

Abstract
White light (5 klux for 2 hr) induces apoptosis of rod photoreceptors in wild-type mice (c-fos+/+) within 24 hr, whereas rods of c-fos knock-out mice (c-fos−/−) are protected (Hafezi et al., 1997b).
The range of this protection was tested by analyzing retinas ofc-fos+/+ andc-fos−/− mice up to 10 d after exposure to threefold increased light intensities (15 klux for 2 hr). In c-fos−/− mice, rods were unaffected, whereas they were destroyed inc-fos+/+ mice. After light exposure, mitochondrial damage in rods was observed exclusively inc-fos+/+ mice. Electroretinograms recorded 48 hr after exposure revealed a decrease of all components inc-fos+/+ mice but indicated no light-induced loss of function inc-fos−/− mice. Thus, inc-fos−/− mice, light-induced apoptosis is blocked or its threshold is elevated more than threefold.
Increased activity of the transcription factor activator protein-1 (AP-1) in retinas of light-exposedc-fos+/+ mice indicated an acute contribution of AP-1 to apoptosis induction. AP-1 activity increased already during exposure and peaked ∼6 hr thereafter, coinciding with the appearance of major morphological signs of apoptosis. Activated AP-1 mainly consisted of c-Fos/Jun heterodimers. Inc-fos−/− mice, AP-1 activity remained unchanged, indicating that no other Jun- or Fos-family member could substitute for c-Fos. Like damaging light,N-methyl-N-nitrosourea (MNU) induced AP-1 containing c-Fos in c-fos+/+ mice and did not induce AP-1 in c-fos−/−mice. In contrast to light, however, MNU induced apoptosis in rods ofc-fos−/− mice. Thus, c-Fos is essential for a specific premitochondrial “private apoptotic pathway” induced by light but not for the execution of apoptosis induced by other stimuli.
Keywords: retinal degeneration, photoreceptor, light damage, apoptosis, mitochondria, c-fos, AP-1, N-methyl-N-nitrosourea, mouse
Apoptosis is the gene-regulated, energy-dependent suicide of individual cells. In contrast to necrosis, cells dying by apoptosis do not affect the adjacent tissue. Apoptosis is involved in morphogenesis and tissue remodeling (programmed cell death) and in the removal of surplus, diseased, or malignant cells. Apoptosis also implies the secondary and therefore “unwanted” death of cells after ischemia reperfusion or excitotoxic lesions (Henderson, 1996; Uren and Vaux, 1996; Jacobson et al., 1997; Stefanis et al., 1997).
Apoptosis is the main mechanism of cell loss in induced (Abler et al., 1996; Hafezi et al., 1997a) or inherited retinal degeneration (Portera Cailliau et al., 1994; Wong, 1997; Remé et al., 1998) in animal models, and it may represent the mechanism of cell death in many human retinal diseases. Excessive light may enhance the progression and severity of human age-related macular degeneration and perhaps some forms of retinitis pigmentosa (Cruickshanks et al., 1993; Simons, 1993;Cideciyan et al., 1998). Likewise, several animal models with inherited retinal degeneration show an increased susceptibility to light damage compared with control animals (summarized in LaVail et al., 1999). Exposure to excessive levels of white light induces photoreceptor apoptosis, thus providing an excellent model to analyze degenerative photoreceptor loss (Remé et al., 1995).
Recently, it was shown that rod photoreceptors of mice lacking the c-Fos component of the transcription factor activator protein-1 (AP-1) (c-fos−/−) are resistant to light (5 klux for 2 hr) that induces apoptosis in wild-type (c-fos+/+) mice (Hafezi et al., 1997b). The basis for this protection and the role of c-Fos in light-induced rod apoptosis are primarily unknown. Rods in retinas of c-fos−/− mice are functional and, during a period of light exposure sufficient to induce apoptotic death of rods in wild-type mice, can absorb similar amounts of photons as rods of wild-type mice (Kueng-Hitz et al., 2000). However, light-induced apoptosis only occurs inc-fos+/+ mice. This suggests that an acute lack of c-Fos in light-induced apoptosis, rather than deficits in rod function induced by its lack, provide the basis for the resistance in c-fos−/− mice.
To test this assumption and to analyze the role of c-Fos in light-induced apoptosis of rods, (1) the intensity of the damaging light was increased threefold to reveal whether the lack of c-Fos resulted in an elevated threshold or even a complete inhibition of apoptosis, (2) the postexposure period was extended to test whether the onset of apoptotic processes was delayed inc-fos−/− mice, (3) retinal function of light-exposed mice was recorded by electroretinography to reveal whether the conservation of rod morphology reflects the conservation of function, (4) the DNA binding activity of AP-1 was examined to test c-Fos function during apoptosis, and (5)N-methyl-N-nitrosourea (MNU), a potent inducer of photoreceptor apoptosis (Yuge et al., 1996; Nambu et al., 1997), was applied to analyze whether apoptosis of rods inc-fos−/− mice is impaired in general or more specifically after light exposure.
MATERIALS AND METHODS
All procedures concerning animals adhered to the Association for Research in Vision and Ophthalmology statement for the use of animals in ophthalmic and vision research.c-fos+/− orc-fos+/+ breeding pairs (129sv/C57Bl/6) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were kept under a 12 hr (6:00 A.M. to 6:00 P.M.) light/dark cycle (60 lux at bottom of cages). For experiments, offspring at the age of 4–8 weeks were used. Reagents were obtained from Sigma-Aldrich Corp. (St. Louis, MO) if not indicated otherwise.
Light-exposure. Before light exposure, animals were dark-adapted for 16 hr overnight. The pupils of the animals were dilated under dim red light (1% Cyclogyl, Alcon, Cham, Switzerland; and 5% Phenylephrine, Ciba Vision, Niederwangen, Switzerland), and the mice were exposed to diffuse white fluorescent light (UV-impermeable diffuser; TLD-36 W/965 tubes; Philips, Hamburg, Germany) for 2 hr (lights on at 10:00 A.M.) with an intensity of 60 lux (habitat intensity), 5 klux, or 15 klux in cages with a reflective interior. After light exposure, animals were analyzed immediately or after a period in darkness.
Microscopy. Tissue preparation was performed as described previously (Hafezi et al., 1998). Briefly, eyes were enucleated and fixed in 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.3, at 4°C overnight. For each eye, the superior central and the inferior central retina adjacent to the optic nerve were trimmed, washed in cacodylate buffer, incubated in osmium tetroxide for 1 hr, dehydrated in increasing ethanol concentrations, and embedded in Epon 812. For light microscopy, sections (0.5 μm) were prepared from the lower central retina (most affected in our light damage model), counterstained with methylene blue, and analyzed using an Axiophot microscope (Zeiss, Oberkochen, Germany). For electron microscopy, sections (50–60 nm) were prepared from the lower central retina and contrasted with 4% uranyl acetate in 50% EtOH and 2.6% lead nitrate in 1 m NaOH. Sections were analyzed using a Hitachi 7000 electron microscope (Hitachi, Tokyo, Japan).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay. Eyes were fixed in 2% paraformaldehyde for 2 hr at 4°C, followed by dehydration and paraffin embedding. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed with modifications using thein situ cell death detection kit (Boehringer Mannheim, Mannheim, Germany) on 5 μm paraffin sections. DNA strand breaks were labeled with fluorescein and visualized with a FITC filter as described by Hafezi et al. (1998).
DNA fragmentation analysis. Retinas were removed rapidly through a slit in the cornea and frozen in liquid nitrogen. Total retinal DNA was extracted as described previously (Hafezi et al., 1997b). Total DNA (10 μg) was analyzed on a 1.8% agarose gel. DNA was visualized at 254 nm by staining with SYBR GREEN (Molecular Probes, Leiden, The Netherlands) and compared with a 100 bp DNA ladder (Amersham Pharmacia Biotech, Uppsala, Sweden).
Electroretinograms. Full-field electroretinograms (ERGs) were recorded from dark-adaptedc-fos+/+ andc-fos−/− mice and from mice kept in darkness for 48 hr after exposure to 15 klux for 2 hr. Anesthetized (xylazine, 20 mg/kg, and ketamine, 40 mg/kg, i.p.) animals were placed on a heating pad (37°C). A silk-AgAgCl electrode was placed on the center of the cornea of the pupil-dilated left eye, an identical reference electrode was placed in the mouth, and a platinum ground electrode was inserted subcutaneously in the tail (Niemeyer and Kueng, 1998). Light stimuli from a halogen source (white light) were presented as pulses of 20 msec over a range of 6 logarithmic units of intensity (8 × 102 to 8 × 104 cd/m2). A bandpass filter of 0.03–500 Hz was used to record the a- and b-waves.
Electrophoretic mobility shift assay. Nuclear extracts were prepared as described previously (Hafezi et al., 1999a). Briefly, one retina was homogenized in 400 μl of 10 mmHEPES-KOH, pH 7.9, 1 mm β-mercaptoethanol, and 1 mm DTT in the presence of protease inhibitors. After incubation on ice for 10 min, the homogenate was vortexed for 10 sec and centrifuged. The pellet was resuspended in 50 μl of 20 mm HEPES-KOH, pH 7.9, 25% glycerol, 420 mm NaCl, 1.5 mmMgCl2, 0.2 mm EDTA, 1 mm β-mercaptoethanol, and 1 mm DTT in the presence of protease inhibitors and incubated on ice for 10 min. Cellular debris was removed by centrifugation at 23,000 × g for 30 min at 4°C. Protein concentrations were determined using the Bradford protein assay (Bio-Rad, Hercules, CA) with BSA as standard.
Electrophoretic mobility shift assays (EMSAs) were performed as described previously (Marti et al., 1994). Briefly, the oligonucleotides coding for an AP-1-specific (5′-AAG CAT GAG TCA GAC AC-3′) DNA binding sequence [TPA response element (TRE)] were labeled using polynucleotide kinase (Boehringer Mannheim) and32P-γATP (Hartmann Analytic GmbH, Braunschweig, Germany). For EMSA, 2–5 μg (5 μl) of protein of nuclear extract were incubated on ice for 20 min with 19 μl of 5 mm MgCl2, 0.1 mm EDTA, 0.75 mm DTT, 7.5% glycerol, and 0.05% NP-40 containing 24 μg of BSA and 2 μg of poly(dI·dC) (Boehringer Mannheim). Radiolabeled oligonucleotide (1 μl) was added, and incubation was continued for another 20 min. Protein–DNA complexes were resolved at 150 V on a 0.75 or 1.5 mm 6% polyacrylamide gel using 0.25× Tris Borat EDTA buffer and visualized on x-ray film. For competition assays, an excess of unlabeled (cold) oligonucleotide corresponding to the DNA binding sites of AP-1, specific protein-1 (SP-1) (5′-TCA CGG GGC GGG TCA A-3′), and the retinoic acid receptor (RAR) (5′-ATC AGG TCA TGA CCT TAA-3′) was used in combination with labeled AP-1 oligonucleotide.
For antibody interference analyses, rabbit polyclonal antibodies directed against c-Fos (2 μl) or a mixture of antibodies directed against c-Jun, JunB, and JunD (each 2 μl) (catalog # sc-045, sc-074, sc-052, sc-046; Santa Cruz Biotechnology, Santa Cruz, CA) were added to nuclear extracts 20 min before the oligonucleotides.
MNU. To establish a dose–response relationship for MNU, 22, 45, 67, and 90 mg/kg MNU in 0.9% NaCl were applied in a single intraperitoneal injection after 16 hr of overnight dark adaptation. Injected animals were kept in darkness for 72 hr. Their retinal morphology was analyzed by light microscopy.
For the comparison of MNU retinal toxicity inc-fos+/+ andc-fos−/− mice, the effect of a single dose of 45 mg/kg was analyzed 24 and 48 hr after the injection. For EMSA, c-fos+/+ andc-fos−/− mice were injected with 45 mg/kg MNU, and retinal nuclear extracts were prepared after 6 hr in darkness.
RESULTS
Light microscopy after exposure to 15 klux
Immediately after lights off
Compared with retinas of dark-adapted c-fos+/+mice, retinas of c-fos+/+ mice, analyzed immediately after 2 hr of exposure to 15 klux of white light, showed condensed rod inner segments (RIS) and disarranged and vesiculated rod outer segments (ROS). These initial signs of light damage were confined to the central retina. Some rods already displayed apoptosis as indicated by condensed nuclear chromatin. Retinas ofc-fos−/− mice showed light-induced lesions, such as vesiculation of ROS, which were absent in dark-adaptedc-fos−/− retinas. Contrary toc-fos+/+ mice, condensation of RIS or nuclear chromatin was rarely seen inc-fos−/− retinas (Fig.1).
Fig. 1.
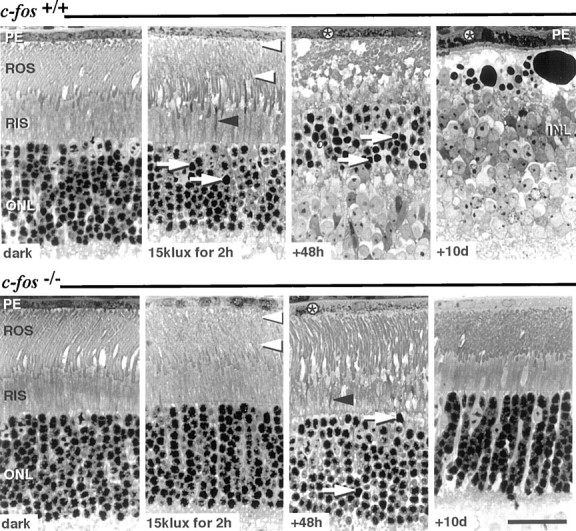
Light microscopy of retinal sections fromc-fos+/+ (top panel) and c-fos−/−(bottom panel) mice: after dark-adaptation (dark), immediately after exposure to 15 klux for 2 hr, and 48 hr and 10 d after light exposure. Retinas of dark-adaptedc-fos+/+ andc-fos−/− mice displayed comparable photoreceptor morphology. Immediately after exposure to 15 klux of white light, ROS showed vesiculation (white arrowheads) in both genotypes, and few RIS appeared condensed (black arrowhead). Forty-eight hours after light exposure, ROS and RIS of c-fos+/+ mice had disintegrated, and the majority of rod nuclei inc-fos+/+ mice showed condensed nuclear chromatin (arrow). Although few condensed RIS (black arrowhead) and darkly stained rod nuclei (arrow) indicated the preceding light exposure, ROS appeared recovered in c-fos−/− mice 48 hr after light exposure. In both genotypes, the presence of phagosomes (*) in the PE indicated the uptake of ROS fragments. Ten days after light exposure, the ONL ofc-fos+/+ mice was amputated, and the INL was in close proximity to the swollen PE. In contrast, photoreceptor and PE morphology inc-fos−/− mice were normal. Stained with methylene blue. Scale bar, 25 μm.
Forty-eight hours after lights off
The outer nuclear layer (ONL) ofc-fos+/+ mice was dramatically reduced in thickness. Most of the remaining rod nuclei displayed condensed chromatin. RIS and ROS were entirely disintegrated, and cystic spaces adjacent to the retinal pigment epithelium (PE) were observed. The PE contained large amounts of phagosomes, most likely indicating the uptake of apoptotic bodies, resulting from the decay of photoreceptors. In contrast, almost no loss of photoreceptors was observed in c-fos−/− mice, and the signs of acute light damage, such as ROS vesiculation, had mostly disappeared. Only few condensed RIS and nuclei containing condensed chromatin indicated the preceding light exposure (Fig. 1).
Ten days after light exposure
Retinas of c-fos+/+ mice lost the entire ONL in the central retinal area. Large darkly stained bodies, presumably containing degradation products of photoreceptors, separated the inner nuclear layer (INL) from the PE. Inc-fos−/− mice, ROS had recovered completely within the regular ROS renewal cycle, and retinas of light-exposed c-fos−/− mice were indistinguishable from control retinas after 10 d (Fig. 1).
Detection of light-induced photoreceptor apoptosis
Forty-eight hours after light exposure (15 klux for 2 hr), DNA fragmentation was detected in the ONL of the central retina ofc-fos+/+ mice by the TUNEL method. Internucleosomal DNA fragmentation was demonstrated by gel electrophoresis of retinal DNA (DNA ladder). No TUNEL signal or DNA ladder was observed in c-fos−/−retinas (Fig. 2).
Fig. 2.
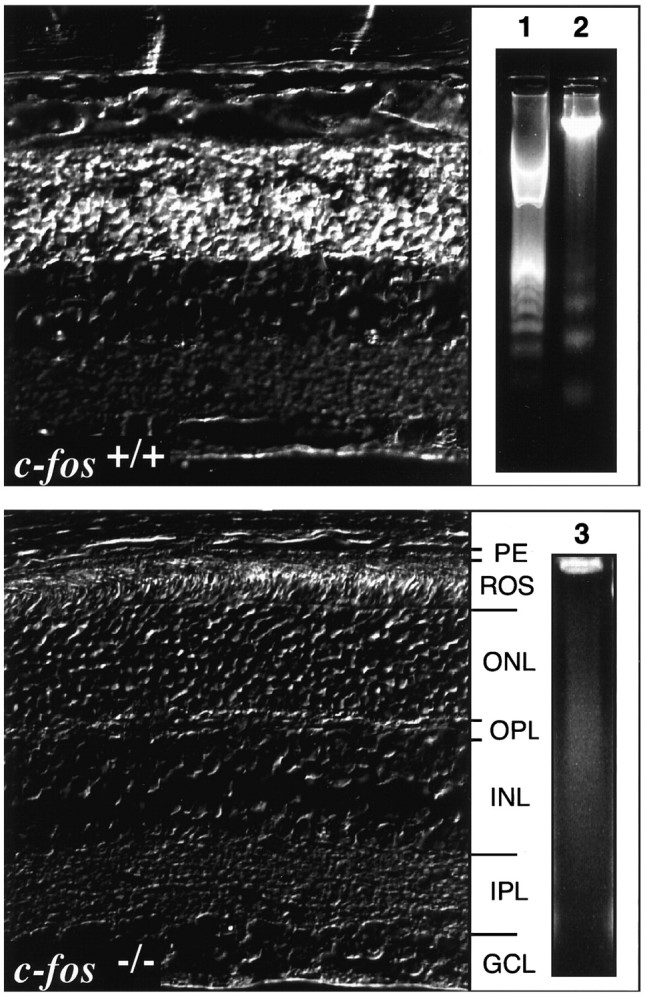
Test for DNA fragmentation in retinas ofc-fos+/+ andc-fos−/− mice 48 hr after exposure to 15 klux for 2 hr. Left, Phase-contrast image of TUNEL staining in paraffin sections of both genotypes revealed DNA fragmentation (white stain) exclusively in the ONL ofc-fos+/+ mice. Right, Electrophoresis of retinal DNA produced the typical DNA ladder only inc-fos+/+ mice (lane 2). No internucleosomal DNA fragmentation was detected inc-fos−/− mice (lane 3). Lane 1, 100 bp DNA standard.OPL, Outer plexiform layer; IPL, inner plexiform layer; GCL, ganglion cell layer.
Electron microscopy after exposure to 5 and 15 klux
Electron microscopy of RIS from dark-adaptedc-fos+/+ andc-fos−/− mice showed mitochondria with regular morphology. Immediately after exposure to 5 or 15 klux for 2 hr, mitochondria in RIS ofc-fos+/+ mice were distinctly swollen and contained disrupted cristae. In contrast, the morphology of mitochondria in RIS of c-fos−/−mice was virtually unaffected (Fig.3).
Fig. 3.
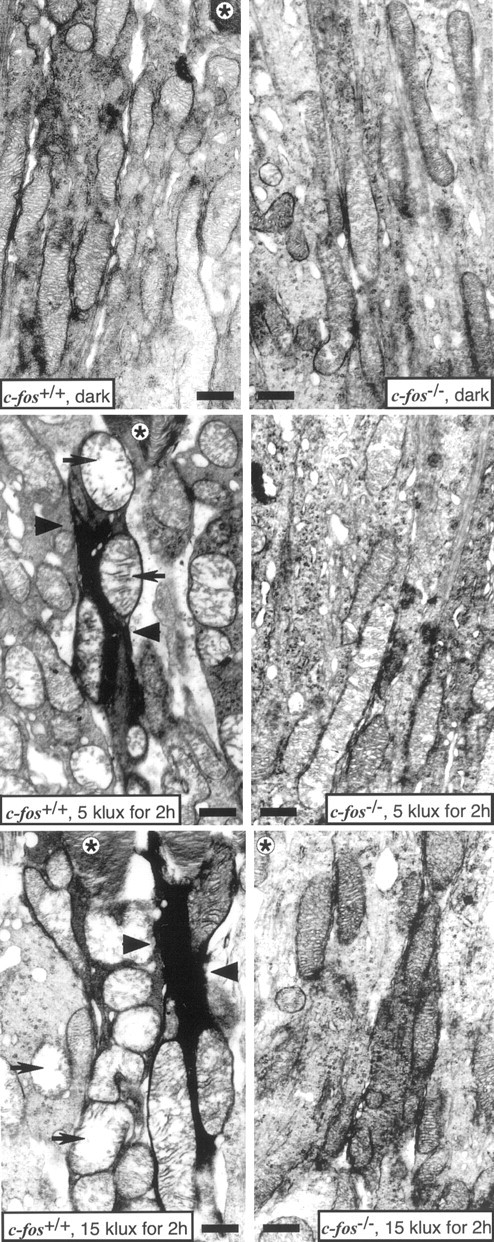
Electron micrographs of RIS ofc-fos+/+ andc-fos−/− mice after dark adaptation (dark) and after 2 hr of exposure to 5 or 15 klux. The morphology of RIS and their mitochondria was comparable in dark-adapted animals of both genotypes. After light exposure, morphology remained unaffected in c-fos−/−mice. In c-fos+/+ mice, light exposure caused the condensation of RIS (arrowheads), which contained swollen mitochondria with disrupted cristae (arrows). *, ROS; Scale bar, 1 μm.
ERG after 15 klux
Luminance–response functions of full-field ERGs inc-fos+/+ mice 48 hr after exposure to 15 klux for 2 hr revealed markedly decreased mean amplitudes of the a- and b-wave compared with untreatedc-fos+/+ animals. The reduction of the mean values of both parameters was ∼50%. In contrast, the mean a- and b-wave amplitudes inc-fos−/− mice after light exposure were comparable with those in untreatedc-fos−/− animals (Fig.4A–D).
Fig. 4.

Luminance–response functions of full-field ERGs of c-fos+/+ (A,C) and c-fos−/−(B, D) mice after dark adaptation (black squares) and 48 hr after exposure to 15 klux for 2 hr (white squares). No reduction of the mean a- and b-wave was caused by light exposure inc-fos−/− animals, both were markedly reduced in c-fos+/+ mice after 48 hr. c-fos+/+, No light,n = 8, mean ± SD;c-fos−/−, no light,n = 8, mean − SD;c-fos+/+, after light,n = 7, mean ± SD;c-fos−/−, after light,n = 7, mean + SD.
AP-1 DNA binding activity after light exposure
AP-1 DNA binding activity in EMSA was specific as shown by competition experiments with unrelated unlabeled oligonucleotides (Fig.5A). AP-1 DNA binding activity in nuclear extracts of c-fos+/+retinas was strongly increased after 1 hr of light exposure (5 klux) and reached a peak of activity between lights off and 6–12 hr thereafter (Fig. 5B). In contrast, AP-1 DNA binding activity in nuclear extracts of c-fos−/−mice remained virtually unchanged after light exposure (5 klux) compared with untreated c-fos−/−mice (Fig. 5C). After exposure ofc-fos+/+ mice to 60 lux, only a very small increase in AP-1 DNA binding activity was observed (Fig.5D).
Fig. 5.

Electrophoretic mobility shift assays of proteins in retinal nuclear extracts (NE) obtained fromc-fos+/+ andc-fos−/− mice. A, Specificity of the protein–DNA interaction. No shift was observed in the absence of NE. Among the unlabeled (cold) oligonucleotides tested, only an excess of the AP-1 binding DNA sequence TRE competed for the binding to radioactively labeled TRE. *, AP-1–DNA complex; **, free probe. B, Induction of AP-1 activity by light (5 klux). NEs from c-fos+/+ mice were prepared from dark-adapted animals and at different time points during and after light exposure. C, Analysis of AP-1 activity inc-fos+/+ (6 hr after 5 klux) andc-fos−/− mice in the same assay. NEs of c-fos−/− mice were prepared from dark-adapted animals (dark), at lights off (120′), and 6 hr thereafter. D, Analysis of AP-1 activity in c-fos+/+ mice dark-adapted, at lights off (120′), and 6 hr thereafter (60 lux). E, AP-1 DNA binding, 6 hr after light-exposure (5 klux), was inhibited by antisera (α) directed against c-Fos and a mixture of anti-Jun antisera (pan Jun: c-Jun, JunD, and JunB). Normal rabbit serum had no effect on the mobility of the AP-1–DNA complex.
Complex composition of light-induced AP-1
Adding an anti-c-Fos antibody to nuclear extracts ofc-fos+/+ retinas 6 hr after exposure to 5 klux (peak of activity) led to an almost complete disappearance of the AP-1-specific band in EMSA. The AP-1–DNA complex was partially supershifted and partially competed by the anti-c-Fos serum. Likewise, the addition of a mixture of three Jun-specific antibodies (c-Jun, JunD, and JunB) resulted in an almost complete competition for AP-1 DNA binding. This indicates that the complex consisted mainly of c-Fos/Jun heterodimers. Addition of rabbit nonimmune serum or unrelated antisera (data not shown) had no effect on the intensity of the AP-1-specific band, demonstrating the specificity of the immunoreaction (Fig. 5E).
Induction of apoptosis byN-methyl-N-nitrosourea
MNU dose-dependently induced photoreceptor degeneration inc-fos+/+ mice. At a dose of 22 mg/kg, no morphological damage was detected, whereas 45 mg/kg induced a distinct degeneration in photoreceptors and PE. With increasing doses of MNU (67 and 90 mg/kg), the destruction of the ONL and the PE was more pronounced and appeared accelerated (Fig.6A).
Fig. 6.
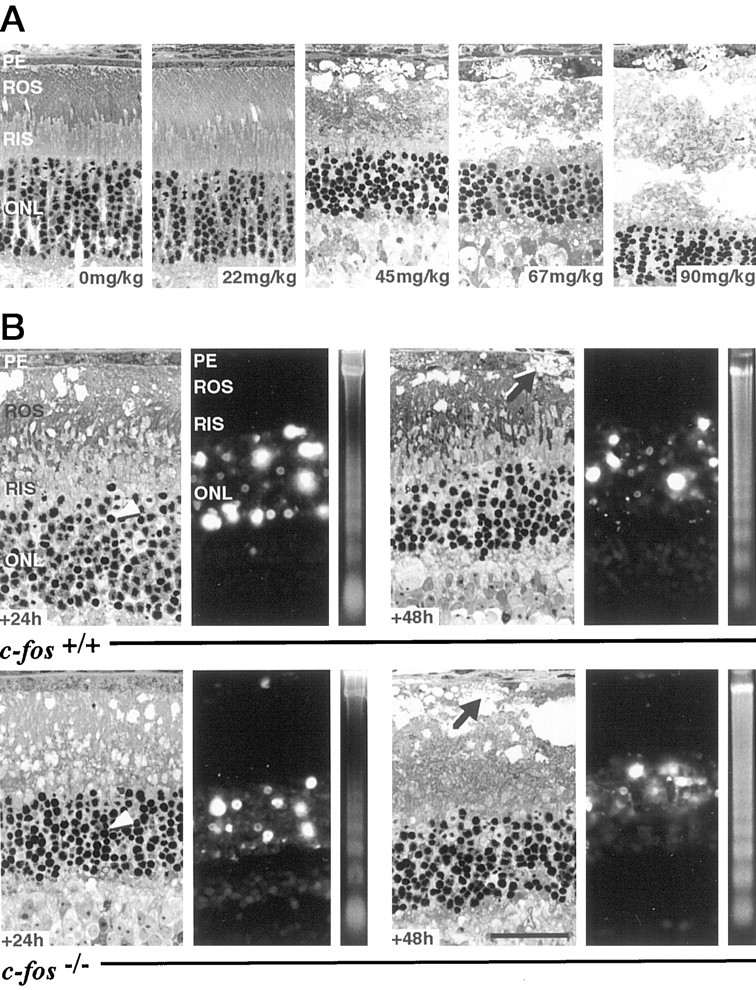
A, MNU induces degeneration of photoreceptors in a dose-dependent manner. Retinas were prepared 72 hr after MNU injection. Only doses higher than 22 mg/kg induced morphological damage to photoreceptors and PE. With increasing doses, damage became more severe and appeared accelerated. (The 90 mg/kg panel is a montage. At this dose of MNU, the ONL consistently was separated from the PE resulting in a large space between both layers.)B, Induction of apoptosis in the retina ofc-fos+/+ andc-fos−/− mice by a single intraperitoneal injection of MNU (45 mg/kg). Retinas were prepared 24 or 48 hr after injection. Light microscopy revealed severe damage to ROS and, at 48 hr, to the PE (arrows). Large cystic spaces were present, and nuclei of rods showed condensed chromatin (arrowheads) at both time points. The ONL was strongly positive for the TUNEL stain, and the presence of internucleosomal DNA fragmentation was indicated by the appearance of the typical DNA ladder. Scale bar, 25 μm.
In contrast to light exposure, there appeared to be no major difference between both genotypes after application of a subsaturating dose of MNU (45 mg/kg). Photoreceptors of bothc-fos+/+ andc-fos−/− mice revealed the characteristic features of apoptosis 24 hr after injection. ROS showed vesiculations, RIS were condensed, and the vast majority of nuclei in the ONL displayed chromatin condensation. The PE was swollen but still viable. Forty-eight hours after injection, the number of rod nuclei in the ONL was reduced, and ROS and RIS were destroyed. In addition, the PE showed intracellular cystic spaces. Although not studied systematically, these signs of degeneration appeared slightly more distinct in retinas of c-fos−/−mice.
The positive TUNEL assay and the presence of a DNA ladder confirmed that photoreceptors of retinas treated with MNU died by apoptosis (Fig.6B). TUNEL-positive nuclei in the PE were detected very rarely, possibly indicating that PE cells may die by a mechanism different from apoptosis.
AP-1 DNA binding activity after injection of MNU
Intraperitoneal injection of MNU (45 mg/kg) increased AP-1 DNA binding activity in retinas ofc-fos+/+ mice but not inc-fos−/− mice 6 hr after the injection (Fig. 7). Increased AP-1 DNA binding activity in c-fos+/+ mice lasted for at least 24 hr (data not shown).
Fig. 7.

MNU induces retinal AP-1 activity only in wild-type mice. No increase in AP-1 DNA binding activity after MNU was detected in c-fos−/− mice. Nuclear extracts (2.5 μg) from saline-injected and MNU-injected (45 mg/kg) mice were analyzed 6 hr after the treatment.
DISCUSSION
After exposure to high doses of light, retinal function and morphology were preserved inc-fos−/− mice but were dramatically disturbed in c-fos+/+mice. In c-fos+/+ mice, an increasing AP-1 DNA binding activity paralleled early phases of apoptosis. No increase in AP-1 activity was observed in retinas of mice lacking c-Fos. Consequently, an essential apoptotic pathway could not be initiated by light in c-fos−/−mice. However, the lack of c-Fos did not inhibit apoptosis in general because apoptosis of rods could be induced by MNU inc-fos−/− mice. Thus, c-Fos seems to be specifically involved in the “private pathway” of light-induced apoptosis.
Range of protection against light-induced apoptosis inc-fos−/− mice
Apoptosis is a process that proceeds within an individual cell. In light damage, absorption of photons by rhodopsin is the trigger for apoptosis (C. Grimm, A. Wenzel, F. Hafezi, S. Yu, T. M. Redmond, C. E. Remè, unpublished observation). Rods ofc-fos+/+ andc-fos−/− mice absorb similar amounts of photons during a light exposure to 5 klux for 60 min (Kueng-Hitz et al., 2000). Whereas rods of wild-type mice undergo apoptosis after this treatment (A. Wenzel C. Grimm, F. Hafezi, C. E. Remé, unpublished observation), those ofc-fos−/− mice survive. Thus, a decreased photon catch capacity is not the basis for the observed protection.
After exposure to a threefold higher intensity (15 klux for 2 hr) and prolonged postexposure periods, no photoreceptor apoptosis was observed in c-fos−/− mice. This indicates that apoptosis in these mice cannot be induced by light or that the threshold for its induction is elevated at least threefold. The possibility of alternate c-Fos-independent pathways, which might induce apoptosis with a delay can be ruled out; 10 d after light exposure, no cell loss or signs of apoptosis were observed inc-fos−/− mice.
Importantly, in c-fos−/−, mice the excessive absorption of photons clearly induced threshold lesions, which were confined to ROS. However, the damage was reversible, and affected cells were able to regain morphology and function completely. In contrast, the ONL in the central retina ofc-fos+/+ mice was practically amputated 10 d after exposure to 15 klux, and total retinal function, as determined by a-wave (photoreceptor function) and b-wave (inner retinal function) recordings, was reduced by ∼50%. Because light damage mainly affected the central retina, the remaining electrical activity may have been triggered by photoreceptors in the periphery. In conclusion, the transition from reversible to irreversible light-induced damage culminating in cell death appeared to depend on functional c-Fos.
It might thus be speculated that rods ofc-fos+/+ mice would be able to survive light exposure and regain function if apoptosis could be blocked. Consequently, therapeutic interference at the level of apoptosis after light damage or other stimuli inducing photoreceptor apoptosis might be beneficial and even rescue vision.
c-Fos is indispensable for the induction of AP-1 by damaging light
The time course of biochemical and morphological changes in retinas after exposure to 5 klux for 2 hr indicates a function of c-Fos in the private pathway of light-induced apoptosis. As soon as after 60 min in light, the activity of AP-1 inc-fos+/+ mice increased. This increase clearly preceded the major synchronized burst of apoptosis (Fig. 1) (Hafezi et al., 1997b). In retinas ofc-fos−/− mice, AP-1 activity remained consistently low. In addition, exposure of wild-type mice to 60 lux for 2 hr induced only a very weak increase in AP-1 activity. In both cases, apoptosis did not occur. Thus, it appears likely that transcriptional activity of AP-1 sufficient to influence the expression of apoptosis-related genes may only be induced after exposure to damaging light doses. The potential pro-apoptotic gene products presumably act at the premitochondrial level (see below). However, preliminary data on the expression of different Bcl-2 family members revealed no changes in response to light exposure depending on thec-fos genotype (Grimm et al., 1999).
From EMSA of retinal nuclear extracts, it is impossible to identify individual cell types with increased AP-1 activity. During exposure to 5 klux, c-fos mRNA is induced in ganglion cells first and appears in the ONL 6 hr after light exposure (Hafezi et al., 1997b). Absence of c-fos transcription in the ONL, however, does not exclude increasing AP-1 activity in photoreceptors. Instead, AP-1 activity may be induced at the post-translational level by phosphorylation of pre-existing Fos or Jun proteins (Deng and Karin, 1994; Kim and Kahn, 1994).
The dimeric transcription factor AP-1 can be assembled differentially from members of two families of proteins: the Fos-family, comprising c-Fos, Fra-1, Fra-2, and Fos-B, and the Jun-family, including c-Jun, Jun-D, and Jun-B (Angel and Karin, 1991). In the retina ofc-fos+/+ mice, a large proportion of the light-induced AP-1 consisted of c-Fos/c-Jun and c-Fos/JunD heterodimers (Hafezi et al., 1999a). However, because light damage in JunD knock-out mice proceeds as in wild-type mice (Hafezi et al., 1999b), it appears that light-induced apoptosis predominantly involves activation of AP-1 composed of c-Fos and c-Jun.
In c-fos−/− mice, no other member of the Fos- or Jun-family could substitute for the lack of c-Fos. Indeed, among the AP-1 proteins, c-Fos appears to play a particular role in apoptosis, as evidenced by studies on castration-induced apoptosis of the prostate. Castration induced similar increases in AP-1 activity in the prostate of c-fos+/+and c-fos−/− mice, but apoptosis only occurred when AP-1 contained c-Fos (Feng et al., 1998).
c-fos is not involved in mediating apoptosis induced by nonlight stimuli
Developmental apoptosis, also referred to as programmed cell death, in the retina (Young, 1984; Penfold and Provis, 1986; Blaschke et al., 1998) proceeds primarily unimpaired in the absence of c-Fos as indicated by the grossly normal retinal morphology and normal, although attenuated, function in c-fos−/−mice (Kueng-Hitz et al., 2000). Likewise, the relatively slow elimination of immature rods by apoptosis in the retinal degeneration (rd)-mouse (Caley et al., 1972; LaVail and Sidman, 1974; Portera Cailliau et al., 1994) is not affected by the lack of c-Fos (Hafezi et al., 1998). Thus, at least before maturity of the retina, photoreceptors of c-fos−/− mice are able to undergo apoptosis.
MNU induces apoptosis via the methylation of genomic DNA (Kokkinakis et al., 1997; Tominaga et al., 1997), and methyl adducts to genomic DNA of photoreceptors have been detected in MNU-induced retinal degeneration (Ogino et al., 1993). Here, MNU was tested as a light-independent stimulus capable to induce degeneration in mature photoreceptors (Yuge et al., 1996; Nambu et al., 1997). MNU induced a strong apoptotic response in photoreceptors ofc-fos+/+ andc-fos−/− mice, indicating that MNU, in contrast to light, involves a pro-apoptotic pathway not depending on c-Fos. Thus, fully developed rods ofc-fos−/− mice are able to undergo apoptosis. Therefore, c-Fos is dispensable for the “common pathway” of rod apoptosis.
Light exposure and MNU both induced retinal AP-1 activity inc-fos+/+ mice (Figs. 5,7). In both cases, AP-1 contained c-Fos (Fig. 5 and data not shown). Depending on the system and stimulus studied, c-Fos may (Feng et al., 1998; Hafezi et al., 1997b; Pruschy et al., 1997) or may not (Gajate et al., 1996;Hafezi et al., 1998) be involved in mediating apoptosis. c-Fos may also be involved in the defense against cell death (Ivanov and Nicolic-Zugic, 1997) induced by, for example, methylating agents, such as MNU (Dosch and Kaina, 1996). Thus, in photoreceptors, AP-1 containing c-Fos might have opposite functions depending on the stimulus; it mediates light-induced apoptosis but may protect against MNU. In c-fos−/− mice, neither stimulus induced AP-1. Therefore, the light stimulus could not be mediated to the apoptotic machinery, and the defense against MNU may have been deficient. Supporting this view, we found preliminary evidence for more severe morphological lesions in retinas ofc-fos−/− mice compared withc-fos+/+ mice after application of MNU. Interestingly, c-fos−/− mouse fibroblasts are hypersensitive to methylating agents (Kaina et al., 1997).
c-fos acts in the private pathway of light-induced apoptosis
The signaling of pro-apoptotic stimuli involves so-called private pathways: a multitude of different signal cascades, which may converge at the level of mitochondria (Kluck et al., 1997a; Green and Reed, 1998). Release of cytochrome c and/or an apoptosis-inducing factor (Liu et al., 1996; Susin et al., 1996; Zhivotovsky et al., 1998) attributable to changes in the mitochondrial membrane appears to represent a “point of no return” in apoptosis (Marchetti et al., 1996; Zamzami et al., 1995) (but see Jaattela et al., 1998). Upon their presence in the cytosol, these factors can trigger the common pathway of apoptosis, involving a tightly regulated cascade of caspases (Kluck et al., 1997b; Bossy Wetzel et al., 1998; Green and Kroemer, 1998;Thornberry and Lazebnik, 1998). Preliminary evidence indicates that light-induced photoreceptor apoptosis might involve the activation of cytochrome c-responsive caspases (Chang et al., 1999; Peng et al., 1999). In contrast to c-fos+/+ mice, the morphology of mitochondria inc-fos−/− mice appeared unaffected by light exposure. One may assume that the morphological changes of mitochondria in c-fos+/+ mice reflect their involvement in the induction of apoptosis. Thus, in the absence of c-Fos, the private pathway that signals the stimulus “excessive light” appears to be interrupted before mitochondrial morphology is affected.
In conclusion, strong evidence accumulates for an acute and specific contribution of AP-1 containing c-Fos to the induction of light-induced apoptosis in rods. Although all collected evidence strongly supports this model (this work; Kueng-Hitz et al., 2000), yet undetected subtle changes of retinal development inc-fos−/− mice might also contribute to an increased light tolerance.
To further elucidate the molecular aspects of light-induced photoreceptor apoptosis, it will be important to reveal mechanisms that convert absorption of photons into activation of AP-1 and to identify genes controlled by AP-1 containing c-Fos, which respond to light. It might then be possible to delineate the private pathway of light-induced apoptosis, leading from photon absorption to mitochondrial damage.
Footnotes
This work was supported by the Swiss National Science Foundation (Zürich, Switzerland), the Bruppacher Foundation (Zürich, Switzerland), and the Ernst & Berta Grimmke Foundation (Düsseldorf, Germany). We thank G. Hoegger, C. Imsand, and K. Munz for skilled technical assistance.
Correspondence should be addressed to A. Wenzel, University Hospital Zürich, Department of Ophthalmology, Frauenklinikstrasse 24, CH-8091 Zürich, Switzerland. E-mail: awenzel@opht.unizh.ch.
Dr. Marti's present address: Department for Clinical Research, Inselspital, University of Bern, Murtenstrasse 35, CH-3010 Bern, Switzerland.
REFERENCES
- 1.Abler AS, Chang CJ, Ful J, Tso MO, Lam TT. Photic injury triggers apoptosis of photoreceptor cells. Res Commun Mol Pathol Pharmacol. 1996;92:177–189. [PubMed] [Google Scholar]
- 2.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 3.Blaschke AJ, Weiner JA, Chun J. Programmed cell death is a universal feature of embryonic and postnatal neuroproliferative regions throughout the central nervous system. J Comp Neurol. 1998;396:39–50. doi: 10.1002/(sici)1096-9861(19980622)396:1<39::aid-cne4>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 4.Bossy Wetzel E, Newmeyer DD, Green DR. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 1998;17:37–49. doi: 10.1093/emboj/17.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caley DW, Johnson C, Liebelt RA. The postnatal development of the retina in the normal and rodless CBA mouse: a light and electron microscopic study. Am J Anat. 1972;133:179–212. doi: 10.1002/aja.1001330205. [DOI] [PubMed] [Google Scholar]
- 6.Chang CJ, Lai HF, Lee WH, Liao CL. Activation of protease CPP32 as an early indicator of apoptosis in photoreceptor degeneration. Invest Ophthalmol Vis Sci [Abstr] 1999;40:225. [Google Scholar]
- 7.Cideciyan AV, Hood DC, Huang Y, Banin E, Li ZY, Stone EM, Milam AH, Jacobson SG. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci USA. 1998;95:7103–7108. doi: 10.1073/pnas.95.12.7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cruickshanks KJ, Klein R, Klein BE. Sunlight and age-related macular degeneration. The beaver dam eye study. Arch Ophthalmol. 1993;111:514–518. doi: 10.1001/archopht.1993.01090040106042. [DOI] [PubMed] [Google Scholar]
- 9.Deng T, Karin M. c-Fos transcriptional activity stimulated by H-Ras-activated protein kinase distinct from JNK and ERK. Nature. 1994;371:171–175. doi: 10.1038/371171a0. [DOI] [PubMed] [Google Scholar]
- 10.Dosch J, Kaina B. Induction of c-fos, c-jun, junB and junD mRNA and AP-1 by alkylating mutagens in cells deficient and proficient for the DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) and its relationship to cell death, mutation induction and chromosomal instability. Oncogene. 1996;13:1927–1935. [PubMed] [Google Scholar]
- 11.Feng Z, Joos HJ, Vallan C, Mühlbauer R, Altermatt HJ, Jaggi R. Apoptosis during castration-induced regression of the prostate is Fos dependent. Oncogene. 1998;17:2593–2600. doi: 10.1038/sj.onc.1202195. [DOI] [PubMed] [Google Scholar]
- 12.Gajate C, Alonso MT, Schimmang T, Mollinedo F. C-Fos is not essential for apoptosis. Biochem Biophys Res Commun. 1996;218:267–272. doi: 10.1006/bbrc.1996.0047. [DOI] [PubMed] [Google Scholar]
- 13.Green DR, Kroemer G. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biology. 1998;8:267–271. doi: 10.1016/s0962-8924(98)01273-2. [DOI] [PubMed] [Google Scholar]
- 14.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 15.Grimm C, Wenzel A, Hafezi F, Remé CE. Gene expression during light-induced apoptosis of photoreceptors in the retina of c-fos knock-out and wildtype mice. Invest Ophthalmol Vis Sci [Abstr] 1999;40:161. [Google Scholar]
- 16.Hafezi F, Marti A, Munz K, Reme CE. Light-induced apoptosis: differential timing in the retina and pigment epithelium. Exp Eye Res. 1997a;64:963–970. doi: 10.1006/exer.1997.0288. [DOI] [PubMed] [Google Scholar]
- 17.Hafezi F, Steinbach JP, Marti A, Munz K, Wang ZQ, Wagner EF, Aguzzi A, Reme CE. The absence of c-fos prevents light-induced apoptotic cell death of photoreceptors in retinal degeneration in vivo. Nat Med. 1997b;3:346–349. doi: 10.1038/nm0397-346. [DOI] [PubMed] [Google Scholar]
- 18.Hafezi F, Abegg M, Grimm C, Wenzel A, Stuermer J, Farber DB, Remé CE. Retinal degeneration in the rd mouse in the absence of c-fos. Invest Ophthalmol Vis Sci. 1998;38:2239–2244. [PubMed] [Google Scholar]
- 19.Hafezi F, Marti A, Grimm C, Wenzel A, Remé C E. Differential DNA binding activities of the transcription factors AP-1 and Oct-1 during light-induced apoptosis of photoreceptors. Vision Res. 1999a;39:2511–2518. doi: 10.1016/s0042-6989(98)00313-7. [DOI] [PubMed] [Google Scholar]
- 20.Hafezi F, Grimm C, Wenzel A, Weitzman J, Abegg M, Yaniv M, Remé CE. Retinal photoreceptors are apoptosis-competent in the absence of JunD/AP-1. Cell Death Differ. 1999b;10:934–937. doi: 10.1038/sj.cdd.4400574. [DOI] [PubMed] [Google Scholar]
- 21.Henderson CE. Programmed cell death in the developing nervous system. Neuron. 1996;17:579–585. doi: 10.1016/s0896-6273(00)80191-9. [DOI] [PubMed] [Google Scholar]
- 22.Ivanov VN, Nicolic-Zugic J. Transcription factor activation during signal-induced apoptosis of immature CD4(+)CD8(+) thymocytes. A protective role for c-Fos. J Biol Chem. 1997;272:8558–8566. [PubMed] [Google Scholar]
- 23.Jaattela M, Wissing D, Kokholm K, Kallunki T, Egeblad M. Hsp70 exerts anti-apoptotic function downstream of caspase-3-like proteases. EMBO J. 1998;17:6124–6134. doi: 10.1093/emboj/17.21.6124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 25.Kaina B, Haas S, Kappes H. A general role for c-Fos in cellular protection against DNA-damaging carcinogens and cytostatic drugs. Cancer Res. 1997;57:2721–2731. [PubMed] [Google Scholar]
- 26.Kim SJ, Kahn CR. Insulin stimulates phosphorylation of c-Jun, c-Fos, and Fos-related proteins in cultured adipocytes. J Biol Chem. 1994;269:11887–11892. [PubMed] [Google Scholar]
- 27.Kluck RM, Bossy Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997a;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 28.Kluck RM, Martin SJ, Hoffman BM, Zhou JS, Green DR, Newmeyer DD. Cytochrome c activation of CPP32-like proteolysis plays a critical role in a Xenopus cell-free apoptosis system. EMBO J. 1997b;16:4639–4649. doi: 10.1093/emboj/16.15.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kokkinakis DM, Ahmed MM, Delgado R, Fruitwala MM, Mohiuddin M, Albores-Saavedra J. Role of O6-methylguanine-DNA methyltransferase in the resistance of pancreatic tumors to DNA alkylating agents. Cancer Res. 1997;57:5360–5368. [PubMed] [Google Scholar]
- 30.Kueng-Hitz N, Grimm C, Lansel N, Hafezi F, He L, Fox D, Remé CE, Niemeyer G, Wenzel A (2000) The retina ofc-fos+/+ and c-fos−/−mice: electrophysiological, morphological and biochemical aspects. Invest Ophthalmol Vis Sci, in press. [PubMed]
- 31.LaVail MM, Sidman RL. C57BL-6J mice with inherited retinal degeneration. Arch Ophthalmol. 1974;91:394–400. doi: 10.1001/archopht.1974.03900060406015. [DOI] [PubMed] [Google Scholar]
- 32.LaVail MM, Gorrin GM, Yasumura D, Matthes MT. Increased susceptibility to constant light in nr and pcd mice with inherited retinal degeneration. Invest Ophthalmol Vis Sci. 1999;40:1020–1024. [PubMed] [Google Scholar]
- 33.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 34.Marchetti P, Castedo M, Susin SA, Zamzami N, Hirsch T, Macho A, Haeffner A, Hirsch F, Geuskens M, Kroemer G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med. 1996;184:1155–1160. doi: 10.1084/jem.184.3.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marti A, Jehn B, Costello E, Keon E, Ke G, Martin F, Jaggi R. Protein kinase A and AP-1 (c-Fos/JunD) are induced during apoptosis of mouse mammary epithelial cells. Oncogene. 1994;9:1213–1223. [PubMed] [Google Scholar]
- 36.Nambu H, Yuge K, Nakajima M, Shikata N, Takahashi K, Miki H, Uyama M, Tsubura A. Morphologic characteristics of N-methyl-N-nitrosourea-induced retinal degeneration in C57BL mice. Pathol Int. 1997;47:377–383. doi: 10.1111/j.1440-1827.1997.tb04511.x. [DOI] [PubMed] [Google Scholar]
- 37.Niemeyer G, Kueng N. A simple and stable d.c. electrode for ocular electrophysiology. Doc Ophthalmol. 1998;95:55–61. doi: 10.1023/a:1001776428445. [DOI] [PubMed] [Google Scholar]
- 38.Ogino H, Ito M, Matsumoto K, Yagyu S, Tsuda H, Hirono I, Wild CP, Montesano R. Retinal degeneration induced by N-methyl-N-nitrosourea and detection of 7-methyldeoxyguanosine in the rat retina. Toxicol Pathol. 1993;21:21–25. doi: 10.1177/019262339302100103. [DOI] [PubMed] [Google Scholar]
- 39.Penfold PL, Provis JM. Cell death in the development of the human retina: phagocytosis of pyknotic and apoptotic bodies by retinal cells. Graefes Arch Clin Exp Ophthalmol. 1986;224:549–553. doi: 10.1007/BF02154744. [DOI] [PubMed] [Google Scholar]
- 40.Peng M, Liu C, Li Y, Laties AM, Wen R. Activation of caspase-3 in rat retina during photoreceptor degeneration induced by exposure to constant light. Invest Ophthalmol Vis Sci [Abtsr] 1999;40:225. [Google Scholar]
- 41.Portera Cailliau C, Sung CH, Nathans J, Adler R. Apoptotic photoreceptor cell death in mouse models of retinitis pigmentosa. Proc Natl Acad Sci USA. 1994;91:974–978. doi: 10.1073/pnas.91.3.974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pruschy M, Shi YQ, Crompton NE, Steinbach J, Aguzzi A, Glanzmann C, Bodis S. The proto-oncogene c-fos mediates apoptosis in murine T-lymphocytes induced by ionizing radiation and dexamethasone. Biochem Biophys Res Commun. 1997;241:519–524. doi: 10.1006/bbrc.1997.7846. [DOI] [PubMed] [Google Scholar]
- 43.Remé CE, Weller M, Szczesny P, Munz K, Hafezi F, Reinboth JJ, Clausen M. Light-induced apoptosis in the rat retina in vivo. In: Anderson RE, LaVail MM, Hollyfield JG, editors. Degenerative diseases of the retina. Plenum; New York: 1995. pp. 19–25. [Google Scholar]
- 44.Remé CE, Grimm C, Hafezi F, Marti A, Wenzel A. Apoptotic cell death in retinal degenerations. Prog Ret Eye Res. 1998;17:443–464. doi: 10.1016/s1350-9462(98)00009-3. [DOI] [PubMed] [Google Scholar]
- 45.Simons K. Artificial light and early-life exposure in age-related macular degeneration and in cataractogenic phototoxicity. Arch Ophthalmol. 1993;111:297–298. doi: 10.1001/archopht.1993.01090030015002. [DOI] [PubMed] [Google Scholar]
- 46.Stefanis L, Burke RE, Greene LA. Apoptosis in neurodegenerative disorders. Curr Opin Neurol. 1997;10:299–305. doi: 10.1097/00019052-199708000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med. 1996;184:1331–1341. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 49.Tominaga Y, Tsuzuki T, Shiraishi A, Kawate H, Sekiguchi M. Alkylation induced apoptosis of embryonic stem cells in which the gene for DNA-repair, methyltransferase, had been disrupted by gene targeting. Carcinogenesis. 1997;18:889–896. doi: 10.1093/carcin/18.5.889. [DOI] [PubMed] [Google Scholar]
- 50.Uren AG, Vaux DL. Molecular and clinical aspects of apoptosis. Pharmacol Ther. 1996;72:37–50. doi: 10.1016/s0163-7258(96)00098-8. [DOI] [PubMed] [Google Scholar]
- 51.Wong F. Investigating retinitis pigmentosa: a laboratory scientist's perspective. Prog Ret Eye Res. 1997;16:353–373. [Google Scholar]
- 52.Young RW. Cell death during differentiation of the retina in the mouse. J Comp Neurol. 1984;229:362–373. doi: 10.1002/cne.902290307. [DOI] [PubMed] [Google Scholar]
- 53.Yuge K, Nambu H, Senzaki H, Nakao I, Miki H, Uyama M, Tsubura A. N-Methyl-N-nitrosourea-induced photoreceptor apoptosis in the mouse retina. In Vivo. 1996;10:483–488. [PubMed] [Google Scholar]
- 54.Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere JL, Petit PX, Kroemer G. Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med. 1995;181:1661–1672. doi: 10.1084/jem.181.5.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhivotovsky B, Orrenius S, Brustugun OT, Doskeland SO. Injected cytochrome c induces apoptosis. Nature. 1998;391:449–450. doi: 10.1038/35060. [DOI] [PubMed] [Google Scholar]