

Abstract
Spinal cord injury involves a component of glutamate-mediated white matter damage, but the cellular targets, receptors, and ions involved are poorly understood. Mechanisms of excitotoxicity were examined in anin vitro model of isolated spinal dorsal columns. Compound action potentials (CAPs) were irreversibly reduced to 43% of control after 3 hr of 1 mm glutamate exposure at 37°C. AMPA (100 μm) and kainate (500 μm) had similar effects. Antagonists (1 mm kynurenic acid, 10 μm NBQX, 30 μm GYKI52466) were each equally protective against a glutamate challenge, improving mean CAP amplitude to ∼80% versus ∼40% without antagonist. Joro spider toxin (0.75 μm), a selective blocker of Ca2+-permeable AMPA receptors, was also protective to a similar degree. Ca2+-free perfusate virtually abolished glutamate-induced injury (∼90% vs ∼40%). MK-801 (10 μm) had no effect. Glutamate caused damage (assayed immunohistochemically by spectrin breakdown products) to astrocytes and oligodendrocytes consistent with the presence of GluR2/3 and GluR4 in these cells. Myelin was also damaged by glutamate likely mediated by GluR4 receptors detected in this region; however, axon cylinders were unaffected by glutamate, showing no increase in the level of spectrin breakdown. These data may guide the development of more effective treatment for acute spinal cord injury by addressing the additional excitotoxic component of spinal white matter damage.
Keywords: glutamate, excitotoxicity, AMPA receptor, spinal cord white matter, myelin, axon, glia, oligodendrocyte, astrocyte, spectrin, Joro spider toxin, GYKI52466, NBQX, MK-801, kainate
White matter tracts play the very important role of transmitting signals between neurons in the CNS. In the case of the spinal cord, disruption of axonal connections spanning even a small segment can result in severe and widespread disability involving functions distal to the lesion. Previous studies indicate that voltage-gated Na+ channels play an important role in anoxic and traumatic cellular injury of myelinated central axons (Agrawal and Fehlings, 1996; Stutzmann et al., 1996;Imaizumi et al., 1997; Teng and Wrathall, 1997; Stys et al., 1992;Stys, 1998). In addition, more recent reports indicate that CNS white matter injury is also dependent on excitotoxic mechanisms involving glutamate receptors of the AMPA/kainate class (Agrawal and Fehlings, 1997; Wrathall et al., 1997; Rosenberg et al., 1999). However, neither the mechanisms nor the cellular and subcellular targets of glutamate excitotoxicity in CNS white matter are well understood.
Oligodendrocytes and astrocytes have been shown to possess glutamate receptors of AMPA and kainate subtypes (Jensen and Chiu, 1993; Matute and Miledi, 1993; Garcia-Barcina and Matute, 1996; Steinhauser and Gallo, 1996; Agrawal and Fehlings, 1997; Matute et al., 1997). Persistent activation of these receptors causes injury to oligodendrocytes both in cell culture and in vivo (Yoshioka et al., 1995, 1996; Matute et al., 1997; Matute, 1998; McDonald et al., 1998). Similarly, overactivation of AMPA receptors is very toxic and even lethal to astrocytes when receptor desensitization is blocked (David et al., 1996). In this study, using isolated rat spinal cord dorsal column slices, we examined the pharmacological features of excitotoxic injury in in vitro spinal cord white matter at physiological temperature and found that spinal cord white matter is markedly damaged by activation of AMPA receptors. We also explored the subcellular loci of injury and observed that oligodendrocytes, astrocytes, and particularly the myelin sheath are targets for excitotoxic injury, with little evidence of damage to the axon cylinder per se. Our data extend previous observations that blockade of glutamate receptors is protective against white matter trauma or anoxia (Wrathall et al., 1994; Agrawal and Fehlings, 1997; Li et al., 1999) by elucidating which elements may be spared by, and which are unlikely to benefit from, glutamate receptor antagonists.
MATERIALS AND METHODS
Electrophysiology. Experimental procedures have been described previously (Li et al., 1999). Briefly, adult Long–Evans male rats (200–250 gm) were deeply anesthetized with sodium pentobarbital, and a thoracic laminectomy was performed. Rats were then perfused intra-aortically with 500 ml zero-Na+/zero-Ca2+solution containing (in mm): choline chloride 135, choline bicarbonate 26, KCl 1, KH2PO4 1.2, dextrose 10, and EGTA 1.0, bubbled with 95% O2/5%CO2. A 30 mm section of spinal cord was rapidly removed and placed in cold (4–6°C) zero-Na+/zero-Ca2+solution bubbled with 95% O2/5%CO2. The spinal cord section was hemisected, and the dorsal columns were gently excised and placed in an interface recording chamber bathed in normal-Na+/zero-Ca2+solution containing (in mmm): NaCl 126, KCl 3.0, MgSO4 2.0, NaHCO3 26, NaH2PO4 1.25, MgCl2 2.0, dextrose 10, and EGTA 0.5, at room temperature bubbled with 95% O2/5%CO2. The bath temperature was slowly raised to and maintained at 37°C with a temperature controller (Model TC-102, Medical Systems Corp, Greenvale, NY), then the perfusate was switched to artificial CSF (aCSF) containing normal [Ca] (in mm): NaCl 126, KCl 3.0, MgSO4 2.0, NaHCO3 26, NaH2PO4 1.25, CaCl2 2.0, dextrose 10. Control recordings were taken 30 min after the temperature reached 37°C in aCSF.
Propagated compound action potentials (CAPs) were evoked using a bipolar silver wire stimulating electrode placed on one end of the dorsal column and a constant voltage pulse (50 μsec and typically 70 V) delivered once every 30 min. CAPs were recorded extracellularly at the opposite end using large-tipped glass microelectrodes filled with 150 mm NaCl. To allow recording of multiple slices during a single experiment, the stimulation and recording sites were marked with a small amount of neutral red dye to allow accurate repositioning of the electrodes. Evoked CAPs were digitized, stored, and analyzed using WaveTrak software (Stys, 1994). The functional integrity of the dorsal column was quantitated by measuring peak CAP amplitude.
Pharmacological agents.l-glutamic acid, MK801, NBQX (Sigma, St. Louis, MO), kainic acid, kynurenic acid, and Joro spider toxin (JSTX-3 tristrifluoroacetate, RBI) were dissolved directly into aCSF. AMPA, cyclothiazide (Sigma), and NMDA (Tocris) were first dissolved in NaOH (0.1N for AMPA and cyclothiazide; 1N for NMDA), and GYKI52466 (RBI) was dissolved in 0.1N HCI, then added to aCSF to the desired final concentration. The pH of the solutions was maintained at 7.4. Glutamate receptor antagonists (MK801, NBQX, kynurenic acid, GYKI52466, JSTX-3) were applied beginning 30 min before addition of agonist (glutamate, kainate, AMPA).
Immunohistochemistry. We used quantitative confocal immunofluorescence to directly examine which dorsal column white matter elements are damaged by glutamate receptor activation. Rabbit antiserum raised against degenerated myelin basic protein (anti-EP; a generous gift from Dr. Pat McGeer, University of British Columbia) was used to assay damage to the myelin sheath. This antibody stains myelin only in damaged, but not intact, white matter regions (Matsuo et al., 1997). Antiserum against spectrin breakdown products (a generous gift from Dr. Jon Durkin, National Research Council, Ottawa, Canada) was used to examine Ca2+-dependent calpain-mediated degradation of the structural protein spectrin in axons (Isayama et al., 1991; Hewitt et al., 1998) and cell bodies and processes of oligodendrocytes and astrocytes. Spectrin is a ubiquitous cytoskeletal protein that is cleaved by calpain, itself activated by a rise in cytosolic [Ca2+]. Thus calpain-cleaved spectrin breakdown is a reliable indicator of Ca2+-dependent tissue injury in CNS ischemia and trauma (Roberts-Lewis et al., 1994; Buki et al., 1999). After surgical preparation and spinal cord dissection as above, dorsal column slices were incubated in normal aCSF or 1 mmglutamate for 3 hr, and then fixed in 4% paraformaldehyde for 24 hr and cryoprotected for 48 hr in PBS, pH 7.4, containing 20% glycerol at 4°C. Ends were sometimes gently teased to allow imaging of individual axons (see Fig. 5C). Slices were then dissected into smaller pieces (∼3 × 2 × 0.5 mm) and preincubated in 10% Triton X-100 for 30 min, followed by 4% normal goat serum (NGS) with 0.1% Triton X-100, and PBS for blocking for 1 hr at room temperature. After a single quick rinse in PBS, the sections were incubated for 24 hr at 4°C with primary antiserum diluted in 2% NGS with 0.1% Triton X-100, PBS at a concentration of 1:100 for anti-EP, anti-spectrin breakdown, anti-mouse neurofilament 160 (Sigma) (marker for axon cylinders), anti-glial fibrillary acidic protein (GFAP; Boehringer Mannheim, Indianapolis, IN) [astrocytes (Dusart et al., 1991)], and anti-2′,3′-cyclic-nucleotide 3′-phosphodiesterases (CNPase; Promega, Madison, WI), a known cytoplasmic marker for oligodendrocytes and their putative progenitors (Braun et al., 1988; Trapp et al., 1988). Antibodies against GluR1, GluR2/3, GluR4 (Chemicon, Temecula, CA) and GluR2 (Oncogene Research Products, Cambridge, MA) receptor subunits were used at 2–4 μg/ml. After the primary antibody incubation, slices were rinsed three times in PBS for 30 min, then incubated for 1 hr with Alexa 594 goat anti-rabbit (1:200) and Alexa 488 goat anti-mouse (1:400, Molecular Probes) diluted in PBS with 2% NGS and 0.1% Triton X-100. Control sections were incubated with either the primary antisera omitted or secondary antibodies omitted. Images were collected on a Bio-Rad 1024 confocal laser scanning microscope with a 60× oil-immersion objective (Olympus Optical, Tokyo, Japan). A minimum of 10 images collected from two to three sections were examined for each combination of markers, and representative images are shown. Digitized images were analyzed using NIH Image 1.61 (http://rsb.info.nih.gov/nih-image/default.html) on a Macintosh Power PC.
Fig. 5.

Confocal microscopic images of dorsal columns stained immunohistochemically with standard markers (neurofilament, CNPase, and GFAP to identify axon cylinders, oligodendrocytes, and astrocytes, respectively) and markers of cellular injury after a 3 hrin vitro incubation in normal CSF (Ctrlimage column) or 1 mm glutamate (Glut).A–C, Tissue double-stained for neurofilament (green) and degenerated myelin basic protein (red) (see Results). Control images show no myelin damage, whereas exposure to glutamate caused marked injury to the myelin sheath surrounding most axon cylinders (arrowheads). D and Eidentify oligodendrocytes using CNPase staining (green), showing cytoskeletal damage demonstrated by a marked increase in spectrin breakdown products (SBP, red) in glutamate-treated versus control slices. GFAP (green in Fand G) identifies astrocytes that also sustained cytoskeletal damage, as shown by increased spectrin breakdown (red) in cells exposed to glutamate. In contrast, axon cylinders showed no appreciable increase in spectrin breakdown products after a 3 hr glutamate treatment (H,I). Scale bars, 10 μm.
Statistics. All data are expressed as means ± SD. Statistical differences were calculated by ANOVA with Dunnett's test for multiple comparisons, with a common control group in the case of electrophysiological data. Student's t test was used for quantitative immunofluorescence data when only two groups were compared. Reported n values represent number of individual dorsal column slices studied electrophysiologically, or the number of confocal image frames analyzed for fluorescence intensity.
RESULTS
Spinal cord white matter is vulnerable to excitotoxins
In control dorsal column slices perfused with normal aCSF, electrophysiological recording of CAPs showed <5% change in mean peak CAP amplitude during 3 hr of in vitro monitoring at physiological temperature (Fig.1B) (Li et al., 1999). Slices incubated in 1 mm glutamate exhibited CAP amplitudes that were significantly decreased 90 min after the start of glutamate exposure in comparison with time-matched controls. At the end of a 3 hr glutamate application, mean CAP amplitude was reduced to 43 ± 18% of baseline CAP amplitude recorded at time 0 and was significantly reduced compared with time-matched controls (p < 0.01) (Fig.1A,B). Moreover, the glutamate-induced conduction failure did not recover after 1 hr wash with glutamate-free perfusate (Fig.2A,B). These results indicate that glutamate caused functional impairment ofin vitro dorsal column white matter tracts at physiological temperature, which appears irreversible at least in the acute period. Similarly, the non-NMDA receptor agonists kainate (500 μm) and AMPA (100 μm) caused a significant attenuation of peak CAP amplitude to a degree similar to that induced by glutamate (Fig. 2). This functional injury was also irreversible after 1 hr of wash. In contrast, a 3 hr exposure to 500 μm NMDA (with 20 μm glycine and in the absence of Mg2+ to maximize activation of NMDA receptors) had no effect (Fig.1A,B).
Fig. 1.

Effect of glutamate (Glut), kainate (KA), or NMDA on excitability of in vitrodorsal column slices. A, Representative CAP tracings after 180 min of exposure to agonist. B, Bar graph showing quantitative changes in mean peak CAP amplitudes (normalized to 100% at time 0) under various treatment conditions. Controls remained stable for 180 min at 37°C. Exposure to glutamate (1 mm) or kainate (500 μm) significantly reduced CAP amplitude to ∼40% of control after 180 min. In contrast, NMDA (500 μm) had no effect. *p < 0.05, **p < 0.01 compared with time-matched controls.C, Addition of cyclothiazide, an inhibitor of AMPA receptor desensitization, caused a more rapid decay of mean CAP amplitude, although the final degree of injury was not different at the end of 180 min. *p < 0.05, **p< 0.01 compared with time-matched readings in glutamate alone.
Fig. 2.

Effect of glutamate (Glut), kainate (KA), or AMPA followed by wash on excitability ofin vitro dorsal column slices. A, Representative CAP tracings after a 120 min exposure to agonist followed by a 60 min wash. B, Bar graph showing reduction of normalized mean peak CAP amplitude during exposure to glutamate (1 mm), kainate (500 μm), or AMPA (100 μm). Impaired conduction was evident as early as 30 min. All three agents reduced excitability to the same degree after a 120 min exposure. No evidence of recovery was observed after 60 min of wash, indicating irreversible excitotoxic injury to the tissue. *p < 0.05, **p < 0.01 compared with time-matched controls.
Excitotoxicity in dorsal columns is mediated via AMPA receptors and is Ca2+ dependent
Kynurenic acid (1 mm), a broad spectrum blocker of both NMDA and AMPA/kainate receptors, applied 30 min before glutamate exposure, significantly protected the dorsal column slices from glutamate toxicity (Fig.3A,B), supporting the notion that glutamate-induced injury to spinal cord white matter is mediated via ionotropic glutamate receptors. Coapplication of MK-801 (10 μm), a noncompetitive NMDA receptor antagonist, with glutamate did not prevent glutamate-induced damage (47 ± 16 vs 43 ± 18%) (Fig.3A,B), further supporting the notion that NMDA receptors play little if any role in glutamate toxicity. It is likely then that AMPA/kainate receptors mediate excitotoxic injury in spinal cord white matter. Enhancing AMPA receptor activation by reducing desensitization with cyclothiazide (100 μm) (Mosbacher et al., 1994) in addition to glutamate caused a more rapid decline in CAP amplitude over time, although the final degree of injury after 3 hr of exposure was not significantly different from glutamate alone (Fig. 1C). The protective effect of the competitive AMPA/kainate receptor antagonist NBQX (10 μm) (Sheardown et al., 1990) [CAP reduction to 79 ± 23 vs 49 ± 17% without NBQX,p < 0.01 (Fig. 3C)] lends further support to the idea that glutamate-induced excitotoxicity in dorsal column white matter occurs primarily via overactivation of this subtype of ionotropic glutamate receptors. Figure 4illustrates the protective effect of GYKI52466 (30 μm), a specific AMPA receptor antagonist (Paternain et al., 1995). This agent provided robust neuroprotection against AMPA/kainate receptor activation (CAP reduction to 82 ± 10 vs 40 ± 19% without antagonist, p < 0.01) to a degree similar to that observed with the broader spectrum blockers NBQX and kynurenic acid. Taken together, these findings indicate that AMPA, rather than NMDA or kainate receptors, are most responsible for glutamate-mediated dorsal column injury.
Fig. 3.

Effects of ionotropic glutamate receptor antagonists on glutamate toxicity in dorsal columns. A, Representative CAP tracings after a 180 min exposure to glutamate or glutamate + antagonist. B, Glutamate (1 mm) alone causes significant functional injury to isolated dorsal columns, as shown by the reduction of normalized mean CAP amplitudes (white bars). The broad spectrum ionotropic glutamate receptor antagonist kynurenic acid (KYN, 1 mm) or Joro spider toxin (JSTX, 0.75 μm), a selective inhibitor of Ca2+-permeable AMPA receptors, each significantly protected the tissue from glutamate toxicity. Blocking of NMDA receptors with MK-801 (10 μm) was not protective.C, The AMPA/kainate receptor blocker NBQX (10 μm) was also protective against glutamate and the desensitization inhibitor cyclothiazide (CTZ, 100 μm). *p < 0.05, **p < 0.01 compared with time-matched readings without antagonist.
Fig. 4.

Protective effects of zero-Ca2+perfusate and selective AMPA receptor blockade against kainate toxicity. A, Representative CAP tracings after a 180 min exposure to kainate (500 μm), kainate in zero-Ca2+ (+100 μm EGTA), or kainate + GYKI52466 (30 μm). B, Bar graph of normalized mean CAP amplitudes showing significant protection against kainate toxicity by removal of Ca2+ from the perfusate or application of the selective AMPA receptor antagonist GYKI52466. These data, together with those of Figure 3, indicate that dorsal column excitotoxicity is largely dependent on influx of extracellular Ca2+ triggered by activation of AMPA receptors. *p < 0.05, **p < 0.01 compared with time-matched readings in 500 μmkainate and normal [Ca2+].
Certain AMPA receptors have been shown to display substantial permeability to Ca2+ (Ozawa et al., 1998;Dingledine et al., 1999), and influx of this divalent cation through these receptors contributes to neuronal death in several pathophysiological conditions, such as anoxia/ischemia and trauma (Pellegrini-Giampietro et al., 1997). Removal of Ca2+ from the perfusate or blocking Ca2+ influx with Joro spider toxin (0.75 μm), a blocker of Ca2+-permeable AMPA receptors (Iino et al., 1996), protected dorsal columns from excitotoxicity (Figs. 3, 4) to virtually the same degree as the AMPA antagonist GYKI52466 (mean CAP amplitude reduction to 89 ± 16% in Ca2+-free, 83 ± 4.6% in JSTX-3 vs 40 ± 18% in normal Ca2+-containing aCSF, p < 0.01). These observations suggest that Ca2+ influx through Ca2+-permeable AMPA receptors plays a major role in the genesis of excitotoxic damage in dorsal columns, probably by activation of Ca2+-dependent degradative pathways such as calpains and phospholipases.
Glutamate-induced cellular damage is localized to myelin and glia
The previous results provide pharmacological evidence that glutamate, kainate, and AMPA are toxic to spinal cord white matter, and this excitotoxicity is associated with Ca2+ influx principally through AMPA receptors. Physiological studies, although providing reliable functional measures, do not give detailed information about the subcellular loci of injury. We used immunocytochemistry with specific antiserum raised against degenerated myelin basic protein (Matsuo et al., 1997) and calpain-cleaved breakdown products of the structural protein spectrin (Hewitt et al., 1998) to examine which white matter elements are vulnerable to excitotoxins. Figure5 shows representative confocal images of dorsal column white matter incubated with normal aCSF (left panels) or 1 mm glutamate for 3 hr (right panels). Double staining with neurofilament (green) to outline axon cylinders, and degenerated myelin basic protein (red), showed that a 3 hr glutamate exposure induced significant damage to the myelin sheath (Fig.5B,C, arrowheads,red signal surrounding axon cylinders). In contrast, time-matched control sections without glutamate displayed virtually no myelin damage (Fig. 5A).
Immunostaining for spectrin breakdown products (SBPs) allows detection of structural damage to cytoskeleton in axon cylinders and glia. Astrocytes and oligodendrocytes were distinguished using GFAP (Dusart et al., 1991) and CNPase (Trapp et al., 1998), respectively. Double staining for cellular marker proteins (i.e., CNPase, GFAP, and neurofilament) and SBP revealed that control sections displayed low levels of SBP in the cell bodies and processes of oligodendrocytes, axon cylinders, and astrocytes, best seen in the separated gray scale images in Figure6C,G,K. These degenerated spectrin products may represent normal basal turnover of structural proteins, induction of mild injury by the procedure of tissue isolation and in vitro incubation, and mild nonspecific staining by the antiserum. In contrast, a 3 hr exposure to 1 mm glutamate induced significant damage to oligodendroglial and astrocytic cell bodies and processes, as shown by the red staining for SBP or yellow signal colocalizing SBP and CNPase/GFAP (Fig. 5E,G). Notably, there was no detectable rise of SBP in axoplasm after glutamate treatment (Figs. 5I, 6H). Figure 6illustrates separated gray scale images of sections double-stained for standard markers and SBP in control and glutamate-treated groups. The increase in SBP fluorescence in oligodendrocytes and astrocytes in the glutamate-treated slices is shown quantitatively in the accompanying bar graphs, with significant increases observed in cytosolic regions of oligodendrocytes (48 ± 9 in glutamate group vs 38 ± 7 in controls, p < 0.01) (Fig. 6, left bar graph) and astrocytes (58 ± 11 vs 31 ± 2 in controls,p < 0.001) (Fig. 6, right bar graph). In contrast, glutamate did not induce any detectable structural injury, as estimated by spectrin breakdown, in axon cylinders even after 3 hr of exposure; SBP fluorescence was identical in treated versus control sections (36 ± 2 vs 36 ± 1, respectively, p= 0.879) (Fig. 6, middle bar graph).
Fig. 6.

Quantitative estimates of structural injury in dorsal columns exposed to 3 hr of 1 mm glutamate in vitro. Immunohistochemistry showing separated gray scale images (e.g., A and C are two channels from the same image) of sections double-stained with standard markers (neurofilament, CNPase, and GFAP) and spectrin breakdown products, in control and glutamate-treated tissue. Regions of interest outlined by each of the standard markers were analyzed for fluorescence intensity from the spectrin breakdown channel, thus producing a semiquantitative estimate of spectrin degradation in oligodendrocytes, axon cylinders, and astrocytes. Mean spectrin breakdown fluorescence from each of these three white matter elements is plotted in the bar graphs. Control sections show detectable levels of spectrin breakdown in all three cell types (C, G,K, and bar graphs). Glutamate significantly increased the levels of spectrin degradation in oligodendrocytes and especially astrocytes (D,L) but had no effect on axon cylinders (compareG and H; middle bar graph). n values represent number of image pairs analyzed for each bar. Scale bars, 10 μm.
AMPA receptors are expressed in oligodendrocytes, astrocytes, and myelin of spinal cord white matter
Our data suggest that overactivation of Ca2+-permeable AMPA receptors by excitotoxins in dorsal column white matter results in significant functional and structural damage to various cellular components including oligodendrocytes, astrocytes, and the myelin sheath. Figure7 shows representative immunohistochemical sections stained for different AMPA receptor subunits. GluR4 was the most ubiquitous subunit, present in oligodendrocytes (Fig. 7G,H) and astrocytes (F), with astrocytic processes associated with capillaries being particularly rich in this receptor subunit. Axoplasm also contained GluR4 (Fig. 7E), and interestingly, the myelin sheath displayed noticeable GluR4 label as well (H). The weaker GluR4 signal in the axon cylinder inH is obscured by the much stronger neurofilament label, with a strong green MBP signal obscuring GluR4 in the myelin inE; separated images (data not shown), and particularly counterstains that are not overlapping (such as MBP in Fig.7E and neurofilament in H), clearly indicate the presence of this subunit in both the axoplasm and myelin. The latter was devoid of isoforms other than GluR4. With the exception of astrocytes, GluR1 was largely absent to any significant degree in dorsal column white matter elements. The combination of GluR2 and GluR2/3 antisera allowed us to distinguish between these two subunits. Given that GluR2 was not seen in glia, myelin, or axon cylinders (results not shown), GluR2/3 positivity in oligodendrocytes and astrocytes (Fig. 7C,D) indicates the presence of GluR3 in these cells. To ensure that the total absence of GluR2 label was not artifactual, positive controls were performed in spinal gray matter, known to contain GluR2-positive neurons (Grossman et al., 1999), where unequivocal neuronal staining was observed (data not shown). GluR1 was weakly present in astrocytes and possibly in axon cylinders as well [faint signal outside of GFAP-positive regions in Fig. 7B; supported by neurofilament double staining (data not shown)]. Table 1 contains a summary of AMPA receptor subunit distributions in dorsal columns observed in our experiments.
Fig. 7.
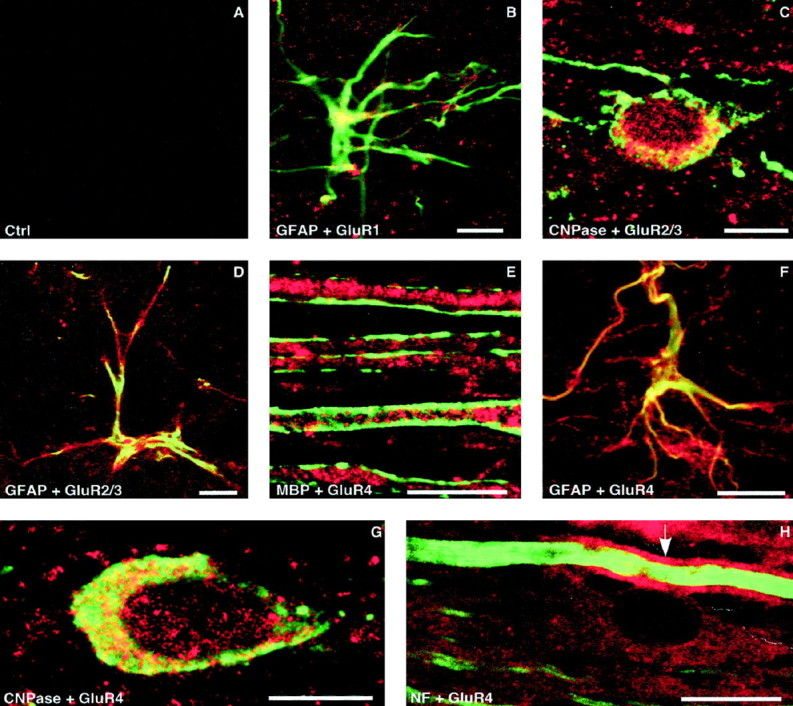
Immunohistochemical localization of AMPA receptor subunits in dorsal columns. Tissue was double-stained with GluR1, GluR2/3, or GluR4 (red), and a standard marker (green). GluR2 staining was consistently absent in dorsal column white matter (data not shown). A, Control section with primary antibody omitted. B, GluR1 was only faintly detected in astrocytes as shown by the yellow hue indicating colocalization of the GluR1 and GFAP signals.C, D, Moderate levels of GluR2/3 were observed in oligodendrocytes and astrocytes, respectively, representing GluR3, given that GluR2 was absent. E, Myelin basic protein label (green) outlines myelinated dorsal column axons whose axoplasm displays strong immunoreactivity to GluR4. GluR4 was also detected in astrocytes (F) and oligodendrocytes (G). H, Neurofilament stain (green) delineates an axon cylinder that is surrounded by easily detectable GluR4 signal (red) in the myelin sheath (arrow). A GluR4-positive oligodendrocyte is seen adjacent to this fiber. Scale bars, 10 μm.
Table 1.
Distribution of AMPA receptor subunits in dorsal columns
| GluR1 | GluR2 | GluR2/3 | GluR4 | |
|---|---|---|---|---|
| Axoplasm | −/+ | − | ++ | +++ |
| Myelin | − | − | − | ++ |
| Oligo | − | − | ++ | ++ |
| Astrocyte | + | − | ++ | +++1-a |
Absent, −; weak, +; moderate, ++; strong, +++.
Especially pronounced around capillaries.
DISCUSSION
White matter tracts of the mammalian spinal cord serve the critical function of conducting signal traffic to and from the brain. Traumatic and ischemic damage to the cord often results in major clinical disability, most of which is attributable to dysfunction of white matter tracts rather than gray matter regions (Blight and Decrescito, 1986; Noble and Wrathall, 1989). Recent reports suggest, perhaps surprisingly, that glutamate-dependent mechanisms appear to play a role in this tissue that is devoid of synaptic elements. However, the source of glutamate, release mechanism, and cellular targets are not known. In this study, using an isolated in vitro spinal white matter tract, we studied the glutamate receptors involved, the cellular targets of excitotoxicity, and the ionic dependence. We observed that overactivation of mainly the AMPA receptor subclass by glutamate or related agonists caused significant irreversible functional injury. Excitatory amino acids were directly toxic to dorsal column slices, and this damage was not dependent on adjacent gray matter or vascular supply, which were absent in our model.
Electrophysiological recordings showed that CAP amplitude was irreversibly reduced to less than half of control after 2–3 hr of agonist exposure at physiological temperature, with conduction impairment appearing as early as 30 min when AMPA receptor desensitization was blocked (Fig. 1). In an earlier study, Agrawal and Fehlings (1997) reported that both AMPA and kainate reduced the amplitude of CAPs recorded from in vitro spinal white matter, but the degree of conduction impairment was much more modest compared to our results and was reversible, in contrast to our findings in which no recovery was detectable after wash of the excitotoxin (Fig.2). We believe that the elevated temperature at which we conducted our experiments induced significantly more excitotoxic injury compared with the hypothermic conditions used previously, indicating that glutamate-dependent injury may be far more important in white matter than previously appreciated.
Using selective agonists and antagonists, we concluded that the AMPA, and not the NMDA, receptor class is largely responsible for glutamate-mediated injury to spinal cord white matter. This is consistent with previous observations of neuroprotection afforded by AMPA receptor antagonists in models of in vitro and in vivo spinal cord injury and anoxia (Wrathall et al., 1994; Agrawal and Fehlings, 1997; Li et al., 1999), but is in contrast to neurons, where both classes of receptors are known to cause damage (Choi and Hartley, 1993; Choi, 1994). Our data also indicate that the AMPA receptor-mediated injury is highly dependent on influx of extracellular Ca2+: removal of this ion from the perfusate allowed dorsal column slices to withstand a 3 hr kainate challenge with virtually no noticeable reduction in excitability (Fig.4). Together with the highly protective effect of Joro spider toxin, a selective blocker of Ca2+-permeable AMPA receptors (Iino et al., 1996), our data strongly suggest that Ca2+ influx directly through AMPA receptors plays an important role. However, alternate Ca2+ entry routes cannot be ruled out. For instance, AMPA receptor-mediated cellular Na+ loading and depolarization may secondarily induce Ca2+ entry through voltage-gated Ca2+ channels or reverse Na+–Ca2+exchange (Hack and Balazs, 1995; Liu et al., 1997).
Electrophysiological measurements yield quantitative information about the overall functional state of tissue but do not provide insight into which white matter elements are vulnerable to glutamate-mediated toxicity. We used confocal microscopy and immunohistochemistry with antisera raised against damaged but not intact structural proteins, including myelin basic protein and spectrin. As shown in Figures 5 and6, glutamate caused significant structural injury to glial elements, with no detectable effect on axon cylinders. Thus only myelin, astrocytes, and oligodendrocytes appeared to be vulnerable to excitotoxins. Our findings are consistent with recent reports showing that both oligodendrocytes and astrocytes are damaged by overactivation of AMPA/kainate receptors. AMPA or kainate causes rapid cell death in cultured oligodendrocytes in a Ca2+-dependent manner (Matute et al., 1997; McDonald et al., 1998). Similarly, application of AMPA or kainate to white matter in vivo causes widespread death of oligodendrocytes (Matute et al., 1997; Matute, 1998). Cultured astrocytes are relatively resistant to excitatory amino acid toxins (Choi et al., 1987; Koh et al., 1990). However, Ca2+-permeable AMPA and kainate subunits of glutamate receptors have been identified in these cells (Burnashev et al., 1992; Seifert and Steinhauser, 1995; Garcia-Barcina and Matute, 1996; Agrawal and Fehlings, 1997), and damage can be induced when receptor desensitization is pharmacologically blocked (David et al., 1996). Our results demonstrate that damage to astrocytes can be detected after a 3 hr exposure to glutamate even without inhibition of receptor desensitization (Figs. 5, 6). Previous reports indicate incompletely desensitizing responses in cultured astrocytes to glutamate application (Blankenfeld et al., 1995). It is therefore likely that the residual permeability induced by glutamate in our tissue was sufficient to induce injury after a 3 hr exposure. Although we believe that AMPA receptors play a prominent role, a component of kainate receptor activation in astrocytic and/or oligodendroglial injury cannot be excluded, because we did not examine the effects of agonists selective for this receptor subtype.
An interesting observation from our studies was glutamate-induced structural damage to the myelin sheath itself (Fig.5B,C). Myelin plays a critical role in sustaining saltatory conduction, with damage to the sheath resulting in slowing or complete failure of conduction (Waxman, 1992). In this study, excitotoxins caused impairment of conduction beginning as early as 30 min after drug application, in the absence of any detectable injury to the axon cylinder per se. This raises the strong possibility that functional white matter impairment secondary to excitotoxic exposure is largely, if not exclusively, caused by damage to glial elements, particularly the myelin sheath, given the rapidity of the effect on conduction. Although we cannot exclude the possibility that myelin damage was secondary to injury of the parent oligodendrocyte, the rapid disturbance of dorsal column excitability by glutamate suggests that excitotoxins exerted a direct effect on the myelin sheath. This hypothesis is further supported by the finding that GluR4, but not GluR2, receptor subunits are present in myelin, indicating that the sheath itself may respond to ambient glutamate, and if excessively stimulated they may suffer a toxic Ca2+influx directly through Ca2+-permeable AMPA receptors.
As summarized in Table 1, we also found GluR3 and GluR4 subunits in astrocytes, oligodendrocytes, and axoplasm. Our findings in glia are consistent with observations from other groups, where PCR studies and immunocytochemistry have directly demonstrated the expression of AMPA/kainate receptors in white matter glia (Jensen and Chiu, 1993;Matute and Miledi, 1993; Patneau et al., 1994; Garcia-Barcina and Matute, 1996; Steinhauser and Gallo, 1996; Agrawal and Fehlings, 1997;Matute et al., 1997; Matute, 1998; McDonald et al., 1998). For example, oligodendrocytes from optic nerve express GluR3 and GluR4 subunits of the AMPA receptor and GluR6–7 and KA1–2 subunits of the kainate receptor, but not GluR2 (Matute et al., 1997), which is congruent with our findings of positive immunoreactivity for GluR4 and GluR2/3, but not GluR2, in this cell. The faint GluR1 immunoreactivity seen in dorsal column astrocytes is consistent with localization exclusively to this cell type in bovine corpus callosum (Garcia-Barcina and Matute, 1998). The significance of GluR2/3 and GluR4 immunoreactivity in axoplasm is unclear, and it is possible that subunits are being transported for insertion at the terminals; however, we cannot exclude the possibility that some receptors are by inference present in the axolemma. If so, unlike glia and myelin, the density must be low enough so that activation of these receptors did not cause any detectable damage to the axon cylinder.
In summary, we have shown that isolated spinal dorsal columns are vulnerable to irreversible excitotoxic injury that is dependent on AMPA receptor activation and Ca2+ influx from the extracellular space. The physiological role of glutamate receptors in white matter is not known but may involve activity-dependent signaling between axons and surrounding glia (Chiu and Kriegler, 1994). Our finding of AMPA receptor subunits directly on myelin raises the intriguing possibility that axonal activity might directly modulate the metabolism and structure of the sheath itself, independently of or in addition to effects from the parent soma. During anoxia/ischemia or trauma, this mechanism, overdriven by ionic deregulation, may lead to irreversible injury. We have recently shown that glutamate is released from axon cylinders via reverse Na+-dependent glutamate transport duringin vitro anoxia and trauma, causing disruption of the myelin sheath in an AMPA receptor-dependent manner (Li et al., 1999). It is very likely that studies demonstrating neuroprotective effects of AMPA/kainate antagonists in models of spinal cord injury conferred functional protection by sparing glia and myelin (Wrathall et al., 1994; Agrawal and Fehlings, 1997; Wrathall et al., 1997); this is supported by a recent morphological study showing sparing of glial elements by the AMPA/kainate antagonist NBQX after spinal cord injury (Rosenberg et al., 1999). An additional important injury mechanism in myelinated axons also involves axoplasmic Ca2+ overload mediated by reverse Na+–Ca2+exchange (Imaizumi et al., 1997; Stys, 1998; Stys and LoPachin, 1998). Taken together, it is possible that injury to central myelinated axons proceeds along two parallel routes, with Ca2+ influx through reverse Na+–Ca2+exchange causing damage to the axon cylinder, whereas glia and myelin suffer Ca2+-dependent damage that is mediated instead by an excitotoxic mechanism.
Footnotes
This work was supported by a grant from the Ontario Neurotrauma Foundation (ONRO-31). S.L. is supported by a scholarship from the Natural Sciences and Engineering Research Council of Canada. P.K.S. is supported by a Career Investigator Award from the Heart and Stroke Foundation of Ontario. We thank Elaine Coderre for assistance with immunohistochemistry.
Correspondence should be addressed to Dr. Peter K. Stys, Loeb Health Research Institute, Division of Neuroscience, 725 Parkdale Avenue, Ottawa, Ontario, Canada K1Y 4K9. E-mail:pstys@lri.ca.
REFERENCES
- 1.Agrawal SK, Fehlings MG. Mechanisms of secondary injury to spinal cord axons in vitro: role of Na+, Na+-K+-ATPase, the Na+-H+ exchanger, and the Na+-Ca2+ exchanger. J Neurosci. 1996;16:545–552. doi: 10.1523/JNEUROSCI.16-02-00545.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agrawal SK, Fehlings MG. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J Neurosci. 1997;17:1055–1063. doi: 10.1523/JNEUROSCI.17-03-01055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blankenfeld GV, Enkvist K, Kettenman H. Gamma-aminobutyric acid and glutamate receptors. In: Kettenman H, Ransom BR, editors. Neuroglia. Oxford; New York: 1995. pp. 335–345. [Google Scholar]
- 4.Blight AR, Decrescito V. Morphometric analysis of experimental spinal cord injury in the cat: the relation of injury intensity to survival of myelinated axons. Neuroscience. 1986;19:321–41. doi: 10.1016/0306-4522(86)90025-4. [DOI] [PubMed] [Google Scholar]
- 5.Braun PE, Sandillon F, Edwards A, Matthieu JM, Privat A. Immunocytochemical localization by electron microscopy of 2′3′-cyclic nucleotide 3′-phosphodiesterase in developing oligodendrocytes of normal and mutant brain. J Neurosci. 1988;8:3057–3066. doi: 10.1523/JNEUROSCI.08-08-03057.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buki A, Siman R, Trojanowski JQ, Povlishock JT. The role of calpain-mediated spectrin proteolysis in traumatically induced axonal injury. J Neuropathol Exp Neurol. 1999;58:365–375. doi: 10.1097/00005072-199904000-00007. [DOI] [PubMed] [Google Scholar]
- 7.Burnashev N, Khodorova A, Jonas P, Helm PJ, Wisden W, Monyer H, Seeburg PH, Sakmann B. Calcium-permeable AMPA-kainate receptors in fusiform cerebellar glial cells. Science. 1992;256:1566–1570. doi: 10.1126/science.1317970. [DOI] [PubMed] [Google Scholar]
- 8.Chiu SY, Kriegler S. Neurotransmitter-mediated signaling between axons and glial cells. Glia. 1994;11:191–200. doi: 10.1002/glia.440110213. [DOI] [PubMed] [Google Scholar]
- 9.Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- 10.Choi DW, Hartley DM. Calcium and glutamate-induced cortical neuronal death. Res Publ Assoc Res Nerv Ment Dis. 1993;71:23–34. [PubMed] [Google Scholar]
- 11.Choi DW, Maulucci GM, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–368. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.David JC, Yamada KA, Bagwe MR, Goldberg MP. AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci. 1996;16:200–209. doi: 10.1523/JNEUROSCI.16-01-00200.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 14.Dusart I, Marty S, Peschanski M. Glial changes following an excitotoxic lesion in the CNS—II. Astrocytes. Neuroscience. 1991;45:541–549. doi: 10.1016/0306-4522(91)90269-t. [DOI] [PubMed] [Google Scholar]
- 15.Garcia-Barcina JM, Matute C. Expression of kainate-selective glutamate receptor subunits in glial cells of the adult bovine white matter. Eur J Neurosci. 1996;8:2379–2387. doi: 10.1111/j.1460-9568.1996.tb01201.x. [DOI] [PubMed] [Google Scholar]
- 16.Garcia-Barcina JM, Matute C. AMPA-selective glutamate receptor subunits in glial cells of the adult bovine white matter. Brain Res Mol Brain Res. 1998;53:270–276. doi: 10.1016/s0169-328x(97)00318-5. [DOI] [PubMed] [Google Scholar]
- 17.Grossman SD, Wolfe BB, Yasuda RP, Wrathall JR. Alterations in AMPA receptor subunit expression after experimental spinal cord contusion injury. J Neurosci. 1999;19:5711–5720. doi: 10.1523/JNEUROSCI.19-14-05711.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hack N, Balazs R. Properties of AMPA receptors expressed in rat cerebellar granule cell cultures: Ca2+ influx studies. J Neurochem. 1995;65:1077–1084. doi: 10.1046/j.1471-4159.1995.65031077.x. [DOI] [PubMed] [Google Scholar]
- 19.Hewitt KE, Lesiuk HJ, Tauskela JS, Morley P, Durkin JP. Selective coupling of μ-calpain activation with the NMDA receptor is independent of translocation and autolysis in primary cortical neurons. J Neurosci Res. 1998;54:223–232. doi: 10.1002/(SICI)1097-4547(19981015)54:2<223::AID-JNR10>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 20.Iino M, Koike M, Isa T, Ozawa S. Voltage-dependent blockage of Ca2+-permeable AMPA receptors by joro spider toxin in cultured rat hippocampal neurones. J Physiol (Lond) 1996;496:431–437. doi: 10.1113/jphysiol.1996.sp021696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Imaizumi T, Kocsis JD, Waxman SG. Anoxic injury in the rat spinal cord: pharmacological evidence for multiple steps in Ca2+-dependent injury of the dorsal columns. J Neurotrauma. 1997;14:299–311. doi: 10.1089/neu.1997.14.299. [DOI] [PubMed] [Google Scholar]
- 22.Isayama T, Goodman SR, Zagon IS. Spectrin isoforms in the mammalian retina. J Neurosci. 1991;11:3531–3538. doi: 10.1523/JNEUROSCI.11-11-03531.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen AM, Chiu SY. Expression of glutamate receptor genes in white matter: developing and adult rat optic nerve. J Neurosci. 1993;13:1664–1675. doi: 10.1523/JNEUROSCI.13-04-01664.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koh JY, Goldberg MP, Hartley DM, Choi DW. Non-NMDA receptor-mediated neurotoxicity in cortical culture. J Neurosci. 1990;10:693–705. doi: 10.1523/JNEUROSCI.10-02-00693.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, Mealing GA, Morley P, Stys PK. Novel injury mechanism in anoxia and trauma of spinal cord white matter: glutamate release via reverse Na(+)-dependent glutamate transport. J Neurosci. 1999;19:RC16. doi: 10.1523/JNEUROSCI.19-14-j0002.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu HN, Molina-Holgado E, Almazan G. Glutamate-stimulated production of inositol phosphates is mediated by Ca2+ influx in oligodendrocyte progenitors. Eur J Pharmacol. 1997;338:277–287. doi: 10.1016/s0014-2999(97)81931-0. [DOI] [PubMed] [Google Scholar]
- 27.Matsuo A, Lee GC, Terai K, Takami K, Hickey WF, McGeer EG, McGeer PL. Unmasking of an unusual myelin basic protein epitope during the process of myelin degeneration in humans: a potential mechanism for the generation of autoantigens. Am J Pathol. 1997;150:1253–1266. [PMC free article] [PubMed] [Google Scholar]
- 28.Matute C. Characteristics of acute and chronic kainate excitotoxic damage to the optic nerve. Proc Natl Acad Sci USA. 1998;95:10229–10234. doi: 10.1073/pnas.95.17.10229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matute C, Miledi R. Neurotransmitter receptors and voltage-dependent Ca2+ channels encoded by mRNA from the adult corpus callosum. Proc Natl Acad Sci USA. 1993;90:3270–3274. doi: 10.1073/pnas.90.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matute C, Sanchez-Gomez MV, Martinez-Millan L, Miledi R. Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc Natl Acad Sci USA. 1997;94:8830–8835. doi: 10.1073/pnas.94.16.8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4:291–297. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- 32.Mosbacher J, Schoepfer R, Monyer H, Burnashev N, Seeburg PH, Ruppersberg JP. A molecular determinant for submillisecond desensitization in glutamate receptors. Science. 1994;266:1059–1062. doi: 10.1126/science.7973663. [DOI] [PubMed] [Google Scholar]
- 33.Noble LJ, Wrathall JR. Correlative analyses of lesion development and functional status after graded spinal cord contusive injuries in the rat. Exp Neurol. 1989;103:34–40. doi: 10.1016/0014-4886(89)90182-9. [DOI] [PubMed] [Google Scholar]
- 34.Ozawa S, Kamiya H, Tsuzuki K. Glutamate receptors in the mammalian central nervous system. Prog Neurobiol. 1998;54:581–618. doi: 10.1016/s0301-0082(97)00085-3. [DOI] [PubMed] [Google Scholar]
- 35.Paternain AV, Morales M, Lerma J. Selective antagonism of AMPA receptors unmasks kainate receptor-mediated responses in hippocampal neurons. Neuron. 1995;14:185–189. doi: 10.1016/0896-6273(95)90253-8. [DOI] [PubMed] [Google Scholar]
- 36.Patneau DK, Wright PW, Winters C, Mayer ML, Gallo V. Glial cells of the oligodendrocyte lineage express both kainate- and AMPA-preferring subtypes of glutamate receptor. Neuron. 1994;12:357–371. doi: 10.1016/0896-6273(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 37.Pellegrini-Giampietro DE, Gorter JA, Bennett MV, Zukin RS. The GluR2 (GluR-B) hypothesis: Ca(2+)-permeable AMPA receptors in neurological disorders. Trends Neurosci. 1997;20:464–470. doi: 10.1016/s0166-2236(97)01100-4. [DOI] [PubMed] [Google Scholar]
- 38.Roberts-Lewis JM, Savage MJ, Marcy VR, Pinsker LR, Siman R. Immunolocalization of calpain I-mediated spectrin degradation to vulnerable neurons in the ischemic gerbil brain. J Neurosci. 1994;14:3934–3944. doi: 10.1523/JNEUROSCI.14-06-03934.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenberg LJ, Teng YD, Wrathall JR. 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(f)quinoxaline reduces glial loss and acute white matter pathology after experimental spinal cord contusion. J Neurosci. 1999;19:464–475. doi: 10.1523/JNEUROSCI.19-01-00464.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seifert G, Steinhauser C. Glial cells in the mouse hippocampus express AMPA receptors with an intermediate Ca2+ permeability. Eur J Neurosci. 1995;7:1872–1881. doi: 10.1111/j.1460-9568.1995.tb00708.x. [DOI] [PubMed] [Google Scholar]
- 41.Sheardown MJ, Neilsen EO, Hansen AJ, Jacobsen P, Honoré T. 2,3-Dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science. 1990;247:571–574. doi: 10.1126/science.2154034. [DOI] [PubMed] [Google Scholar]
- 42.Steinhauser C, Gallo V. News on glutamate receptors in glial cells. Trends Neurosci. 1996;19:339–345. doi: 10.1016/0166-2236(96)10043-6. [DOI] [PubMed] [Google Scholar]
- 43.Stutzmann JM, Pratt J, Boraud T, Gross C. The effect of riluzole on post-traumatic spinal cord injury in the rat. NeuroReport. 1996;7:387–392. doi: 10.1097/00001756-199601310-00003. [DOI] [PubMed] [Google Scholar]
- 44.Stys PK. WaveTrak: a data acquisition system and waveform database for the Macintosh. Sci Comput Autom. 1994;10:19–24. [Google Scholar]
- 45.Stys PK. Anoxic and ischemic injury of myelinated axons in CNS white matter: from mechanistic concepts to therapeutics. J Cereb Blood Flow Metab. 1998;18:2–25. doi: 10.1097/00004647-199801000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Stys PK, LoPachin RM. Mechanisms of ion flux in anoxic myelinated CNS axons. Neuroscience. 1998;82:21–32. doi: 10.1016/s0306-4522(97)00230-3. [DOI] [PubMed] [Google Scholar]
- 47.Stys PK, Waxman SG, Ransom BR. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+-Ca2+ exchanger. J Neurosci. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Teng YD, Wrathall JR. Local blockade of sodium channels by tetrodotoxin ameliorates tissue loss and long-term functional deficits resulting from experimental spinal cord injury. J Neurosci. 1997;17:4359–4366. doi: 10.1523/JNEUROSCI.17-11-04359.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Trapp BD, Bernier L, Andrews SB, Colman DR. Cellular and subcellular distribution of 2′,3′-cyclic nucleotide 3′-phosphodiesterase and its mRNA in the rat central nervous system. J Neurochem. 1988;51:859–868. doi: 10.1111/j.1471-4159.1988.tb01822.x. [DOI] [PubMed] [Google Scholar]
- 50.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 51.Waxman SG. Demyelination in spinal cord injury and multiple sclerosis: what can we do to enhance functional recovery? J Neurotrauma. 1992;9[Suppl 1]:S105–117. [PubMed] [Google Scholar]
- 52.Wrathall JR, Choiniere D, Teng YD. Dose-dependent reduction of tissue loss and functional impairment after spinal cord trauma with the AMPA/kainate antagonist NBQX. J Neurosci. 1994;14:6598–6607. doi: 10.1523/JNEUROSCI.14-11-06598.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wrathall JR, Teng YD, Marriott R. Delayed antagonism of AMPA/kainate receptors reduces long-term functional deficits resulting from spinal cord trauma. Exp Neurol. 1997;145:565–573. doi: 10.1006/exnr.1997.6506. [DOI] [PubMed] [Google Scholar]
- 54.Yoshioka A, Hardy M, Younkin DP, Grinspan JB, Stern JL, Pleasure D. Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors mediate excitotoxicity in the oligodendroglial lineage. J Neurochem. 1995;64:2442–2448. doi: 10.1046/j.1471-4159.1995.64062442.x. [DOI] [PubMed] [Google Scholar]
- 55.Yoshioka A, Bacskai B, Pleasure D. Pathophysiology of oligodendroglial excitotoxicity. J Neurosci Res. 1996;46:427–437. doi: 10.1002/(SICI)1097-4547(19961115)46:4<427::AID-JNR4>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]