Grant K01DA00291. I thank Samuel Madamba for technical assistance and George Siggins for support (Grant DA03665 from NIDA).
Correspondence should be addressed to Dr. Paul Schweitzer, Neuropharmacology-CVN 12, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, CA 92037. E-mail:pschweitzer@scripps.edu.
REFERENCES
- 1.Aiken SP, Lampe BJ, Murphy PA, Brown BS. Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 1995;115:1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ameri A. The effects of cannabinoids on the brain. Prog Neurobiol. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- 3.Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. J Physiol (Lond) 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- 5.Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- 6.Chan PK, Yung WH. Occlusion of the presynaptic action of cannabinoids in rat substantia nigra pars reticulata by cadmium. Neurosci Lett. 1998;249:57–60. doi: 10.1016/s0304-3940(98)00396-6. [DOI] [PubMed] [Google Scholar]
- 7.Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like K+ channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- 8.Colino A, Halliwell JV. Differential modulation of three separate K+ conductances in hippocampal CA1 neurons by serotonin. Nature. 1987;328:73–77. doi: 10.1038/328073a0. [DOI] [PubMed] [Google Scholar]
- 9.Collins DR, Pertwee RG, Davies SN. The action of synthetic cannabinoids on the induction of long-term potentiation in the rat hippocampal slice. Eur J Pharmacol. 1994;259:R7–R8. doi: 10.1016/0014-2999(94)90666-1. [DOI] [PubMed] [Google Scholar]
- 10.Cruzblanca H, Koh DS, Hille B. Bradykinin inhibits M-current via phospholipase C and Ca2+ release from IP3 -sensitive Ca2+ stores in rat sympathetic neurons. Proc Natl Acad Sci USA. 1998;95:7151–7156. doi: 10.1073/pnas.95.12.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deadwyler SA, Hampson RE, Bennett BA, Edwards TA, Mu J, Pacheco MA, Ward SJ, Childers SR. Cannabinoids modulate potassium current in cultured hippocampal neurons. Receptors Channels. 1993;1:121–134. [PubMed] [Google Scholar]
- 12.Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharmacol Exp Ther. 1995;273:734–743. [PubMed] [Google Scholar]
- 13.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 14.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz J-C, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 15.Dutar P, Nicoll RA. Classification of muscarinic responses in hippocampus in terms of receptor subtypes and second-messenger systems: electrophysiological studies in vitro. J Neurosci. 1988;8:4214–4224. doi: 10.1523/JNEUROSCI.08-11-04214.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.French ED, Dillon K, Wu X. Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. NeuroReport. 1997;8:649–652. doi: 10.1097/00001756-199702100-00014. [DOI] [PubMed] [Google Scholar]
- 17.Garcia DE, Brown S, Hille B, Mackie K. Protein kinase C disrupts cannabinoid actions by phosphorylation of the CB1 cannabinoid receptor. J Neurosci. 1998;18:2834–2841. doi: 10.1523/JNEUROSCI.18-08-02834.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gifford AN, Ashby CR., Jr Electrically evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN 55212–2, and is potentiated by the cannabinoid antagonist, SR 141716A. J Pharmacol Exp Ther. 1996;277:1431–1436. [PubMed] [Google Scholar]
- 19.Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- 20.Henry DJ, Chavkin C. Activation of inwardly rectifying potassium channels (GIRK1) by co-expressed rat brain cannabinoid receptors in Xenopus oocytes. Neurosci Lett. 1995;186:91–94. doi: 10.1016/0304-3940(95)11289-9. [DOI] [PubMed] [Google Scholar]
- 21.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, De Costa BR, Rice KC. Cannabinoid receptor localization in brain. Proc Natl Acad Sci USA. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Himmi T, Perrin J, El Ouazzani T, Orsini JC. Neuronal responses to cannabinoid receptor ligands in the solitary tract nucleus. Eur J Pharmacol. 1998;359:49–54. doi: 10.1016/s0014-2999(98)00630-x. [DOI] [PubMed] [Google Scholar]
- 23.Johnston D, Brown TH. Interpretation of voltage-clamp measurements in hippocampal neurons. J Neurophysiol. 1983;50:464–486. doi: 10.1152/jn.1983.50.2.464. [DOI] [PubMed] [Google Scholar]
- 24.Keros S, McBain CJ. Arachidonic acid inhibits transient potassium currents and broadens action potentials during electrographic seizures in hippocampal pyramidal and inhibitory interneurons. J Neurosci. 1997;17:3476–3487. doi: 10.1523/JNEUROSCI.17-10-03476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lévénès C, Daniel H, Soubrié P, Crépel F. Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar Purkinje cells. J Physiol (Lond) 1998;510:867–879. doi: 10.1111/j.1469-7793.1998.867bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lichtman AH, Martin BR. Delta9-tetrahydrocannabinol impairs spatial memory through a cannabinoid receptor mechanism. Psychopharmacology. 1996;126:125–131. doi: 10.1007/BF02246347. [DOI] [PubMed] [Google Scholar]
- 27.Mackie K, Lai Y, Westenbroek R, Mitchell R. Cannabinoids activate an inwardly rectifying potassium conductance and inhibit Q-type calcium currents in AtT20 cells transfected with rat brain cannabinoid receptor. J Neurosci. 1995;15:6552–6561. doi: 10.1523/JNEUROSCI.15-10-06552.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marrion NV. Control of M-current. Annu Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- 29.Martin BR. Cellular effects of cannabinoids. Pharmacol Rev. 1986;38:45–74. [PubMed] [Google Scholar]
- 30.Matsuda LA, Lolait J, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346:561–564. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 31.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z. Identification of an endogenous 2-mono-glyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50:83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 32.Mechoulam R, Fride E, Di Marzo V. Endocannabinoids. Eur J Pharmacol. 1998a;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- 33.Mechoulam R, Fride E, Ben-Shabat S, Meiri U, Horowitz M. Carbachol, an acetylcholine receptor agonist, enhances production in rat aorta of 2-arachidonoyl glycerol, a hypotensive endocannabinoid. Eur J Pharmacol. 1998b;362:R1–R3. doi: 10.1016/s0014-2999(98)00777-8. [DOI] [PubMed] [Google Scholar]
- 34.Meves H. Modulation of ion channels by arachidonic acid. Prog Neurobiol. 1994;43:175–186. doi: 10.1016/0301-0082(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 35.Miller AS, Walker JM. Effects of a cannabinoid on spontaneous and evoked neuronal activity in the substantia nigra pars reticulata. Eur J Pharmacol. 1995;279:179–185. doi: 10.1016/0014-2999(95)00151-a. [DOI] [PubMed] [Google Scholar]
- 36.Moore SD, Madamba SG, Joëls M, Siggins GR. Somatostatin augments the M-current in hippocampal neurons. Science. 1988;239:278–280. doi: 10.1126/science.2892268. [DOI] [PubMed] [Google Scholar]
- 37.Netzeband JG, Conroy SM, Parsons KL, Gruol DL. Cannabinoids enhance NMDA-elicited Ca2+ signals in cerebellar granule neurons in culture. J Neurosci. 1999;19:8765–8777. doi: 10.1523/JNEUROSCI.19-20-08765.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pan XH, Ikeda SR, Lewis DL. Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol Pharmacol. 1996;49:707–714. [PubMed] [Google Scholar]
- 39.Piomelli D. Eicosanoids in synaptic transmission. Crit Rev Neurobiol. 1994;8:65–83. [PubMed] [Google Scholar]
- 40.Rinaldi-Carmona M, Barth F, Héaulme F, Shire D, Calandra B, Congy C, Martinez S, Marauni J, Néliat G, Caput D, Ferrara P, Soubrié P, Breliere JC, LeFur G. SR 141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett. 1994;350:240–244. doi: 10.1016/0014-5793(94)00773-x. [DOI] [PubMed] [Google Scholar]
- 41.Schmitt H, Meves H. Protein kinase C as mediator of arachidonic acid-induced decrease of neuronal M current. Pflügers Arch. 1993;425:134–139. doi: 10.1007/BF00374513. [DOI] [PubMed] [Google Scholar]
- 42.Schweitzer P, Madamba SG, Siggins GR. Arachidonic acid metabolites as mediators of somatostatin- induced increase of neuronal M-current. Nature. 1990;346:464–467. doi: 10.1038/346464a0. [DOI] [PubMed] [Google Scholar]
- 43.Schweitzer P, Madamba SG, Champagnat J, Siggins GR. Somatostatin inhibition of hippocampal CA1 pyramidal neurons: mediation by arachidonic acid and its metabolites. J Neurosci. 1993;13:2033–2049. doi: 10.1523/JNEUROSCI.13-05-02033.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen MX, Thayer SA. The cannabinoid agonist Win55,212–2 inhibits calcium channels by receptor-mediated and direct pathways in cultured rat hippocampal neurons. Brain Res. 1998;783:77–84. doi: 10.1016/s0006-8993(97)01195-5. [DOI] [PubMed] [Google Scholar]
- 45.Stella N, Schweitzer P, Piomelli D. A second endogenous cannabinoid that modulates long-term potentiation. Nature. 1997;388:773–778. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 46.Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- 47.Sugiura T, Kodaka T, Kondo S, Nakane S, Kondo H, Waku K, Ishima Y, Watanabe K, Yamamoto I. Is the cannabinoid CB1 receptor a 2-arachidonoylglycerol receptor? Structural requirements for triggering a Ca2+ transient in NG108–15 cells. J Biochem. 1997;122:890–895. doi: 10.1093/oxfordjournals.jbchem.a021838. [DOI] [PubMed] [Google Scholar]
- 48.Szabo B, Dörner L, Pfreundtner C, Nörenberg W, Starke K. Inhibition of GABAergic inhibitory postsynaptic currents by cannabinoids in rat corpus striatum. Neuroscience. 1998;85:395–403. doi: 10.1016/s0306-4522(97)00597-6. [DOI] [PubMed] [Google Scholar]
- 49.Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM. Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- 50.Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- 51.Wang HS, Pan ZM, Shi WM, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 52.Williams JH, Errington ML, Lynch MA, Bliss TVP. Arachidonic acid induces a long-term activity-dependent enhancement of synaptic transmission in the hippocampus. Nature. 1989;341:739–742. doi: 10.1038/341739a0. [DOI] [PubMed] [Google Scholar]
- 53.Xue BG, Belluzzi JD, Stein L. In vitro reinforcement of hippocampal bursting by the cannabinoid receptor agonist (−)CP-55,940. Brain Res. 1993;626:272–277. doi: 10.1016/0006-8993(93)90587-d. [DOI] [PubMed] [Google Scholar]