

Abstract
Apoptosis is an important mechanism of physiological and pathological cell death. It is regulated by several gene products, including caspases and the bcl-2-like proteins, whose roles have been demonstrated in numerous systems. One of these is a model of cerebellar granule cells (CGCs) in which apoptosis is induced by acute removal of serum and depolarizing concentrations of potassium. Previous work by several authors showed that benzyloxycarbonyl-DEVD-fluoromethylketone, a somewhat selective caspase inhibitor, significantly protected CGCs from apoptosis; however, because this molecule targets multiple caspases, it is not known whether a single caspase is primarily responsible for effecting cell death in this model. We attempted to answer this question by cotransfecting CGCs with green fluorescent protein reporter and a hammerhead ribozyme directed against caspase-3 mRNA. Maximal protection by this ribozyme was observed after 24 hr of deprivation, at which time apoptosis was 18 ± 0.7% compared with 32 ± 2% in control cells. Significant protection was also observed with human inhibitor of apoptosis (IAP)-like protein–X-linked IAP, a specific inhibitor of caspase-3, -7, and -9, and with p35, a general caspase inhibitor. Overexpression of bcl-2 produced almost complete protection from apoptosis after 24 hr of serum–K+ deprivation (5 ± 2 vs 44 ± 2% in control cells). These results confirm that caspases play an important role in CGC apoptosis and indicate that caspase-3 itself is a significant mediator of this process.
Keywords: apoptosis, caspase, cerebellar granule cells, ribozyme, IAP, transfection
Apoptosis is a morphologically distinct form of cell death characterized by cellular fragmentation, chromatin condensation and margination, and internucleosomal cleavage of genomic DNA (Bredesen, 1995). Many of the gene products involved in apoptosis have been identified in the nematode Caenorhabditis elegans, including ced-9, which inhibits apoptosis, andced-3 and ced-4, which promote apoptosis (Ellis and Horvitz, 1986). In mammals, the ced-9 homologs comprise a growing family of proteins related to bcl-2 (Adams and Cory, 1998). One mammalian homolog of ced-4 is apoptotic protease activating factor-1 (APAF-1) (Zou et al., 1997). The mammalian homologs of ced-3 are known as caspases, a family of at least 14 aspartate-specific proteases (Thornberry and Lazebnik, 1998;Van de Craen et al., 1998).
Caspase activity may lead to many morphological and biochemical features of apoptosis. Cellular fragmentation and plasma membrane blebbing may result from actin depolymerization caused by caspase-3-mediated cleavage of gelsolin (Kothakota et al., 1997). Cleavage of nuclear lamins by caspase-6 may result in chromatin condensation and margination (Orth et al., 1996; Takahashi et al., 1996). Changes in chromatin structure may also be related to internucleosomal cleavage of DNA. Caspase-activated deoxyribonuclease (CAD), homologous to DNA fragmentation factor-45, digests genomic DNA after cleavage of its inhibitor, ICAD, by caspase-3 (Liu et al., 1997; Enari et al., 1998; Sakahira et al., 1998).
Caspases are inhibited by natural anti-apoptotic proteins, such as the p35 protein of Autographica californica nuclear polyhedrosis virus (Bump et al., 1995) or members of the inhibitor of apoptosis (IAP) family (Miller, 1997). One such member, X-linked IAP (XIAP) [or IAP-like protein (ILP)], specifically inhibits caspase-3, -7, and -9 (Deveraux et al., 1997, 1998).
Caspases play an important role in neuronal apoptosis, both in vitro and in vivo. Peptide inhibitors of caspases are protective in cultures of cerebellar granule cells (CGCs) (Armstrong et al., 1997; Taylor et al., 1997), a model of neuronal apoptosis induced by glutamate (Du et al., 1997) or acute deprivation of serum and/or depolarizing concentrations of K+(D'Mello et al., 1993; Miller and Johnson, 1996; Eldadah et al., 1997). Similar protective effects have been shown in animal models of traumatic brain injury (Yakovlev et al., 1997) and stroke (Endres et al., 1998). The caspase inhibitors examined in these systems are peptide-fluoromethylketones, modified oligopeptides that bind irreversibly at the caspase active site. Benzyloxycarbonyl-DEVD-fluoromethylketone, for example, targets multiple caspases, including caspase-3 (Talanian et al., 1997), and protection with this inhibitor is highly indicative of caspase involvement in cell death. However, involvement of specific caspases in apoptosis cannot be determined with this inhibitor.
An alternative approach to identifying which particular caspases may be responsible for mediating apoptosis involves selective downregulation by ribozyme targeting. Ribozymes are enzymatic RNA molecules that suppress specific gene expression by selective cleavage of other RNA species. We sought to determine whether ribozyme-mediated inhibition of caspase-3 could protect against apoptosis in CGCs. This caspase has been identified in several studies as a mediator of many morphological and biochemical changes associated with apoptosis (Kothakota et al., 1997; Enari et al., 1998; Sakahira et al., 1998).
MATERIALS AND METHODS
Preparation of cerebellar granule cells. CGCs were prepared essentially as described previously (Thangnipon et al., 1983). Briefly, cerebella from 8-d-old Sprague Dawley rat pups were dissected from their meninges and minced. The tissue was then trypsinized for 20 min, triturated, and centrifuged through a 4% bovine serum albumin layer to remove cellular debris. Cells were seeded onto poly-l-lysine-coated tissue culture plates (24-well; Corning, Corning, NY) at 3 × 105 cells per well in seeding media consisting of 2 mm glutamine, 50 μg/ml gentamicin sulfate, 10% fetal bovine serum (Hyclone, Logan, UT), and 20 mm supplemented KCl in Basal Medium Eagle (Life Technologies, Gaithersburg, MD). Twenty to 24 hr after seeding, 10 μmcytosine-β-d-arabinofuranoside was added to prevent overproliferation of non-neuronal cells.
Construction of plasmid DNA. Coding sequences for β-galactosidase, human bcl-2, p35, and RZ101 were cloned into pCR3.1 (Invitrogen, San Diego, CA). β-Galactosidase was excised from pIND-lacZ (Invitrogen) using HindIII and XhoI; human bcl-2 was excised from pB4 (American Type Culture Collection, Manassas, VA) using EcoRI; and p35 was excised from pBluescriptII KS−-p35 (courtesy of Winnie Wong, BASF Corporation, Worcester, MA) using BamHI andXbaI. The coding sequence for pEBB-human ILP–XIAP and empty vector control were kindly provided by Colin Duckett (National Cancer Institute, Bethesda, MD) (Duckett et al., 1998). RZ101 is a hammerhead ribozyme construct prepared from synthetic oligonucleotides (Life Technologies) and ligated between HindIII andXhoI restriction sites. The flanking arms of RZ101 are complementary to bases 93–113 of rat caspase-3 mRNA, respectively (GenBank accession number U49930).
Transfection of CGCs. CGCs were cotransfected by calcium phosphate precipitation essentially as described previously (Dudek et al., 1997). Briefly, plasmid DNA from each pCR3.1 expression construct was mixed with pGreenLantern-1 (Life Technologies), a green fluorescent protein (GFP)-expressing vector, in a 2–3:1 weight ratio and precipitated in 2× HEPES-buffered saline (Promega, Madison, WI) for 25 min at room temperature. The original media from CGCs was removed and saved, and CGCs were washed twice with Minimal Essential Medium (MEM) (Life Technologies). DNA precipitates (3–4 μg total) were added dropwise to the media, and cells were incubated for 45 min at room temperature under ambient CO2. Cells were then washed twice with MEM, the original seeding media was replaced, and cells were returned to the incubator. GFP was used as a reporter because it allows identification of transfected cells in the living state and is stable under the apoptotic conditions examined.
Terminal deoxynucleotidyl transferase-mediated biotinylated dUTP-nick end labeling. CGCs were fixed for 10 min at room temperature in PBS containing 4% paraformaldehyde and 4% sucrose. Cells were then permeabilized in 0.1% saponin for 10 min, and DNA ends were labeled for 1 hr at 37°C in the following reaction mixture: Tris-saline buffer, pH 7.6, 0.3 U/μl recombinant terminal deoxynucleotidyl transferase (Life Technologies), TACS dNTPs (Trevigen), and 2.5 mmCoCl2. Cells were then incubated in 1:50 FITC-avidin DN (Vector Laboratories, Burlingame, CA) for 1 hr at room temperature. After terminal deoxynucleotidyl transferase-mediated biotinylated dUTP-nick end labeling (TUNEL) was completed, Hoechst 33258 was added to the same samples at 10 μg/ml.
In vitro ribozyme cleavage. RNA transcripts were generated using T7 RNA polymerase (Ambion, Austin, TX) and purified by gel filtration chromatography (Clontech, Cambridge, UK). Each 10 μl reaction contained 50 mm Tris-HCl, pH 8.0, 5 mm MgCl2, 40 U RNasin, 5 pmol of rat caspase-3 RNA, and 20 pmol of ribozyme (or water for the control reaction). Mixtures were incubated at 37°C for 1 hr and stopped by the addition of 2× RNA loading buffer. Reaction products were heat-denatured, run on a 6% polyacrylamide gel containing 7m urea, and silver stained as described previously (Palfner et al., 1995).
Assessment of ribozyme functional activity. To demonstrate the ability of the ribozyme to downregulate caspase-3 expression, rat pheochromocytoma PC12 cells were transiently transfected with RZ101 or β-galactosidase by lipofection (Lipofectamine; Life Technologies). Downregulation of caspase-3 mRNA was assessed by quantitative reverse transcription (RT)-PCR. mRNA was prepared from total cell lysates by standard phenol–chloroform extraction, and 0.5 μg from each condition was subjected to 35 cycles of RT-PCR using the following program: initial denaturation at 95°C for 2 min, 94°C for 30 sec, 55°C for 15 sec, 72°C for 45 sec, and a final primer extension at 72°C for 2 min. A standard curve demonstrated linearity with these conditions. Primers for caspase-3 and cyclophilin have been published previously (Eldadah et al., 1997 and Haendler et al., 1987, respectively). After electrophoresis and staining with ethidium bromide, bands were analyzed by densitometry with Multi Analyst software (Bio-Rad, Hercules, CA). Each value represents the ratio of the adjusted volumes of caspase-3 signal to cyclophilin internal control signal. These ratios were then standardized to the lacZ-transfected values.
For Western blot analysis, total cellular protein was extracted 48 hr after transfection by incubating in lysis solution containing 1% Triton X-100, 0.1% SDS, 0.1 m NaCl, 0.5% deoxycholic acid, 1 mm PMSF, and 10 mm Tris-HCl, pH 8.0. Protein concentrations for each sample were quantified using the Bradford method (Bio-Rad), and twofold dilutions of each sample were run on a 12% Tris–glycine–SDS polyacrylamide gel. After transfer to polyvinylidene difluoride membrane, Western blotting was performed using 2 μg/ml anti-pro-caspase-3 antibody (catalog #06–538; Upstate Biotechnology, Lake Placid, NY) and enhanced chemiluminescent signal detection (Pierce, Rockford, IL).
Caspase-3-like activity was measured essentially as described byNicholson et al. (1995). Briefly, total cellular protein was extracted in a solution containing 10 mm HEPES-KOH, 2 mmEDTA, 0.1% 3-([3-cholamidoprophyl]dimethylammonio)-1-propane sulfonate, and 5 mm dithiothreitol, pH 7.4. Twenty micrograms were incubated at room temperature with 20 μmacetyl-DEVD-aminomethylcoumarin (Ac-DEVD-AMC), a fluorogenic caspase-3 substrate. Cleavage products were measured on a fluorometer (PerSeptive Biosystems, Framingham, MA) using 360/460 nm excitation/emission filters. Activity was calculated by linear regression analysis of free aminomethylcoumarin fluorescence over time.
Induction and assessment of apoptosis in CGCs. The method ofDudek et al. (1997) was used to assess apoptosis in transfected CGCs. At 1–1.5 d after transfection, CGCs were placed in fresh media containing either 5 mm KCl and no serum (apoptotic induction) or 25 mm KCl and 10% dialyzed fetal bovine serum (Hyclone) (control). Twenty-four to 48 hr later, Hoechst 33258 was added at 2 μg/ml, and GFP-positive cells were scored blindly for morphological features indicative of apoptosis. These included cytoplasmic blebbing and dendritic fragmentation (assessed by GFP fluorescence) and nuclear condensation and margination (assessed by Hoechst 33258 fluorescence).
RESULTS
CGCs were transfected with plasmid vectors expressing human bcl-2, baculoviral p35, human ILP–XIAP, or a ribozyme against rat caspase-3. Transfected cells were induced to undergo apoptosis by deprivation of serum and supplemented KCl and were assessed for apoptotic morphology after 24 hr, and in the case of ILP–XIAP and ribozyme, after 36 and 48 hr as well.
Figure 1 shows GFP fluorescence and 5-bromo-4-chloro-3-indolyl-β-d-galactopyranosidase(X-gal) staining for β-galactosidase in the same cells to demonstrate colocalization of reporter molecule with experimental vector. All GFP-positive CGCs selected randomly for analysis were also X-gal-positive, indicating that GFP can serve as an adequate reporter molecule in this model despite low transfection efficiencies (0.1–0.3% on average).
Fig. 1.
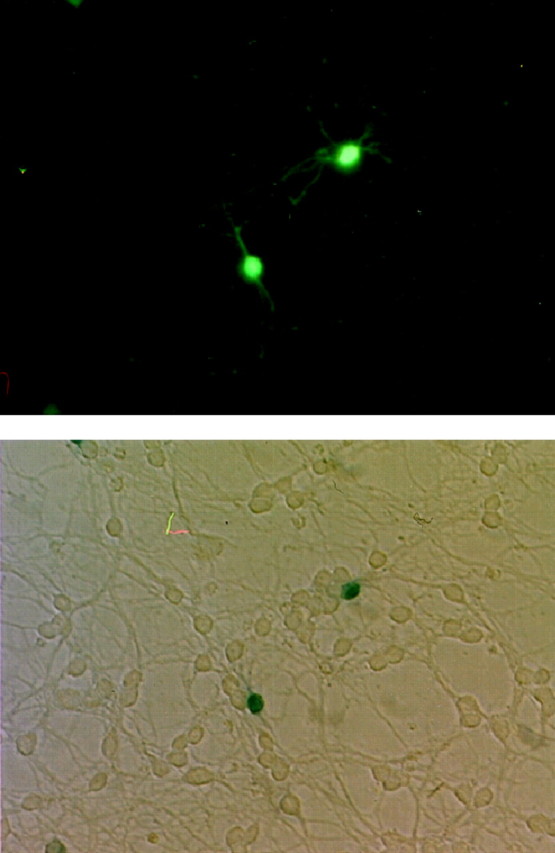
Colocalization of GFP fluorescence with β-galactosidase activity. CGCs were cotransfected with pGreenLantern-1 and pCR3.1-β-galactosidase. Two cells were imaged for GFP positivity using a standard fluorescein filter set (top) and for β-galactosidase reactivity by X-gal staining with light microscopy (bottom). Every GFP-positive cell observed was also X-gal-positive. Transfection efficiency was ∼0.1–0.3%.
Figure 2 shows examples of typical morphological and fluorescent patterns of healthy and apoptotic CGCs as assessed by GFP and Hoechst 33258. Left panels depict a healthy CGC transfected with anti-caspase-3 ribozyme after 24 hr of serum–potassium deprivation. The cell has a small, slightly oval cell body and intact processes. After staining chromatin with Hoechst 33258, the nucleus is dim and occupies virtually the entire cell body volume.Right panels show an apoptotic CGC transfected with control plasmid after 24 hr of serum–K+deprivation. The cell body is larger and rounder than that of its healthy counterpart, its processes have undergone extensive fragmentation, and its border is blurry, probably because of membrane blebbing. In addition, the nucleus is intensely fluorescent, condensed to a pinpoint, and shifted to the extreme periphery of the cell body.
Fig. 2.
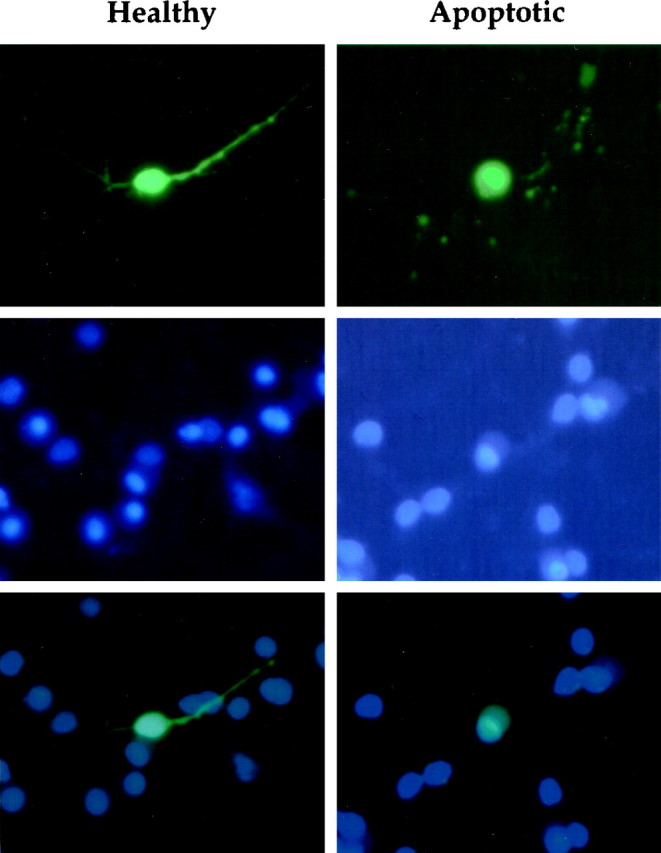
Morphological patterns of healthy and apoptotic CGC transfectants. Fluorescence micrographs of the same cells with GFP (top), Hoechst 33258 (middle), or both (bottom). Left column, A healthy cell transfected with anti-caspase-3 ribozyme after 24 hr of serum–K+ deprivation. The cell has an oval-shaped cell body and intact processes, and its chromatin stains dimly and occupies the majority of the cell body volume. Right column, An apoptotic cell transfected with control plasmid after 24 hr of serum–K+ deprivation. This cell has a large irregularly round cell body, blurred borders, and fragmented processes. Furthermore, its chromatin is condensed, intensely staining, and shifted to the periphery of the cell body.
Figure 3 compares Hoechst 33258 with calcein or TUNEL as measures of cell viability and apoptosis. Calcein is a derivative of fluorescein diacetate that stains only living cells (Miller and Johnson, 1996). Left panels demonstrate dim Hoechst 33258 staining in calcein-positive cells, which suggests that there is a low probability of identifying a Hoechst 33258-positive cell as healthy. Right panels show similar staining patterns between Hoechst 33258 and TUNEL. With both methods, there is nuclear condensation as in Figure 2, which indicates that TUNEL and Hoechst 33258 are comparable methods for detecting apoptosis.
Fig. 3.

Comparison of methods for assessing apoptosis or cell viability. Left column, Apoptotic CGCs were costained with Hoechst 33258 (top) and 20 μm calcein (middle), which stains only living cells. Superimposition of these images (bottom) demonstrates no colocalization of positive signals, which indicates that Hoechst 33258 does not identify living cells under the conditions used here. Right column, Apoptotic features as assessed by Hoechst 33258 or TUNEL. Hoechst 33258-stained nuclei are condensed as in Figure 2 (top). The same cells labeled by TUNEL show a similar staining pattern (bottom), indicating that both methods provide comparable estimates of apoptosis.
Positive controls for protection against apoptosis are shown in Figure4, which compares the level of apoptosis in unprotected cells transfected with β-galactosidase with that in cells transfected with human bcl-2 or baculoviral p35. Because bcl-2 protects against apoptosis in several other models, it was used to demonstrate the validity of the method used in this study. Cells expressing human bcl-2 were almost completely protected from death with a mean of 5 ± 2% apoptosis versus 44 ± 2% for β-galactosidase-expressing negative control cells (p < 0.001). Overexpressed p35 was used as an additional control to demonstrate apoptotic protection with a general caspase inhibitor. The level of apoptosis in cells expressing p35 was 29 ± 1% (p < 0.001).
Fig. 4.

Apoptosis after 24 hr of serum–K+ deprivation in CGCs expressing positive and negative control vectors. β-Galactosidase, a negative control, and human bcl-2 or p35, positive controls, were expressed transiently in CGCs. After 24 hr of deprivation of serum and K+, the percentage of apoptotic cells was determined as described in Materials and Methods. Apoptosis measured 44 ± 2% in negative control cells (average cells counted, x = 241;n = 10), 5 ± 2% with human bcl-2 (x = 88; n = 3), and 29 ± 1% with p35 (x = 186; n = 7). ***p < 0.001 compared with negative control by ANOVA and one-tailed Dunnett's post hoc test.
Human ILP–XIAP, an inhibitor of caspase-3, -7, and -9, was overexpressed to narrow the field of possible candidate caspases responsible for apoptosis. As shown in Figure5, apoptosis in ILP–XIAP-expressing cells was 12 ± 0.4% at 24 hr, 20 ± 0.6% at 36 hr, and 47 ± 1.1% at 48 hr after induction of apoptosis. These values are significantly less than the level of apoptosis in control cells at the same time points, which were 32 ± 0.3, 36 ± 1.3, and 64 ± 1.1%, respectively (p < 0.001).
Fig. 5.

Apoptosis in transfected CGCs expressing human ILP–XIAP. Apoptosis was induced by serum–K+deprivation, and cells were analyzed after 24, 36, or 48 hr. Apoptosis in negative control cells expressing empty pEBB vector measured 32 ± 0.3% (average cells counted, x =164), 36 ± 1.3% (x = 171), and 64 ± 1.1% (x = 79), respectively. In human ILP–XIAP-expressing cells, apoptosis was 12 ± 0.4% (x = 277), 20 ± 0.6% (x= 185), and 47 ± 1.1% (x = 103), respectively. ***p < 0.001 by one-tailed Student's t test (n = 5).
RZ101 was tested for its ability to cleave rat caspase-3 mRNA in vitro (Fig. 6). The ribozyme was designed to cleave at a kinetically preferred GUC triplet site that would generate fragments of 289 and 804 bases (A andB). To confirm that this specific cleavage ability could affect protein production, rat PC12 cells were transfected with the ribozyme construct or β-galactosidase control, and the resulting levels of endogenous caspase-3 mRNA or protein were analyzed. It was necessary to use PC12 cells instead of CGCs for these analyses because the transfection efficiency of the latter was very low, i.e., 0.1–0.3%. Transfection efficiency in PC12 cells was ∼20%. In Figure 6, C and D, quantitative RT-PCR was used to measure levels of caspase-3 mRNA. Cultures transfected with RZ101 showed a 13 ± 1% decrease in caspase-3 message compared with β-galactosidase controls. Furthermore, Western blot analysis of cultures transfected with RZ101 had significantly less protein levels of caspase-3 than control cultures. As shown in Figure6E, whereas all analyzed dilutions of protein from β-galactosidase-transfected cells generated prominent bands, 5 μg of protein from RZ101-transfected cells produced a faint band, and 2.5 μg of protein was almost undetectable.
Fig. 6.

Efficacy of ribozyme RZ101 against rat caspase-3.A, Expected cleavage fragments with RZ101.B, In vitro cleavage of rat caspase-3 RNA by RZ101. Ribozyme or caspase-3 RNA were generated by in vitro transcription using T7 RNA polymerase. Caspase-3 RNA was incubated with either ribozyme or water for 1 hr at 37°C and then analyzed by electrophoresis and silver staining. Specific cleavage fragments migrated at their expected positions. C, Quantitative RT-PCR of endogenous caspase-3 in rat PC12 cells transfected with RZ101 or β-galactosidase. After 24 hr of serum deprivation, total mRNA was amplified for caspase-3 (323 bp) and cyclophilin (330 bp). D, After staining with ethidium bromide, the bands were quantified by densitometry. The ratio of caspase-3 to cyclophilin band volumes were calculated, and data were expressed as a percent value of the β-galactosidase-transfected control result. RZ101 produced a 13 ± 1% reduction in signal. Given a 20% transfection rate, the calculated reduction in caspase-3 mRNA in RZ101-transfected cells alone is ∼65%. E, Western blot for endogenous caspase-3 in PC12 cells transiently transfected with β-galactosidase or RZ101. For each condition, 10, 5, and 2.5 μg of protein dilutions were analyzed. RZ101 effectively downregulates caspase-3 protein expression.
The effect of RZ101 on caspase-3 expression was also analyzed functionally. As shown in Figure 7, PC12 cells were transfected with RZ101 or β-galactosidase, and protein extracts were analyzed for caspase-3-like enzyme activity using a fluorogenic tetrapeptide substrate. In control cells, constitutive caspase-3-like activity with RZ101 was reduced to ∼67 ± 2% of that in β-galactosidase-transfected cells. Forty-eight hours after induction of apoptosis, caspase-3-like activity increased to 205 ± 14% of control with β-galactosidase but was reduced to 114% ± 2% with RZ101 (p < 0.001).
Fig. 7.

Caspase-3-like activity in PC12 cells after transfection with RZ101 or β-galactosidase. Total protein lysates were assayed for cleavage of Ac-DEVD-AMC, a fluorogenic caspase-3-like substrate. In control cells not undergoing apoptosis, caspase-3-like activity with RZ101 was reduced to 67 ± 2% of that in β-galactosidase-transfected cells. After 48 hr of serum deprivation, caspase-3-like activity increased to 205 ± 14% of control, but with RZ101, activity was reduced to 114 ± 2%. Activity in untransfected cells (118 ± 4%) was not significantly different from that in β-galactosidase control cells. ***p< 0.001 by one-tailed Student's t test (n = 5).
Finally, RZ101 was overexpressed to determine whether specific inhibition of a single caspase could protect against apoptosis. As shown in Figure 8, the level of apoptosis in cells expressing RZ101 was 18 ± 0.7% at 24 hr, 32 ± 0.5% at 36 hr, and 68 ± 1.7% at 48 hr. Apoptosis at 24 and 36 hr was significantly reduced from control levels at the same time points, which were 32 ± 1.7 and 45 ± 1.0%, respectively (p < 0.001).
Fig. 8.

Apoptosis in transfected CGCs expressing a ribozyme against rat caspase-3. Apoptosis was assessed after 24, 36, or 48 hr of serum–K+ deprivation. In negative control cells expressing β-galactosidase, apoptosis measured 32 ± 2% (average cells counted, x = 69), 45 ± 1% (x = 89), and 71 ± 3% (x= 38), respectively. In cells expressing RZ101, apoptosis at the same times points was reduced to 18 ± 0.7% (x = 158), 32 ± 0.5% (x = 151), and 68 ± 1.7% (x = 41), respectively. ***p < 0.001 by one-tailed Student'st test (n = 5).
DISCUSSION
Ribozymes have been used in several studies to suppress the expression of specific genes and thereby modify cellular behavior (Dolnikov et al., 1996; Dorai et al., 1997). Most of these studies have used hammerhead ribozymes, which cleave at the site NUH, where N is any ribonucleotide, and H is A, U, or C (Birikh et al., 1997). Here, we describe the use of a ribozyme against caspase-3, RZ101, to protect against apoptosis after serum–K+deprivation in CGCs. In addition, we show that overexpression of p35, a general caspase inhibitor, and ILP–XIAP, a specific inhibitor of caspase-3, -7, and -9, also provide significant protection. Together, these data implicate caspase-3 as an important mediator of apoptosis in CGCs.
The specificity of RZ101 for caspases was determined by Blast 2 sequence analysis (National Center for Biotechnology Information, Bethesda, MD) of all the rat caspase sequences currently in GenBank (caspases-1, -2, -3, -6, and -7). The only other caspase beside caspase-3 with significant homology to the ribozyme was caspase-1 at an eight nucleotide region complementary to part of the 5′ ribozyme recognition arm. However, there was no homology at all with the 3′ arm. Moreover, CGCs do not express caspase-1 (Eldadah et al., 1997; Ni et al., 1997); thus, RZ101 is probably specific for caspase-3 alone.
The ability of RZ101 to downregulate caspase-3 expression was determined by quantitative RT-PCR, Western blot, and enzyme activity analysis. The reduction in mRNA signal in ribozyme-transfected cultures was 13%, which represents the average value from total culture extracts (i.e., transfected plus nontransfected cells). Given an estimated transfection efficiency of 20% in PC12 cells, the calculated reduction in the transfected proportion is ∼65%. For caspase activity in which ribozyme-transfected cultures exhibited a one-third to one-half reduction in signal, the calculated reduction for transfected cells is closer to 100%. The disparity between these values and those obtained with RT-PCR may be caused by the higher sensitivity of PCR.
Caspases may be divided into three main groups based on their DNA coding sequences and substrate specificities (Alnemri et al., 1996;Talanian et al., 1997; S. Hu et al., 1998; Thornberry and Lazebnik, 1998). The caspase-1-like proteases consist of caspase-1, -4, -5, -11, -12, -13, and -14; caspase-2-like proteases include caspase-2 and -9; and caspase-3-like proteases consist of caspase-3, -6, -7, -8, and -10. The role of caspase-3-like proteases in CGC apoptosis has been described previously (Armstrong et al., 1997; Eldadah et al., 1997; Ni et al., 1997; Taylor et al., 1997). After serum–K+ deprivation, cell death is accompanied by increasing caspase-3-like activity. In addition, cell death is diminished after treatment with z-DEVD-fmk, an irreversible inhibitor of caspase-3-like proteases. At the same time, there is no detectable activity of caspase-1-like proteases, no protection with benzyloxycarbonyl-YVAD-fluoromethylketone, an inhibitor of caspase-1-like proteases, and no amplification of caspase-1 mRNA by RT-PCR.
Caspases -1, -2, -3, and -4 are all inhibited by p35 with similar potencies. Because these caspases represent the three main subdivisions, p35 could be regarded as a general caspase inhibitor (Bump et al., 1995). p35 protects against apoptosis induced by a variety of stimuli in several different cell culture models (for review, see Miller, 1997), implicating one or more caspases in the apoptotic pathways of these systems. In CGCs, p35 also conferred protection against apoptosis, increasing cell viability by approximately one-third of control levels. To narrow the number of candidate caspases responsible for CGC apoptosis, human ILP–XIAP was overexpressed. ILP–XIAP is a member of the IAP family of proteins, characterized by repeated zinc finger-like motifs and a C-terminal RING domain (Miller, 1997). ILP–XIAP specifically inhibits caspase-3, -7, and -9 (Deveraux et al., 1997, 1998); however, because caspase-7 mRNA is not expressed in brain (Juan et al., 1997), ILP–XIAP may function even more specifically in CGCs.
Because bcl-2 is known to exert protective effects in many models of apoptosis, it was used as a positive control to validate the methods used in this study. Overexpression of bcl-2 conferred virtually complete protection in CGCs. Similar resistance to apoptosis has been observed in CGCs cultured from transgenic mice overexpressing bcl-2 (Tanabe et al., 1997) and in PC12 cells stably transfected with bcl-2 (Batistatou and Greene, 1993). The protective effects of p35, ILP–XIAP, and RZ101 were not as complete as those observed with overexpression of bcl-2. This partial protection most likely stems from the different locations within the apoptotic pathway in which each molecule exerts its effects. Bcl-2 is thought to inhibit apoptosis at multiple levels, one of which is the bcl–APAF-1–caspase-9 complex trigger point (Y. Hu et al., 1998; Pan et al., 1998). The fine balance between pro-apoptotic and anti-apoptotic bcl-2-like proteins may be easily tipped with only a small excess of either protein. As such, an inhibitory signal at this upstream event would probably produce more profound effects on apoptosis than at more downstream events in which several caspases, including caspase-3, are believed to act. In addition, there may be apoptotic pathways in CGCs that are caspase-independent, as has been proposed previously (Miller et al., 1997; Taylor et al., 1997). Therefore, complete caspase inhibition may not necessarily provide the same degree of protection as expression of bcl-2. However, the viability conferred by p35, ILP–XIAP, and RZ101 may be underestimated because of variable plasmid expression. This is particularly important in light of the finding that caspase-3 transcription is upregulated during apoptosis (Eldadah et al., 1997;Miller et al., 1997; Ni et al., 1997). Furthermore, variability of expression may have led the ribozyme construct to protect significantly after 24 and 36 hr but not at 48 hr. Expression of transiently expressed products would be expected to diminish over time, and because the functional product of the ribozyme expression vector is RNA, the effects of changes in ribozyme expression levels may appear earlier than that of protein products.
That caspase-3 is an important mediator of CGC apoptosis is consistent with the findings of other authors. As reported by Kuida et al. (1996), gross developmental abnormalities from insufficient apoptosis are found in mice deficient in caspase-3. Among other defects in these mice, both the internal and external granule cell layers of the cerebellum are markedly hyperplastic. Nicholson et al. (1995) found that treatment of isolated liver nuclei with cytosolic extracts from apoptotic cells failed to induce apoptosis when the extracts were deprived of caspase-3-like proteases. However, upon replenishing the extracts with purified caspase-3, the nuclei regained their apoptotic phenotype. Finally, MCF-7 breast carcinoma cells, which are naturally deficient in caspase-3, fail to exhibit membrane blebbing and DNA fragmentation after an apoptotic stimulus. However, ectopic expression of caspase-3 in these cells restores a normal apoptotic phenotype (Janicke et al., 1998).
The current results do not exclude the possibility that other caspases may also be involved in CGC apoptosis. Indeed, others have proposed a cascade of proteolytic activation in which caspase-3 may cleave, or be cleaved by, other caspases (Martin and Green, 1995; Srinivasula et al., 1996). However, at this time it appears that, if other caspases are involved, they would likely comprise a serial pathway rather than multiple parallel pathways. This is suggested by the fact that p35, ILP–XIAP, and an anti-caspase-3 ribozyme all afford similar levels of protection. If other caspases were acting in parallel with caspase-3, then ribozyme- or ILP–XIAP-transfected cells would be expected to respond more like negative control cells than those expressing p35, a general caspase inhibitor. Of course, this hypothesis may need revision if other caspases insensitive to p35 inhibition are identified. For such caspases, as well as those already identified, determining whether they may play a role in CGC apoptosis will require the development of specific inhibitors at the protein and/or nucleic acid levels.
Our results differ from those of Miller et al. (1997), who concluded that caspases may not be necessary for apoptosis in CGCs. These authors used a similar method of transfection with GFP reporter but observed no protection with p35 and only modest protection with bcl-2. An explanation for this disparity may lie in the method by which cell viability and apoptosis were determined in each system. Whereas Miller et al. used loss of GFP positivity as a marker of cell death, we assessed apoptosis by GFP and Hoechst 33258 fluorescence. This method was used previously in a similarly aimed study (Dudek et al., 1997) and allows identification of specific apoptotic features. The staining pattern observed with this method was similar to that produced by TUNEL, indicating that both techniques may provide a similar estimate of apoptosis. However, we quantified apoptosis with only Hoechst 33258 because it is far less expensive than TUNEL for the large volumes and number of replications used in this study. In addition, apoptotic cells are much more discernable because of their condensed Hoechst 33258-stained nuclei on a GFP background. Costaining of apoptotic cells with Hoechst 33258 and calcein, a marker of cell viability, demonstrated no colocalization. This indicates that false positive assessment of apoptosis with Hoechst 33258 is unlikely in this model, not only for apoptosis in particular but also for cell death in general.
Footnotes
This study was supported by National Institutes of Health Grant RO1 NS36537–0 and Department of Defense Grant DAMD 17–93-V-3018. We are grateful to Randyll Goodnight for her expert technical assistance. In addition, we thank Henryk Dudek, Colin Duckett, Winnie Wong, and Nancy Bump for their helpful advice and kind provision of materials used in this study.
Correspondence should be addressed to Dr. Alan I. Faden, Research Building, Room EP-04, Georgetown University Medical Center, 3970 Reservoir Road NW, Washington, DC 20007. E-mail:fadena@giccs.georgetown.edu.
REFERENCES
- 1.Adams JM, Cory S. The bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- 2.Alnemri ES, Livingston DJ, Nicholson DW, Salvesen G, Thornberry NA, Wong WW, Yuan J. Human ICE/CED-3 protease nomenclature. Cell. 1996;87:171. doi: 10.1016/s0092-8674(00)81334-3. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong RC, Aja TJ, Hoang KD, Gaur S, Bai X, Alnemri ES, Litwack G, Karanewsky DS, Fritz LC, Tomaselli KJ. Activation of the CED3/ICE-related protease caspase-3 in cerebellar granule neurons undergoing apoptosis but not necrosis. J Neurosci. 1997;17:553–562. doi: 10.1523/JNEUROSCI.17-02-00553.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Batistatou A, Greene LA. Internucleosomal DNA cleavage and neuronal cell survival/death. J Cell Biol. 1993;122:523–532. doi: 10.1083/jcb.122.3.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birikh KR, Heaton PA, Echstein F. The structure, function and application of the hammerhead ribozyme. Eur J Biochem. 1997;245:1–16. doi: 10.1111/j.1432-1033.1997.t01-3-00001.x. [DOI] [PubMed] [Google Scholar]
- 6.Bredesen DE. Neural apoptosis. Ann Neurol. 1995;38:839–851. doi: 10.1002/ana.410380604. [DOI] [PubMed] [Google Scholar]
- 7.Bump NJ, Hackett M, Hugunin M, Seshagiri S, Brady K, Chen P, Ferenz C, Franklin S, Ghayur T, Li P, Licari P, Mankovich J, Shi L, Greenberg AH, Miller LK, Wong WW. Inhibition of ICE family proteases by baculovirus antiapoptotic protein p35. Science. 1995;269:1885–1888. doi: 10.1126/science.7569933. [DOI] [PubMed] [Google Scholar]
- 8.D'Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deveraux Q, Takahashi R, Salvesen GS, Reed JC. X-linked IAP is a direct inhibitor of cell death proteases. Nature. 1997;388:300–303. doi: 10.1038/40901. [DOI] [PubMed] [Google Scholar]
- 10.Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998;17:2215–2223. doi: 10.1093/emboj/17.8.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolnikov A, King A, Luxford C, Symonds G, Sun LQ. Ribozyme-mediated suppression of v-myc expression abrogates apoptosis in transformed monocytes. Cancer Gene Ther. 1996;3:289–295. [PubMed] [Google Scholar]
- 12.Dorai T, Goluboff ET, Olsson CA, Buttyan R. Development of a hammerhead ribozyme against BCL-2. II. Ribozyme treatment sensitizes hormone-resistant prostate cancer cells to apoptotic agents. Anticancer Res. 1997;17:3307–3312. [PubMed] [Google Scholar]
- 13.Du Y, Bales KR, Dodel RC, Hamilton-Byrd E, Horn JW, Czilli DL, Simmons LK, Ni B, Paul SM. Activation of a caspase 3-related cysteine protease is required for glutamate-mediated apoptosis of cultured cerebellar granule neurons. Proc Natl Acad Sci USA. 1997;94:11657–11662. doi: 10.1073/pnas.94.21.11657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duckett CS, Li F, Wang Y, Tomaselli KJ, Thompson CB, Armstrong RC. Human IAP-like protein regulates programmed cell death downstream of Bcl-xL and cytochrome c. Mol Cell Biol. 1998;18:608–615. doi: 10.1128/mcb.18.1.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dudek H, Datta SR, Franke TF, Brinbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 16.Eldadah BA, Yakovlev AG, Faden AI. The role of CED-3-related cysteine proteases in apoptosis of cerebellar granule cells. J Neurosci. 1997;17:6105–6113. doi: 10.1523/JNEUROSCI.17-16-06105.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellis HM, Horvitz HR. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- 18.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. A caspase-activated DNase that degrades during apoptosis and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 19.Endres M, Namura S, Shimizu-Sasamata M, Waeber C, Zhang L, Gomez-Isla T, Hyman BT, Moskowitz MA. Attenuation of delayed neuronal death after mild focal ischemia in mice by inhibition of the caspase family. J Cereb Blood Flow Metab. 1998;18:238–247. doi: 10.1097/00004647-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Haendler B, Hofer-Warbinek R, Hofer E. Complementary DNA for human T-cell cyclophilin. EMBO J. 1987;6:947–950. doi: 10.1002/j.1460-2075.1987.tb04843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu S, Snipas SJ, Vincenz C, Salvesen G, Dixit VM. Caspase-14 is a novel developmentally regulated protease. J Biol Chem. 1998;273:29648–29653. doi: 10.1074/jbc.273.45.29648. [DOI] [PubMed] [Google Scholar]
- 22.Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA. 1998;95:4386–4391. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janicke RU, Sprengart ML, Wati MR, Porter AG. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem. 1998;17:9357–9360. doi: 10.1074/jbc.273.16.9357. [DOI] [PubMed] [Google Scholar]
- 24.Juan TS, McNiece IK, Argento JM, Jenkins NA, Gilbert DJ, Copeland NG, Fletcher FA. Identification and mapping of Casp7, a cysteine protease resembling CPP32β, interleukin-1β converting enzyme, and CED-3. Genomics. 1997;40:86–93. doi: 10.1006/geno.1996.4548. [DOI] [PubMed] [Google Scholar]
- 25.Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K, McGarry TJ, Kirschner MW, Koths K, Kwiatkowski DJ, Williams LJ. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science. 1997;278:294–298. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- 26.Kuida K, Zheng TS, Na S, Kuan C-Y, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 27.Liu X, Zou H, Slaughter C, Wang X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell. 1997;89:175–184. doi: 10.1016/s0092-8674(00)80197-x. [DOI] [PubMed] [Google Scholar]
- 28.Martin SJ, Green DR. Protease activation during apoptosis: death by a thousand cuts? Cell. 1995;82:349–352. doi: 10.1016/0092-8674(95)90422-0. [DOI] [PubMed] [Google Scholar]
- 29.Miller LK. Baculovirus interaction with host apoptotic pathways. J Cell Physiol. 1997;173:178–182. doi: 10.1002/(SICI)1097-4652(199711)173:2<178::AID-JCP17>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 30.Miller TM, Johnson EM. Metabolic and genetic analyses of apoptosis in potassium/serum-deprived rat cerebellar granule cells. J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ni B, Wu X, Du Y, Su Y, Hamilton-Byrd E, Rockey PK, Rosteck P, Jr, Poirier GG, Paul SM. Cloning and expression of a rat brain interleukin-1β-converting enzyme (ICE)-related protease (IRP) and its possible role in apoptosis of cultured cerebellar granule neurons. J Neurosci. 1997;17:1561–1569. doi: 10.1523/JNEUROSCI.17-05-01561.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nicholson DW, Ali A, Thornberry NA, Vaillancourt JP, Ding CK, Gallant M, Gareau Y, Griffin PR, Labelle M, Lazebnik YA, Munday NA, Raju SM, Smulson ME, Yamin T-T, Yu VL, Miller DK. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature. 1995;376:37–43. doi: 10.1038/376037a0. [DOI] [PubMed] [Google Scholar]
- 34.Orth K, Chinnaiyan AM, Garg M, Froelich CJ, Dixit VM. The CED-3/ICE-like protease Mch2 is activated during apoptosis and cleaves the death substrate lamin A. J Biol Chem. 1996;271:16443–16446. [PubMed] [Google Scholar]
- 35.Palfner K, Kneba M, Hiddemann W, Bertram J. Quantification of ribozyme-mediated RNA cleavage using silver-stained polyacrylamide gels. BioTechniques. 1995;19:926–929. [PubMed] [Google Scholar]
- 36.Pan G, O'Rourke K, Dixit VM. Caspase-9, Bcl-XL, and Apaf-1 form a ternary complex. J Biol Chem. 1998;273:5841–5845. doi: 10.1074/jbc.273.10.5841. [DOI] [PubMed] [Google Scholar]
- 37.Sakahira H, Enari M, Nagata S. Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature. 1998;391:96–99. doi: 10.1038/34214. [DOI] [PubMed] [Google Scholar]
- 38.Srinivasula SM, Fernandes-Alnemri T, Zangrilli J, Robertson N, Armstrong RC, Wang L, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES. The Ced-3/interleukin-1β converting enzyme-like homolog Mch6 and the lamin-cleaving enzyme Mch2α are substrates for the apoptotic mediator CPP32. J Biol Chem. 1996;271:27099–27106. doi: 10.1074/jbc.271.43.27099. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi A, Musy PY, Martins LM, Poirier GG, Moyer RW, Earnshaw WC. CrmA/SPI-2 inhibition of an endogenous ICE-related protease responsible for lamin A cleavage and apoptotic nuclear fragmentation. J Biol Chem. 1996;271:32487–32490. doi: 10.1074/jbc.271.51.32487. [DOI] [PubMed] [Google Scholar]
- 40.Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J Biol Chem. 1997;272:9677–9682. doi: 10.1074/jbc.272.15.9677. [DOI] [PubMed] [Google Scholar]
- 41.Tanabe H, Eguchi Y, Kamada S, Martinou JC, Tsujimoto Y. Susceptibility of cerebellar granule neurons derived from Bcl-2-deficient and transgenic mice to cell death. Eur J Neurosci. 1997;9:848–856. doi: 10.1111/j.1460-9568.1997.tb01434.x. [DOI] [PubMed] [Google Scholar]
- 42.Taylor J, Gatchalian CL, Keen G, Rubin LL. Apoptosis in cerebellar granule neurones: involvement of interleukin-1β converting enzyme-like proteases. J Neurochem. 1997;68:1598–1605. doi: 10.1046/j.1471-4159.1997.68041598.x. [DOI] [PubMed] [Google Scholar]
- 43.Thangnipon W, Kingsbury A, Webb M, Balazs R. Observations on rat cerebellar cells in vitro: influence of substratum, potassium concentration and relationship between neurones and astrocytes. Dev Brain Res. 1983;11:177–189. doi: 10.1016/0165-3806(83)90215-8. [DOI] [PubMed] [Google Scholar]
- 44.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 45.Van de Craen M, Van Loo G, Pype S, Van Criekinge W, Van den brande I, Molemans F, Fiers W, Declercq W, Vandenabeele P. Identification of a new caspase homologue: caspase-14. Cell Death Differ. 1998;5:838–846. doi: 10.1038/sj.cdd.4400444. [DOI] [PubMed] [Google Scholar]
- 46.Yakovlev AG, Knoblach SM, Fan L, Fox GB, Goodnight R, Faden AI. Activation of CPP32-like caspases contributes to neuronal apoptosis and neurological dysfunction after traumatic brain injury. J Neurosci. 1997;17:7415–7424. doi: 10.1523/JNEUROSCI.17-19-07415.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4: participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]