

Abstract
Ca2+ entry into nerve terminals through clusters of voltage-dependent Ca2+ channels (VDCCs) at active zones creates a microdomain of elevated intracellular free Ca2+ concentration ([Ca2+]i) that stimulates exocytosis. We show that this VDCC-mediated [Ca2+]i elevation has no specific role in stimulating endocytosis but can inhibit endocytosis evoked by three different methods in isolated mammalian nerve terminals. The inhibition can be relieved by using either VDCC antagonists or fast, but not slow, binding intracellular Ca2+ chelators. The Ca2+-dependent inhibition of endocytosis is mimickedin vitro by a low-affinity inhibition of dynamin I vesiculation of phospholipids. Increased [Ca2+]i also inhibits dynamin II GTPase activity and receptor-mediated endocytosis in non-neuronal cells. VDCC-meditated Ca2+ entry inhibits dynamin-mediated endocytosis at the active zone and provides neurons with a mechanism to clear recycling vesicles to nonactive zone regions during periods of high activity.
Keywords: endocytosis, exocytosis, FM2–10, dynamin, calcium, nerve terminal
Synaptic transmission relies on the stimulation of neurotransmitter release by exocytosis of synaptic vesicles and subsequent vesicle retrieval by endocytosis. Exocytosis and endocytosis are assumed to be intimately coupled in the CNS (Neher, 1998) because synaptic transmission can be maintained by a very small pool of recycling synaptic vesicles (Ryan, 1996). One possible mechanism by which the two processes could be coupled is influx of extracellular Ca2+ through voltage-dependent Ca2+ channels (VDCCs). Exocytosis is stimulated by VDCC-mediated Ca2+ influx in nerve terminals at subsynaptic regions called active zones in which there is a higher density of VDCCs than the surrounding nonactive zones (Wu et al., 1999). VDCC activation evokes a large localized increase in intracellular free Ca2+ concentration ([Ca2+]i) of at least 200 μm within the active zone that stimulates exocytosis (Heidelberger et al., 1994; Neher, 1998). The microdomains of high [Ca2+]icreated by VDCC clustering are essential for exocytosis because the Ca2+ receptor that stimulates the process, thought to be synaptotagmin, possesses a very low affinity for Ca2+ (Brose et al., 1992).
Increases in [Ca2+]i also stimulate endocytosis in excitable cells (Thomas et al., 1994; Artalejo et al., 1995, 1996; Engisch and Nowycky, 1998) and central neurons (Cousin and Robinson, 1998; Guatimosim et al., 1998; Klingauf et al., 1998; Marks and McMahon, 1998). The Ca2+receptor responsible for triggering endocytosis has been proposed to be calmodulin (Artalejo et al., 1996; Cousin and Robinson, 1998; Marks and McMahon, 1998), which stimulates a dephosphorylation pathway mediated by calcineurin in nerve terminals (Liu et al., 1994; Marks and McMahon, 1998). This pathway has a much higher affinity for Ca2+ (in the nanomolar range) (Nichols and Suplick, 1996) than the low-affinity receptor for exocytosis. Therefore, the observed close coupling between exocytosis and endocytosis might be attributable to the fact that much lower increases in [Ca2+]i are required to stimulate synaptic vesicle retrieval during exocytosis, ensuring that endocytosis is always activated with all stimuli.
Not all studies show that Ca2+ is required for synaptic vesicle retrieval. Ca2+ has been proposed to play no role in endocytosis at central and neuromuscular synapses (Ramaswami et al., 1994; Ryan et al., 1996; Wu and Betz, 1996) and elevated [Ca2+]i inhibits synaptic vesicle retrieval in goldfish ribbon synapses (Von Gersdorff and Matthews, 1994, 1997). Therefore, the role for Ca2+ in endocytosis still requires clarification. One unifying explanation is that Ca2+ may have multiple roles in synaptic vesicle retrieval. This could be achieved by taking advantage of the different [Ca2+]igradients within the neuron upon stimulation. For example, microdomains of high [Ca2+]i at the active zone may have a specific effect on synaptic vesicle retrieval or, alternatively, different routes of Ca2+ entry may differentially modulate endocytosis. We have addressed these questions in isolated nerve terminals by investigating the role in endocytosis of VDCC-mediated Ca2+ influx and the high localized Ca2+ microdomains they generate. We report that VDCC-mediated Ca2+ influx does not specifically play a role in stimulating synaptic vesicle retrieval but that it inhibits endocytosis at the active zone during prolonged stimulation. This inhibitory pathway may provide a feedback mechanism for uncoupling exocytosis and endocytosis, which may relocate synaptic vesicle retrieval to nonactive zone regions under conditions of intense stimulation.
MATERIALS AND METHODS
Materials. FM2–10, BAPTA-AM, EGTA-AM, fura-2 AM, and Texas Red-conjugated transferrin were obtained from Molecular Probes (Eugene, OR). ω-Agatoxin-IVA was obtained from the Peptide Institute (Osaka, Japan). ω-Conotoxin-GVIA and ω-Conotoxin-MVIIC were obtained from Bachem (Saffron-Walden, UK). Ionomycin was obtained from Calbiochem/Novabiochem (Alexandria, Australia). [γ-32P]GTP was obtained from NEN (Boston, MA). All other reagents were obtained from Sigma (Poole, UK).
Glutamate release assay. Synaptosomes were prepared from rat cerebral cortex by centrifugation on discontinuous percoll gradients (Dunkley et al., 1986). The glutamate release assay was performed using enzyme-linked fluorescent detection of released glutamate (Nicholls and Sihra, 1986; Cousin and Robinson, 1998). Briefly, synaptosomes (0.6 mg in 2 ml) were resuspended in either plus (1.2 mmCaCl2) or minus (1 mm EGTA) Ca2+ Krebs'-like solution (in mm: 118.5 NaCl, 4.7 KCl, 1.18 MgCl2, 0.1 Na2HPO4, 20 HEPES, and 10 glucose, pH 7.4) at 37°C. Experiments were started after addition of 1 mmNADP+ and, after 1 min, 50 U of glutamate dehydrogenase was added and the synaptosome suspension was stimulated after 4 min with either KCl, 4-aminopyridine (4-AP) or ionomycin, respectively. Increases in fluorescence caused by production of NADPH were monitored in a Perkin-Elmer (Emeryville, CA) LS-50B spectrofluorimeter at 340 nm excitation and 460 nm emission. Experiments were standardized by the addition of 4 nmol of glutamate. Data are presented as Ca2+-dependent glutamate release, calculated as the difference between release in plus and minus Ca2+ solution for identical stimulation conditions. In experiments using antagonists, synaptosomes were preincubated for 2 min with VDCC inhibitors (plus 100 μg/ml bovine serum albumin) or for 30 min with varying concentrations of BAPTA-AM or EGTA-AM before stimulation.
Endocytosis assay. Endocytosis was measured using uptake of the fluorescent dye FM2–10 as described previously (Cousin and Robinson, 1998). Synaptosomes (0.6 mg in 2 ml) were incubated for 5 min at 37°C in plus or minus Ca2+Krebs'-like solution. FM2–10 (100 μm) was added 1 min before stimulation with KCl, 4-AP, or ionomycin (S1). FM2–10 is taken up inside vesicles by endocytosis at the S1 phase of stimulation; therefore, synaptosomes were incubated with antagonists during this phase (synaptosomes were incubated with VDCC antagonists for 2 min plus 100 μg/ml bovine serum albumin, and for 30 min with BAPTA-AM and EGTA-AM). After 2 min of stimulation with KCl, 4-AP, or ionomycin, synaptosomes were washed twice in plus Ca2+ solution containing 1 mg/ml bovine serum albumin. The washing steps remove noninternalized FM2–10 and extracellular antagonists. Washed synaptosomes were resuspended in plus Ca2+ solution at 37°C, transferred to a fluorimeter cuvette, and stimulated with a standard addition of 30 mm KCl (S2). The standard S2 stimulation releases all accumulated FM2–10 (Marks and McMahon, 1998), and therefore endocytosis can be measured as the decrease in FM2–10 fluorescence caused by dye release into solution (excitation 488 nm, emission 540 nm). The kinetics of FM2–10 release are slower than observed using perfused neuronal cultures (Klingauf et al., 1998), because the dye is sill present in the cuvette after its release and is able to rebind lipid more readily then in a perfusion system.
Endocytosis was calculated as the decrease in absolute fluorescence from FM2–10-labeled synaptosomes stimulated by 30 mm KCl. The displayed traces in all figures represent the release of FM2–10 from synaptosomes after subtraction of background traces acquired from synaptosomes loaded with FM2–10 in the absence of Ca2+. Retrieval efficiency is a more accurate measure of endocytosis because it takes into account the amount of previous exocytosis (Cousin and Robinson, 1998). Retrieval efficiency was calculated as endocytosis/exocytosis in which endocytosis is defined as above and exocytosis as Ca2+-dependent glutamate release after 2 min of stimulation. The retrieval efficiency value was normalized to a ratio of 1.0 for 30 mm KCl.
[Ca2+]idetermination. Synaptosome [Ca2+]i levels were monitored using fura-2 fluorescence as described previously (Cousin et al., 1993). Briefly, synaptosomes were incubated for 30 min in plus Ca2+ Krebs' solution supplemented with 5 μm fura-2 AM and 1 mg/ml bovine serum albumin. Synaptosomes were washed and resuspended in plus Ca2+ Krebs' solution. Fura-2 responses were recorded using a Perkin-Elmer LS-50B spectrophotometer monitoring excitation at 340 and 380 nm and emission at 505 nm. Fura-2-loaded synaptosomes were stimulated with KCl, 4-AP, or ionomycin. Synaptosomal [Ca2+]i values were taken 20 sec after stimulation and were calculated using the Grynkiewicz equation (Grynkiewicz et al., 1985).Rmax was obtained by addition of 10% SDS (final concentration), andRmin was obtained by addition of 15 mm EGTA (final concentration).
Lipid vesiculation assay. Dynamin I was purified from ovine brain (Robinson et al., 1993). The lipid vesiculation assay was performed as described previously (Sweitzer and Hinshaw, 1998) with minor modifications. Dynamin I (0.25 mg/ml) was incubated with phosphatidylserine liposomes (0.75 mg/ml) in assembly buffer [in mm: 20 Tris-HCl, 20 NaCl, 1 EGTA, 1 PMSF, 1 dithiothreitol, and complete protease inhibitors (Roche Products, Hertforshire, UK), pH 7.4] for 1 hr at room temperature. This allows dynamin I to assemble around the lipid and form long tubules. After 1 hr, the dynamin–lipid tubules were diluted 10-fold in assembly buffer and transferred to an LS50-B spectrofluorimeter. After 2 min, 1 mm GTP-MgCl2 was added (final concentration), which causes dynamin to produce vesicles from the tubulated lipid. Vesicle production was monitored as a decrease in 90° light scattering at 450 nm with a 4% screen. CaCl2 was added 2 min before addition of GTP.
Transferrin internalization assay. Transferrin internalization in HeLa cells was performed as described previously (van der Bliek et al., 1993). Briefly, HeLa cells (plated to 60% confluency) were removed from culture medium (DMEM plus 10% fetal calf serum) and incubated in DMEM minus fetal calf serum for 30 min at 37°C. Medium was then removed and replaced with medium supplemented with 50 μg/ml Texas Red-conjugated transferrin for 10 min. The cells were washed three times with ice-cold PBS supplemented with 1.2 mmCaCl2 and 1.2 mmMgCl2. Cells were immediately fixed using 4% formaldehyde in PBS for 30 min and then washed three times with PBS. Cells were mounted on slides using Dabco (Sigma), and fluorescence was monitored using a Leica (Nussloch, Germany) confocal microscope. Ionomycin was added simultaneously with Texas Red-conjugated transferrin where indicated.
GTPase assay. The GTPase activity of baculovirus-expressed dynamin II was determined as described previously (Liu et al., 1996). Briefly, dynamin II (80 μg/ml) was incubated with phosphatidylserine liposomes (10 μg/ml) for 1 hr in GTPase buffer (10 mm NaCl, 1 mmMgCl2, 0.05% Tween 80, 1 μg/ml leupeptin, 2 mm PMSF, and 10 mm Tris, pH 7.4) at 4°C. After 1 hr and at 30°C, CaCl2 was added to the dynamin–lipid mixture and, 5 min later, [γ-32P]GTP was added. After 30 min, hydrolysis of [γ-32P]GTP by dynamin II was terminated by addition of 2% formic acid–8% acetic acid. Activated charcoal was added to bind the guanosine nucleotides, the samples were centrifuged in a microfuge, and the supernatant was counted for released32Pi. Results are normalized to control and are presented as stimulation above basal activity.
RESULTS
Increasing [Ca2+]i inhibits endocytosis in isolated nerve terminals
Endocytosis occurs in both the active zone and nonactive zones of the nerve terminal (Heuser and Reese, 1973; Koenig and Ikeda, 1996). Therefore, our first aim was to determine whether a component of endocytosis may be specifically coupled to activation of VDCCs at the active zone. Nerve terminals were stimulated with agents that increase [Ca2+]i by three distinct mechanisms. KCl depolarizes excitable membranes, greatly increasing the probability of VDCC opening, and produces localized increases in [Ca2+]i at the pore of VDCCs, which are clustered at active zones (Verhage et al., 1991). 4-AP blocks a presynaptic K+channel that regulates plasma membrane excitability and thereby induces transient, Na+ channel-generated membrane depolarizations similar to action potentials (Tibbs et al., 1989). 4-AP also activates VDCCs and, like KCl, produces localized increases in [Ca2+]i at the active zone but without the prolonged depolarization characteristic of KCl. The ionophore ionomycin raises [Ca2+]i from all points across the plasma membrane of the nerve terminal without the localized increases (Verhage et al., 1991). All three stimuli produced concentration-dependent increases in Ca2+-dependent glutamate release (Fig.1A,D,G) (McMahon and Nicholls, 1991; Verhage et al., 1991; Sihra et al., 1992).
Fig. 1.

Endocytosis is inversely proportional to exocytosis with increasing stimulation. A,D, G, Ca2+-dependent glutamate release from nerve terminals was stimulated with increasing concentrations of KCl (A), 4-AP (D), or ionomycin (IONO;G) as indicated. The period of stimulation is indicated by the bar; n = 3–7 (±SEM).B, E, H, Nerve terminals were loaded for 2 min with FM2–10 during S1 stimulation, using increasing concentrations of KCl (B), 4-AP (E), or ionomycin (H) in plus Ca2+medium, with minus Ca2+ medium (1 mmEGTA) serving as a control. A representative trace of the subsequent release of loaded FM2–10 (S2) by a standard stimulus of 30 mm KCl is displayed. All traces are corrected for background uptake by subtracting the minus Ca2+ trace. Exocytosis is constant during the S2 stimulation; therefore, endocytosis is calculated as the evoked Ca2+-dependent decrease in absolute fluorescence units. The period of KCl (30 mm) stimulation is indicated by the bar; n = 4–5.C, F, I, Retrieval efficiency (endocytosis/exocytosis, normalized to 1.0 for 30 mm KCl) for increasing concentrations of KCl (C), 4-AP (F), or ionomycin (I). Endocytosis is defined as above, and exocytosis is Ca2+-dependent glutamate release after 2 min; n = 3–7 (±SEM).
KCl, 4-AP, and ionomycin all stimulated endocytosis (Fig.1B,E,H). However, unlike exocytosis, evoked endocytosis was not always concentration-dependent. For example, endocytosis decreased with increasing 4-AP (Fig. 1E) or ionomycin (Fig.1H) concentrations. This was not because of a reduced ability of synaptosomes to release the accumulated FM2–10, because Ca2+-dependent glutamate release was identical in the S2 phase for all concentrations of either KCl, 4-AP, or ionomycin (data not shown).
To accurately quantify the amount of endocytosis, the preceding amount of exocytosis must also be considered, because endocytosis of synaptic vesicles is dependent on the availability of fused vesicles for retrieval. A parameter termed retrieval efficiency serves as a more quantitative measure of endocytosis because it takes this into account by dividing the amount of endocytosis by exocytosis for the same stimulus (Cousin and Robinson, 1998). In all experiments, retrieval efficiency is arbitrarily set to 1.0 for loading with 30 mmKCl. If endocytosis was unaffected by increasing concentrations of KCl, 4-AP, or ionomycin, then retrieval efficiency would be constant for these different stimuli. However, increasing concentrations of KCl, 4-AP, or ionomycin all decreased retrieval efficiency (Fig.1C,F,I). Thus, endocytosis is reduced 4- to 20-fold by increasing the strength of the stimuli.
We next examined whether a particular route of Ca2+ entry was more efficient in stimulating either exocytosis or endocytosis. To achieve this, exocytosis and endocytosis were correlated with the average increases in [Ca2+]iproduced by KCl, 4-AP, or ionomycin. When Ca2+-dependent glutamate release was correlated with the evoked increase in nerve terminal [Ca2+]i measured using fura-2, two observations were made (Fig.2A). First, the amount of exocytosis correlated with increasing concentrations of [Ca2+]i. Second, KCl and 4-AP were more efficient than ionomycin in supporting exocytosis. This is because much lower [Ca2+]i increases evoked by KCl or 4-AP were required to stimulate release over the whole range of average [Ca2+]i than ionomycin. Thus, VDCC-mediated Ca2+ influx stimulated vesicle fusion more efficiently than the ionomycin-induced global increase in [Ca2+]i (McMahon and Nicholls, 1991; Verhage et al., 1991; Sihra et al., 1992; Neher, 1998). This was expected, because exocytosis is dependent on high localized concentrations of [Ca2+]i binding to a low-affinity Ca2+ sensor (Brose et al., 1992; Heidelberger et al., 1994). The KCl- and 4-AP-evoked [Ca2+]i increase localized to the active zone is probably much larger than that detected here, because fura-2 only measures the average [Ca2+]i in the nerve terminal (Grynkiewicz et al., 1985) and not the microdomain of high [Ca2+]i at the pore of the VDCCs.
Fig. 2.
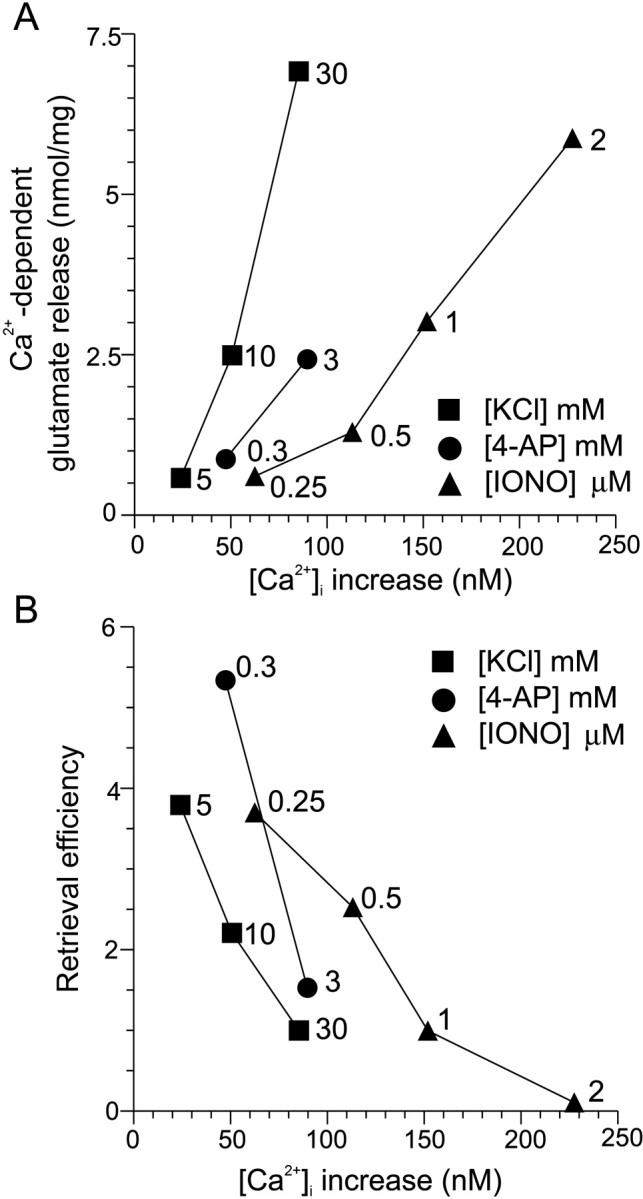
VDCC-mediated Ca2+ influx stimulates exocytosis and inhibits endocytosis more efficiently than a uniform [Ca2+]i increase.A, Ca2+-dependent glutamate release after 2 min evoked by KCl (squares), 4-AP (circles), or ionomycin (IONO;triangles) is plotted against the stimulated [Ca2+]i increase for each agent measured using fura-2. [Ca2+]i was sampled after 20 sec stimulation. For all experiments,n = 3–7 (±SEM). B, Retrieval efficiency for increasing concentrations of KCl (squares), 4-AP (circles), or ionomycin (triangles) is plotted against evoked [Ca2+]i increases for each agent. For all experiments, n = 3–7 (±SEM). In bothA and B, error bars are smaller than thesymbols.
When retrieval efficiency was correlated with evoked [Ca2+]i increases, a relative decrease in endocytosis was observed with increasing [Ca2+]iconcentrations for KCl, 4-AP, and ionomycin stimulation (Fig.2B). The result is the opposite of the Ca2+-dependence of exocytosis (Fig.2A) and suggests that higher levels of Ca2+ inhibit endocytosis (Von Gersdorff and Matthews, 1994). VDCC-mediated Ca2+influx was more efficient in inhibiting endocytosis than a global increase in [Ca2+]i. This is because much lower [Ca2+]i increases evoked by KCl or 4-AP were required to inhibit endocytosis than ionomycin (Fig. 2B). These results reveal a Ca2+-mediated inhibition of endocytosis occurring with larger stimuli and suggests that the inhibition may be mediated by Ca2+ entry through VDCCs and the resultant high localized [Ca2+]i at the active zone. The brief depolarizations evoked by 4-AP were approximately as efficient at inhibiting endocytosis as the more prolonged depolarization evoked by KCl.
VDCC-mediated Ca2+ entry inhibits endocytosis
To examine whether Ca2+ entry through VDCCs inhibits synaptic vesicle retrieval, we examined the effect of specific VDCC antagonists on KCl-evoked endocytosis. At most central synapses, N-, P-, and Q-type VDCCs are coupled to exocytosis (Dunlap et al., 1995). These channels can be inhibited specifically by the peptide toxins ω-conotoxin-GVIA (N-type), ω-agatoxin-IVA (P-type), and ω-conotoxin-MVIIC (N/P/Q-type), respectively (McCleskey et al., 1987;Hillyard et al., 1992; Mintz et al., 1992). KCl-evoked Ca2+-dependent glutamate release was substantially blocked by ω-agatoxin-IVA (30 nm) and ω-conotoxin-MVIIC (5 μm), but ω-conotoxin-GVIA (1 μm) was not as effective (Fig.3A). All three VDCC antagonists blocked KCl-evoked [Ca2+]i increases to a similar extent (data not shown), suggesting that P- and Q-type VDCCs predominate at the active zone in our system. KCl-evoked endocytosis was apparently unaffected by any of the antagonists when FM2–10 fluorescence was examined, with the exception of ω-agatoxin-IVA, which increased endocytosis (Fig. 3B). Retrieval efficiency was also greatly increased by ω-agatoxin-IVA and ω-conotoxin-MVIIC (Fig. 3C). Therefore, Ca2+ influx through those VDCCs that are coupled to transmitter release at the active zone (P/Q-type) also inhibits endocytosis. This suggests that KCl-induced inhibition of endocytosis occurs primarily at the active zone in which Ca2+ channels are clustered rather than nonactive zones. This also indicates that endocytosis is not always strictly coupled to exocytosis, because the amount of endocytosis was unaffected by a reduction in exocytosis and Ca2+ influx.
Fig. 3.

Activation of VDCCs coupled to exocytosis also inhibits endocytosis. A, Ca2+-dependent glutamate release evoked by 30 mm KCl in the presence of 1 μmω-conotoxin-GVIA (N), 30 nmω-agatoxin-IVA (P), or 5 μmω-conotoxin-MVIIC (NPQ); n = 4 (±SEM). B, Nerve terminals were loaded for 2 min with FM2–10 during S1 stimulation using KCl (30 mm) in the presence of N-, P-, or N/P/Q-type VDCC blockers. A representativetrace of the subsequent Ca2+-dependent release of loaded FM2–10 (S2) by a standard stimulus of 30 mm KCl is displayed. Exocytosis is constant during S2 stimulation; therefore, endocytosis is calculated as Ca2+-dependent fluorescence decrease in the S2 stimulation; n = 3–4. In all experiments, VDCC antagonists were present 2 min before stimulation. KCl stimulation is represented by the bar. C, KCl-stimulated retrieval efficiency in the presence of VDCC antagonists normalized to control [n = 4 (±SEM)].
Next, we determined whether the inhibition of endocytosis at the active zone by P/Q-type VDCCs requires [Ca2+]imicrodomains that are both localized and of high concentration, such as required for exocytosis (Adler et al., 1991; Heidelberger et al., 1994). The effects of the membrane-permeable intracellular Ca2+ chelators EGTA-AM and BAPTA-AM were compared. These chelators share a similarKd for Ca2+, but BAPTA-AM can bind Ca2+ with much faster kinetics (Adler et al., 1991) and is thus able to block localized Ca2+ responses in most nerve terminals with relative specificity. BAPTA-AM loading of nerve terminals inhibited KCl-stimulated glutamate release in a concentration-dependent manner (Fig. 4A), and EGTA-AM had a smaller effect (Fig. 4C). Presumably, intracellular BAPTA can better compete for Ca2+ with the low-affinity Ca2+ receptor for exocytosis because of its faster binding kinetics compared with EGTA (Adler et al., 1991;Nichols and Suplick, 1996). BAPTA-AM loading did not block KCl-evoked endocytosis as effectively as exocytosis (Fig. 4B), and EGTA-AM again had little effect (Fig. 4D). When retrieval efficiency was calculated for BAPTA-AM-loaded nerve terminals, relative endocytosis was increased in a concentration-dependent manner, whereas EGTA-AM-loaded terminals were unaffected (Fig. 4E). The lack of effect of EGTA-AM at any concentration illustrates that some agents that affect exocytosis have a parallel effect on endocytosis. This emphasizes the importance of retrieval efficiency as a more quantitative index of endocytosis. These results demonstrate that, by rapidly chelating Ca2+ entering via VDCCs, the inhibition of endocytosis by high [Ca2+]i can be relieved. This provides evidence for the existence of a relatively low-affinity Ca2+-binding receptor that blocks endocytosis and that resides in close proximity to the VDCCs at the active zone. Additionally, the relative increase in endocytosis observed with BAPTA-AM-loaded nerve terminals reflects an uncoupling of exocytosis and endocytosis.
Fig. 4.

Fast chelation of VDCC-mediated Ca2+ influx increases endocytosis. A,C, Ca2+-dependent glutamate release from nerve terminals evoked by 30 mm KCl after previous loading with increasing concentrations of either BAPTA-AM or EGTA-AM;n = 4–5 (±SEM). B,D, Nerve terminals were loaded for 2 min with FM2–10 during S1 stimulation with KCl (30 mm) after previous loading with increasing concentrations of either BAPTA-AM or EGTA-AM in either plus or minus Ca2+ medium. A representativetrace of the subsequent Ca2+-dependent release of loaded FM2–10 (S2) by a standard stimulus of 30 mm KCl is displayed. In all experiments, either BAPTA-AM or EGTA-AM were preincubated with the synaptosomes for 30 min before stimulation. KCl stimulation is represented by the bar; n = 4.E, KCl-stimulated retrieval efficiency in BAPTA-AM- (triangles) and EGTA-AM- (squares) loaded synaptosomes. Exocytosis was constant during S2 stimulation for EGTA-AM-loaded synaptosomes but was greatly reduced for BAPTA-AM-loaded synaptosomes (data not shown). Therefore, retrieval efficiency is an underestimate with respect to BAPTA-AM-loaded synaptosomes. For all experiments, n = 3–4 (±SEM). Error bars are smaller than the symbols.
Ca2+ blocks dynamin I vesiculation of phospholipid
By blocking entry of extracellular Ca2+ and by chelating intracellular Ca2+, our results show that VDCC-mediated, localized Ca2+ microdomains inhibit endocytosis at the active zone in nerve terminals via a proposed low-affinity receptor for Ca2+. To determine the identity of this proposed receptor, we examined the presynaptic GTPase dynamin I, which can bind Ca2+ with low affinity (Liu et al., 1996). High concentrations of free Ca2+ inhibit dynamin I (Liu et al., 1996), whose GTPase activity is essential for the fission of a synaptic vesicle from the plasma membrane in the final stages of endocytosis (Takei et al., 1995; Stowell et al., 1999). We therefore investigated whether dynamin I may be the proposed low-affinity receptor for Ca2+. To directly test whether high [Ca2+]i inhibits endocytosis via an interaction with dynamin I, we used an in vitro assay of dynamin-mediated endocytosis (Sweitzer and Hinshaw, 1998). In this assay, purified dynamin I is incubated with large phosphatidylserine liposomes, which dynamin I can vesiculate upon addition of GTP. Dynamin-dependent production of vesicles is monitored as a decrease in light scattering upon addition of 1 mm GTP (Fig.5A). Vesicle production is dependent on GTP hydrolysis because a nonhydrolyzable analog of GTP (1 mm GTP-γS) was without effect (Fig.5A). Free Ca2+ decreased dynamin-dependent vesiculation in a concentration-dependent manner (IC50 of 21 μm) (Fig.5B). These Ca2+ concentrations are well below those observed at the active zone during stimulation of exocytosis (∼200 μm) (Heidelberger et al., 1994; Neher, 1998). Therefore the inhibition of endocytosis by VDCC-mediated Ca2+ microdomains in nerve terminals correlates with a low-affinity inhibition of dynamin I by Ca2+.
Fig. 5.

Ca2+ inhibition of phospholipid vesiculation by dynamin I. A, Purified dynamin I and phosphatidylserine liposomes were incubated for 1 hr to allow dynamin assembly into helices around the liposomes. Addition of 1 mm GTP (bar), but not 1 mmGTP-γS, produced a decrease in light scattering corresponding to production of small vesicles from long tubule assemblies. Free Ca2+ was added 2 min before GTP addition.B, Inhibition of lipid vesiculation by Ca2+ is shown as a percentage of control [n = 4 (±SEM)].
Ca2+ blocks dynamin II GTPase activity and receptor-mediated endocytosis
To determine whether Ca2+ can inhibit other forms of dynamin-dependent endocytosis, we examined receptor-mediated endocytosis, which is mediated by the ubiquitously expressed dynamin II. High concentrations of free Ca2+ blocked the GTPase activity of dynamin II (IC50 of 150 μm) (Fig.6A). We examined the effect of increased [Ca2+]i on receptor-mediated endocytosis by observing uptake of fluorescently conjugated transferrin into HeLa cells (van der Bliek et al., 1993). Addition of 2 μm ionomycin severely reduced transferrin uptake into HeLa cells (Fig.6B,C). Thus, the same concentration of ionomycin that blocked endocytosis in nerve terminals also inhibited receptor-mediated endocytosis in non-neuronal cells. Ionomycin increased [Ca2+]iand blocked transferrin internalization in a concentration-dependent manner (data not shown). The inhibition by ionomycin was not caused by cell damage because it could be reversed upon washout of the ionophore (data not shown). Therefore, the inhibition of GTPase activity by low-affinity Ca2+ binding is a property of both dynamins, and this inhibition can block both dynamin I- and II-dependent endocytosis.
Fig. 6.

Ca2+ inhibition of dynamin II GTPase activity and transferrin endocytosis. A, Purified recombinant dynamin II and phosphatidylserine liposomes were incubated for 1 hr. Increasing concentrations of free Ca2+were added as indicated, and the hydrolysis reaction was stimulated by the addition of substrate, [γ-32P]GTP. [γ-32P]GTP hydrolysis is shown as a percentage of control [n = 6 (±SEM)]. B,C, Images display the uptake of Texas Red-conjugated transferrin into cultured HeLa cells in either the absence (B) or presence (C) of 2 μm ionomycin. HeLa cells were incubated with transferrin with or without ionomycin for 10 min before fixing.
DISCUSSION
VDCC-mediated Ca2+ influx inhibits endocytosis in nerve terminal active zones
Because synaptic vesicle retrieval in nerve terminals is triggered by Ca2+ influx, we aimed to determine whether the route of Ca2+ entry was important for endocytosis, because it is critical for exocytosis. In this study, we demonstrate that VDCC-mediated, localized increases in [Ca2+]i do not specifically stimulate endocytosis but inhibit endocytosis at the active zone in nerve terminals, probably by a low-affinity inhibition of dynamin I. Therefore, the Ca2+inhibition of endocytosis shares similar properties to Ca2+-triggered exocytosis in that the route of Ca2+ entry and the generation of Ca2+ microdomains in nerve terminals are essential for their function.
Endocytosis must be monitored in parallel with exocytosis in nerve terminals, because the extent of endocytosis is dependent on the number of fused synaptic vesicles produced by exocytosis (Murthy and Stevens, 1998). Therefore, the retrieval efficiency index was used to provide a better estimate of endocytosis by taking into account the variable amounts of exocytosis evoked by different stimulation methods. Although retrieval efficiency provides a value for the relative proportion of endocytosis to exocytosis, it does not provide information on the absolute number of synaptic vesicles released and retrieved. It therefore cannot discount the possibility that endocytosis may be increased at lower [Ca2+]i levels rather than endocytosis being inhibited at high [Ca2+]i (Marks and McMahon, 1998). We resolved this question by showing that high [Ca2+]i inhibits receptor-mediated endocytosis in HeLa cells. This demonstrated that [Ca2+]i can inhibit endocytosis in a cell in which endocytosis is not dependent on previous exocytosis. One known mechanism by which endocytosis can increase is a process called “excess retrieval” in neuroendocrine cells (Engisch and Nowycky, 1998). This occurs when more surface membrane is retrieved from the plasma membrane than was originally incorporated by exocytosis. However, excess retrieval requires higher [Ca2+]i levels than exocytosis for stimulation (Engisch and Nowycky, 1998), which is the opposite of our results in nerve terminals.
An alternative explanation for our data is that mild stimulation preferentially labels the readily releasable pool of synaptic vesicles, resulting in greater dye release with the second stimulation. If this were true, during strong stimulation, FM2–10-labeled synaptic vesicles should be sequestered into a reserve pool after endocytosis, rendering vesicles inaccessible for immediate re-release, giving the impression of an inhibition. However, imaging of single FM2–10-labeled synaptosomes revealed that all accumulated dye was fully released after loading and unloading with 30 mm KCl (Marks and McMahon, 1998). Also, no decrease in the amount of glutamate release was detected during the S2 stimulation in the present study, indicating that exocytosis is unaffected.
Recent studies in cultured neurons and chromaffin cells have proposed that increased extracellular Ca2+ either during or without stimulation causes a transition to a rapid form of synaptic vesicle recycling called “kiss-and-run” (Klingauf et al., 1998; Alés et al., 1999). In cultured hippocampal neurons, FM2–10-loaded nerve terminals released less dye when stimulated in the presence of elevated extracellular Ca2+(Klingauf et al., 1998). This was interpreted as a Ca2+-stimulated transition to kiss-and-run because the vesicle was retrieved from the plasma membrane before it could empty all of its dye content. A transition to kiss-and-run recycling could also explain the reduced uptake of FM2–10 we observe upon VDCC activation, because this would allow less time for FM2–10 to become incorporated inside retrieving synaptic vesicles. However, we found that glutamate and FM2–10 release increased linearly with increasing concentrations on KCl. This indicates that KCl-evoked increases in [Ca2+]i do not cause a transition to kiss-and-run in isolated nerve terminals (our unpublished observations). This agrees with previous studies using nerve terminals, which show a linear relationship between FM1–43 release and stimulus intensity (Meffert et al., 1994). Therefore, the decrease in FM2–10 uptake during VDCC activation in our study is caused by an inhibition of endocytosis rather than by a transition to kiss-and-run.
Dual roles for Ca2+ in endocytosis
Ca2+ also stimulates synaptic vesicle endocytosis. Ca2+-dependent stimulation is mediated by a high-affinity Ca2+interaction with a calmodulin–calcineurin dephosphorylation pathway (Artalejo et al., 1996; Marks and McMahon, 1998; Cousin and Robinson, 1998). This stimulatory pathway is distinct from the low-affinity inhibitory pathway, because antagonism of the low-affinity inhibitory pathway does not inhibit endocytosis (Figs. 3B,4B). This is because sufficient [Ca2+]i is still present to fully stimulate calmodulin–calcineurin and endocytosis. Because calmodulin–calcineurin has a high affinity for Ca2+, endocytosis will always be maximally activated upon nerve terminal stimulation. This has led to previous studies in central neurons and neuromuscular preparations concluding that endocytosis was a Ca2+-independent process (Ryan et al., 1996; Wu and Betz, 1996).
Ca2+ blocks dynamin GTPase and dynamin-dependent endocytosis
The GTPase activity of dynamin I, which is expressed primarily in neurons, is inhibited in vitro by relatively high concentrations of Ca2+(IC50 of 30 μm) (Liu et al., 1996). The vesiculation assay further reveals that essentially the same concentration of Ca2+ also inhibits the ability of dynamin I to produce vesicles from phospholipid (IC50 of 21 μm). These IC50 values are well within the range of [Ca2+]i proposed to be present at the active zone during stimulation (Heidelberger et al., 1994). This strongly suggests, but does not prove, that micromolar [Ca2+]i in microdomains at the active zone would inhibit endocytosis locally in neurons by blocking the GTPase activity of dynamin I. This is supported by the observation that point mutations of dynamin in the GTP-binding domain in the temperature-sensitive Drosophila mutantshibire and mammalian cells overexpressing dominant negative mutant dynamin with deficient GTPase activity exhibit a block of endocytosis in vivo (Koenig and Ikeda, 1989; van der Bliek et al., 1993). The effect of Ca2+ on dynamin-dependent endocytosis is not restricted to neurons because the GTPase activity of the ubiquitously expressed dynamin II and the form of endocytosis it controls are also inhibited by high [Ca2+]i.
High [Ca2+]i at active zones shunts endocytosis to nonactive zones
The number of sites available for synaptic vesicle docking and fusion is severely limited at the active zone (Schikorski and Stevens, 1997); therefore, the nerve terminal needs to ensure that these sites are rapidly cleared of vesicle membrane. A Ca2+-mediated inhibition of endocytosis may provide a mechanism for neurons to help clear the active zone of recycling synaptic vesicles during periods of highly active exocytosis. The net result of the inhibition will be that endocytosis is effectively “shunted” from active to nonactive zones in which it is still able to proceed because of the high-affinity stimulatory pathway (Cousin and Robinson, 1998; Marks and McMahon, 1998) (Fig.7). Ca2+-dependent shunting would therefore allow the active zone to be reserved more exclusively for exocytosis during intense stimulation. In our model, shunting of endocytosis is not an active process but would result from the size of the Ca2+ microdomains generated by the active zone VDCCs (Neher, 1998). Once stimulation is terminated, [Ca2+]i will decrease, and endocytosis should occur at both active and nonactive zone regions as demonstrated in Drosophila nerve terminals (Koenig and Ikeda, 1996).
Fig. 7.

Model for the control of endocytosis by VDCCs in nerve terminals. Upon VDCC activation, exocytosis is stimulated and endocytosis is blocked by high localized [Ca2+]i concentrations at the active zone. Vesicle membrane can be retrieved once it clears the active zone, because the block of endocytosis is relieved and endocytosis is stimulated by lower [Ca2+]i levels in the nonactive zone. Once stimulation is terminated and [Ca2+]i decreases, endocytosis can occur from either location within the nerve terminal.
Ca2+-dependent shunting of endocytosis challenges the concept that exocytosis and endocytosis are always strictly coupled in central neurons. Under mild stimulation conditions, exocytosis and endocytosis are closely coupled, because the amount of FM1–43 uptake equals its release in cultured hippocampal neurons (Murthy and Stevens, 1998). However, in this paper, we have shown that endocytosis can be uncoupled from exocytosis by high [Ca2+]i.Therefore, we propose that, during intense stimulation, shunting uncouples exocytosis and endocytosis in time (by delaying the retrieval of vesicles) and in space (by, in effect, relocating endocytosis to nonactive zones). Because high [Ca2+]i shunts endocytosis to the nonactive zone, it may also play a role in determining where proposed “hot spots” for endocytosis can occur in these regions (Koenig and Ikeda, 1996; Teng et al., 1999).
Shunting of endocytosis by elevated [Ca2+]i may have a key role in the short-term regulation of synaptic transmission. During high-frequency stimulation, synaptic transmission is rapidly depressed because of depletion of the readily releasable synaptic vesicle pool [comprised of vesicles docked or primed for release at the active zone (Stevens and Wesseling, 1998)]. This may in part be because of the time taken for fused vesicle membrane to clear the active zone for retrieval at the nonactive zone. After high-frequency stimulation and synaptic depression, the readily releasable vesicle pool is replenished by new vesicles at much faster rates, which are dependent on stimulated Ca2+ influx (Stevens and Wesseling, 1998;Wang and Kaczmarek, 1998). Ca2+-dependent shunting of endocytosis may facilitate this process by preventing endocytosis in congested active zones, allowing unimpeded access for new incoming vesicles. Refilling rates of the readily releasable pool after synaptic depression are not reduced upon inhibition of endocytosis in retinal bipolar neurons (Von Gersdorff and Matthews, 1997), supporting the hypothesis that shunting contributes toward the increased refilling rates. Shunting cannot fully explain this phenomenon, however, because the increased refilling rate can be reduced by intracellular EGTA, which does not affect shunting (Stevens and Wesseling, 1998; Wang and Kaczmarek, 1998). Thus, shunting may depress synaptic transmission in the short term but will maintain its efficiency by allowing a more rapid refilling of release sites by incoming vesicles. Ca2+-dependent shunting therefore provides a new modulatory mechanism for the control of short-term synaptic signaling and regulation of synaptic vesicle traffic.
Ca2+ inhibition of receptor-mediated endocytosis
The finding that receptor-mediated endocytosis in non-neuronal cells is inhibited by Ca2+ suggests that high [Ca2+]iinhibits all forms of endocytosis mediated by the dynamins and has wider implications for cell biology. The inhibition is not likely to play a role in normal receptor-mediated endocytosis, which has no known Ca2+-dependence, and nonexcitable cells do not have clustered VDCCs that generate microdomains. However, it may have consequences for the mechanisms underlying cellular injury and repair. Upon membrane damage, high localized [Ca2+]i gradients are created, and large membranous vesicles are formed in a variety of cell types, including cut axons (De Mello, 1973; Nishiye, 1977; Severs et al., 1990; Eddleman et al., 1997). The vesicles originate from the plasma membrane and mediate repair by patching the cell membrane using the exocytosis protein machinery (Eddleman et al., 1998; Togo et al., 1999). In cut giant axons, no vesiculation or membrane repair occurs in the absence of extracellular Ca2+ but is reactivated by internal perfusion of the axon with 100 μmCa2+ (Eddleman et al., 1998). The process of membrane repair is morphologically analogous to the formation of large vacuoles in the dynamin-defective Drosophila shibiremutant (Koenig and Ikeda, 1989, 1996). Our results raise the possibility that a block of dynamin-dependent membrane vesiculation by high [Ca2+]i may contribute to the large membrane structures that are created in cells to facilitate membrane repair after injury.
Footnotes
This research was supported by a grant from the Australian National Health and Medical Research Council. M.A.C. is supported by a fellowship from the Human Frontiers of Science Program. We thank Sandra Schmid for the gift of purified dynamin II and Chandra Malladi for the purification of dynamin I. We thank John Bekkers, Bruce Walmsley, Max Bennett, and Peter Rowe for helpful comments.
Correspondence should be addressed to Michael A. Cousin, Cell Signaling Unit, Children's Medical Research Institute, Locked Bag 23, Wentworthville 2145, Sydney, New South Wales, Australia. E-mail:mcousin@cmri.usyd.edu.au.
REFERENCES
- 1.Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alés E, Tabares L, Poyato JM, Valero V, Lindau M, Alvarez de Toledo G. High calcium concentrations shift the mode of exocytosis to the kiss-and-run mechanism. Nat Cell Biol. 1999;1:40–44. doi: 10.1038/9012. [DOI] [PubMed] [Google Scholar]
- 3.Artalejo CR, Henley JR, McNiven MA, Palfrey HC. Rapid endocytosis coupled to exocytosis in adrenal chromaffin cells involves Ca2+, GTP, and dynamin but not clathrin. Proc Natl Acad Sci USA. 1995;92:8328–8332. doi: 10.1073/pnas.92.18.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Artalejo CR, Elhamdani A, Palfrey HC. Calmodulin is the divalent cation receptor for rapid endocytosis, but not exocytosis, in adrenal chromaffin cells. Neuron. 1996;16:195–205. doi: 10.1016/s0896-6273(00)80036-7. [DOI] [PubMed] [Google Scholar]
- 5.Brose N, Petrenko AG, Südhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- 6.Cousin MA, Robinson PJ. Ba2+ does not support synaptic vesicle retrieval in rat isolated presynaptic nerve terminals. Neurosci Lett. 1998;253:1–4. doi: 10.1016/s0304-3940(98)00610-7. [DOI] [PubMed] [Google Scholar]
- 7.Cousin MA, Nicholls DG, Pocock JM. Flunarizine inhibits both calcium-dependent and -independent release of glutamate from synaptosomes and cultured neurones. Brain Res. 1993;606:227–236. doi: 10.1016/0006-8993(93)90989-z. [DOI] [PubMed] [Google Scholar]
- 8.De Mello WC. Membrane sealing in frog skeletal-muscle fibers. Proc Natl Acad Sci USA. 1973;70:982–984. doi: 10.1073/pnas.70.4.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dunkley PR, Jarvie PE, Heath JW, Kidd GJ, Rostas JA. A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res. 1986;372:115–129. doi: 10.1016/0006-8993(86)91464-2. [DOI] [PubMed] [Google Scholar]
- 10.Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- 11.Eddleman CS, Ballinger ML, Smyers ME, Godell CM, Fishman HM, Bittner GD. Repair of plasmalemmal lesions by vesicles. Proc Natl Acad Sci USA. 1997;94:4745–4750. doi: 10.1073/pnas.94.9.4745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eddleman CS, Ballinger ML, Smyers ME, Fishman HM, Bittner GD. Endocytotic formation of vesicles and other membranous structures induced by Ca2+ and axolemmal injury. J Neurosci. 1998;18:4029–4041. doi: 10.1523/JNEUROSCI.18-11-04029.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engisch KL, Nowycky MC. Compensatory and excess retrieval: two types of endocytosis following single step depolarizations in bovine adrenal chromaffin cells. J Physiol (Lond) 1998;506:591–608. doi: 10.1111/j.1469-7793.1998.591bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 15.Guatimosim C, Romano-Silva MA, Gomez MV, Prado MA. Recycling of synaptic vesicles at the frog neuromuscular junction in the presence of strontium. J Neurochem. 1998;70:2477–2483. doi: 10.1046/j.1471-4159.1998.70062477.x. [DOI] [PubMed] [Google Scholar]
- 16.Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- 17.Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hillyard DR, Monje VD, Mintz IM, Bean BP, Nadasdi L, Ramachandran J, Miljanich G, Azimi-Zoonooz A, McIntosh JM, Cruz LJ. A new Conus peptide ligand for mammalian presynaptic Ca2+ channels. Neuron. 1992;9:69–77. doi: 10.1016/0896-6273(92)90221-x. [DOI] [PubMed] [Google Scholar]
- 19.Klingauf J, Kavalali ET, Tsien RW. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature. 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- 20.Koenig JH, Ikeda K. Disappearance and reformation of synaptic vesicle membrane upon transmitter release observed under reversible blockage of membrane retrieval. J Neurosci. 1989;9:3844–3860. doi: 10.1523/JNEUROSCI.09-11-03844.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koenig JH, Ikeda K. Synaptic vesicles have two distinct recycling pathways. J Cell Biol. 1996;135:797–808. doi: 10.1083/jcb.135.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu JP, Sim ATR, Robinson PJ. Calcineurin inhibition of dynamin I GTPase activity coupled to nerve terminal depolarization. Science. 1994;265:970–973. doi: 10.1126/science.8052858. [DOI] [PubMed] [Google Scholar]
- 23.Liu JP, Zhang Q-X, Baldwin G, Robinson PJ. Calcium binds dynamin I and inhibits its GTPase activity. J Neurochem. 1996;66:2074–2081. doi: 10.1046/j.1471-4159.1996.66052074.x. [DOI] [PubMed] [Google Scholar]
- 24.Marks B, McMahon HT. Calcium triggers calcineurin-dependent synaptic vesicle recycling in mammalian nerve terminals. Curr Biol. 1998;8:740–749. doi: 10.1016/s0960-9822(98)70297-0. [DOI] [PubMed] [Google Scholar]
- 25.McCleskey EW, Fox AP, Feldman DH, Cruz LJ, Olivera BM, Tsien RW, Yoshikami D. Omega-conotoxin: direct and persistent blockade of specific types of calcium channels in neurons but not muscle. Proc Natl Acad Sci USA. 1987;84:4327–4331. doi: 10.1073/pnas.84.12.4327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McMahon HT, Nicholls DG. Transmitter glutamate release from isolated nerve terminals: evidence for biphasic release and triggering by localized Ca2+. J Neurochem. 1991;56:86–94. doi: 10.1111/j.1471-4159.1991.tb02566.x. [DOI] [PubMed] [Google Scholar]
- 27.Meffert MK, Premack BA, Schulman H. Nitric oxide stimulates Ca(2+)-independent synaptic vesicle release. Neuron. 1994;12:1235–1244. doi: 10.1016/0896-6273(94)90440-5. [DOI] [PubMed] [Google Scholar]
- 28.Mintz IM, Venema VJ, Swiderek KM, Lee TD, Bean BP, Adams ME. P-type calcium channels blocked by the spider toxin omega-Aga-IVA. Nature. 1992;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- 29.Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- 30.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 31.Nicholls DG, Sihra TS. Synaptosomes possess an exocytotic pool of glutamate. Nature. 1986;321:772–773. doi: 10.1038/321772a0. [DOI] [PubMed] [Google Scholar]
- 32.Nichols RA, Suplick GR. Rapid chelation of calcium entering isolated rat brain nerve terminals during stimulation inhibits neurotransmitter release. Neurosci Lett. 1996;211:135–137. doi: 10.1016/0304-3940(96)12728-2. [DOI] [PubMed] [Google Scholar]
- 33.Nishiye H. The mechanism of Ca2+ action on the healing-over process in mammalian cardiac muscles: a kinetic analysis. Jpn J Physiol. 1977;27:451–466. doi: 10.2170/jjphysiol.27.451. [DOI] [PubMed] [Google Scholar]
- 34.Ramaswami M, Krishnan KS, Kelly RB. Intermediates in synaptic vesicle recycling revealed by optical imaging of Drosophila neuromuscular junctions. Neuron. 1994;13:363–375. doi: 10.1016/0896-6273(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 35.Robinson PJ, Sontag J-M, Liu JP, Fykse EM, Slaughter C, McMahon HT, Südhof TC. Dynamin GTPase regulated by protein kinase C phosphorylation in nerve terminals. Nature. 1993;365:163–166. doi: 10.1038/365163a0. [DOI] [PubMed] [Google Scholar]
- 36.Ryan TA. Endocytosis at nerve terminals: timing is everything. Neuron. 1996;17:1035–1037. doi: 10.1016/s0896-6273(00)80236-6. [DOI] [PubMed] [Google Scholar]
- 37.Ryan TA, Smith SJ, Reuter H. The timing of synaptic vesicle endocytosis. Proc Natl Acad Sci USA. 1996;93:5567–5571. doi: 10.1073/pnas.93.11.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schikorski T, Stevens CF. Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci. 1997;17:5858–5867. doi: 10.1523/JNEUROSCI.17-15-05858.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Severs NJ, Slade AM, Powell T, Twist VW, Green CR. Integrity of the dissociated adult cardiac myocyte: gap junction tearing and the mechanism of plasma membrane resealing. J Muscle Res Cell Motil. 1990;11:154–166. doi: 10.1007/BF01766494. [DOI] [PubMed] [Google Scholar]
- 40.Sihra TS, Bogonez E, Nicholls DG. Localized Ca 2+ entry preferentially effects protein dephosphorylation, phosphorylation, and glutamate release. J Biol Chem. 1992;267:1983–1989. [PubMed] [Google Scholar]
- 41.Stevens CF, Wesseling JF. Activity-dependent modulation of the rate at which synaptic vesicles become available to undergo exocytosis. Neuron. 1998;21:415–424. doi: 10.1016/s0896-6273(00)80550-4. [DOI] [PubMed] [Google Scholar]
- 42.Stowell MH, Marks B, Wigge P, McMahon HT. Nucleotide-dependent conformational changes in dynamin: evidence for a mechanochemical molecular spring. Nat Cell Biol. 1999;1:27–32. doi: 10.1038/8997. [DOI] [PubMed] [Google Scholar]
- 43.Sweitzer SM, Hinshaw JE. Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell. 1998;93:1021–1029. doi: 10.1016/s0092-8674(00)81207-6. [DOI] [PubMed] [Google Scholar]
- 44.Takei K, McPherson PS, Schmid SL, De Camilli P. Tubular membrane invaginations coated by dynamin rings are induced by GTP-γS in nerve terminals. Nature. 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- 45.Teng H, Cole JC, Roberts RL, Wilkinson RS. Endocytic active zones: hot spots for endocytosis in vertebrate neuromuscular terminals. J Neurosci. 1999;19:4855–4866. doi: 10.1523/JNEUROSCI.19-12-04855.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas P, Lee AK, Wong JG, Almers W. A triggered mechanism retrieves membrane in seconds after Ca2+-stimulated exocytosis in single pituitary cells. J Cell Biol. 1994;124:667–675. doi: 10.1083/jcb.124.5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tibbs GR, Barrie AP, Van Mieghem FJE, McMahon HT, Nicholls DG. Repetitive action potentials in isolated nerve terminals in the presence of 4-aminopyridine: effects on cytosolic free Ca2+ and glutamate release. J Neurochem. 1989;53:1693–1699. doi: 10.1111/j.1471-4159.1989.tb09232.x. [DOI] [PubMed] [Google Scholar]
- 48.Togo T, Alderton JM, Bi GQ, Steinhardt RA. The mechanism of facilitated cell membrane resealing. J Cell Sci. 1999;112:719–731. doi: 10.1242/jcs.112.5.719. [DOI] [PubMed] [Google Scholar]
- 49.van der Bliek AM, Redelmeier TE, Damke H, Tisdale EJ, Meyerowitz EM, Schmid SL. Mutations in human dynamin block an intermediate stage in coated vesicle formation. J Cell Biol. 1993;122:553–563. doi: 10.1083/jcb.122.3.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Verhage M, McMahon HT, Ghijsen WEJM, Boomsma F, Scholten G, Wiegant VM, Nicholls DG. Differential release of amino acids, neuropeptides, and catecholamines from isolated nerve terminals. Neuron. 1991;6:517–524. doi: 10.1016/0896-6273(91)90054-4. [DOI] [PubMed] [Google Scholar]
- 51.Von Gersdorff H, Matthews G. Inhibition of endocytosis by elevated internal calcium in a synaptic terminal. Nature. 1994;370:652–655. doi: 10.1038/370652a0. [DOI] [PubMed] [Google Scholar]
- 52.Von Gersdorff H, Matthews G. Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. J Neurosci. 1997;17:1919–1927. doi: 10.1523/JNEUROSCI.17-06-01919.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- 54.Wu LG, Betz WJ. Nerve activity but not intracellular calcium determines the time course of endocytosis at the frog neuromuscular junction. Neuron. 1996;17:769–779. doi: 10.1016/s0896-6273(00)80208-1. [DOI] [PubMed] [Google Scholar]
- 55.Wu LG, Westenbroek RE, Borst JGG, Catterall WA, Sakmann B. Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci. 1999;19:726–736. doi: 10.1523/JNEUROSCI.19-02-00726.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]