

Abstract
The reason for the selective vulnerability of motor neurons in amyotrophic lateral sclerosis (ALS) is primarily unknown. A possible factor is the expression by motor neurons of Ca2+-permeable AMPA/kainate channels, which may permit rapid Ca2+ influx in response to synaptic receptor activation. However, other subpopulations of central neurons, most notably forebrain GABAergic interneurons, consistently express large numbers of these channels but do not degenerate in ALS. Indeed, when subjected to identical excitotoxic exposures, motor neurons were more susceptible than GABAergic neurons to AMPA/kainate receptor-mediated neurotoxicity. Microfluorimetric studies were performed to examine the basis for the difference in vulnerability. First, AMPA or kainate exposures appeared to trigger substantial mitochondrial Ca2+ loading in motor neurons, as indicated by a sharp increase in intracellular Ca2+after addition of the mitochondrial uncoupler carbonyl cyanidep-(trifluoromethoxy)phenyl hydrazone (FCCP) after the agonist exposure. The same exposures caused little mitochondrial Ca2+ accumulation in GABAergic cortical neurons. Subsequent experiments examined other measures of mitochondrial function to compare sequelae of AMPA/kainate receptor activation between these populations. Brief exposure to either AMPA or kainate caused mitochondrial depolarization, assessed using tetramethylrhodamine ethylester, and reactive oxygen species (ROS) generation, assessed using hydroethidine, in motor neurons. However, these effects were only seen in the GABAergic neurons after exposure to the nondesensitizing AMPA receptor agonist kainate. Finally, addition of either antioxidants or toxins (FCCP or CN−) that block mitochondrial Ca2+ uptake attenuated AMPA/kainate receptor-mediated motor neuron injury, suggesting that the mitochondrial Ca2+ uptake and consequent ROS generation are central to the injury process.
Keywords: glutamate, kainate, hydroethidine, oxidative stress, mitochondria, GABA, ALS
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the progressive loss of upper (Betz cells) and lower (ventral horn) motor neurons. Although of unknown cause, abnormalities in glutamate uptake (Rothstein et al., 1992) and metabolism (Plaitakis and Caroscio, 1987; Hugon et al., 1989a; Rothstein et al., 1990) suggest that excitotoxic injury may contribute (Leigh and Meldrum, 1996; Rothstein, 1996; Shaw and Ince, 1997). Although NMDA receptors likely contribute critically to neuronal injury in various acute conditions, several observations support the hypothesis that AMPA/kainate receptors may be of greater importance to the neurodegenerative process seen in ALS. First, three syndromes with prominent motor system manifestations, lathyrism (Spencer et al., 1986), domoic acid toxicity (Teitelbaum et al., 1990), and β-N-methylamino-l-alanine toxicity (Spencer et al., 1987), are linked to the consumption of AMPA/kainate receptor agonists found in the environment (Ross et al., 1987; Bridges et al., 1989; Debonnel et al., 1989; Weiss et al., 1989). In addition, kainate exposures preferentially injure motor neurons both in vivo (Hugon et al., 1989b) and in vitro (Carriedo et al., 1995, 1996; Rothstein and Kuncl, 1995), and AMPA/kainate receptor antagonists protect against motor neuron degeneration caused by chronic blockade of glutamate uptake in both spinal cord slice cultures (Rothstein et al., 1993) and dissociated cultures (Carriedo et al., 1996).
Considerable evidence supports a link between Ca2+ influx and glutamate receptor-mediated neurodegeneration. Brief periods of activation of highly Ca2+-permeable NMDA channels can result in substantial intracellular Ca2+accumulation and widespread neuronal injury (Hartley et al., 1993; Lu et al., 1996; Hyrc et al., 1997). Although mitochondria can buffer these large Ca2+ loads (Wang et al., 1994;Werth and Thayer, 1994; White and Reynolds, 1995; Babcock et al., 1997;Peng et al., 1998), they do so at the expense of triggering injurious reactive oxygen species (ROS) production (Lafon-Cazal et al., 1993;Dugan et al., 1995; Reynolds and Hastings, 1995; Bindokas et al., 1996). In contrast to NMDA receptors, AMPA/kainate receptors are generally Ca2+ impermeable and trigger injury more slowly, with prolonged (several hours) periods of activation needed before significant neuronal injury occurs (Koh et al., 1990). Subpopulations of central neurons, however, are highly vulnerable to AMPA/kainate receptor-mediated injury (Koh and Choi, 1988; Weiss et al., 1994; Yin et al., 1994), likely attributable in part to the expression of large numbers of AMPA/kainate channels with high Ca2+ permeability (Ca2+-A/K channels) (Iino et al., 1990;Pruss et al., 1991; Brorson et al., 1992; Turetsky et al., 1994; Lu et al., 1996). Although mechanisms of Ca2+-A/K channel-dependent neuronal injury have been studied relatively little, we found recently that brief kainate exposures trigger high [Ca2+]i elevations and subsequent mitochondrial ROS production in GABAergic cortical neurons (Carriedo et al., 1998), a well defined neuronal population that strongly expresses these channels (Jonas et al., 1994; Yin et al., 1994; Geiger et al., 1995).
Recent studies indicating that motor neurons often possess Ca2+-A/K channels (Carriedo et al., 1995,1996; Launey et al., 1998; Terro et al., 1998; Bar-Peled et al., 1999) support the idea that the presence of these channels contributes to their high vulnerability. However, GABAergic cortical neurons, which also possess Ca2+-A/K channels and are vulnerable to rapidly triggered AMPA/kainate receptor-mediated injury (Yin et al., 1994), do not degenerate in ALS. The broad aim of the present study was to compare the vulnerability of GABAergic cortical neurons with that of spinal motor neurons to AMPA/kainate receptor-mediated injury and to examine downstream sequelae of Ca2+ entry through Ca2+-A/K channels expressed on these populations that might account for differences in vulnerability. Specifically, because of evidence that Ca2+ influx interferes with mitochondrial function, our studies focused on assessments of mitochondrial Ca2+ buffering, mitochondrial depolarization, and ROS generation.
MATERIALS AND METHODS
Chemicals and reagents. Hydroethidine (HEt) and tetramethylrhodamine ethylester (TMRE) were purchased from Molecular Probes (Eugene, OR). Fura-2FF was purchased from Texas Fluorescence Lab (Austin, TX). MK-801 was purchased from Research Biochemicals (Natick, MA). Tissue culture media and serum were from Life Technologies (Grand Island, NY). 2,3-Dihydroxy-6-nitro-7-sulfamoylbenzo(F)quinoxaline (NBQX) was kindly provided by Novo Nordisk (Malov, Denmark). NMDA, kainate, rotenone, cyanide, trolox, and carbonyl cyanidep-(trifluoromethoxy)phenyl hydrazone (FCCP) were obtained from Sigma (St. Louis, MO). U74500 was kindly provided by Upjohn (Kalamazoo, MI). All other chemicals and reagents were obtained from common commercial sources.
Dissociated cultures. Dissociated cortical and spinal cord cultures were prepared primarily as described previously. Neocortical cell suspensions were prepared from 15- to 16-d-old embryonic Swiss–Webster mice and plated 1–2 × 105 cells/cm2(Yin et al., 1994). Spinal cord suspensions (removed of both meninges and dorsal root ganglia) were prepared from 13 d mouse embryos and plated at a density of 3 × 105cells/cm2 (approximately “4 spinal cords” per 24-well plate) (Carriedo et al., 1996). Cells were plated on a previously established layer of cortical astrocytes grown on either 15 mm Primaria-coated culture plates (Falcon, Franklin Lake, NJ) or poly-l-lysine-coated glass-bottomed dishes. Plating medium consisted of Eagle's minimal essential medium (MEM; Earle's salts supplied glutamine free; Life Technologies), supplemented with 10% heat-inactivated horse serum (Life Technologies), 10% fetal bovine serum (Life Technologies), glutamine (2 mm; Life Technologies), and glucose (total, 25 mm). Cultures were maintained in a 37°C, 5% CO2 incubator. After 4–6 d in vitro, non-neuronal cell division was halted by exposure to 10−5mcytosine arabinoside for 1–3 d. The cells were then shifted into a maintenance medium identical to the plating medium but lacking fetal serum. Subsequent media replacement occurred twice a week. Cultures were studied after 13–17 d in vitro.
Glial cultures were prepared similarly except that tissue was obtained from early postnatal (1–3 d) mice and plating media was supplemented with epidermal growth factor (10 ng/ml). Cell suspensions were plated directly on Primaria tissue culture-treated multiwell plates or poly-l-lysine-coated glass-bottomed dishes.
Identification of motor neurons and GABAergic cortical neurons. Motor neurons were identified using both morphological criteria (soma > 20 μm and extensive dendritic arborization) (Schaffner et al., 1987) and labeling for Sternberger Monoclonal Inc. (SMI)-32, an antibody to nonphosphorylated neurofilaments that has been found to label motor neurons in dissociated spinal cultures (Gotow and Tanaka, 1994; Carriedo et al., 1996; Bar-Peled et al., 1999). GABAergic cortical neurons were labeled using glutamic acid decarboxylase (GAD; Developmental Studies Hybridoma Bank at the University of Iowa, Iowa City, IA) immunohistochemistry (Yin et al., 1994). For staining, cultures were fixed for 40 min in 4% paraformaldehyde, washed three times with PBS, and incubated for 30 min with “blocking solution” (10% heat-inactivated horse serum in PBS) to minimize background staining. For SMI-32, the blocking solution also included 0.2% Triton X-100. Primary antibody exposures (in blocking solution at 1:6000 for SMI-32 and 1:500 for GAD) were performed for 48–72 hr at 4°C. Biotinylated secondary antibody (Vector Laboratories, Burlingame, CA), ABC solution (Vector Laboratories), and 3-amino-9-ethyl-carbazole (Sigma) were used to visualize stained cells.
Neurotoxicity experiments. Brief (5–20 min) toxic exposures were performed in room air, using a HEPES-buffered salt solution (HSS) with the following composition (in mm): Na+ 130, K+5.4, Mg2+ 0.8, Ca2+ 1.8 (except where indicated), Cl− 130.6, HEPES, pH 7.4 at 25°C, 20, and glucose 15. Exposures were terminated by replacing the exposure solution with MEM + glucose, along with the ionotropic glutamate receptor antagonists MK-801 and NBQX (both at 10 μm), and returning the cultures to the 37°C, 5% CO2incubator. Longer (24 hr) exposures were in MEM + glucose in the 37°C, 5% CO2 incubator. MK-801 (10 μm) was added during all toxicity exposures to prevent activation of NMDA receptors from endogenously released glutamate and thereby to ensure a pure AMPA/kainate receptor-mediated injury mechanism.
Overall neuronal injury was assessed 20–24 hr after the start of the exposure both by morphological examination and by quantitative measurement of lactate dehydrogenase (LDH) in the bathing medium, an index that is proportional to the total number of neurons damaged by excitotoxic exposure (Koh and Choi, 1987). LDH values were scaled to the near maximal mean value found in sister cultures exposed to 300 μm kainate for 24 hr (equal to 100% cell loss).
Damage to motor neurons or GABAergic neurons was assessed as the difference between the mean number of intact labeled cells (determined by direct counts) in neuronal cultures exposed to an excitotoxic agonist and the mean number in sister cultures exposed to sham wash alone (typically 200–500 labeled cells counted per culture well), expressed as a percentage of the latter.
Imaging studies. Spinal or cortical cultures were plated on poly-l-lysine-coated glass-bottomed dishes (Mattek Cultureware, Ashland, MA) and mounted to a stage adapter on an inverted microscope (Nikon Diaphot). All agonist exposures were performed at room temperature (25°C) in HSS either constantly perfused or in a 1.5 ml static bath. Preselected fields (typically containing 20–50 healthy-appearing nonoverlapping neurons) were illuminated by a xenon light source using a Nikon 40× magnification and 1.3 numerical aperture epifluorescence oil immersion objective, and the emitted fluorescence was collected using a Hamamatsu intensified CCD camera. In all experiments, the electronic background signals (obtained with the camera shutter closed) were obtained at each wavelength and subtracted from measured signals. To analyze experiments, we outlined neuronal somata and gathered data on an 80486-based computer using Image-1/Fluor software from Universal Imaging Corporation (West Chester, PA).
For intracellular Ca2+([Ca2+]i) measurements, cultures were loaded in the dark with 5 μmFura-2FF AM in HSS containing 0.2% pluronic acid and 1.5% dimethylsulfoxide (DMSO) for 30–45 min at 25°C. Cultures were then washed in HSS (three times) and kept in the dark for an additional 30 min to allow for complete dye deesterification. Cells were alternately illuminated at 340 and 380 nm, and fluorescence was monitored at 510 nm. [Ca2+]i was determined by the equation: [Ca2+]i = KD(Fmin/Fmax) [(R −Rmin)/(Rmax− R)], where R is the observed 340:380 fluorescence ratio (Grynkiewicz et al., 1985) and theKD used was 35 μm(Golovina and Blaustein, 1997). Fminindicates 380 nm fluorescence at Rmin, and Fmax indicates 380 nm fluorescence at Rmax. Because of the very low affinity of Fura-2FF for Ca2+,Rmin is the 340:380 fluorescence ratio determined at the start of each experiment. Control experiments revealed a negligible difference (<0.1 ratio unit) between this value and the Rmin obtained in the presence of 2 mm EGTA, 0 Ca2+, and 2 μmionomycin. Rmax is the 340:380 fluorescence ratio value determined in the same neurons exposed to 10 μm ionomycin in the presence of 30 mm Ca2+.Rmin (and baseline ratios) ranged from 0.5 to 0.8, whereas Rmax ranged from 5.5 to 7.5. The system was recalibrated after any adjustments to the apparatus.
ROS production and changes in mitochondrial polarization (ΔΨm) were monitored using the oxidation-sensitive dye HEt (Bindokas et al., 1996; Carriedo et al., 1998) and the ΔΨm-sensitive dye TMRE (Farkas et al., 1989; Schinder et al., 1996; Carriedo et al., 1998), respectively. Advantages of HEt over other oxidation-sensitive dyes include its relative resistance to both auto- and photo-oxidation (permitting fluorescence monitoring for prolonged periods) and the increasing intensity of the dye fluorescence seen after intercalation of ethidium within nuclear DNA (increasing sensitivity for ROS detection). Cultures were loaded in the dark with 5 μmHEt in HSS (45 min; 25°C) or 0.5 μm TMRE in MEM (30 min; 37°C). After loading, cultures were washed (four times) into a static bath of HSS containing either probe. Cells were excited at 510–560 nm, and emission was monitored at >590 nm. To minimize photobleaching, we attentuated the fluorescence intensity with neutral density filters (Omega Optical, Battleboro, VT). Camera gain was adjusted to give baseline maximal florescence levels of 20–40 (HEt experiments) or of 150–200 (TMRE experiments) arbitrary units of a maximal eight-bit signal output of 256. Fluorescence measurements for each cell (Fx) were normalized to the fluorescence intensity for that cell at the beginning of the experiment (F0). In the TMRE experiments, fluorescence changes were monitored only in “mitochondria-rich” perinuclear regions of the soma, which undergo sharp decreases in fluorescence after mitochondrial depolarization. In HEt experiments, ROS production causes an increase in somatic and nuclear fluorescence. Because HEt fluorescence is cumulative, the rate of ROS generation was assessed as the rate of increase (or slope) of theFx/F0curves over time, and net ROS production was assessed as the increase in Fx/F0over baseline.
After completion of imaging experiments, cultures were then fixed and stained for either SMI-32 (for identifying spinal motor neurons) or GAD (for identifying GABAergic cortical neurons). Only imaged fields containing labeled neurons were analyzed.
Experiment replication. All experiments reported represent at least four independent replications. All imaging studies represent at least 7 motor and 150 other spinal neurons or 15 GABAergic and 150 other cortical neurons.
RESULTS
Motor neurons are more vulnerable than GABAergic cortical neurons to AMPA/kainate receptor-mediated neurotoxicity
Before comparing downstream injury mechanisms, we first set out to compare directly the vulnerability of motor neurons with that of GABAergic cortical neurons after identical exposures to kainate or to the rapidly desensitizing agonist AMPA. To perform these studies, we prepared spinal cord cultures as described (Carriedo et al., 1996) and used the cultures after 15 d in vitro. Previous characterization of these cultures indicated that ∼1.5% of the spinal cells in our culture system were motor neurons as assessed by morphological appearance (soma > 20 μm and extensive dendritic arborization) (Schaffner et al., 1987) and intense staining with the neurofilament marker SMI-32, which has been found to identify motor neurons in culture and in slice (Gotow and Tanaka, 1994; Carriedo et al., 1996). Using this culture system we find that >80% of the motor neurons express Ca2+-A/K channels (Carriedo et al., 1996), as indicated by histochemical labeling for kainate-stimulated Co2+ uptake (Pruss et al., 1991). Previous characterization of our cortical neuronal cultures indicated that ∼9% of the neurons are GABAergic, as indicated by glutamic acid decarboxylase immunohistochemistry, and that 90% of these GABAergic neurons express Ca2+-A/K channels (Yin et al., 1994; Carriedo et al., 1998).
Initial neurotoxicity studies examined injury induced by prolonged (24 hr) kainate (5–10 μm) or AMPA (2.5–5 μm) exposures. After each of these exposures, both motor neurons and GABAergic cortical neurons (identified by glutamic acid decarboxylase immunohistochemistry) (Yin et al., 1994) were preferentially damaged in comparison with the overall neuronal population (see Materials and Methods). However, in each case, the damage to the motor neurons was significantly greater than was the damage to the GABAergic neurons (Fig. 1A). Further studies used a brief exposure paradigm with AMPA as the agonist. Although 10 min exposures to AMPA alone caused little injury, motor neurons and GABAergic cortical neurons were preferentially damaged if the AMPA receptor desensitization inhibitor cyclothiazide (100 μm) (Yamada and Tang, 1993) was added during the exposure. Again, the damage to the motor neurons was greater than was the damage to the GABAergic neurons (Fig. 1B). The injury to both of these populations was Ca2+ dependent; removal of extracellular Ca2+ substantially prevented injury.
Fig. 1.

Spinal motor neurons are more vulnerable than GABAergic cortical neurons to AMPA/kainate receptor-mediated injury.A, Slowly triggered neurotoxicity. Spinal or cortical cultures were exposed to kainate (5–10 μm + 10 μm MK-801) or AMPA (2.5–5 μm + 10 μm MK-801) for 24 hr, followed by assessment of injury. Identification of motor neurons was based on morphology and staining for SMI-32; GABAergic cortical neurons were identified by staining for GAD. Injury to the overall spinal population (gray bars) and cortical population (hatched bars) was assessed by LDH measurement, whereas injury to motor neurons (black bars) and GABAergic cortical neurons (white bars) was assessed by direct cell counts (see Materials and Methods). B, Rapidly triggered neurotoxicity. Spinal or cortical cultures were exposed to AMPA (50 μm + 10 μm MK-801; 10 min), either alone (left) or in the presence of the AMPA receptor desensitization inhibitor cyclothiazide (100 μm;right), in either Ca2+-containing or Ca2+-free buffer as indicated. Twenty-four hours after the exposure, injury was assessed as described above. Values represent the means ± SEM compiled from at least four experiments; n = 10–12 cultures per condition. Anasterisk indicates motor neuron or GABAergic cell loss significantly different from overall cell loss; anampersand indicates motor neuron cell loss significantly different from GABAergic cell loss (p < 0.01 by two-tailed t test).
Brief kainate exposure triggers large [Ca2+]i elevations and mitochondrial Ca2+ loading in motor neurons
In previous microfluorimetric studies we found that AMPA/kainate receptor activation triggers high [Ca2+]i rises (to tens of micromoles per liter) in GABAergic cortical neurons (Carriedo et al., 1998), consistent with their frequent expression of Ca-A/K channels. Therefore, we used similar microfluorimetric techniques to assess kainate-triggered [Ca2+]i rises in motor neurons. [Ca2+]i levels were measured using the low-affinity Ca2+-sensitive fluorescent dye Fura-2FF (KD = 35 μm), because recent studies indicate that high-affinity dyes, like fura-2, may markedly underestimate true [Ca2+]i levels (Hyrc et al., 1997; Carriedo et al., 1998; Khodorov et al., 1999; Stout and Reynolds, 1999). After dye loading, a field containing one to three neurons with the morphological features of motor neurons was selected, and basal [Ca2+]ilevels were recorded before exposing cultures to kainate (100 μm + 10 μm MK-801) for 5 min. After imaging, the phenotype of the putative motor neurons was further assessed by staining with SMI-32. Immediately after exposure, large increases in [Ca2+]i levels were seen in motor neurons (with lesser increases seen in other spinal neurons; Fig. 2).
Fig. 2.

Brief kainate exposure triggers large [Ca2+]i rises in motor neurons. In Fura-2FF-loaded spinal cultures, [Ca2+]i levels were monitored for 5 min before and 25 min after a 5 min exposure to kainate (100 μm + 10 μm MK-801;horizontalbar). Fields selected for imaging contained morphologically identified putative motor neurons (large soma size and extensive dendritic tree); after imaging their identity was verified by immunostaining for SMI-32.Traces represent the means ± SEM of 9 motor neurons and >150 other spinal neurons derived from six experiments.
Because mitochondria are important buffers of intracellular Ca2+ (Wang et al., 1994; Werth and Thayer, 1994; White and Reynolds, 1995; Peng et al., 1998), we next compared their role in buffering AMPA/kainate receptor-mediated Ca2+ loads in motor neurons and GABAergic neurons. These studies made use of the protonophore FCCP, which dissipates the mitochondrial membrane potential and releases Ca2+ from mitochondria, while preventing further Ca2+ uptake (Wang et al., 1994;White and Reynolds, 1995; Stout et al., 1998). Fura-2FF-loaded cultures were subjected to a control pulse of AMPA (50 μm + 10 μm MK-801) or kainate (100 μm + 10 μm MK-801) for 15 sec and allowed to recover for 20 min. Cultures were then given a second identical pulse of agonist followed immediately by exposure to FCCP (750 nm + 10 μm MK-801 for 2 min) (Fig.3). In both motor neurons and GABAergic cortical neurons, exposure to either agonist triggered immediate [Ca2+]i rises, which returned to baseline within 5 min. However, addition of FCCP after exposure to either agonist resulted in dramatically increased [Ca2+]i rises in the motor neurons but only a mild prolongation in the [Ca2+]i rises in GABAergic neurons (Figs. 3, 4), suggesting that these exposures cause particularly high levels of mitochondrial Ca2+ loading in motor neurons.
Fig. 3.

AMPA exposure triggers mitochondrial Ca2+ loading in motor neurons. Spinal cultures were loaded with Fura-2FF, and basal [Ca2+]i levels were recorded before cultures were exposed to an initial AMPA pulse (50 μm + 10 μm MK-801 for 15 sec). After a 20 min recovery period, the cultures were reexposed to AMPA (50 μm + 10 μm MK-801 for 15 sec), followed immediately by a 2 min pulse of the mitochondrial protonophore FCCP (750 nm + 10 μm MK-801). Fluorescent images were taken before AMPA exposure (A), at the peak [Ca2+]i rise seen during the initial AMPA exposure (B), and at the peak [Ca2+]i rise seen after the AMPA/FCCP exposure (C). After imaging, the cultures were fixed and stained for SMI-32 (D). Note that exposure to AMPA followed by FCCP (C) causes higher [Ca2+]i rises than occur with AMPA exposure alone (B). Thepseudocolorbar shows the Fura-2FF fluorescence ratio scale. Scale bar, 50 μm.
Fig. 4.

AMPA/kainate receptor activation triggers greater degrees of mitochondrial Ca2+ loading in motor neurons than in GABAergic cortical neurons. Fura-2FF-loaded spinal (A, B) and cortical (C, D) cultures were exposed for 15 sec to either AMPA (50 μm + 10 μm MK-801; A, C) or kainate (100 μm + 10 μm MK-801; B, D) (thin lines) and allowed to recover for 20 min. After recovery, the cultures were given an identical 15 sec agonist exposure followed immediately by FCCP (750 nm + 10 μmMK-801 for 2 min; thick lines). Tracesrepresent means ± SEM from 10 to 20 motor neurons and >20 GABAergic cortical neurons from at least seven experiments.
To examine the duration of mitochondrial Ca2+ loading in motor neurons after AMPA exposure, we exposed spinal cultures to AMPA for 15 sec as described above, but with the addition of FCCP (750 nm + 10 μm MK-801 for 2 min) at various intervals (0, 3, or 10 min) after the exposure (FCCP alone caused no [Ca2+]i rise). The magnitude of the FCCP-induced [Ca2+]i rise decreased with the increasing interval after the AMPA exposure (Fig.5). By 20 min after the AMPA exposure FCCP no longer triggered a rise in [Ca2+]i, indicating probable clearance of the mitochondrial Ca2+ load.
Fig. 5.

Time course of recovery of AMPA-triggered mitochondrial Ca2+ loading in motor neurons. Fura-2FF-loaded spinal cultures were exposed to AMPA (50 μm + 10 μm MK-801 for 15 sec;A; thick line) and allowed to recover for 20 min. After recovery, the cultures were given identical 15 sec AMPA exposures followed either immediately after (B;filled circles), 3 min after (C;open circles), or 10 min after (D;opensquares) by FCCP (750 nm+ 10 μm MK-801 for 2 min). Note the progressive decrease in the size of the FCCP-induced [Ca2+]i rise with increasing duration between AMPA and FCCP exposures. Control studies showed that in the absence of AMPA preexposures, the FCCP exposures caused no change in [Ca2+]i (E; thin line). Traces represent means ± SEM from 7 to 20 motor neurons from at least five experiments.
A possible additional affect of FCCP may be that uncoupling of electron transport from ATP production could result in reversal of the ATP synthetase with consequent depletion of cellular ATP and impairment of ATPase-driven extrusion of Ca2+ from the cell (Budd and Nicholls, 1996). However, recent studies have found that FCCP exposures of several minutes at levels higher than those used here caused little ATP depletion (White and Reynolds, 1995; Wang and Thayer, 1996). In addition, our conclusions are not substantively altered even if ATP levels are depleted. Specifically, because FCCP-triggered [Ca2+]i rises only occur within a 10 min period after agonist-triggered Ca2+ loading, the FCCP-triggered rise must represent cytosolic accumulation of Ca2+ that had been sequestered during the brief agonist exposure.
AMPA exposure causes strong mitochondrial depolarization and ROS generation in motor neurons
Because above studies suggest that the degree of mitochondrial Ca2+ loading may be a key factor differentiating the responses of motor neurons from those of GABAergic neurons to AMPA/kainate receptor activation, subsequent experiments focused on the comparison of other indexes of mitochondrial function. To examine the AMPA/kainate receptor-mediated changes in the mitochondrial membrane potential (ΔΨm) that might occur in response to mitochondrial Ca2+ uptake, we used the dye TMRE that rapidly equilibrates between cellular compartments as a function of potential differences; the rapid loss of fluorescence from cellular domains rich in mitochondria is indicative of loss of ΔΨm (Farkas et al., 1989; Schinder et al., 1996). In a previous study we reported that rapid kainate exposure triggers abrupt loss of ΔΨm in GABAergic cortical neurons, reflecting the rapid Ca2+ influx through Ca2+-A/K channels expressed on these cells (Carriedo et al., 1998).
In TMRE-loaded spinal or cortical cultures, the fluorescence in mitochondrial-rich perinuclear regions was measured before, during, and for 30 min after a 10 min exposure to either AMPA (50 μm + 10 μm MK-801) or kainate (100 μm + 10 μm MK-801). Although motor neurons typically showed strong basal TMRE fluorescence, presumably indicative of large numbers of mitochondria in the perinuclear region, AMPA and kainate exposures triggered substantial decreases in TMRE fluorescence in motor neurons that persisted long after the end of the exposure (Figs. 6,7). In contrast, in GABAergic cortical neurons, kainate exposures triggered a moderate decrease in TMRE fluorescence, while little change was seen in response to AMPA exposures. It is possible that slow decreases in TMRE fluorescence could partially reflect Na+-dependent plasma membrane depolarization with redistribution of dye from the cytoplasmic space, in addition to more rapid fluorescence decreases resulting from Ca2+-dependent loss of ΔΨm. However, the lack of comparable fluorescence changes in the non-GABAergic cortical neurons and the nonmotor spinal neurons provides an internal control indicating that the signal is caused by loss of ΔΨm, because kainate induces Na+-dependent neuronal depolarization in virtually all neurons. In further control studies, removal of Ca2+ from the media during kainate exposures (10 min; 100 μm) prevented TMRE fluorescence decreases in cortical neurons expressing Ca2+-A/K channels (normalized TMRE fluorescence at the end of the exposure, 1.02 ± 0.02 in the absence of Ca2+; 0.81 ± 0.04 with Ca2+; n > 28 neurons from four experiments in each condition).
Fig. 6.

AMPA exposure causes mitochondrial depolarization in motor neurons. TMRE-loaded spinal cultures were exposed to AMPA (50 μm for 10 min). Fluorescent images were obtained before (A), during (8 min; B), and 30 min after termination of (C) the exposure. After imaging, the culture was stained for SMI-32 (D). Note the relatively selective and long-lasting decrease in TMRE fluorescence seen in the mitochondrial-rich perinuclear regions of the motor neuron, indicative of significant mitochondrial depolarization. The pseudocolorbar shows the eight-bit intensity scale used for TMRE fluorescence. Scale bar, 50 μm.
Fig. 7.

AMPA/kainate receptor activation causes greater mitochondrial depolarization in motor neurons than in GABAergic cortical neurons. In TMRE-loaded spinal (A, B) and cortical (C, D) cultures, the fluorescence in mitochondrial-rich regions was measured for 10 min before, during, and for 30 min after a 10 min exposure to AMPA (50 μm + 10 μm MK-801; A, C) or kainate (100 μm + 10 μm MK-801; B, D). Note the substantially greater decrease in fluorescence in motor neurons compared with GABAergic neurons after AMPA exposure in contrast to the similar fluorescence changes after kainate exposures.Traces represent the means ± SEM compiled from 10 to 17 motor and 150 other spinal neurons and from 40 to 70 GABAergic and 200 other cortical neurons from at least seven experiments.KA, Kainate.
To address further the possible differences between motor neurons and GABAergic cortical neurons in downstream responses to AMPA/kainate receptor activation, we examined kainate-induced ROS production using the oxidation-sensitive dye HEt that readily permeates living cells and is reported to be oxidized selectively by superoxide radicals into the highly fluorescent compound ethidium (Bindokas et al., 1996) (see Materials and Methods).
We reported previously that kainate exposure triggers HEt oxidation in GABAergic neurons. Thus before comparing HEt responses between motor neurons and GABAergic neurons during and after brief agonist exposures, we first characterized changes in HEt fluorescence in motor neurons triggered by prolonged kainate exposures. In HEt-loaded cultures, fluorescence readings were acquired for 10 min before and for 25 min after addition of kainate (100 μm + 10 μmMK-801). Basal ROS production was minimal as evidenced by the stable but small increases in neuronal HEt fluorescence seen during the baseline period (Fig. 8). As we observed previously in GABAergic cortical neurons, motor neurons displayed noticeable increases in fluorescence within 3 min of kainate exposure. At the end of the exposure, motor neurons showed substantially greater increases in HEt fluorescence than do most other spinal neurons (normalized fluorescence increase over baseline of 1.94 ± 0.49 in motor neurons vs 0.40 ± 0.04 in other neurons; Fig. 8). In further studies, the electron transport chain inhibitor rotenone was used to examine the role of mitochondria in kainate-triggered ROS production. In agreement with our previous findings in GABAergic cortical neurons, addition of rotenone (10 μm) for 40 min before and during the kainate exposure almost completely prevented the increase in HEt fluorescence (Fig. 8).
Fig. 8.

Kainate exposure triggers preferential ROS generation in motor neurons. HEt-loaded cultures were imaged for 10 min before and 25 min after addition of kainate (100 μm + 10 μm MK-801). To assess involvement of mitochondria in the ROS generation, in some cultures, we added the electron transport chain inhibitor rotenone (10 μm) for 40 min before and during the kainate exposure (broken line). After exposure, cultures were fixed and stained for SMI-32. Fluorescence changes for each neuron are expressed as the ratio of fluorescence at each time point (FX) to its own baseline (F0). Traces represent the means ± SEM of 7–10 motor neurons and >100 other spinal neurons derived from at least six experiments.
To address possible differences between motor neurons and GABAergic cortical neurons in downstream responses to AMPA/kainate receptor activation, we used HEt to compare directly the ROS generation triggered by identical 10 min AMPA or kainate exposures (Fig.9). Paralleling effects on mitochondrial potential, exposures to either agonist triggered sharp increases in HEt fluorescence in motor neurons (normalized fluorescence increase over baseline of 3.46 ± 1.02 for AMPA; 4.43 ± 1.71 for kainate), with little increase in most spinal neurons (0.51 ± 0.03 for AMPA; 0.96 ± 0.12 for kainate). In contrast, although kainate triggered comparable selective increases in HEt fluorescence in GABAergic neurons (3.55 ± 0.28 vs 0.73 ± 0.05 in most cortical neurons), AMPA exposures caused only a moderate increase in GABAergic neurons (1.54 ± 0.2) compared with most other cortical neurons (0.61 ± 0.03).
Fig. 9.

AMPA/kainate receptor activation causes greater ROS generation in motor neurons than in GABAergic cortical neurons. HEt-loaded spinal (A, B) and cortical (C, D) cultures were imaged for 5 min before and 30 min after a 10 min exposure to AMPA (50 μm + 10 μm MK-801; A, C) or kainate (100 μm + 10 μm MK-801; B, D).Traces represent the means ± SEM compiled from 10 to 12 motor and 200 other spinal neurons and from 60 GABAergic and 200 other cortical neurons from at least six experiments. Note the substantially greater fluorescence increase in motor neurons than in GABAergic neurons after AMPA exposure in contrast to the comparable fluorescence increases seen after kainate exposure (p < 0.01 by two-tailed ttest).
Because a recent report by Budd et al. (1997) suggested that mitochondrial depolarization (loss of ΔΨm) might cause a voltage-dependent release of oxidized ethidium from the mitochondria, we previously performed control studies to examine the degree to which observed kainate-triggered HEt signals reflect ROS production. First, we found that qualitatively similar increases in fluorescence were seen after kainate exposures in cells loaded with only 1 μm HEt, a concentration at which Budd et al. (1997) found ethidium to remain bound within the mitochondria after loss of ΔΨm (Carriedo et al., 1998). In addition, although rotenone completely blocked the agonist-triggered increase in HEt fluorescence (as above), it had no effect on the loss of ΔΨm assessed using the ΔΨm-sensitive dye TMRE (Carriedo et al., 1998; Sensi et al., 1999). Thus, the HEt signal that is blocked by rotenone cannot simply be explained as a loss of ΔΨm and most likely reflects ROS production.
AMPA/kainate receptor-mediated injury to motor neurons is dependent on mitochondrial Ca2+ loading and ROS generation
Finally, we examined the hypothesis that Ca2+ accumulation within the mitochondria and consequent ROS generation contribute directly to AMPA/kainate receptor-mediated motor neuron injury. To assess the role of mitochondrial Ca2+ accumulation in the injury, we made use of either the protonophore FCCP (750 nm) or the electron transport chain inhibitor cyanide (CN−; 3 mm). In agreement with previous studies of electron transport blockers (Budd and Nicholls, 1996; Khodorov et al., 1996; Stout et al., 1998), in control experiments we found that, like FCCP, CN−also appears to trigger release of mitochondrial Ca2+ (as evidenced by a sharp increase in [Ca2+]i when added after AMPA/kainate receptor activation; data not shown). To maximize motor neuron injury while minimizing the neurotoxicity associated with prolonged application of FCCP or CN−, we induced toxicity by a 5 min exposure to AMPA (50 μm + 10 μm MK-801) in the presence of the AMPA receptor desensitization inhibitor cyclothiazide (100 μm) and elevated extracellular Ca2+ (10 mm). Under these conditions, AMPA triggered substantial motor neuron cell loss. The presence of either FCCP or CN− during the exposure attenuated this injury (Fig.10A).
Fig. 10.
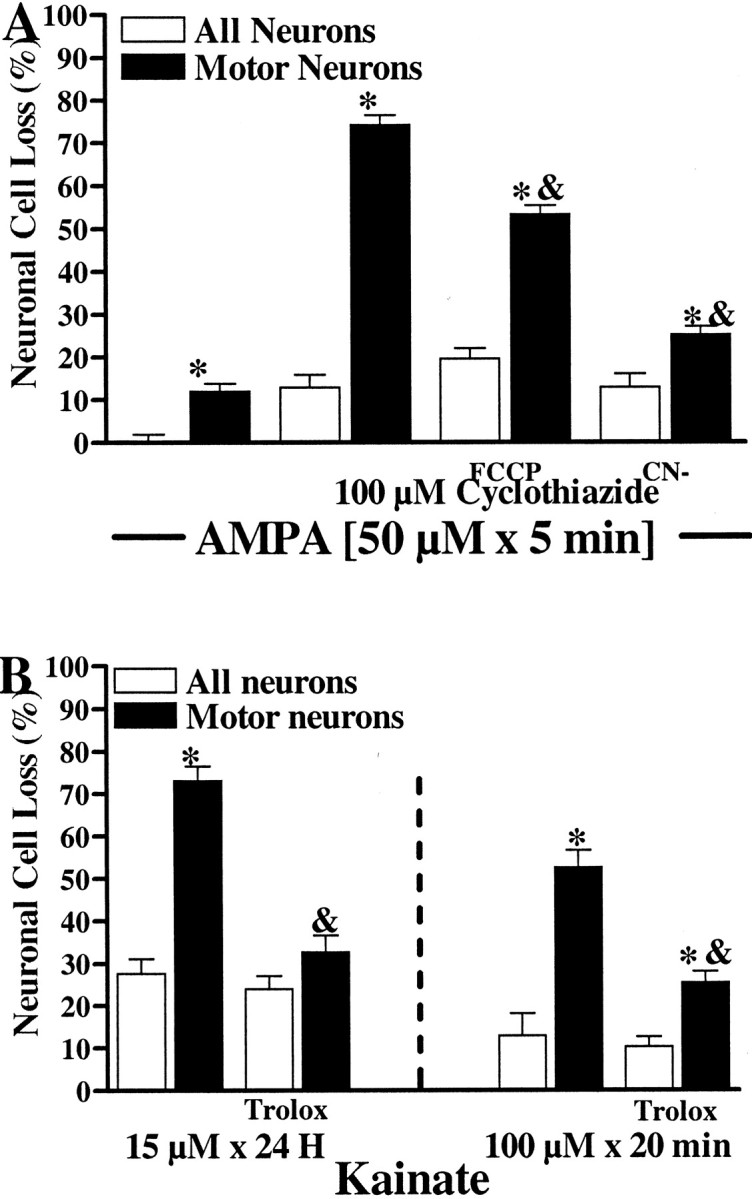
Mitochondrial Ca2+ uptake inhibitors and antioxidants attenuate AMPA/kainate receptor-mediated motor neuron injury. A, Effects of mitochondrial toxins. Cultures were exposed to AMPA for 5 min (50 μm + 10 μm MK-801, 100 μm cyclothiazide [CYZ], and 10 mm extracellular Ca2+) either alone or in the additional presence of FCCP (750 nm) or cyanide (CN−; 3 mm). CYZ and elevated extracellular Ca2+ were included during the exposure to induce substantial injury in brief exposures that minimized the direct toxic effects of FCCP and CN−. The following day, injury to the overall spinal population (white bars) and the motor neuron population (black bars) was assessed as described. Values represent the means ± SEM compiled from at least four experiments;n = 16–20 cultures per condition. Anasterisk indicates motor neuron cell loss significantly different from overall cell loss; an ampersand indicates motor neuron cell loss significantly different from that caused by AMPA/CYZ exposure alone (p < 0.01 by two-tailed t test). B, Effects of the antioxidant trolox. Cultures were subjected to either a prolonged (15 μm for 24 hr) or brief (100 μm for 20 min) kainate exposure (+ 10 μm MK-801), in the presence or absence of the antioxidant trolox (3 mm). (For the 20 min exposures, trolox was present for 1 hr before, during, and after the exposure.) The following day, injury to the overall spinal population (white bars) and the motor neuron population (black bars) was assessed as described (see Materials and Methods). Values represent the means ± SEM compiled from at least four experiments; n = 13–19 cultures per condition. An asterisk indicates motor neuron cell loss significantly different from overall cell loss; anampersand indicates motor neuron cell loss significantly different from that caused by kainate exposure alone (p < 0.01 by two-tailed ttest).
To test the role of ROS in AMPA/kainate receptor-mediated motor neuron injury, we performed brief (100 μm for 20 min) and prolonged (15 μm for 24 hr) kainate exposures alone or with the antioxidant trolox (a vitamin E derivative) (Chow et al., 1994). For the brief exposures, trolox (3 mm) was added for 1 hr before and for 24 hr after as well as during the exposure. Injury was assessed 24 hr after the start of the exposures (see Material and Methods). In both toxicity paradigms, trolox significantly attenuated motor neuron cell loss (Fig. 10B).
DISCUSSION
Expression of Ca2+-A/K channels by motor neurons likely contributes to their high vulnerability in ALS. However, GABAergic cortical neurons, which also strongly express these channels, are not conspicuously damaged in human disease. The broad aim of this study thus has been to compare AMPA/kainate receptor-mediated injury between these two populations with the hope of uncovering clues to the particularly high vulnerability of motor neurons in disease. We find that motor neurons are indeed more vulnerable than GABAergic cortical neurons to AMPA/kainate receptor-mediated injury and have identified two differences in the injury pathway downstream from Ca2+ entry through Ca2+-A/K channels that may pertain to these differences in vulnerability. First, brief AMPA or kainate exposures triggered substantial mitochondrial Ca2+ loading in motor neurons but not in GABAergic cortical neurons. Further differences concern the agonist specificity of mitochondrial effects. In motor neurons, brief exposures to either AMPA or kainate triggered abrupt mitochondrial depolarization and ROS generation. In GABAergic cortical neurons, however, these effects were only seen with kainate exposures (which, unlike AMPA, cause nondesensitizing activation of AMPA channels). Finally, observations that both mitochondrial poisons (which prevent Ca2+ uptake) and ROS scavengers are protective suggest that the mitochondrial Ca2+ uptake and consequent ROS generation contribute directly to AMPA/kainate receptor-mediated motor neuron injury.
Ca2+ ion entry and vulnerability of motor neurons to AMPA/kainate receptor-mediated injury
The greater vulnerability of motor neurons than GABAergic cortical neurons to AMPA/kainate receptor-mediated injury could in part reflect a greater quantity of Ca2+ entering the motor neurons. Differences in agonist-triggered Ca2+ entry would most likely reflect differences in AMPA/kainate receptor expression, either in terms of numbers of channels or their subunit composition. There is considerable evidence of strong AMPA subunit expression on both motor neurons and GABAergic cortical neurons (Furuyama et al., 1993; Yin et al., 1994;Williams et al., 1996). Furthermore, recent studies suggesting that both motor neurons (Bar-Peled et al., 1999; Shaw et al., 1999) and GABAergic cortical neurons (Bochet et al., 1994; Jonas et al., 1994;Yin et al., 1994) may express low levels of the GluR2 AMPA subunit (which confers Ca2+ impermeability to heteromeric AMPA channels) (Hollmann et al., 1991) support the idea that Ca2+-A/K channels expressed on both of these populations are comprised of AMPA subunits lacking GluR2. Further support to this idea is provided by the present observation that the selective AMPA receptor desensitization inhibitor cyclothiazide (Yamada and Tang, 1993) strongly increased Ca2+-dependent AMPA-mediated injury to both of these populations.
Numerous studies have confirmed that the behavior of AMPA channels is dependent both on the agonist activating the channel and on the subunit splice variants of which the channel is comprised. Although AMPA channels generally display rapid desensitization after activation by AMPA or the endogenous agonist glutamate, these channels undergo a relatively nondesensitizing response after activation by kainate (Kiskin et al., 1986; Trussell et al., 1988). Thus, the substantial mitochondrial depolarization and ROS generation seen with kainate but not AMPA exposures in GABAergic cortical neurons likely reflect the rapid channel desensitization after AMPA exposures, resulting in less Ca2+ influx.
Splice variant-dependent changes in the function of channels comprised of AMPA subunits depend primarily on the sequence at the “flip-flop” site of the subunits; channels with the flip splice form show much slower desensitization rates and larger steady-state currents than do channels composed of subunits in the flop splice form (Sommer et al., 1990; Lambolez et al., 1996). In agreement with the idea that slowly desensitizing AMPA channels may be expressed on motor neurons, AMPA receptor subunits present in GABAergic cortical neurons primarily exist in the flop splice form (Bochet et al., 1994; Geiger et al., 1995; Lambolez et al., 1996), whereas those on motor neurons may primarily be in the flip splice form (Tölle et al., 1993, 1995;Jakowec et al., 1995; Temkin et al., 1997). Thus, expression of such slowly desensitizing AMPA receptors could in part account for the observed relative lack of difference between the effects of AMPA and kainate on motor neurons.
Intracellular Ca2+ handling and the vulnerability of motor neurons to AMPA/kainate receptor-mediated injury
In addition to potential differences in the magnitude of Ca2+ entry, the greater vulnerability of motor neurons to AMPA/kainate receptor-mediated injury could also reflect a greater intrinsic susceptibility to Ca2+-dependent injury mechanisms, downstream from the Ca2+ entry. Neuronal sensitivity to Ca2+-mediated injury may primarily reflect the cellular mechanisms used to neutralize heavy intracellular Ca2+ loads. One likely relevant factor that may differentiate the sensitivity of motor neurons and GABAergic cortical neurons to [Ca2+]i elevations is the presence of Ca2+-binding proteins (CBPs). Indeed, GABAergic interneurons are characterized by the strong expression of CBPs including parvalbumin (Celio, 1986), calretinin (Rogers, 1992), and calbindin-D28K (Hendry and Jones, 1991). Recent studies suggest that CBPs may serve important roles in protecting neurons from intracellular Ca2+ loads by directly chelating free intracellular Ca2+(Mattson et al., 1991; Lledo et al., 1992; Lukas and Jones, 1994; Roy et al., 1998). Motor neuron subpopulations that are relatively resistant to degeneration in ALS express high levels of CBPs, suggesting that the expression of CBPs may modulate motor neuron vulnerability (Alexianu et al., 1994; Elliott and Snider, 1995; Reiner et al., 1995). Thus, a lack of CBPs in most motor neurons may compel them to resort to other mechanisms, such as mitochondrial uptake, for buffering Ca2+ loads. Indeed, mitochondria have been found to take up Ca2+ after AMPA/kainate receptor activation (Hoyt et al., 1998) as well as NMDA receptor activation (Wang et al., 1994; White and Reynolds, 1997; Peng et al., 1998).
Although mitochondrial Ca2+ uptake may have a physiological role in the coupling of neuronal activity to mitochondrial energy production (McCormack and Denton, 1990), there is compelling evidence that mitochondrial Ca2+ overload plays a central role in mediating excitotoxic injury (Schinder et al., 1996). Mitochondrial Ca2+ loading has been shown to cause loss of ΔΨm with the consequent cessation of ATP production (Beatrice et al., 1980; Wang et al., 1994). In addition, Ca2+ entry through either NMDA channels (Lafon-Cazal et al., 1993; Dugan et al., 1995; Reynolds and Hastings, 1995; Bindokas et al., 1996) or Ca2+-A/K channels (Carriedo et al., 1998) can trigger mitochondrial ROS production. Recent studies demonstrating that NMDA receptor-mediated toxicity is attenuated by mitochondrial toxins that prevent mitochondrial Ca2+ uptake strengthen the idea that mitochondrial Ca2+ loading may be a key step in the translation of excitotoxic exposure into neuronal injury (Budd and Nicholls, 1996; Khodorov et al., 1996; Stout et al., 1998). Indeed, the present finding that either inhibitors of mitochondrial Ca2+ uptake or ROS scavengers can protect motor neurons against injury resulting from Ca2+ influx through Ca2+-A/K channels indicates that perturbations in mitochondrial function are central to selective AMPA/kainate receptor-mediated motor neuron injury.
Are the present findings of relevance to ALS pathogenesis?
Mounting evidence implicates a role for excitotoxicity in the pathogenesis of sporadic ALS (Leigh and Meldrum, 1996; Rothstein, 1996;Shaw and Ince, 1997). Although one must be careful in extrapolating findings in murine cultures to human disease, it is notable that, even across species, motor neurons in vitro and in vivo seem to express similar glutamate receptor profiles and vulnerabilities to AMPA/kainate receptor-mediated injury. Thus, to the degree that AMPA/kainate receptor-mediated injury contributes to the loss of motor neurons in ALS, the present findings may well pertain to their high susceptibility in the disease.
These studies suggest that a critical factor underlying the selective vulnerability of motor neurons may be their propensity to mitochondrial Ca2+ overload in response to Ca2+ entry through the Ca2+-A/K channels that they strongly express. Two considerations support the idea that these mechanisms may be well suited for the induction of cumulative motor neuron injury that may occur in ALS. First, unlike NMDA channels, which are blocked by Mg2+ ions in the absence of postsynaptic depolarization, Ca2+-A/K channels permit direct Ca2+ entry whenever activated. In addition, the observation that substantial mitochondrial Ca2+ uptake occurs after brief exposures to the physiologically desensitizing agonist AMPA supports the possibility that similar mitochondrial Ca2+ loading may readily occur in response to physiological synaptic activity in vivo. Disruption of mitochondrial function by Ca2+ may in itself be injurious to cells such as motor neurons with high metabolic rates. In addition, repeated mitochondrial Ca2+ loading and consequent ROS generation could be relevant to the findings of mitochondrial dysfunction (Siklós et al., 1996; Beal, 1998; Kong and Xu, 1998; Swerdlow et al., 1998), oxidative tissue damage (Mecocci et al., 1993; Shaw et al., 1995; Ferrante et al., 1997), and deficiencies in glutamate uptake (Rothstein et al., 1992, 1995; Volterra et al., 1994; Trotti et al., 1999) seen in ALS.
Footnotes
This work was supported by National Institutes of Health Grants NS36548 and AG00836 (J.H.W.) and AG00919 (S.L.S.), a grant from the ALS Association (J.H.W.), and the National Research Service Award Predoctoral Fellowship HG00179 (S.G.C.). We thank Simin Amindari for her assistance in cell culture and Shyam Rao and Jade Jeng for their thoughtful comments on this manuscript.
Correspondence should be addressed to Dr. John H. Weiss, Department of Neurology, University of California, Irvine, Irvine, CA 92697-4292. E-mail: jweiss@uci.edu.
REFERENCES
- 1.Alexianu ME, Ho BK, Mohamed AH, La Bella V, Smith RG, Appel SH. The role of calcium-binding proteins in selective motoneuron vulnerability in amyotrophic lateral sclerosis. Ann Neurol. 1994;36:846–858. doi: 10.1002/ana.410360608. [DOI] [PubMed] [Google Scholar]
- 2.Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. J Cell Biol. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bar-Peled O, O'Brien RJ, Morrison JH, Rothstein JD. Cultured motor neurons possess calcium-permeable AMPA/kainate receptors. NeuroReport. 1999;10:855–859. doi: 10.1097/00001756-199903170-00034. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta. 1998;1366:211–223. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- 5.Beatrice MC, Palmer JW, Pfeiffer DR. The relationship between mitochondrial membrane permeability, membrane potential, and the retention of Ca2+ by mitochondria. J Biol Chem. 1980;255:8663–8671. [PubMed] [Google Scholar]
- 6.Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bochet P, Audinat E, Lambolez B, Crθpel F, Rossier J, Iino M, Tsuzuki K, Ozawa S. Subunit composition at the single-cell level explains functional properties of a glutamate-gated channel. Neuron. 1994;12:383–388. doi: 10.1016/0896-6273(94)90279-8. [DOI] [PubMed] [Google Scholar]
- 8.Bridges RJ, Stevens DR, Kahle JS, Nunn PB, Kadri M, Cotman CW. Structure–function studies on N-oxaly-diamino-dicarboxylic acids and excitatory amino acid receptors: evidence that beta-l-ODAP is a selective non-NMDA agonist. J Neurosci. 1989;9:2073–2079. doi: 10.1523/JNEUROSCI.09-06-02073.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brorson JR, Bleakman D, Chard PS, Miller RJ. Calcium directly permeates kainate/alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors in cultured cerebellar Purkinje neurones. Mol Pharmacol. 1992;41:603–608. [PubMed] [Google Scholar]
- 10.Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- 11.Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997;415:21–24. doi: 10.1016/s0014-5793(97)01088-0. [DOI] [PubMed] [Google Scholar]
- 12.Carriedo SG, Yin HZ, Lamberta R, Weiss JH. In vitro kainate injury to large, SMI-32(+) spinal neurons is Ca2+ dependent. NeuroReport. 1995;6:945–948. doi: 10.1097/00001756-199504190-00030. [DOI] [PubMed] [Google Scholar]
- 13.Carriedo SG, Yin HZ, Weiss JH. Motor neurons are selectively vulnerable to AMPA/kainate receptor-mediated injury in vitro. J Neurosci. 1996;16:4069–4079. doi: 10.1523/JNEUROSCI.16-13-04069.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carriedo SG, Yin HZ, Sensi SL, Weiss JH. Rapid Ca2+ entry through Ca2+-permeable AMPA/kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J Neurosci. 1998;18:7727–7738. doi: 10.1523/JNEUROSCI.18-19-07727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Celio MR. Parvalbumin in most gamma-aminobutyric acid-containing neurons of the rat cerebral cortex. Science. 1986;231:995–997. doi: 10.1126/science.3945815. [DOI] [PubMed] [Google Scholar]
- 16.Chow HS, Lynch JJ, Rose K, Choi DW. Trolox attenuates cortical neuronal injury induced by iron, ultraviolet light, glucose deprivation, or AMPA. Brain Res. 1994;639:102–108. doi: 10.1016/0006-8993(94)91769-8. [DOI] [PubMed] [Google Scholar]
- 17.Debonnel G, Beauchesne L, de Montigny C. Domoic acid, the alleged “mussel toxin” might produce its neurotoxic effect through kainate receptor activation: an electrophysiological study in dorsal hippocampus. Can J Physiol Pharmacol. 1989;67:29–33. doi: 10.1139/y89-005. [DOI] [PubMed] [Google Scholar]
- 18.Dugan LL, Sensi SL, Canzoniero LMT, Handran SD, Rothman SM, Lin T-S, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elliott JL, Snider WD. Parvalbumin is a marker of ALS-resistant motor neurons. NeuroReport. 1995;6:449–452. doi: 10.1097/00001756-199502000-00011. [DOI] [PubMed] [Google Scholar]
- 20. Farkas DL, Wei MD, Febbroriello P, Carson JH, Loew LM. Simultaneous imaging of cell and mitochondrial membrane potentials. Biophys J 56 1989. 1053 1069[Erratum(1990)57:684]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U, Kowall NW, Brown RH, Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- 22.Furuyama T, Kiyama H, Sato K, Park HT, Maeno H, Takagi H, Tohyama M. Region-specific expression of subunits of ionotropic glutamate receptors (AMPA-type, KA-type and NMDA receptors) in the rat spinal cord with special reference to nociception. Mol Brain Res. 1993;18:141–151. doi: 10.1016/0169-328x(93)90183-p. [DOI] [PubMed] [Google Scholar]
- 23.Geiger JR, Melcher T, Koh DS, Sakmann B, Seeburg PH, Jonas P, Monyer H. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 24.Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–1648. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- 25.Gotow T, Tanaka J. Phosphorylation of neurofilament H subunit as related to arrangement of neurofilaments. J Neurosci Res. 1994;37:691–713. doi: 10.1002/jnr.490370604. [DOI] [PubMed] [Google Scholar]
- 26.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 27.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced 45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hendry SH, Jones EG. GABA neuronal subpopulations in cat primary auditory cortex: co-localization with calcium binding proteins. Brain Res. 1991;543:45–55. doi: 10.1016/0006-8993(91)91046-4. [DOI] [PubMed] [Google Scholar]
- 29.Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–853. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- 30.Hoyt KR, Stout AK, Cardman JM, Reynolds IJ. The role of intracellular Na+ and mitochondria in buffering of kainate-induced intracellular free Ca2+ changes in rat forebrain neurones. J Physiol (Lond) 1998;509:103–116. doi: 10.1111/j.1469-7793.1998.103bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hugon J, Tabaraud F, Rigaud M, Vallat JM, Dumas M. Glutamate dehydrogenase and aspartate aminotransferase in leukocytes of patients with motor neuron disease. Neurology. 1989a;39:956–958. doi: 10.1212/wnl.39.7.956. [DOI] [PubMed] [Google Scholar]
- 32.Hugon J, Vallat JM, Spencer PS, Leboutet MJ, Barthe D. Kainic acid induces early and late delayed degenerative neuronal changes in rat spinal cord. Neurosci Lett. 1989b;104:258–262. doi: 10.1016/0304-3940(89)90585-5. [DOI] [PubMed] [Google Scholar]
- 33.Hyrc K, Handran SD, Rothman SM, Goldberg MP. Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. J Neurosci. 1997;17:6669–6677. doi: 10.1523/JNEUROSCI.17-17-06669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iino M, Ozawa S, Tsuzuki K. Permeation of calcium through excitatory amino acid receptor channels in cultured rat hippocampal neurons. J Physiol (Lond) 1990;424:151–165. doi: 10.1113/jphysiol.1990.sp018060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jakowec MW, Yen L, Kalb RG. In situ hybridization analysis of AMPA receptor subunit gene expression in the developing rat spinal cord. Neuroscience. 1995;67:909–920. doi: 10.1016/0306-4522(95)00094-y. [DOI] [PubMed] [Google Scholar]
- 36.Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential Glur-B subunit expression. Neuron. 1994;12:1281–1289. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- 37.Khodorov B, Pinelis V, Storozhevykh T, Vergun O, Vinskaya N. Dominant role of mitochondria in protection against a delayed neuronal Ca2+ overload induced by endogenous excitatory amino acids following a glutamate pulse. FEBS Lett. 1996;393:135–138. doi: 10.1016/0014-5793(96)00873-3. [DOI] [PubMed] [Google Scholar]
- 38.Khodorov B, Pinelis V, Storozhevykh T, Yuravichus A, Khaspekhov L. Blockade of mitochondrial Ca(2+) uptake by mitochondrial inhibitors amplifies the glutamate-induced calcium response in cultured cerebellar granule cells. FEBS Lett. 1999;458:162–166. doi: 10.1016/s0014-5793(99)01130-8. [DOI] [PubMed] [Google Scholar]
- 39.Kiskin NI, Krishtal OA, Tsyndrenko A. Excitatory amino acid receptors in hippocampal neurons: kainate fails to desensitize them. Neurosci Lett. 1986;63:225–230. doi: 10.1016/0304-3940(86)90360-5. [DOI] [PubMed] [Google Scholar]
- 40.Koh J, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 41.Koh J, Choi DW. Vulnerability of cultured cortical neurons to damage by excitotoxins: differential susceptibility of neurons containing NADPH-diaphorase. J Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koh J, Goldberg MP, Hartley DM, Choi DW. Non-NMDA receptor-mediated neurotoxicity in cortical culture. J Neurosci. 1990;10:693–705. doi: 10.1523/JNEUROSCI.10-02-00693.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- 45.Lambolez B, Ropert N, Perrais D, Rossier J, Hestrin S. Correlation between kinetics and RNA splicing of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors in neocortical neurons. Proc Natl Acad Sci USA. 1996;93:1797–1802. doi: 10.1073/pnas.93.5.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Launey T, Ivanov A, Ferrand N, Gueritaud JP. Developing rat brainstem motoneurones in organotypic culture express calcium permeable AMPA-gated receptors. Brain Res. 1998;781:148–158. doi: 10.1016/s0006-8993(97)01225-0. [DOI] [PubMed] [Google Scholar]
- 47.Leigh PN, Meldrum BS. Excitotoxicity in ALS. Neurology. 1996;47:S221–S227. doi: 10.1212/wnl.47.6_suppl_4.221s. [DOI] [PubMed] [Google Scholar]
- 48.Lledo PM, Somasundaram B, Morton AJ, Emson PC, Mason WT. Stable transfection of calbindin-D28k into the GH3 cell line alters calcium currents and intracellular calcium homeostasis. Neuron. 1992;9:943–954. doi: 10.1016/0896-6273(92)90246-a. [DOI] [PubMed] [Google Scholar]
- 49.Lu YM, Yin HZ, Chiang J, Weiss JH. Ca2+-permeable AMPA/kainate and NMDA channels: high rate of Ca2+ influx underlies potent induction of injury. J Neurosci. 1996;16:5457–5465. doi: 10.1523/JNEUROSCI.16-17-05457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lukas W, Jones KA. Cortical neurons containing calretinin are selectively resistant to calcium overload and excitotoxicity in vitro. Neuroscience. 1994;61:307–316. doi: 10.1016/0306-4522(94)90233-x. [DOI] [PubMed] [Google Scholar]
- 51.Mattson MP, Rychlik B, Chu C, Christakos S. Evidence for calcium-reducing and excito-protective roles for the calcium-binding protein calbindin-D28k in cultured hippocampal neurons. Neuron. 1991;6:41–51. doi: 10.1016/0896-6273(91)90120-o. [DOI] [PubMed] [Google Scholar]
- 52.McCormack JG, Denton RM. Intracellular calcium ions and intramitochondrial Ca2+ in the regulation of energy metabolism in mammalian tissues. Proc Nutr Soc. 1990;49:57–75. doi: 10.1079/pns19900009. [DOI] [PubMed] [Google Scholar]
- 53.Mecocci P, Macgarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- 54.Peng TI, Jou MJ, Sheu SS, Greenamyre JT. Visualization of NMDA receptor-induced mitochondrial calcium accumulation in striatal neurons. Exp Neurol. 1998;149:1–12. doi: 10.1006/exnr.1997.6599. [DOI] [PubMed] [Google Scholar]
- 55.Plaitakis A, Caroscio JT. Abnormal glutamate metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1987;22:575–579. doi: 10.1002/ana.410220503. [DOI] [PubMed] [Google Scholar]
- 56.Pruss RM, Akeson RL, Racke MM, Wilburn JL. Agonist-activated cobalt uptake identifies divalent cation-permeable kainate receptors on neurons and glial cells. Neuron. 1991;7:509–519. doi: 10.1016/0896-6273(91)90302-g. [DOI] [PubMed] [Google Scholar]
- 57.Reiner A, Medina L, Figueredo-Cardenas G, Anfinson S. Brainstem motoneuron pools that are selectively resistant in amyotrophic lateral sclerosis are preferentially enriched in parvalbumin: evidence from monkey brainstem for a calcium-mediated mechanism in sporadic ALS. Exp Neurol. 1995;131:239–250. doi: 10.1016/0014-4886(95)90046-2. [DOI] [PubMed] [Google Scholar]
- 58.Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rogers JH. Immunohistochemical markers in rat cortex: co-localization of calretinin and calbindin-D28k with neuropeptides and GABA. Brain Res. 1992;587:147–157. doi: 10.1016/0006-8993(92)91439-l. [DOI] [PubMed] [Google Scholar]
- 60.Ross SM, Seelig M, Spencer PS. Specific antagonism of excitotoxic action of “uncommon” amino acids assayed in organotypic mouse cortical cultures. Brain Res. 1987;425:120–127. doi: 10.1016/0006-8993(87)90490-2. [DOI] [PubMed] [Google Scholar]
- 61.Rothstein JD. Advances in amyotrophic lateral sclerosis: excitotoxicity hypothesis. Neurology. 1996;47[Suppl 2]:s19–s26. doi: 10.1212/wnl.47.4_suppl_2.19s. [DOI] [PubMed] [Google Scholar]
- 62.Rothstein JD, Kuncl RW. Neuroprotective strategies in a model of chronic glutamate-mediated motor neuron toxicity. J Neurochem. 1995;65:643–651. doi: 10.1046/j.1471-4159.1995.65020643.x. [DOI] [PubMed] [Google Scholar]
- 63.Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis [see comments]. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- 64.Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis [see comments]. New Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- 65.Rothstein JD, Jin L, Dykes-Hoberg M, Kuncl RW. Chronic inhibition of glutamate uptake produces a model of slow neurotoxicity. Proc Natl Acad Sci USA. 1993;90:6591–6595. doi: 10.1073/pnas.90.14.6591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rothstein JD, Vankammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter Glt-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 67.Roy J, Minotti S, Dong L, Figlewicz DA, Durham HD. Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J Neurosci. 1998;18:9673–9684. doi: 10.1523/JNEUROSCI.18-23-09673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaffner AE, St John PA, Barker JL. Fluorescence-activated cell sorting of embryonic mouse and rat motoneurons and their long-term survival in vitro. J Neurosci. 1987;7:3088–3104. doi: 10.1523/JNEUROSCI.07-10-03088.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schinder AF, Olsen EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sensi SL, Yin HZ, Carriedo SG, Rao SS, Weiss JH. Preferential Zn2+ influx through Ca2+-permeable AMPA/kainate channels triggers prolonged mitochondrial superoxide production. Proc Natl Acad Sci USA. 1999;96:2414–2419. doi: 10.1073/pnas.96.5.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shaw PJ, Ince PG. Glutamate, excitotoxicity and amyotrophic lateral sclerosis. J Neurol. 1997;244[Suppl 2]:S3–S14. doi: 10.1007/BF03160574. [DOI] [PubMed] [Google Scholar]
- 72.Shaw PJ, Ince PG, Falkous G, Mantle D. Oxidative damage to protein in sporadic motor neuron disease spinal cord. Ann Neurol. 1995;38:691–695. doi: 10.1002/ana.410380424. [DOI] [PubMed] [Google Scholar]
- 73.Shaw PJ, Williams TL, Slade JY, Eggett CJ, Ince PG. Low expression of GluR2 AMPA receptor subunit protein by human motor neurons. NeuroReport. 1999;10:261–265. doi: 10.1097/00001756-199902050-00011. [DOI] [PubMed] [Google Scholar]
- 74.Siklós L, Engelhardt J, Harati Y, Smith RG, Joo F, Appel SH. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophic lateral sclerosis. Ann Neurol. 1996;39:203–216. doi: 10.1002/ana.410390210. [DOI] [PubMed] [Google Scholar]
- 75.Sommer B, KeinΣnen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, Köhler M, Takagi T, Sakmann B, Seeburg PH. Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science. 1990;249:1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- 76.Spencer PS, Ludolph A, Dwivedi MP, Roy DN, Hugon J, Schaumberg HH. Lathyrism: evidence for the role of the neuroexcitatory amino acid BOAA. Lancet. 1986;2:1066–1067. doi: 10.1016/s0140-6736(86)90468-x. [DOI] [PubMed] [Google Scholar]
- 77.Spencer PS, Nunn PB, Hugon J, Ludolph J, Ross AC, Ross SM, Roy DN, Robertson RC. Guam amyotrophic lateral sclerosis–parkinsonism–dementia linked to a plant excitant neurotoxin. Science. 1987;237:517–522. doi: 10.1126/science.3603037. [DOI] [PubMed] [Google Scholar]
- 78.Stout AK, Reynolds IJ. High-affinity calcium indicators underestimate increases in intracellular calcium concentrations associated with excitotoxic glutamate stimulations. Neuroscience. 1999;89:91–100. doi: 10.1016/s0306-4522(98)00441-2. [DOI] [PubMed] [Google Scholar]
- 79.Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- 80.Swerdlow RH, Parks JK, Cassarino DS, Trimmer PA, Miller SW, Maguire DJ, Sheehan JP, Maguire RS, Pattee G, Juel VC, Phillips LH, Tuttle JB, Bennett JP, Jr, Davis RE, Parker WD., Jr Mitochondria in sporadic amyotrophic lateral sclerosis. Exp Neurol. 1998;153:135–142. doi: 10.1006/exnr.1998.6866. [DOI] [PubMed] [Google Scholar]
- 81.Teitelbaum JS, Zatorre RJ, Carpenter S, Gendron D, Evans AC, Gjedde A, Cashman NR. Neurologica sequelae of domoic acid intoxication due to the ingestion of contaminated mussels. New Engl J Med. 1990;322:1781–1787. doi: 10.1056/NEJM199006213222505. [DOI] [PubMed] [Google Scholar]
- 82.Temkin R, Lowe D, Jensen P, Hatt H, Smith DO. Expression of glutamate receptor subunits in alpha-motoneurons. Mol Brain Res. 1997;52:38–45. doi: 10.1016/s0169-328x(97)00249-0. [DOI] [PubMed] [Google Scholar]
- 83.Terro F, Yardin C, Esclaire F, AyerLelievre C, Hugon J. Mild kainate toxicity produces selective motoneuron death with marked activation of Ca2+-permeable AMPA/kainate receptors. Brain Res. 1998;809:319–324. doi: 10.1016/s0006-8993(98)00883-x. [DOI] [PubMed] [Google Scholar]
- 84.Tölle TR, Berthele A, ZieglgΣnsberger W, Seeburg PH, Wisden W. The differential expression of 16 NMDA and non-NMDA receptor subunits in the rat spinal cord and in periaqueductal gray. J Neurosci. 1993;13:5009–5028. doi: 10.1523/JNEUROSCI.13-12-05009.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tölle TR, Berthele A, ZieglgΣnsberger W, Seeburg PH, Wisden W. Flip and flop variants of AMPA receptors in the rat lumbar spinal cord. Eur J Neurosci. 1995;7:1414–1419. doi: 10.1111/j.1460-9568.1995.tb01134.x. [DOI] [PubMed] [Google Scholar]
- 86.Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:427–433. doi: 10.1038/8091. [DOI] [PubMed] [Google Scholar]
- 87.Trussell LO, Thio LL, Zorumski CF, Fischbach GD. Rapid desensitization of glutamate receptors in vertebrate central neurons. Proc Natl Acad Sci USA. 1988;85:4562–4566. doi: 10.1073/pnas.85.12.4562-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Turetsky DM, Canzoniero LMT, Sensi SL, Weiss JH, Goldberg MP, Choi DW. Cortical neurones exhibiting kainate-activated Co2+ uptake are selectively vulnerable to AMPA/kainate receptor-mediated injury. Neurobiol Dis. 1994;1:101–110. doi: 10.1006/nbdi.1994.0013. [DOI] [PubMed] [Google Scholar]
- 89.Volterra A, Trotti D, Tromba C, Floridi S, Racagni G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J Neurosci. 1994;14:2924–2932. doi: 10.1523/JNEUROSCI.14-05-02924.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang GJ, Thayer SA. Sequestration of glutamate-induced Ca2+ loads by mitochondria in cultured rat hippocampal neurons. J Neurophysiol. 1996;76:1611–1621. doi: 10.1152/jn.1996.76.3.1611. [DOI] [PubMed] [Google Scholar]
- 91.Wang GJ, Randall RD, Thayer SA. Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. J Neurophysiol. 1994;72:2563–2569. doi: 10.1152/jn.1994.72.6.2563. [DOI] [PubMed] [Google Scholar]
- 92.Weiss JH, Koh JY, Choi DW. Neurotoxicity of beta-N-methylamino-l-alanine (BMAA) and beta-N-oxalylamino-l-alanine (BOAA) on cultured cortical neurons. Brain Res. 1989;497:64–71. doi: 10.1016/0006-8993(89)90970-0. [DOI] [PubMed] [Google Scholar]
- 93.Weiss JH, Yin H-Z, Choi DW. Basal forebrain cholinergic neurons are selectively vulnerable to AMPA/kainate receptor-mediated neurotoxicity. Neuroscience. 1994;60:659–664. doi: 10.1016/0306-4522(94)90494-4. [DOI] [PubMed] [Google Scholar]
- 94.Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.White RJ, Reynolds IJ. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurones. J Physiol (Lond) 1997;498:31–47. doi: 10.1113/jphysiol.1997.sp021839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Williams TL, Ince PG, Oakley AE, Shaw PJ. An immunocytochemical study of the distribution of AMPA selective glutamate receptor subunits in the normal human motor system. Neuroscience. 1996;74:185–198. doi: 10.1016/0306-4522(96)00117-0. [DOI] [PubMed] [Google Scholar]
- 98.Yamada KA, Tang CM. Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J Neurosci. 1993;13:3904–3915. doi: 10.1523/JNEUROSCI.13-09-03904.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yin H, Turetsky D, Choi DW, Weiss JH. Cortical neurons with Ca2+ permeable AMPA/kainate channels display distinct receptor immunoreactivity and are GABAergic. Neurobiol Dis. 1994;1:43–49. doi: 10.1006/nbdi.1994.0006. [DOI] [PubMed] [Google Scholar]