

Abstract
RNU4ATAC pathogenic variants to date have been associated with microcephalic osteodysplastic primordial dwarfism, type 1 and Roifman syndrome. Both conditions are clinically distinct skeletal dysplasias with microcephalic osteodysplastic primordial dwarfism, type 1 having a more severe phenotype than Roifman syndrome. Some of the overlapping features of the two conditions include developmental delay, microcephaly, and immune deficiency. The features also overlap with Lowry Wood syndrome, another rare but well-defined skeletal dysplasia for which the genetic etiology has not been identified. Characteristic features include multiple epiphyseal dysplasia and microcephaly. Here, we describe three patients with Lowry Wood syndrome with biallelic RNU4ATAC pathogenic variants. This report expands the phenotypic spectrum for biallelic RNU4ATAC disorder causing variants and is the first to establish the genetic cause for Lowry Wood syndrome.
Keywords: epiphyseal dysplasia, Lowry Wood syndrome, microcephaly, RNU4ATAC, skeletal dysplasia
1 |. INTRODUCTION
Since the increase in use of molecular genetic testing, there has been a recurring theme wherein a gene found to be associated with a classic phenotype is later identified to be responsible for additional clinical disorders, often with overlapping features, creating a clinical spectrum. Based upon clinical and radiographic characteristics, by 1982, Majewski et al. were able to classify microcephalic osteodysplastic primordial dwarfism (MOPD) into types I, II, and III (Majewski & Goecke, 1982; Majewski, Ranke, & Schinzel, 1982; Majewski, Stoeckenius, & Kemperdick, 1982). Overtime, it was recognized that MOPD I and MOPD III were variants of the same disorder that had previously been described by Drs. Taybi and Linder as cephalo-skeletal dysplasia (Haan et al., 1989; Meinecke & Passarge, 1991; Meinecke, Schaefer, & Wiedemann, 1991; Taybi, 1992a, 1992b; Winter, Wigglesworth, & Harding, 1985) (OMIM #210710). In 2011, several groups identified biallelic RNU4ATAC mutations as causative in the classic severe MOPD I/III phenotype (Abdel-Salam et al., 2011; Edery et al., 2011; He et al., 2011; Nagy et al., 2012). Four years later, RNU4ATAC pathogenic variants were also shown to cause Roifman syndrome (Merico et al., 2015). Roifman Syndrome (OMIM #300258) is a rare disorder characterized by growth retardation, cognitive delay, spondyloepiphyseal dysplasia, immunodeficiency, and retinal dystrophy. While it was surprising that individuals with such varied severity of phenotype were allelic, in hindsight it is clear that there are overlapping features. Individuals with Roifman as well as MOPDI/III syndromes have growth, developmental, and immunological abnormalities, though with differing severity. They can also be differentiated on the basis of skeletal involvement: Roifman syndrome is a spondyloepiphyseal dysplasia (SED), while MOPDI/III is a spondyloe-pimetaphyseal dysplasia (SEMD).
Lowry Wood syndrome (LWS; OMIM #226969) is characterized by multiple epiphyseal dysplasia (MED) without spinal involvement and accompanied by microcephaly and intellectual disability. It has an autosomal recessive pattern of inheritance. Intrauterine growth restriction, short stature, congenital nystagmus, retinitis pigmentosa, elbow contractures, coxa vara, and joint dislocations have also been described (Brunetti-Pierri et al., 2003; Hankenson, Ozonoff, & Cassidy, 1989; Lowry & Wood, 1975; Lowry, Wood, Cox, & Hayden, 1989; Magnani et al., 2009; Nevin, Thomas, Hutchinson, Opitz, & Reynolds, 1986). After the description of RNU4ATAC being associated with Roifman syndrome, we asked whether LWS could be an allelic condition, based on several overlapping features between LWS and Roifman syndrome. Subsequently, RNU4ATAC pathogenic variants were identified as the molecular cause of LWS, and once again expanded the spectrum of phenotypes of the RNU4ATAC-opathies.
2 |. CLINICAL REPORT
Patient 1 (Figures 1a and 1b) was diagnosed clinically with LWS at 3.5 years of age. She was twin born via cesarean section at 35 weeks to her then 38-year-old mother. Family history is unknown because she and her twin were adopted. At birth, her OFC was 26.5 cm (Z-score −3.36), length 37.0 cm (Z-score −3.16), and BW 1.5 kg (Z-score −2.2). Initially, an atrial septal defect was noted on echocardiogram, and over time it spontaneously closed. She had delayed speech with first words at 27 months, and delayed motor milestones, first walking at 22 months. At 10 years and 9 months, she had an OFC of 41.2 cm (Z-score −8.6), heightof 118.2 cm (Z-score −3.5), and weightof 20.5 kg (Z-score −3.6). Additional clinical features included radiographic findings of MED, mild intellectual disability, arachnoid cyst, myopia, brachydactyly with hypoplastic middle phalanges, brachyclinodactyly V, and retinitis pigmentosa (no night vision, with peripheral vision loss and retinal pigmentary clumping). Previous research exome sequencing was not revealing. Based on the newly identified RNU4ATAC mutations in Roifman syndrome and the overlapping features with Lowry Wood, RNU4ATAC sequencing was performed. This testing identified NR_023343.1:n51G>A and NR_023343.1:n5A>C variants in trans, lying in the 5’ stem loop and stem II regions of u4atac respectively, where they are likely to disrupt both internal pairing to form the loop structure and essential base-pairing with u6atac. Based on ACMG standards, 51G>A is pathogenic and 5A>C is likely pathogenic (PS3, PS4 and PM1, PM2, PM3; respectively), (Richards et al., 2015). Given the known immunologic dysfunction associated with other patients with RNU4ATAC-opathies, immunologic testing was pursued, which demonstrated low IgG and IgM levels in the absence of a clear clinical immunodeficiency.
FIGURE 1.
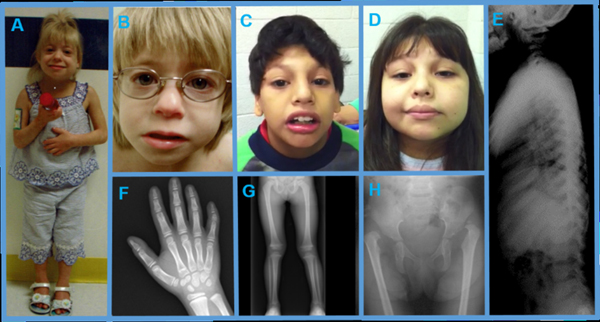
Sloping forehead, prominent nose, and full lips are present in all patients: patient 1 (a and b), patient 2 (c), and patient 3 (d). Representative X-rays demonstrate generalized epiphyseal dysplasia and normal vertebrae, consistent with MED. Findings include mild epiphyseal dysplasia and brachyclinodactyly V (f); epiphyseal dysplasia of the femoral heads, distal femur, and proximal and distal tibia (g); epiphysealdysplasia, shallow acetabula, dislocated femoral heads, and coxa valga (h); and normal vertebrae (e).
Patients 2 and 3 (Figures 1c and 1d) were a brother, age 14 years, and sister, age 15 years, diagnosed with LWS based on microcephaly, intellectual disability, and MED. Family history was otherwise unremarkable. Both were born full-term from uncomplicated pregnancies. Growth parameters at birth were OFC 31 cm (Z-score −2.7), L 45 cm (Z-score −3.1), and BW 2.85 kg (Z-score −1.8) for the brother; and OFC 31cm (Z-score −3.9), L 46 cm (Z-score −2.6), and 2.65 kg (Z-score −2.1) for the sister. At age 8, the sister was 96 cm tall (Z-score −6.3). At age 10, her OFC was 46 cm (Z-score −5.4). The brother had similar growth but recent height measurements are not available. His OFC at age 10 was 43 cm (Z-score −7.2). Both had full lips, prominent nose, sloping forehead, brachyclinodactyly V, contractures of the elbows bilaterally, and bilateral femoral head dislocations. In addition, the brother had scoliosis and coxa vara, while the sister had mild right knee contracture and coxa valga. They appeared to have normal immune system and normal vision, but had not had formal testing. Given the shared clinical diagnosis with patient 1, RNU4ATAC sequencing was performed and demonstrated three variants: NR_023343.1:n.111G>A in trans with NR_023343.1:n46G>A and NR_023343.1:n123G>A. The 111G>A and 46G>A variants were both previously reported in MOPDI (He et al., 2011; Kilic et al., 2015), are in trans, are predicted to disrupt the 5’ stem loop region and a region of importance for protein-binding, and are classified as pathogenic according to ACMG criteria (PS3, PS4 and PS4, PM2, PM3, PP1, PP4, respectively), (Richards et al., 2015) (Table 1).
TABLE 1.
Phenotypic features and RNU4ATAC variants in the presented LWS patients; as compared to reported presence in the three RNU4ATAC-associated disorders
| MOPDI/III | RS | LWS | P1 (10 year) | P2 (14 year) | P3 (15 year) | |
|---|---|---|---|---|---|---|
| U4atac snRNA variant | ||||||
| 51G>A | +* | +^ | − | + | − | − |
| 5A>C | +~* | − | − | + | − | − |
| 111G>A | +~** | − | − | − | + | + |
| 46G>A | − | − | − | + | + | |
| MED | − | − | + | + | + | + |
| SED | − | + | − | − | − | − |
| SEMD | + | − | − | − | − | − |
| Microcephaly | + | + | + | + | + | + |
| DD or ID | Severe | Mild | Mild | Mild | Mild | Mild |
| IUGR | Severe | Mild | Mild | Mild | Mild | Mild |
| Retinal anomalies | − | + | + | + | NE | NE |
| ↙Elbow extension | − | − | + | − | + | + |
| Hip dislocations | + | − | + | − | + | + |
| Irregular vertebrae | + | + | − | − | − | − |
| Immunodeficiency | + | + | − | Sub-clinical | NE | NE |
| Brachydactyly | + | + | − | + | − | − |
| Clinodactyly V | + | + | − | + | + | + |
| Cardiac defects | + | + | − | + | − | − |
| Contractures | + | + | − | − | − | + |
| Brain anomalies | Major | − | − | Minor | NE | NE |
| Restrictive lung disease | − | − | − | + | − | − |
MOPDI/III-microcephalic osteodysplastic dwarfism type I/III; RS-Roifman syndrome; LWS-Lowry Wood syndrome. P1, patient 1; P2, patient 2; P3, patient 3; P4, patient 4; MED, multiple epiphyseal dysplasia; SED, spondyloepiphyseal dysplasia; SEMD, spondyloepimetaphyseal dysplasia; DD, developmental delay; ID, intellectual disability; IUGR, intrauterine growth restriction; NE, not evaluated.
reported;
not reported;
MOPD1 causal, reduced severity;
3 |. DISCUSSION
In this study, we show that biallelic pathogenic variants in RNU4ATAC cause LWS, expanding the phenotypic spectrum of RNU4ATAC-opathies that includes MOPDI/III and Roifman syndrome. RNU4ATAC is located at 2q14.2 and encodes small nuclear RNA U4atac, a component of the minor spliceosome that is required for the correct excision of the U12-dependent class of introns (He et al., 2011).
MOPDI/III, Roifman syndrome, and LWS were historically categorized as separate clinical entities based on clinical and radiographic criteria. MOPDI/III is an SEMD, Roifman syndrome is an SED, and LWS is an MED. However, all are associated with varying degrees of IUGR, poor growth, epiphyseal dysplasia, microcephaly, intellectual disability, retinal abnormalities, and autosomal recessive inheritance. Features common to at least two of the disorders include joint dislocations, contractures, eczema, and immunodeficiency. The conditions are on a spectrum, with the short stature and overall phenotype associated with MOPDI/III being more severe than LWS and Roifman syndrome. Most MOPDI patients do not survive past the first year of life (Pierce & Morse, 2012). The mechanism through which RNU4ATAC variants cause different clinical phenotypes is unclear. In 2015, Merico et al. proposed that two variants which severely disrupt function cause MOPDI, while a mix of a variant severely disrupting function and a variant causing minor disruption to function causes Roifman syndrome (Merico et al., 2015). This was consistent in their cohort and for patients 2 and 3, whose variants were categorized as MOPDI variants with reduced severity and were found in an area of variable importance for splicing. However, both of the variants in patient 1 are in regions predicted by Merico et al. to be important for splicing (Supplemental Figure S1), which is inconsistent with their theory. Presently, at least three distinct clinical diagnoses are associated with RNU4ATAC pathogenic variants, and clinical features from all three overlap. Thus, the syndromes are less clinically distinct than previously considered and the mechanism for which the genotype causes clinical manifestations, although partially explained by their model, appears to be more complex. Promisingly, in 2012, Nagy et al., (2012) observed a possible genotype-phenotype correlation within their MOPDI/III patient cohort. They noted that patients homozygous for the 51G>A variant had a mean survival of 10.4 months compared to a mean survival of 78.75 months in patients with MOPDI/III caused by other mutations, and in compound heterozygotes for the 51G>A variant and another mutation (Nagy et al., 2012). Phenotypic analysis of a larger sample of patients with RNU4ATAC variants may yield more complete information for outcome predictions. Alternatively, modifying genes may be identified which may explain the phenotypic variability observed with RNU4ATAC variants.
This study demonstrates that RNU4ATAC pathogenic variants cause a spectrum of phenotypes involving multiple metaphases, epiphyses, with and without spinal involvement (MED, SED, and SEMD). This results in severe MOPDI/III to milder Roifman and LWS. Many patients are unlikely to fit perfectly into the clinically described MOPDI/III, Roifmann syndrome, or LWS; and instead may have features of all three. Since RNU4ATAC is not consistently targeted in standard exome capture methodologies that only target protein-coding exons, many such cases will be missed. Sanger sequencing of this locus is needed for a complete assessment. As the phenotype may continue to expand, targeted RNU4ATAC sequencing should be considered in undiagnosed patients who have any combination of epiphyseal dysplasia with intellectual disability, microcephaly, immunodeficiency, and/or retinal anomalies.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank the Potentials Foundation and the Walking With Giants Foundation for their support, and all primordial families for their participation in research throughout the world. Their contribution to the knowledge about these rare conditions has provided important information towards the care of these conditions. This work was funded by the Rare Disease Clinical Research Network (1U54NS092090–01) (LSF), Medical Research Council UK (APJ) and the European Research Council (ERC; award 281847, APJ), Walking with Giants Foundation (MB), Potentials Foundation (MB), and Leah Lewis Foundation (JTH).
Funding information
Leah Lewis Foundation; Medical Research Council UK; Rare Disease Clinical Research Network, Grant number: IU54NS092090–01; H2020 European Research Council,
Grant number: 281847; Walking with Giants Foundation; Potentials Foundation
Footnotes
SUPPORTING INFORMATION
Additional Supporting Information may be found online in the supporting information tab for this article.
REFERENCES
- Abdel-Salam GM, Miyake N, Eid MM, Abdel-Hamid MS, Hassan NA, Eid OM, ... Matsumoto N (2011). A homozygous mutation in RNU4ATAC as a cause of microcephalic osteodysplastic primordial dwarfism type I (MOPD I) with associated pigmentary disorder. American Journal of Medical Genetics Part A, 155(11), 2885–2896. [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri N, De Brasi D, Ikegawa S, Camera G, Andria G, & Sebastio G (2003). A new patient with Lowry-Wood syndrome with mild phenotype. American Journal of Medical Genetics Part A, 118(1), 68–70. [DOI] [PubMed] [Google Scholar]
- Edery P, Marcaillou C, Sahbatou M, Labalme A, Chastang J, Touraine R, ... Leutenegger AL (2011). Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science, 332(6026), 240–243. [DOI] [PubMed] [Google Scholar]
- Haan EA, Furness ME, Knowles S, Morris LL, Scott G, Svigos JM, & Vigneswaren R (1989). Osteodysplastic primordial dwarfism: Report of a further case with manifestations similar to those of types I and III. American Journal of Medical Genetics Part A, 33(2), 224–227. [DOI] [PubMed] [Google Scholar]
- Hankenson LG, Ozonoff MB, & Cassidy SB (1989). Epiphyseal dysplasia with coxa vara, microcephaly, and normal intelligence in sibs: Expanded spectrum of Lowry-Wood syndrome? American Journal of Medical Genetics Part A, 33(3), 336–340. [DOI] [PubMed] [Google Scholar]
- He H, Liyanarachchi S, Akagi K, Nagy R, Li J, Dietrich RC,... Singh J (2011). Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science, 332(6026), 238–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilic E, Yigit G, Utine GE, Wollnik B, Mihci E, Nur BG, & Boduroglu K (2015). A novel mutation in RNU4ATAC in a patient with microcephalic osteodysplastic primordial dwarfism type I. American Journal of Medical Genetics Part A, 167, 919–921. [DOI] [PubMed] [Google Scholar]
- Lowry RB, & Wood BJ (1975). Syndrome of epiphyseal dysplasia, short stature, microcephaly, and nystagmus. Clinical Genetics, 8(4), 269–274. [DOI] [PubMed] [Google Scholar]
- Lowry RB, Wood BJ, Cox TA, & Hayden MR (1989). Epiphyseal dysplasia, microcephaly, nystagmus, and retinitis pigmentosa. American Journal of Medical Genetics Part A, 33(3), 341–345. [DOI] [PubMed] [Google Scholar]
- Magnani C, Tedesco SA, Dallaglio S, Sommi M, Bacchini E, Vetro A, ... Bevilacqua G (2009). Multiple joint dislocations: An additional skeletal finding in LowryWood syndrome? American Journal of Medical Genetics Part A, 149(4), 737–741. [DOI] [PubMed] [Google Scholar]
- Majewski F, & Goecke T (1982). Studies of microcephalic primordial dwarfism I: Approach to a delineation of the Seckel syndrome. American Journal of Medical Genetics Part A, 12(1), 7–21. [DOI] [PubMed] [Google Scholar]
- Majewski F, Ranke M, & Schinzel A (1982). Studies of microcephalic primordial dwarfism II: The osteodysplastic type II of primordial dwarfism. American Journal of Medical Genetics Part A, 12(1), 23–35. [DOI] [PubMed] [Google Scholar]
- Majewski F, Stoeckenius M, & Kemperdick H (1982). Studies of microcephalic primordial dwarfism III: An intrauterine dwarf with platyspondyly and anomalies of pelvis and clavicles—osteodysplastic primordial dwarfism type III. American Journal of Medical Genetics Part A, 12(1), 37–42. [DOI] [PubMed] [Google Scholar]
- Meinecke P, & Passarge E (1991). Microcephalic osteodysplastic primordial dwarfism type I/III in sibs. Journal of Medical Genetics, 28(11), 795–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinecke P, Schaefer E, & Wiedemann HR (1991). Microcephalic osteodysplastic primordial dwarfism: Further evidence for identity of the so-called types I and III. American Journal of Medical Genetics Part A, 39(2), 232–236. [DOI] [PubMed] [Google Scholar]
- Merico D, Roifman M, Braunschweig U, Yuen RK, Alexandrova R, Bates A,... Gray P (2015). Compound heterozygous mutations in the noncoding RNU4ATAC cause Roifman syndrome by disrupting minor intron splicing. Nature Communications, 6, 8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy R, Wang H, Albrecht B, Wieczorek D, Gillessen-Kaesbach G, Haan E, ... Westman JA (2012). Microcephalic osteodysplastic primordial dwarfism type I with biallelic mutations in the RNU4ATAC gene. Clinical Genetics, 82(2), 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevin NC, Thomas PS, Hutchinson J, Opitz JM, & Reynolds JF (1986). Syndrome of short stature, microcephaly, mental retardation, and multiple epiphyseal dysplasia-Lowry-Wood syndrome. American Journal of Medical Genetics Part A, 24(1), 33–39. [DOI] [PubMed] [Google Scholar]
- Pierce MJ, & Morse RP (2012). The neurologic findings in Taybi-Linder syndrome (MOPD I/III): Case report and review of the literature. American Journal of Medical Genetics Part A, 158(3), 606–610. [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, ... Voelkerding K (2015). Standards and guidelines for the interpretation of sequence variants: Joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taybi H (1992). Cephalo-skeletal dysplasia and microcephalic osteodysplastic primordial dwarfism. Pediatric Radiology, 22(6), 476–476. [DOI] [PubMed] [Google Scholar]
- Taybi H (1992). Microcephalic osteodysplastic primordial dwarfism and cephalo-skeletal dysplasia (Taybi-Linder syndrome). American Journal of Medical Genetics Part A, 43(3), 628–628. [DOI] [PubMed] [Google Scholar]
- Winter RM, Wigglesworth J, & Harding BN (1985). Osteodysplastic primordial dwarfism: Report of a further patient with manifestations similar to those seen in patients with types I and III. American Journal of Medical Genetics Part A, 21(3), 569–574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.