

Abstract
The ubiquitin–proteasome protein degradation pathway is crucial in controlling intracellular levels of a variety of short-lived proteins and maintaining cellular growth and metabolism. In a previous study, we showed the accumulation of conjugated ubiquitin in CA1 neurons of the gerbil after 5 min of forebrain ischemia (Morimoto et al., 1996; Ide et al., 1999). The accumulation of conjugated ubiquitin may reflect proteasome malfunction. In the present study, we investigated the effects of proteasome inhibitors on primary neuronal cultures to determine whether proteasomal malfunction induces neuronal death. When carbobenzoxy-Leu-Leu-Leu-aldehyde or lactacystin, two different types of proteasome inhibitors, were separately used to suppress proteasome activity, we observed induction of apoptotic neuronal cell death in both cases. During the apoptotic process, mitochondrial membrane potential was disrupted, cytochrome-c was released from mitochondria into the cytosol, and caspase-3-like proteases were activated. Apoptosis was inhibited by pretreatment with acetyl-aspartyl-glutamyl-valyl-aspart-1-aldehyde or overexpression of Bcl-x/L. These results demonstrated that suppression of proteasome function induces neuronal apoptosis via the release of cytochrome c from mitochondria and activation of caspase-3-like proteases.
Keywords: neuron, apoptosis, proteasome, cytochrome c, caspase-3-like proteases, ubiquitin
The ubiquitin–proteasome pathway is predominantly a nonlysosomal protein degradation pathway that is responsible for degrading many critical regulatory proteins that must be rapidly eliminated for normal growth and metabolism. Degradation of a protein by this pathway involves two distinct and successive steps: covalent attachment of multiple ubiquitin molecules to the target protein and degradation of the targeted protein by proteasomes (Ciechanover, 1998). This pathway has multiple cellular targets and is involved in differentiation, development, response to stress, and the pathogenesis of various diseases. Several recent studies have demonstrated that proteasome dysfunction induces apoptosis in various types of cells (Drexler, 1997; Lopes et al., 1997).
Apoptosis is a subtype of cell death that is involved in diverse physiological and pathological processes, including diseases. Various stimuli that induce apoptosis lead to the release of cytochrome c from mitochondria, which plays a key role in a common pathway of activation of caspases (Susin et al., 1998; Thornberry and Lazebnik, 1998). Cytosolic cytochrome c has been shown to bind apoptosis proteases-activating factor-1 (Apaf-1) and mediate activation of caspase-9 and caspase-3 (Green and Reed, 1998; Kuida et al., 1998). Activated caspases cleave a variety of target proteins, thereby disabling important cellular processes and breaking down structural components of the cell, such as lamin, and eventually causing cell death (Nicholson and Thornberry, 1997).
Neuronal cell death is the final pathological consequence of many CNS diseases, including degenerative disease, trauma, chemical poisoning, and brain ischemia. Among the ischemia-induced neuronal deaths, less severe, transient ischemia leads to the selective death of certain neuronal populations in animal models (Kirino, 1982). Recent evidence suggests that the apoptotic process is partly responsible for the selective neuronal death (Bottiger et al., 1998; Chen et al., 1998). Although some pro-apoptotic and anti-apoptotic proteins such as Bax, Bcl-2, and Bcl-x/L, show alterations of protein expression after brain ischemia (Antonawich et al., 1998), none of those alterations are linked to a specific region; thus, the precise mechanism of the selective vulnerability is still unknown.
We recently noticed that accumulation of significant amounts of conjugated ubiquitin in CA1 region of the hippocampus after transient global ischemia is accompanied by depletion of free ubiquitin (Morimoto et al., 1996; Ide et al., 1999). The accumulation of conjugated ubiquitin may reflect hypofunction of downstream proteasome activity that normally degrades ubiquitinated proteins. Moreover, direct injection of proteasome inhibitor into the lateral ventricles of the rat induced DNA fragmentation in various CNS areas, suggesting that suppression of proteasome is able to induce neuronal apoptosis (Taglialatela et al., 1998). Therefore, it is reasonable to speculate that proteasome malfunction may in part underlie the molecular events of the ischemia-induced neuronal death.
In this study, we induced proteasomal hypofunction in cultured rat cortical neurons to investigate whether and how proteasome hypofunction affects neurons. We found suppression of proteasome activity induced apoptotic neuronal death. Mitochondrial membrane potential was disrupted, cytosolic cytochrome c increased, and caspase-3-like proteases were activated in this model of proteasomal hypofunction.
MATERIALS AND METHODS
Primary neuronal culture. Primary cultures of neurons were prepared from the cerebral cortex of fetal Wistar rats (18 d gestation) and cultivated in Neurobasal medium supplemented with 2% B27, 0.5 mm glutamine, 10 μm 2-mercaptoethanol, and 25 μm glutamate (Life Technologies, Rockville, MD). Cells were seeded at a density of 1106 cells per well in six-well plates coated with polyethylenimine and then incubated in a humidified atmosphere of 5% CO2 at 37°C. On day 3, the cultures were incubated with 10 μm cytosine arabinoside for 24 hr to suppress the growth of glial cells. Half of the medium in each well was changed every 4 d. At day 8, glutamate and 2-mercaptoethanol were removed from the medium. Neurons cultured for 2 weeks were used in this study.
Inhibition of proteasome activity. Primarily neuronal cultures were incubated with various concentrations of carbobenzoxy-Leu-Leu-Leu-aldehyde (Z-LLLal), a proteasome inhibitor, and carbobenzoxy-Leu-Leu-Leu-COOH (Z-LLL), a corresponding control or lactacystin. Neurons were harvested for analysis at various times after incubation.
Proteasome and caspase-3-like protease activity assays.Harvested neurons were lysed in proteasome buffer [10 mm Tris-HCl, pH 7.5, 1 mmEDTA, 2 mm ATP, 20% glycerol, and 4 mm dithiothreitol (DTT)] (Tsukahara et al., 1988; Figueiredo-Pereira et al., 1994) or caspase-3 lysis buffer [10 mm HEPES-KOH, pH 7.4, 2 mmEDTA, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 1 mm phenylmethylsulfonyl fluoride (PMSF), and 5 mm DTT], sonicated, and then centrifuged at 13,000 × g at 4°C for 10 min. The supernatants (20 μg of protein) were incubated with proteasome activity assay buffer [0.05 m Tris-HCl, pH 8.0, 0.5 mm EDTA, 40 μmSuc-Leu-Leu-Val-Tyr-4-methyl-coumaryl-7-amide (LLVY-MCA) (Peptide Institute Inc., Osaka, Japan)] or caspase-3-like protease activity assay buffer [20 mm HEPES-KOH, pH 7.5, 2 mm DTT, 10% glyceral, and 40 μm Ac-Asp-Glu-Val-Asp-4-methyl-coumaryl-7-amide (DEVD-MCA) (Peptide Institute Inc. Osaka, Japan)] for 1 hr at 37°C. The reactions were stopped by adding 0.9 ml of cold water and placing the reaction mixtures on ice for at least 10 min. The intensity of fluorescence of each solution was measured by fluorescence spectrophotometry (Hitachi F-2000; Hitachi Instruments, Tokyo, Japan) at 380 nm excitatory (Ex) and 440 nm emission (Em) wavelengths. All readings were standardized using the fluorescence intensity of an equal volume of free 7-amino-4-methyl-coumarin (AMC) solution (40 μm).
Analysis of neuronal cell death. Cell death was assayed by trypan blue dye exclusion test. Cultured neurons were incubated with 0.4% trypan blue solution (Sigma, St. Louis, MO) for 15 min, and cells were counted under a phase-contrast microscope.
For nuclear staining, cultured neurons were incubated with 25 μm Hoechst 33258 (Molecular Probes, Engene, OR) for 15 min at 37°C and were then examined by fluorescence microscopy (BX60; Olympus Opticals, Tokyo, Japan) (Ex/Em: 352 nm/461 nm).
Measurement of mitochondrial membrane potential. After treatment with Z-LLLal, neurons were incubated with 10 μm5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine2++ iodide (JC-1) (Molecular Probes) fluorescence dye for 15 min and then washed three times in PBS. The mitochondrial membrane potential was estimated qualitatively under a fluorescence microscope (Ex/Em: 352 nm/461 nm) (Ankarcrona et al., 1995).
Western blot analysis. For detection of cytochrome c, neurons were lysed in buffer A (in mm: 20 HEPES-KOH, pH 7.5, 10 KCl, 1.5 MgCl2, 1 sodium EDTA, 1 sodium EGTA, 1 dithiothreitol, and 0.1 PMSF) containing 250 mm sucrose and homogenized with a Dounce homogenizer (Yang et al., 1997). Homogenates were centrifuged twice at 750 × g for 10 min at 4°C, and supernatants were centrifuged at 10,000 × g for 15 min at 4°C. The resulting mitochondrial pellets were resuspended in buffer A containing 250 mm sucrose, and supernatants were further centrifuged at 100,000 × g for 1 hr at 4°C. The remaining supernatants (cytosolic fractions) were prepared for Western blotting analysis.
The protein concentration of lysates from cytosolic fraction and mitochondrial fraction was determined with the Bio-Rad protein assay system (Bio-Rad, San Francisco, CA). Total cellular protein lysate was denatured in an equal volume of sample buffer (62.5 mmTris-HCl, pH 6.8, 2% SDS, 5 mm EDTA, 10% glycerol, 35 mm 2-mercaptoethanol, and 0.01% bromophenol blue) at 100°C for 5 min and separated by 12.5% SDS-PAGE. After separation, protein was electrically transferred to nitrocellulose membrane at 1 mA/cm2 in a semi-wet condition (Atto Corporation, Tokyo, Japan). The membrane was incubated with blocking solution [5% skim milk in Tris-buffered saline containing 0.05% Tween 20] at 4 overnight and subsequently incubated with primary antibodies against cytochrome c (clone 7H8.2c12, monoclonal antibody; PharMingen, San Diego, CA). The membranes were then incubated with the appropriate horseradish peroxidase-conjugated secondary antibodies. Detection was achieved using a chemiluminescence system (ECL system; Amersham, Buckinghamshire, UK).
Introduction of Bcl-x/L overexpression in cultured neurons. The EcoRI fragment of human Bcl-x/L cDNA from pSKIIhBcl-XL, provided by Dr. L. Boise (Howard Hughes Medical Institute, University of Chicago, Chicago, IL) (Boise et al., 1993), was inserted into the EcoRI site of pCAcc, which generated pCA-hBcl-x/L. The cosmid pAxCA-hBcl-x/L was generated by inserting theClaI expression cassette from pCA-hBcl-XL into the ClaI site of cosmid pAxcw. Recombinant adenovirus expressing Bcl-x/L (AxCA-hbcl-x/L) was then generated by homologous recombination withEcoT22I/ClaI-digested adenovirus DNA-terminal protein complex in 293 host cells (Miyake et al., 1996; Shinoura et al., 1999).
Neurons were plated on six-well plates at an initial density of 1106 cells per well. To introduce Bcl-x/L overexpression, cultured cortical neurons were incubated with AxCA-hbcl-x/L at a multiplicity of infection (MOI) of 20 pfu/cell, for 3 d at 37°C. As a control, neurons were also incubated with AxcA-lacZ at the same concentration. Bcl-x/L protein level was confirmed by Western blot.
Three days after infection, the neurons were treated with Z-LLLal. Neuronal death was then determined by trypan blue dye exclusion test.
RESULTS
Inhibition of proteasome activity
Because some peptide aldehydes are known to inhibit proteasome activity (Tsubuki et al., 1993), we tested the ability of Z-LLLal and lactacystin to inhibit proteasome activity by monitoring the levels of AMC, the hydrolysate of the fluorogenic substrates Suc-Leu-Leu-Val-Tyr-MCA created by proteasome cleavage. We confirmed that Z-LLLal inhibited proteasome activity in a dose-dependent manner in primary cultures of cortical neurons (Fig.1A). Lactacystin has also suppressed proteasome activity. Z-LLLal suppresses proteasome activity 6 hr after addition to the medium of cultured neurons. Z-LLL, a noninhibitory substrate of the proteasome and also an analog of Z-LLLal peptide aldehyde (Tsubuki et al., 1993), was used as a control. Furthermore, Z-LLL competed the inhibitory effect of Z-LLLal on proteasomal activity in a dose-dependent manner (Fig.1B). When neurons were treated with low-dose (<0.04 μm) Z-LLLal, proteasomal activity was not inhibited, even with prolonged treatment up to 7 d (Fig.1C).
Fig. 1.
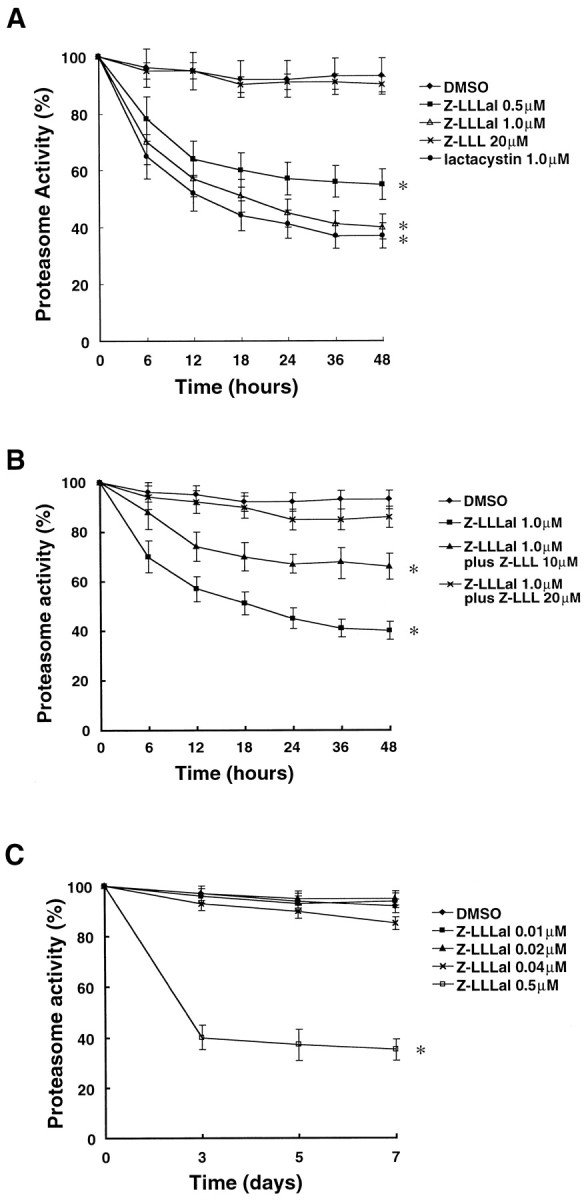
Proteasome activity after treatment with a proteasome inhibitor. Two-week-old cultured cortical neurons were incubated with various concentrations of Z-LLLal or 20 μmZ-LLL as indicated. Neurons were also treated with 0.5% DMSO in 3 ml of medium as a control. The concentration of DMSO in medium is the same as in 1.0 μm Z-LLLal group. After treatment with these agents for 6, 12, 18, 24, 36, and 48 hr, neurons were collected and homogenized in proteasome buffers. Proteasome activity, assayed with 20 μg of total protein, was determined by measuring cleavage of the fluorogenic substrate LLVY-MCA using a Hitachi fluorescence spectrophotometer and is shown as relative proteasome activity. Proteasome activity of neurons was suppressed by a proteasome inhibitor, Z-LLLal, in a dose-dependent manner (A). Inhibition of proteasomal activity by Z-LLLal was competed by Z-LLL, a proteasomal substrate in a dose-dependent manner (B). Prolonged treatment with low concentrations of Z-LLLal (<0.04 μm) minimally lowered proteasomal activity (C). Displayed values are the means ± SD of four independent experiments. *p < 0.01, compared with DMSO group.
Induction of neuronal cell death
We then examined whether inhibition of proteasome function in neurons would induce apoptosis. Cultured cortical neurons were incubated with Z-LLLal or lactacystin and evaluated by trypan blue dye exclusion test (Schwartz and Osborne, 1995). Dead neurons first appeared at 24 hr and markedly increased at 36 hr after treatment with Z-LLLal or lactacystin (Fig.2A). Nuclear staining with Hoechst 33258 showed nuclear condensation and fragmentation in dead cells (Fig. 3).
Fig. 2.
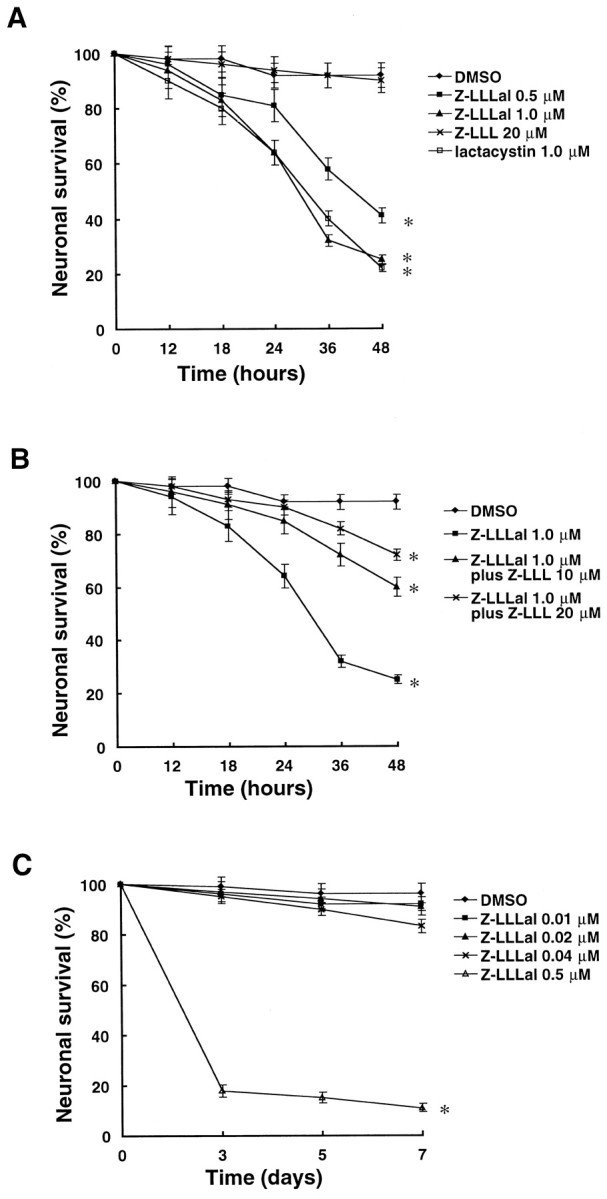
Cell viability after incubation with a proteasome inhibitor. Cortical neurons cultured in polyethylenimine-coated six-well plates were incubated with different concentrations of Z-LLLal, 20 μm Z-LLL, or 0.5% DMSO as indicated for 12, 24, 36, and 48 hr. These neurons were then incubated with 0.4% trypan blue dye for 10 min. Neuronal viability was examined under a light microscope. Cell viability decreased in a dose-dependent manner after treatment with Z-LLLal (A). Z-LLLal-induced cell death was suppressed by Z-LLL competitively (B). Prolonged treatment with low concentrations of Z-LLLal (<0.04 μm) minimally lowered neuronal survival (C). Displayed values are the means ± SD of four independent experiments. *p < 0.01, compared with DMSO group.
Fig. 3.

Photomicrographs of cultured neurons after treatment with Z-LLLal. Cortical neurons were cultured with or without 1 μm Z-LLLal for 48 hr. Then, neurons were stained with 50 μm Hoechst 33258 for 20 min at 37°C and washed three times using PBS. Nuclear condensation was examined under a fluorescence microscope (Ex/Em: 352 nm/461 nm). Nuclear condensation and fragmentation were prominent 48 hr after treatment with 1.0 μm Z-LLLal (right) when compared with those treated with 0.5% DMSO alone (left). Magnification, 500×.
In contrast to Z-LLLal, apoptosis was not induced by treatment with the Z-LLL control (Fig. 2A); furthermore, it rescued neurons from Z-LLLal-induced death in a dose-dependent manner (Fig.2B). Even prolonged exposure to low-dose Z-LLLal did not induce cell death in neurons unless proteasomal function was suppressed (Figs. 1C, 2C).
Activation of caspase-3-like proteases in apoptosis induced by proteasome inhibitors
Caspase-3-like proteases are activated during apoptosis induced by a variety of agents (Thornberry and Lazebnik, 1998). In this study, we observed an increase in caspase-3-like protease activity before neuronal death. Caspase-3-like protease activity increased in a dose-dependent manner to approximately fourfold to sixfold of control levels 48 hr after neurons were incubated with Z-LLLal or lactacystin (Fig. 4).
Fig. 4.

Caspase-3-like protease activity after incubation with proteasome inhibitors. Cultured neurons were incubated with different concentrations of Z-LLLal or 20 μm Z-LLL as indicated for 6, 12, 18, 24, 36, and 48 hr, collected, and lysed in caspase-3 buffer. The protein concentration of these lysates was determined using the Bio-Rad protein assay system. Total protein (20 μg) was incubated with 40 μm DEVD-MCA for 60 min at 37°C. Caspase-3-like protease activity was determined by measuring cleavage of the fluorogenic substrate DEVD-MCA using Hitachi fluorescence spectrophotometer and is shown as relative caspase-3-like protease activity. Caspase-3-like protease activity increased by the treatment with Z-LLLal in a dose-dependent manner. Displayed values are the means ± SD of four independent experiments. *p < 0.01, compared with DMSO group.
To confirm that activated caspase-3-like proteases mediate apoptosis induced by proteasome inhibitors, cultured cortical neurons were pretreated with several concentrations of acetyl-aspartyl-glutamyl-valyl-aspart-1-aldehyde (DEVD-CHO), a caspase-3-like protease inhibitor, for 12 hr before exposure to Z-LLLal. Caspase-3-like protease activity decreased by DEVD-CHO in a dose-dependent manner within a range of 20–80 μm (Fig. 5B). We also found that surviving neurons increased markedly by DEVD-CHO treatment in a dose-dependent manner within the range (Fig.5C). The results indicated that proteasome inhibition induces apoptosis via caspase-3-like protease activation.
Fig. 5.
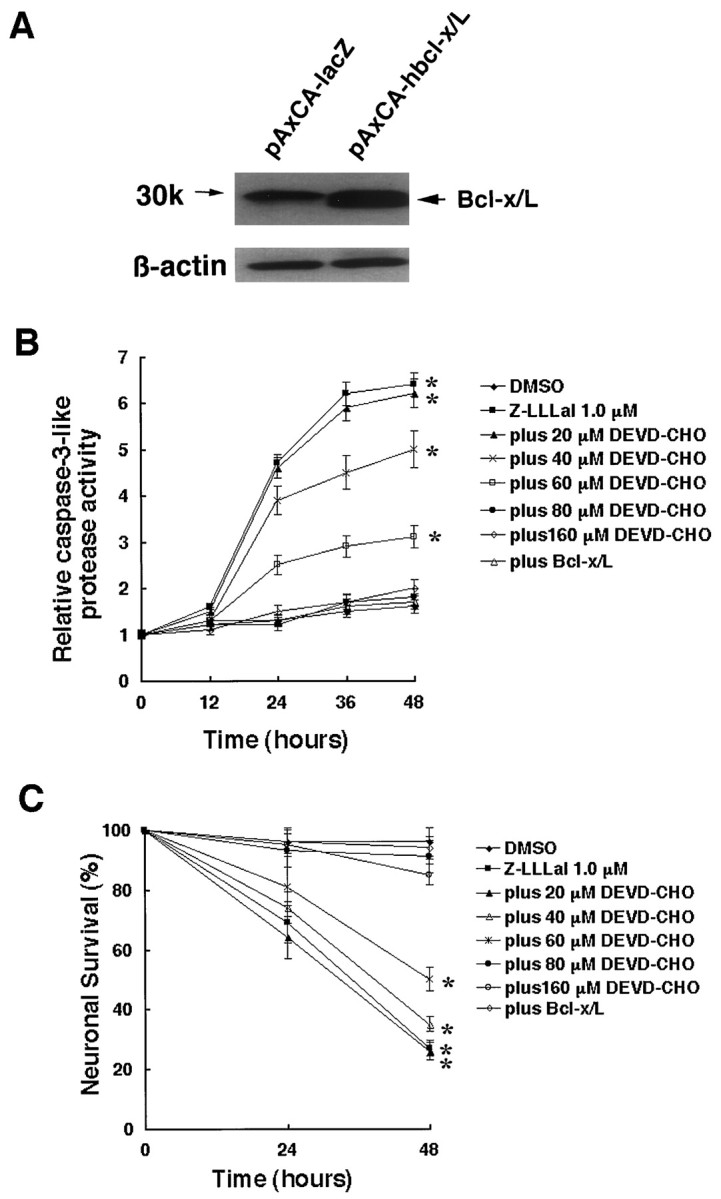
Suppression of proteasome inhibitor-induced apoptosis by pretreatment with DEVD-CHO or overexpression of Bcl-x/L. Cultured neurons were pretreated with 150 μm DEVD-CHO for 12 hr or were infected by AxCA-hbcl-x/L or AxCA–lacZ at an MOI of 20 pfu/cell. Bcl-x/L expression level was examined by Western blot analysis [antibody: anti-Bcl-x (S-18; Santa Cruz, Biotechnology, Santa Cruz, CA)] at day 3 after infection (A). After pretreatment with concentrations of DEVD-CHO or infection with AxCA-hbcl-x/L or pAxCA–lacZ for 3 d, neurons were incubated with 1 μm Z-LLLal or DMSO for 24 and 48 hr. Caspase-3-like protease activity was suppressed by DEVD-CHO in a dose-dependent manner and Bcl-x/L expression (B). Z-LLLal-induced neuronal death was suppressed by DEVD-CHO in a dose-dependent manner and Bcl-x/L expression. Percent of neuronal survival was determined by trypan blue exclusion test. Data shown are the means ± SD of three independent experiments. *p < 0.01, compared with DMSO group.
Inhibition of proteasomal hypofunction-induced apoptosis by Bcl-x/L expression
Most of Bcl-2 family proteins are located on the surface of the outer mitochondrial membrane (Reed et al., 1998). These proteins are assumed to regulate the release of some proteins from mitochondria and subsequent apoptosis. One of these proteins, Bcl-x/L, can also interact with Apaf-1 and inhibit caspase-9 activation (Hu et al., 1998). We investigated whether proteasome inhibitor-induced apoptosis would be suppressed by the anti-apoptotic Bcl-x/L protein. Cultured cortical neurons were forced to overexpress Bcl-x/L by infection with AxCA-hbcl-x/L (Fig.5A), and 3 d later, were incubated with proteasome inhibitor. We assessed caspase-3-like protease activity and cell viability and found that Bcl-x/L strongly inhibited caspase-3-like protease activation and apoptosis induced by Z-LLLal (Fig. 5B,C).
Mitochondrial hypofunction and cytochrome c release induced by proteasome inhibitor
To investigate whether cytochrome c is involved in proteasome inhibitor-induced apoptosis, we examined the distributional change in cytochrome c. In recent studies, cytochrome c has been demonstrated to directly induce activation of some caspases (Mignotte and Vayssiere, 1998). The lysates of cultured cortical neurons treated with Z-LLLal were divided into cytosolic and mitochondrial fractions, as described in Material and Methods. Western blot analysis revealed accumulation and reduction of cytosolic and mitochondrial cytochrome c, respectively (Fig. 6). The fractional change of cytochrome c is inhibited by Bcl-x/Loverexpression, whereas it is not inhibited by DEVD-CHO (Fig. 6).
Fig. 6.

Changes in cytochrome c in cytosolic fraction and mitochondrial membrane potential after treatment with Z-LLLal. Cultured neurons were incubated with 1 μm Z-LLLal for 24 and 48 hr. Then, neurons were lysed in buffer A with 250 mmsucrose by Dounce homogenizer, as described in Materials and Methods. Homogenates were centrifuged and divided into cytosolic and mitochondrial fractions. Cytochrome c was detected in lysates from cytosolic and mitochondrial fractions by Western blot analysis with an anti-cytochrome c monoclonal antibody (PharMingen). Cytosolic fraction of cytochrome c increased, whereas mitochondrial fraction decreased during 48 hr after Z-LLLal treatment. Bcl-x/L expression inhibited cytochrome c release from the mitochondria, whereas DEVD-CHO did not perturb it.
Increases in cytosolic cytochrome c after treatment with proteasome inhibitor were studied by measuring mitochondrial membrane potential using a potential-sensitive voltage-sensitive fluorescent indicator, JC-1, that is incorporated into mitochondria when the mitochondrial membrane potential is normal (Reers et al., 1995). As shown in Figure7, the mitochondrial membrane potential of cortical neurons was disrupted in a time-dependent manner after treatment with Z-LLLal. The mitochondrial membrane potential disruption was inhibited by Bcl-x/L overexpression but not by DEVD-CHO (Fig. 7).
Fig. 7.

Change in mitochondrial membrane potential after treatment with Z-LLLal. Mitochondrial membrane potential of cortical neurons was disrupted in a time-dependent manner after treatment with Z-LLLal. Red fluorescence indicates a higher mitochondrial membrane potential, and green fluorescence indicates disruption of mitochondrial membrane potential. Cultured neurons were treated with 1 μm Z-LLLal for 24 and 48 hr and were then incubated with 10 μm JC-1 for 15 min at 37°C. The mitochondrial membrane potential was observed by fluorescence microscopy. The mitochondrial membrane potential was disrupted during 48 hr after the treatment with Z-LLLal. Bcl-x/L expression inhibited the mitochondrial membrane potential disruption, whereas DEVD-CHO did not suppress it. Magnification, 500×.
DISCUSSION
In this study, we demonstrated that suppression of cellular proteasome activity with Z-LLLal or lactacystin induced apoptotic neuronal death in a dose-dependent manner. The process of apoptosis in this model involved disruption of the mitochondrial membrane potential, increase in cytosolic cytochrome c, and activation of caspase-3-like proteases. Apoptosis was blocked by pretreatment with a caspase-3-like protease inhibitor, DEVD-CHO, or by overexpression of Bcl-x/L.
Z-LLLal is known as a specific proteasome inhibitor and is a useful tool in studying ubiquitin–proteasome protein degradation pathway (Saito et al., 1990; Figueiredo-Pereira et al., 1994; Traenckner et al., 1994; Jensen et al., 1995). It inhibits proteasome activity more strongly than its close analogs N-acetyl-Leu-Leu-morleucinal and N-acetyl-Leu-leu-norleucinal (Tsubuki et al., 1993). The control peptide Z-LLL, lacking the proteasome-inhibiting effect, cannot induce apoptosis. Rather, as we have shown, it rescued neuronal cells from Z-LLLal-induced cell death in a competitive manner. Therefore, we believe that apoptosis is induced not by the direct toxicity of the tetrapeptide itself but mainly by the suppression of proteasome function. Induction of proteasome suppression and subsequent apoptosis by another proteasome inhibitor, lactacystin, also support this notion.
The ubiquitin–proteasome pathway consists of several critical enzymes, including ubiquitin-activating enzyme, ubiquitin-conjugating enzyme, ubiquitin-protein ligase and proteasomes. This pathway degrades many critical proteins that must be rapidly destroyed for normal cell growth and metabolism. Despite its essential role in cellular homeostasis, inhibition of proteasomes leads to varying consequences depending on cell type and condition (Sadoul et al., 1996; Drexler, 1997; Lopes et al., 1997). The mechanism by which proteasomal malfunction induces apoptosis is still controversial. In the present study, we showed that suppression of proteasomal function induced cytochrome c–caspase-3-like protease-mediated apoptosis in cultured neurons. Our data showed that a decrease in mitochondrial cytochrome c was accompanied by an increase in cytosolic cytochrome c. Several mechanisms may contribute to the shift of cytochrome c seen in the present study. Some proteins that are able to promote apoptosis are proven substrates of the ubiquitin–proteasome protein degradation pathway, such as Bax (Chang et al., 1998) and p53 (Scheffner et al., 1993). Hypofunction of proteasomes may result in the accumulation of these pro-apoptotic proteins. The increase in these upstream pro-apoptotic proteins may be responsible for disruption of mitochondrial membrane potential and release of cytochrome c (Finucane et al., 1999). Another possibility is that accumulation of cytochrome c in the cytosol may be caused directly by suppression of proteasomal function, because cytochrome c appears to be degraded by the ubiquitin–proteasome protein degradation pathway (Sokolik and Cohen, 1992; Pearce and Sherman, 1997). It has been demonstrated that cytosolic cytochrome c can bind Apaf-1 and subsequently trigger the sequential activation of caspase-9 and caspase-3 (Green and Reed, 1998;Thornberry and Lazebnik, 1998). Activation of caspase-3 has been recently shown to be a key step in the execution process of apoptosis, and its inhibition can block apoptotic cell death. In the present study, we indicated that the caspase-3-like protease inhibitor DEVD-CHO prevented Z-LLLal-induced apoptosis. DEVD-CHO could not suppress mitochondrial membrane potential disruption or cytochrome c release from mitochondria, suggesting that the inhibitor acts downstream of the mitochondria. Furthermore, overexpression of Bcl-x/L, which can bind Apaf-1 to inhibit caspase-9 activation and prevent mitochondrial permeability transition pore opening and release of cytochrome c (Hu et al., 1998), also inhibited Z-LLLal-induced apoptotic neuronal death. Our data that overexpression of Bcl-x/L inhibited mitochondrial membrane disruption and cytochrome c release from mitochondria are compatible with the notion that Bcl-x/L acts at or upstream of the mitochondria. These data indicate that the cascade involving mitochondria, cytochrome c–Apaf-1, and caspase-3-like proteases mediates proteasome hypofunction-induced apoptosis in neurons.
As we have shown previously, multi-ubiquitin-conjugated proteins are accumulated in dying CA1 neurons that show selective vulnerability after transient global ischemia (Kirino, 1982; Morimoto et al., 1996;Ide et al., 1999). Although the accumulation of conjugated ubiquitin significantly declines in CA3 and the dentate gyrus, its accumulation does not decrease in the CA1 region (Ide et al., 1999), suggesting that proteasomes are not potent enough to degrade target proteins in the CA1 region after transient ischemia. In fact, the activity of proteasomes, especially the activity of 26S proteasome that degrades multi-ubiquitin-conjugated proteins to amino acids, decreases in gerbil cortex neurons after 10 min global ischemia (Kamikubo and Hayashi, 1996). Furthermore, inhibition of proteasome activity induces accumulation of ubiquitin-protein conjugates (Figueiredo-Pereira et al., 1994; Soldatenkov and Dritschilo, 1997) and apoptosis in CNSin vivo (Taglialatela et al., 1998). Taken together, it is reasonable to speculate that ischemia-induced apoptosis is in part via proteasome hypofunction that elicits accumulation of conjugated ubiquitin and unnecessary proteins. Obviously, further studies are required to verify the molecular events that underlie ischemia-induced proteasomal hypofunction and the proteasomal hypofunction-induced neuronal death.
Footnotes
This work was supported by Core Research for Evolutional Science and Technology, Japan Science and Technology Corporation, by a Grant-in-Aid for Scientific Research, and by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Science, and Culture of Japan. We thank Emily Rowell for editorial assistance.
Drs. Qiu and Asai contributed equally to this work.
Correspondence should be addressed to Dr. Akio Asai, Laboratory for Neuroscience and Neurooncology, Department of Neurosurgery, Faculty of Medicine, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8655, Japan. E-mail: asaisan-tky@umin.ac.jp.
REFERENCES
- 1.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 2.Antonawich FJ, Krajewski S, Reed JC, Davis JN. Bcl-x(l) Bax interaction after transient global ischemia. J Cereb Blood Flow Metab. 1998;18:882–886. doi: 10.1097/00004647-199808000-00008. [DOI] [PubMed] [Google Scholar]
- 3.Boise LH, Gonzalez-Garcia M, Postema CE, Ding L, Lindsten T, Turka LA, Mao X, Nunez G. bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death. Cell. 1993;74:597–608. doi: 10.1016/0092-8674(93)90508-n. [DOI] [PubMed] [Google Scholar]
- 4.Bottiger BW, Schmitz B, Wiessner C, Vogel P, Hossmann KA. Neuronal stress response and neuronal cell damage after cardiocirculatory arrest in rats. J Cereb Blood Flow Metab. 1998;18:1077–1087. doi: 10.1097/00004647-199810000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Chang YC, Lee YS, Tejima T, Tanaka K, Omura S, Heintz NH, Mitsui Y, Magae J. mdm2 and bax, downstream mediators of the p53 response, are degraded by the ubiquitin-proteasome pathway. Cell Growth Differ. 1998;9:79–84. [PubMed] [Google Scholar]
- 6.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17:7151–7160. doi: 10.1093/emboj/17.24.7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Drexler HC. Activation of the cell death program by inhibition of proteasome function. Proc Natl Acad Sci USA. 1997;94:855–860. doi: 10.1073/pnas.94.3.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Figueiredo-Pereira ME, Berg KA, Wilk S. A new inhibitor of the chymotrypsin-like activity of the multicatalytic proteinase complex (20S proteasome) induces accumulation of ubiquitin-protein conjugates in a neuronal cell. J Neurochem. 1994;63:1578–1581. doi: 10.1046/j.1471-4159.1994.63041578.x. [DOI] [PubMed] [Google Scholar]
- 10.Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG, Green DR. Bax-induced caspase activation and apoptosis via cytochrome c release from mitochondria is inhibitable by Bcl-xL. J Biol Chem. 1999;274:2225–2233. doi: 10.1074/jbc.274.4.2225. [DOI] [PubMed] [Google Scholar]
- 11.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 12.Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA. 1998;95:4386–4391. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ide T, Takada K, Qiu JH, Saito N, Kawahara N, Asai A, Kirino T. Ubiquitin stress response in postischemic hippocampus under nontolerant and tolerant conditions. J Cereb Blood Flow Metab. 1999;19:750–756. doi: 10.1097/00004647-199907000-00004. [DOI] [PubMed] [Google Scholar]
- 14.Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell. 1995;83:129–135. doi: 10.1016/0092-8674(95)90241-4. [DOI] [PubMed] [Google Scholar]
- 15.Kamikubo T, Hayashi T. Changes in proteasome activity following transient ischemia. Neurochem Int. 1996;28:209–212. doi: 10.1016/0197-0186(95)00071-2. [DOI] [PubMed] [Google Scholar]
- 16.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 17.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 18.Lopes UG, Erhardt P, Yao R, Cooper GM. p53-dependent induction of apoptosis by proteasome inhibitors. J Biol Chem. 1997;272:12893–12896. doi: 10.1074/jbc.272.20.12893. [DOI] [PubMed] [Google Scholar]
- 19.Mignotte B, Vayssiere JL. Mitochondria and apoptosis. Eur J Biochem. 1998;252:1–15. doi: 10.1046/j.1432-1327.1998.2520001.x. [DOI] [PubMed] [Google Scholar]
- 20.Miyake S, Makimura M, Kanegae Y, Harada S, Sato Y, Takamori K, Tokuda C, Saito I. Efficient generation of recombinant adenoviruses using adenovirus DNA-terminal protein complex and a cosmid bearing the full-length virus genome. Proc Natl Acad Sci USA. 1996;93:1320–1324. doi: 10.1073/pnas.93.3.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morimoto T, Ide T, Ihara Y, Tamura A, Kirino T. Transient ischemia depletes free ubiquitin in the gerbil hippocampal CA1 neurons. Am J Pathol. 1996;148:249–257. [PMC free article] [PubMed] [Google Scholar]
- 22.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- 23.Pearce DA, Sherman F. Differential ubiquitin-dependent degradation of the yeast apo-cytochrome c isozymes. J Biol Chem. 1997;272:31829–31836. doi: 10.1074/jbc.272.50.31829. [DOI] [PubMed] [Google Scholar]
- 24.Reed JC, Jurgensmeier JM, Matsuyama S. Bcl-2 family proteins and mitochondria. Biochim Biophys Acta. 1998;1366:127–137. doi: 10.1016/s0005-2728(98)00108-x. [DOI] [PubMed] [Google Scholar]
- 25.Reers M, Smiley ST, Mottola-Hartshorn C, Chen A, Lin M, Chen LB. Mitochondrial membrane potential monitored by JC-1 dye. Methods Enzymol. 1995;260:406–417. doi: 10.1016/0076-6879(95)60154-6. [DOI] [PubMed] [Google Scholar]
- 26.Sadoul R, Fernandez PA, Quiquerez AL, Martinou I, Maki M, Schroter M, Becherer JD, Irmler M, Tschopp J, Martinou JC. Involvement of the proteasome in the programmed cell death of NGF-deprived sympathetic neurons. EMBO J. 1996;15:3845–3852. [PMC free article] [PubMed] [Google Scholar]
- 27.Saito Y, Tsubuki S, Ito H, Kawashima S. The structure-function relationship between peptide aldehyde derivatives on initiation of neurite outgrowth in PC12h cells. Neurosci Lett. 1990;120:1–4. doi: 10.1016/0304-3940(90)90153-z. [DOI] [PubMed] [Google Scholar]
- 28.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz LM, Osborne BA. Cell death. Methods Cell Biol. 1995;46:15–18. [PubMed] [Google Scholar]
- 30.Shinoura N, Yoshida Y, Asai A, Kirino T, Hamada H. Relative level of expression of Bax and Bcl-XL determines the cellular fate of apoptosis/necrosis induced by the overexpression of Bax. Oncogene. 1999;18:5703–5713. doi: 10.1038/sj.onc.1202966. [DOI] [PubMed] [Google Scholar]
- 31.Sokolik CW, Cohen RE. Ubiquitin conjugation to cytochromes c. Structure of the yeast iso-1 conjugate and possible recognition determinants. J Biol Chem. 1992;267:1067–1071. [PubMed] [Google Scholar]
- 32.Soldatenkov VA, Dritschilo A. Apoptosis of Ewing's sarcoma cells is accompanied by accumulation of ubiquitinated proteins. Cancer Res. 1997;57:3881–3885. [PubMed] [Google Scholar]
- 33.Susin SA, Zamzami N, Kroemer G. Mitochondria as regulators of apoptosis: doubt no more. Biochim Biophys Acta. 1998;1366:151–165. doi: 10.1016/s0005-2728(98)00110-8. [DOI] [PubMed] [Google Scholar]
- 34.Taglialatela G, Kaufmann JA, Trevino A, Perez-Polo JR. Central nervous system DNA fragmentation induced by the inhibition of nuclear factor kappa B. NeuroReport. 1998;9:489–493. doi: 10.1097/00001756-199802160-00024. [DOI] [PubMed] [Google Scholar]
- 35.Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281:1312–1316. doi: 10.1126/science.281.5381.1312. [DOI] [PubMed] [Google Scholar]
- 36.Traenckner EB, Wilk S, Baeuerle PA. A proteasome inhibitor prevents activation of NF-kappa B and stabilizes a newly phosphorylated form of I kappa B-α that is still bound to NF-kappa B. EMBO J. 1994;13:5433–5441. doi: 10.1002/j.1460-2075.1994.tb06878.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsubuki S, Kawasaki H, Saito Y, Miyashita N, Inomata M, Kawashima S. Purification and characterization of a Z-Leu-Leu-Leu-MCA degrading protease expected to regulate neurite formation: a novel catalytic activity in proteasome. Biochem Biophys Res Commun. 1993;196:1195–1201. doi: 10.1006/bbrc.1993.2378. [DOI] [PubMed] [Google Scholar]
- 38.Tsukahara T, Ishiura S, Sugita H. An ATP-dependent protease and ingensin, the multicatalytic proteinase, in K562 cells. Eur J Biochem. 1988;177:261–266. doi: 10.1111/j.1432-1033.1988.tb14371.x. [DOI] [PubMed] [Google Scholar]
- 39.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]