

Abstract
In this systematic review article, we aim to summarize the most up-to-date evidence regarding elevations of cardiac troponin, especially in clinical scenarios other than obstructive coronary artery disease. The accurate interpretation of raised cardiac troponin is challenging because it relies on unconfirmed postulations and dogmatic knowledge (e.g., the exclusive provenience of cardiac troponin from cardiac myocytes), based on which every troponin elevation is assumed to definitely indicate myocardial damage. Indeed, the investigation of the pathophysiologic mechanism leading to the release in the bloodstream of cardiac biomarkers should be the first step of the diagnostic process to fully understand the clinical significance of the elevated serum levels and identify the best management. A prominent effort should be put in place to identify the contribution of potential confounding factors, both cardiac and non-cardiac in etiology, with the ability to affect synthesis and clearance of cardiac biomarkers. Regardless of the underlying cause, it is well established that cardiovascular biomarkers are increasingly useful to further risk stratification and prognosticate patients. Accordingly, we sought to clarify the meaning and impact of elevated cardiac troponin in those frequently encountered real-world scenarios presenting clinicians with a diagnostic dilemma, with the final goal of facilitating the diagnosis and help optimize individually tailored treatment strategies.
MeSH Keywords: Biological Markers, Coronary Artery Disease, Myocardial Ischemia, Troponin
Background
Since the landmark discovery of the utility of serum biomarkers in diagnosing myocardial necrosis more than a half century ago [1], numerous advances and evolving concepts in macromolecular biomarkers have emerged. Approximately 40 years ago, the International Society and Federation of Cardiology and World Health Organization (WHO) defined the initial diagnostic criteria for ischemic heart disease [2]. Subsequently, it was recognized by expert consensus opinion that acute myocardial infarction (MI) can be detected by utilization of pertinent clinical history, electrocardiographic (ECG) findings, and highly sensitive biochemical markers for myocardial injury [3]. With the advent of more accurate cardiovascular biomarkers, the term myocardial infarction was updated with a more precise and meaningful definition to assist in immediate treatment directives: revascularization and reperfusion strategies. In 2018, a joint taskforce of the European Society of Cardiology/American College of Cardiology Federation/American Heart Association/World Heart Federation revised the definition of MI as myocardial necrosis secondary to ischemia [4]. It is now a standard practice to first categorize MI, based on ECG findings, as ST-segment elevation myocardial infarction (STEMI) versus non-ST-segment elevation myocardial infarction (NSTEMI), and then further distinguish MI into 5 groups based on clinical, pathological, and prognostic variations, as well as treatment dictums (Table 1). Acute coronary syndrome (ACS) includes unstable angina, NSTEMI, and STEMI; it is a continuum of myocardial ischemia due to an abrupt decrease of oxygen-rich blood flow through the coronary arteries [4]. The measurement of serum cardiac troponin (cTn) levels has become the criterion standard biomarker in diagnosing myocardial ischemia and infarction [5,6]. Indeed, as cardio-specific contractile proteins, cardiac troponin T (cTnT) and I (cTnI) have been shown to be superior to creatine kinase MB (CK-MB) in the detection of myocardial injury, especially in instances of minor damage, due to their high sensitivity and favorable kinetics (Tables 2–4) [7,8].
Table 1.
Classification of myocardial infarction [4].
| Type 1: Spontaneous MI |
| Related to atherosclerotic plaque rupture, ulceration, fissuring, erosion, or dissection leading to intraluminal thrombus in one or more coronary arteries leading to ischemia and resultant myocyte necrosis |
| Type 2: MI secondary to oxygen supply and demand mismatch/ischemia imbalance |
| Ischemia imbalance leading to myocyte necrosis and is resultant from a condition other than CAD |
| Type 3: MI secondary to sudden cardiac death (biomarker values unavailable) |
| Cardiac death associated with symptoms and ECG findings suggestive of ischemia in the absence of cardiac biomarkers |
| Type 4a: MI secondary to percutaneous coronary intervention (PCI) |
| PCI-associated MI defined arbitrarily as an elevation in cTn >5×99th percentile of the upper limit of normal with normal baseline values or a rise of cTn values >20% of previously elevated baseline values that are stable or falling in association with the presence of at least one of the following: symptoms suggestive of cardiac ischemia, new ischemic ECG findings or new LBBB, angiographic loss of patency of a major coronary vessel or a side branch or persistent slow- or no-flow or embolization, or imaging demonstrating new loss of viable myocardium or new regional wall motion abnormality |
| Type 4b: MI secondary to stent thrombosis |
| MI associated with stent thrombosis as detected by coronary angiography or autopsy with a combination of myocardial ischemia and rise and/or fall of cardiac biomarkers with at least one value ≥99th percentile of the upper limit of normal |
| Type 5: MI secondary to coronary artery bypass grafting (CABG) |
| MI associated with CABG defined arbitrarily as an elevation in cardiac biomarker levels >10×99th percentile of the upper limit of normal in patients with normal baseline values in addition to at least one of the following: new pathological Q waves or new LBBB, new graft or new native coronary artery occlusion, or imaging demonstrating new loss of viable myocardium or new regional wall motion abnormality |
Table 2.
| • High concentration in the heart muscle |
| • Absence from other non-cardiac tissue |
| • Rapid release into the blood following myocardial injury |
| • High sensitivity and specificity in laboratory serum samples |
| • Favorable kinetics for detection of myocardial damage and acute coronary syndrome in the hours or days when patients seek medical attention |
| • Correlation between cardiac biomarker and extent of myocardial injury and prognosis |
| • Wide-spread assay availability that are simple, automated, and rapid |
| • Well-defined role in diagnosis and management based on medical peer-reviewed literature and clinical trials |
Table 3.
| Modern markers in use | Cardiac Troponin T |
| Cardiac Troponin I | |
| CK and CK-MB with relative index | |
| Point-of-care Troponin I, CK-MB and myoglobin panel | |
| Obsolete markers | Aspartate transaminase |
| Lactate dehydrogenase | |
| Markers in development or undergoing further study | High-sensitivity troponin assay |
| High-sensitivity C-reactive protein | |
| Urocortin | |
| CK-MB isoforms | |
| B-type natriuretic peptide |
Table 4.
| Marker | Onset | Peak | Return to baseline |
|---|---|---|---|
| Troponin T | 4–9 h | 12–24 h | 7–14 d |
| Troponin I | 4–9 h | 12–24 h | 7–14 d |
| CK/CK-MB | 4–9 h | 24 h | 2–3 d |
| Myoglobin | 1 h | 4–12 h | 24 h |
Conventionally, the term myocardial injury is used in a broad fashion to describe a disorder in which cardiac troponin is elevated in the serum, with at least 1 value above the 99th percentile upper reference limit (URL). Such injury is classified as acute if characterized by a rise and/or fall in cardiac troponin values (Figure 1). Elevated cardiac troponin can also be observed in the absence of acute myocardial ischemia or obstructive CAD in a host of clinical conditions (reviewed in Table 5), confirming that cardiac biomarkers are specific for the heart but not for ACS.
Figure 1.
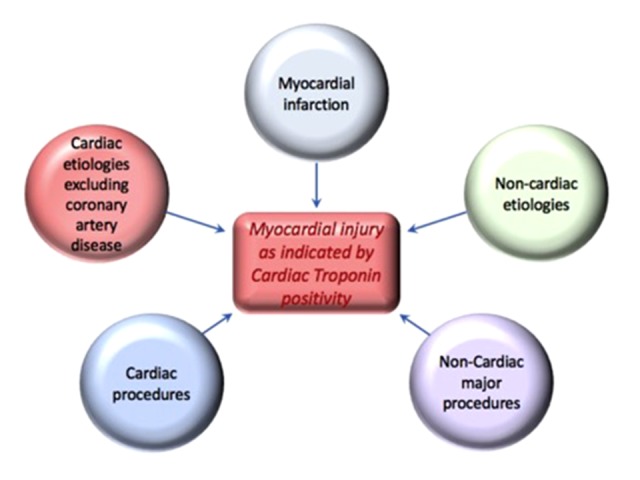
Various clinical scenarios causing an elevation in cardiac troponins.
Table 5.
Various etiologies of cardiac troponin positivity in myocardial injury [4].
| Primary Myocardial Ischemia | Plaque rupture |
| Intraluminal formation of coronary artery thrombus | |
| Supply-Demand Mismatch/Imbalance Leading To/Associated with Myocardial Ischemia | Coronary vasospasm |
| Coronary embolism or vasculitis | |
| Coronary endothelial (microvascular) dysfunction without obstructive (significant) CAD | |
| Hypertension with and without left ventricular hypertrophy | |
| Hypertrophic cardiomyopathy | |
| Cardiogenic, hypovolemic, or septic shock | |
| Aortic dissection | |
| Severe aortic valve disease | |
| Severe anemia | |
| Severe respiratory failure | |
| Tachy-/brady-arrhythmias | |
| Cardiac-related in the Absence of CAD | Cardiac contusion |
| Cardiac procedure such as ablation, pacing, or defibrillator shocks | |
| Cardiac surgery | |
| Myocarditis | |
| Cardiotoxic agents, i.e., anthracyclines, Herceptin | |
| Rhabdomyolysis with cardio-involvement | |
| Multi-factorial or Indeterminate | Heart failure |
| Stress (Takotsubo) cardiomyopathy | |
| Massive pulmonary embolism or pulmonary hypertension | |
| Sepsis or critical illness | |
| Acute kidney injury or chronic kidney disease | |
| Severe acute central nervous system disease, i.e., stroke, subarachnoid hemorrhage | |
| Infiltrative diseases, i.e., amyloidosis, sarcoidosis | |
| Strenuous exercise |
Slight elevations in serum cTn often lead to extensive and costly cardiovascular workups, which may sidetrack clinicians, leading them astray from the true cause of cTn elevation. The present article reviews a) the pathophysiology of cTn release from the heart and different organs, and b) the clinical scenarios leading to cTn elevations, with a special focus on those unrelated to obstructive coronary artery disease.
Physiology of cardiac troponin
Troponins are integral regulatory proteins located on the thin actin filament within the myocytes of striated heart muscle, and are released when cardiac myocyte injury occurs. These proteins are responsible for the intricate contraction-relaxation cycle of myocytes (Figure 2). The cTn complex is composed of 3 different subunits: cardiac troponin-C (cTnC, the calcium-binding component), cTnT (the tropomyosin-binding component), and cTnI (the inhibitory-activity regulator of the myosin-binding sites on actin thin filaments) [9]. During depolarization of the cardiac myocyte, calcium enters the sarcoplasm, binds to cTnC, and induces structural changes in the cTn complex. The resulting shift of tropomyosin away from the active site of actin allows the myosin heads of the thick filament to interact with the now-exposed myosin-binding site of the actin filaments, thereby producing contraction of the sarcomere and, consequently, the myocardium as a syncytium [9,10]. The existence of several isoforms of troponin depends upon differential gene expression in particular tissues. For example, expression of cTnC occurs in both cardiac and skeletal muscle, making it a poor indicator of myocardial injury. In contrast, cTnT and cTnI are expressed almost exclusively in myocardial tissue, making them ideal markers of myocardial damage [10]. Both cTnT and cTnI possess unique N-terminal amino acid sequences, ideal targets for modern serum assays [11], which were developed using antibodies directed against these sequences [12]. Utilization of these assays to detect cTn positivity in serum has proven both highly sensitive and specific for myocardial injury [13]. Prior to their development, clinical decision-making relied on the detection of other molecular biomarkers such as CK-MB, lactate dehydrogenase, and myoglobin, which all lacked specificity, due to their expression in the musculoskeletal tissue (Table 3) [14]. The various biomarkers have different properties, with troponins having the most ideal kinetics, along with the highest sensitivity and specificity, for acute coronary syndromes (Table 4) [1].
Figure 2.
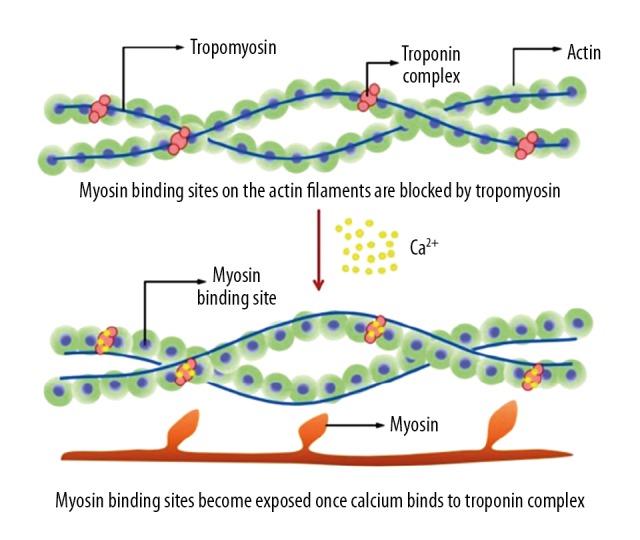
Physiology of contraction-relaxation cycle in cardiac myocytes and role of troponin [10].
Pathophysiology of Troponin Release
Detection of cTn proteolytic products
Accurate interpretation of cTn positivity can be challenging because it relies on unverified assumptions, such as the exclusive provenience of cTn from cardiac myocytes, the freedom from laboratory error, the appropriate time of acquisition of blood samples, and the cognizance of the patient baseline cTn values [15]. Based on these postulations, every incremental change in cTnT, cTnI, and CK-MB, even the smallest, is seen as persuasive evidence for myocardial injury [5,6,15,16]. However, such a dogma seems to be challenged by a fresher revisitation of old findings. It is known that elevated cTn levels can reflect not only the detection of intact cTn, but also of its proteolytic degradation products, generated by the calcium-activated protease, calpain. Such products, due to their reduced size, may egress from cells more easily and rapidly than their parent structural proteins. Calpain activation, eventuating in generation of proteolytic products, may explain the apparent inconsistency of modest, persistent elevations of cTn associated with negative CK-MB, which can occur in some patients suffering from recurrent bouts of short-lasting sublethal myocardial ischemia, or in the setting of systemic inflammatory processes [15]. Furthermore, recent findings indicate that cardiac troponin T (cTnT) is also expressed in human smooth muscle cells of different organs and apparatuses, including aorta, trachea, gut, and urinary bladder [17]. Hence, calpain activation at these sites would result, at least theoretically, in mild troponinemia (i.e., a slight troponin increase), which is totally unrelated to myocardial ischemia or any other cardiac injury.
Detection of intact cTn
Disruption of myocyte cell membrane integrity (occurring in any type of myocardial injury) results in leakage of cytoplasmic proteins into the extracellular serum. The majority of cTn is bound to actin thin filaments; however, a small proportion of approximately 3–8% exists in an unbound state, free in the cytosol [18]. During myocardial injury, this unbound troponin is released first and detected as the initial rise of troponin assays [19]. Troponin elevations from this pool, in cases of short-lasting myocardial ischemia or injury, would be expected to quickly rise and fall over the course of hours due to the relatively short half-life (2–4 hours) of cTnI and cTnT [20]. However, if there is significant injury causing extensive and progressive necrosis, a continued release of the myofibril-bound troponin pool results in elevations of cTnI and cTnT for up to 7–14 days. From a pathophysiological standpoint, release of cTn into the circulation can be caused by 2 major categories of events: 1) myocardial injury due to ischemia (including ACS), and 2) myocardial injury in the absence of ischemia.
Ischemic myocardial injury
The majority of ischemic myocardial injury is caused by obstructive coronary artery disease, which may result in ACS. Ischemia, defined as reduced oxygen availability due to lack of blood flow, causes a series of crucial biochemical and metabolic changes, which decrease mitochondrial oxidative phosphorylation and deplete cellular adenosine triphosphate (ATP). If ischemia is prolonged, metabolism shifts to anaerobic glycolysis with lactate accumulation and consequent intracellular acidosis, which activates the Na+-H+ exchanger. The ensuing acidosis and lack of ATP alters Na+-K+ ATPase function and causes accumulation of cytosolic Na+, which is exchanged with Ca2+ from the sarcoplasmic reticulum, resulting in overload of intracellular Ca2+ [21]. Such alterations in cellular metabolism lead to osmotic cell stress, lysosomal activation, reactive oxygen species production, and infiltration of inflammatory cells, which actively release mediators, promoting both inflammation and increased catabolism [22]. Accordingly, the metabolic state arising from ischemic myocardial injury results in catabolic protein degradation and compromised cell membrane integrity, allowing for leakage of cytoplasmic proteins into the extracellular serum, and subsequent detection of cTn on serum assays.
Clinical encounters associated with ischemic myocardial injury
Besides obstructive CAD and ACS, typically occurring in the setting of thrombotic coronary artery obstruction, elevation of cTn may be caused by a number of other disease states that do not compromise coronary artery patency. Elevation of cTn in the absence of obstructive CAD is often caused by ischemia secondary to an imbalance between demand and supply for oxygen-rich blood flow (demand ischemia). Thus, in the 2018 Joint ESC/ACCF/AHA/WHF Fourth Universal Definition of MI, demand ischemia is referred to as type-2 myocardial infarction, which is cardiac ischemia due to either increased oxygen demand or decreased supply, occurring in the absence of an acute primary coronary thrombotic event [4]. The conditions leading to demand ischemia are very broad and diverse. The most commonly observed disorders in clinical practice include fixed coronary artery atherosclerosis, coronary spasm, coronary embolism caused by thrombi, calcium, or vegetation originated from atria or ventricles, coronary artery dissection with or without intracoronary hematoma, tachyarrhythmias, bradyarrhythmias, severe hypertension with or without left ventricular hypertrophy, hypovolemic shock and other types of hypotension, and severe anemia (Table 5).
Myocardial injury in the absence of ischemia
Numerous clinical conditions other than ACS and ischemic myocardial injury have been recognized to cause elevated cTn, with several occurring independent of myocardial necrosis [23–25]. Different mechanisms for troponin release in non-ACS disorders have been postulated, including, but not limited to, increased cell membrane permeability due to reversible injury or myocardial stretch [26] and increased rates of cell turnover, whether physiologic or pathologic [27]. Such mechanisms seem to have a common denominator in the hypercatabolic state associated with the generation of inflammatory and/or hormonal catabolic molecules.
Indeed, it is well known that inflammatory molecules, like those produced from reperfusion injury, can cause the formation of reactive oxygen species (ROS) via altered mitochondrial oxidation and function. ROS interact with cell membrane phospholipids, which alters membrane permeability and favors the extracellular leakage of cytosolic proteins [28]. Cell membrane dysfunction also causes cytosolic Ca+ overload, which is particularly important because Ca2+ activates specific proteases able to reinforce myofibril degradation [28,29]. Moreover, levels of circulating catecholamines and inflammatory cytokines, like tumor necrosis factor-alpha (TNF-alpha), interleukin-1 (IL-1), and interleukin-6 (IL-6), are known to increase in catabolic conditions such as sepsis, immunological diseases, trauma, and/or chronic diseases. It was recently postulated that such molecules stimulate muscular fiber proteolysis, most notably in skeletal muscle, but also in cardiac myocytes, contributing to the hypercatabolic state [30]. Furthermore, animal studies showed that IL-6, TNF-alpha, and circulating catabolic molecules bind specific cardiomyocyte membrane receptors, engaging an intracellular cascade (likely via the phosphatidylinositol (PI3K)/Aky pathway), whose activation results in protein disarrangement. Protein Kinase B (or Ak strain transforming/AKT), a key molecule in such a pathway, decreases the phosphorylation (and therefore the activity level) of mammalian target of rapamycin (mTOR), which exerts pro-catabolic effects by inhibiting the synthesis of survival proteins such as eukaryotic initiation factor binding protein-1 (4E-BP1) and S6 ribosomal protein, and activating the ubiquitin proteasome system, with ensuing intracellular protein breakdown [31]. The adverse effects of cytokines on cardiac cells and sarcomeres are compounded by other molecular mechanisms responsible for proteolysis and myocyte loss. Indeed, binding of TNF-alpha to its membrane receptor results in direct myocyte apoptosis (elicited by activation of the death receptor, caspase 8-mediated pathway), and activation of other pro-apoptotic molecules such as Bcl-2-associated X protein (BAX) [32]. Figure 3 illustrates the potential mechanisms of contractile protein breakdown associated with hypercatabolic states.
Figure 3.

Postulated biochemical mechanisms responsible for hypercatabolic protein breakdown.
Clinical encounters of elevated cardiac biomarkers in the absence of ischemia
Numerous conditions, varied in presentation and nature, can engender myocardial injury in the absence of ischemia. Such conditions include: sepsis, cerebrovascular accidents, cardiotoxic medications, renal failure, infiltrative diseases, acute respiratory failure, extreme exertion, cardiac contusion, burns affecting >30% of the body surface area, pacing and electrical shock in the setting of cardioversion, carbon monoxide exposure, Takotsubo cardiomyopathy, peripartum cardiomyopathy, heart failure, endocarditis, myocarditis, myopericarditis, rhabdomyolysis, and sequelae of malignancy. Among these, the most studied are sepsis, neurogenic diseases such as stroke and subarachnoid hemorrhage, and cardiotoxic medications such as anthracyclines and trastuzumab.
Sepsis-induced cardiomyopathy
Despite major advances in an organized treatment approach to sepsis over the last 2 decades, sepsis remains the most common cause of death in critically ill patients [33]. Sepsis-induced cardiomyopathy is a common complication of sepsis, defined by left ventricular dilation with normal or low filling pressures, a decrease in LVEF, and normalization of cardiac function, typically within 7–10 days [34]. However, sepsis-induced cardiomyopathy has also been described in the setting of normal LVEF due to complex hemodynamic changes during sepsis, including decreased systemic vascular resistance, tachycardia, and significant alterations in intravascular volume during resuscitation [35]. Septic cardiomyopathy commonly results in elevated serum levels of cardiac troponin, which has significant prognostic value. The pathophysiologic mechanisms underlying septic cardiomyopathy involve the release of cardiac-depressive factors like endotoxin and endogenous inflammatory cytokines (TNF-alpha, IL-1, and IL-6), thereby increasing nitric oxide production. Excess nitric oxide depresses myofibril response to calcium, downregulates beta-adrenergic receptors, and leads to global mitochondrial dysfunction [34]. These cellular alterations cause protein demolition, impair the contractile apparatus, and increase the microvascular permeability of cardiac myocytes [35,36], which can facilitate muscle contractile protein breakdown and troponin release. Inflammatory cytokines like TNF-alpha, IL-1, and IL-6 have been shown to cause degradation of proteins, including free troponin, to lower molecular-weight fragments, which are released into systemic circulation through the highly permeable membranes of cardiac myocytes [30]. The detection of these small troponin fragments by conventional assays would explain troponinemia in the absence of myocyte necrosis. Although this hypothesis is highly contentious and not supported by definitive evidence, the clinical observation that sepsis-related myocardial depression resolves without residual ventricular wall motion abnormalities [34] suggests that cardiac enzymes can be released in the absence of significant cardiomyocyte cell death.
Septic cardiomyopathy is commonly accompanied by an elevation of serum cTn, which is highly correlated with left ventricular dysfunction and increased mortality [34,35,37–39]. In one study of 58 consecutive patients admitted to the medical intensive care unit with sepsis, all patients with LVEF <45% had detectable serum levels of cardiac troponin. The degree of troponin elevation demonstrated a significant inverse correlation with LVEF, and 30-day mortality was 4 times greater in patients with elevated troponin levels [38]. Regardless of the mechanism of troponin leakage, septic cardiomyopathy demonstrates that elevated serum troponin, especially in critically ill patients, should not be assumed to indicate ACS or the presence of obstructive coronary artery disease. Critically ill patients often have multiple non-coronary etiologies of serum cTn elevation, which may be deceivingly heightened or prolonged by concomitant renal impairment. Unfortunately, no specific treatment exists for septic cardiomyopathy except for implementing evidence-based, guideline-directed therapy for the treatment of sepsis [35]. While it is not recommended by guideline-directed therapy, physicians may utilize cardiac enzymes as an adjunctive tool to risk-stratify septic patients in the intensive care unit in an effort to optimize treatment strategies and improve outcomes.
Neurogenic cardiac damage
An elevation of cTn, often associated with electrocardiogram abnormalities and clinical evidence of left ventricular dysfunction, is detected in up to 20% of patients after stroke, particularly subarachnoid hemorrhage [40]. Besides preexisting CAD, another postulated mechanism for cardiac damage in this specific patient population seems to be myocardial injury elicited by centrally mediated release of catabolic molecules like catecholamines in response to hypoperfusion of the posterior hypothalamus. A powerful catecholamine surge may be an alternative mechanism of cardiac damage, because troponinemia is often detected even in young individuals with very low pretest probability for an acute coronary syndrome. Cardiac myofibrillar degeneration, which was described in patients who died due to acute stroke, occurs in the proximity of the cardiac nerves and is histologically indistinguishable from myocyte death caused by catecholamine infusion or reperfusion of transiently ischemic myocardium [41], being characterized by contraction bands necrosis and associated with mononuclear infiltration and early calcification. Mounting evidence suggests that stroke affecting the insular cortex, which plays a central role in the autonomic control of cardiovascular function, is associated with increased risk of adverse cardiac outcomes, including neurogenic cardiac damage. Brain regions associated with cTnT elevation include the right posterior, superior, and medial insula, and the right inferior parietal lobule.
Medication-induced troponin release
Many medications, especially chemotherapeutics and anti-neoplastic agents, have adverse effects on cardiac myocytes and contribute to the development of cardiovascular complications such as hypertension, heart failure, arrhythmias, and ischemia, all of which may be accompanied by elevated serum cardiac biomarkers [42]. Advances in cancer treatment provide a unique opportunity for cardiologists and oncologists to collaborate in an effort to improve treatment outcomes for patients with concomitant heart disease and malignancy [43]. One way to mitigate the cardiovascular complications of cancer treatment is to expand the current guidelines regarding cardiac monitoring during the administration of cardiotoxic chemotherapeutic agents. Besides cardiac imaging, physicians have also discussed the routine monitoring of serum cardiac troponin before and during treatment with cardiotoxic medications, in an effort to diagnose cardiotoxicity at its earliest stages, when it is more amenable to medical treatment [44].
While a comprehensive review of cardiotoxic medications is beyond the scope of this discussion, Table 6 reports some of the medications commonly implicated in the causality of cardiac damage. A brief review of the most studied cardiotoxic medications is presented below.
Table 6.
| Anti-metabolites | Decitabine |
| Clofarabine | |
| Alkylating agents | Cyclophosphamide |
| Iphosphamide | |
| Melphalan | |
| Small molecule tyrosine kinase inhibitors | Sunitinib and sorafenib |
| Pazopanib | |
| Dabrafenib and dasatinib | |
| Lapatinib and trametinib | |
| Microtubule polymerization inhibitors | Paclitaxel |
| Docetaxel | |
| Anthracyclines | Doxorubicin |
| Daunorubicin | |
| Epirubicin | |
| Idarubicin | |
| Proteasome inhibitors | Carfilzomib and bortezomib |
| Monoclonal antibody-based tyrosine kinase inhibitors | Trastuzumab |
| Bevacizumab | |
| Adotrastuzumab emtansine | |
| Pertuzumab |
Anthracyclines injure cardiac myocytes primarily by inhibiting topoisomerase 2-beta, which increases breaks in double-stranded DNA [45] in addition to increasing free radical production via iron deposition in mitochondria [46]. These mechanisms can lead to apoptosis, necrosis, and permanent cardiomyopathy [45,46]. Anthracyclines also increase intracellular calcium levels and cause direct cellular injury through drug metabolism, oxidative damage, and influx of pro-inflammatory molecules. A direct correlation exists between the cumulative dose of anthracycline, the magnitude of troponin elevation, and the subsequent degree of left ventricular dysfunction, which is often permanent [47].
Trastuzumab is a monoclonal antibody that antagonizes the human epidermal growth factor receptor 2 (HER-2), which is expressed in 20–25% of breast cancers. It has reduced disease recurrence and increased survival in patients with breast cancers that express HER-2, especially when it is combined with anthracyclines like doxorubicin [48]. Unfortunately, this combination results in cardiac dysfunction in approximately 25% of patients [49]. Anthracycline-induced oxidative damage to cardiomyocytes results in upregulation of HER-2 receptors as part of cellular repair. While trastuzumab inhibition of HER-2-mediated cell repair plays a role in the loss of cardiac myocytes, the precise mechanism of trastuzumab-induced cardiomyopathy is still unclear [49]. Such cardiomyopathy differs from that caused by anthracyclines in that the cardiac dysfunction is not dose-dependent, and it is reversible in approximately 60% of patients [50]. Elevated cardiac biomarkers in the setting of trastuzumab regimen decrease the chance of cardiac recovery and increase the risk for major adverse cardiac events (MACE) [50]. Patients with elevated troponin who were treated with trastuzumab had a greater risk of developing cardiac dysfunction and a 25-fold increase in MACE [49]. Patients at highest risk were those treated with both anthracyclines and trastuzumab [51], requiring cardiac monitoring with serial echocardiograms or multigated acquisition scans (MUGA), due to the risk of cardiac injury and left ventricular dysfunction. High-sensitivity cTn measured prior to and during the administration of trastuzumab seems to be useful in the early identification of patients at higher risk of developing drug-induced cardiotoxicity, as well as of those whose cardiac function will remain impaired despite an appropriate medical regimen [50].
While anthracyclines and trastuzumab are the most studied cardiotoxic medications used in cancer treatment, a host of anti-neoplastic agents are known to produce cardiac dysfunction and have the potential to cause an elevation of cardiac biomarkers. The major classes of anti-neoplastic agents most commonly associated with troponin elevations are listed in Table 6. As the field of oncology continues to advance, more malignancies are being managed with a variety of novel therapeutics. The evaluation of cardiac biomarkers before and during cancer treatment, in conjunction with serial imaging studies, is currently the earliest and most accurate way to identify cardiotoxicity, and has the potential to be integrated into future cardio-oncology guidelines.
Conclusions
In this review, we described the different pathophysiological mechanisms of cTn release, and appraised a variety of clinical conditions presenting with elevated cTn, giving special attention to those cases of troponinemia unrelated to obstructive CAD.
Ischemic myocardial injury is most commonly caused by obstructive coronary artery disease and ACS, which typically occur in the setting of thrombotic coronary artery occlusion. The 2018 Joint ESC/ACCF/AHA/WHF Fourth Universal Definition of MI placed greater emphasis on the measurement of cardiac enzymes, specifically high-sensitivity cTn, in the diagnostic criteria of acute myocardial infarction [4]. Testing of cardiac enzymes is ordered in 16.9% of patients presenting to emergency departments in the United States, but chest pain only accounts for approximately 5.3% of chief complaints [52]. Current international guidelines regarding chest pain recommend trending serial cTn and EKGs to assess for a potentially evolving ACS, thereby subjecting many patients to prolonged ED observation or hospital admission, and increasing the number of false-positive troponin results in patients with low pretest probability for coronary artery disease [53–55].
The term myocardial infarction in the absence of obstructive coronary artery disease (MINOCA) is an increasingly recognized diagnosis used to describe ischemic myocardial injury resulting in elevated cTn, despite insignificant coronary artery disease on angiography [56]. Mild troponinemia can also reflect the detection of small proteolytic products of cTn, which are released in the circulation following calpain activation in the setting of recurrent bouts of short-lasting sublethal myocardial ischemia or systemic inflammatory processes [16]. Likewise, calpain activation in organs other than the heart (such as smooth muscle cells of the aorta, trachea, gut, and urinary bladder, where cTnT is also expressed) might explain the occurrence of troponinemia in medical conditions totally unrelated to myocardial ischemia or any cardiac injury.
Scoring systems incorporating the first cTn, such as a low HEART score (0–3) or low TIMI score (0–1), are the most helpful in identifying patients less likely to have ACS [57]. In patients with a low pretest probability for ACS, an elevation in cardiac biomarkers can become a confounding factor, delaying the formulation of the correct diagnosis and thereby postponing initiation of adequate medical management.
Regardless of the clinical situation, elevated serum troponin is associated with increased mortality and adverse outcomes [58–60], and may be used to risk-stratify patients. Awareness of the multiple conditions associated with positive troponin, as well as understanding of the pathophysiology of its release, are essential preconditions for minimizing unnecessary, costly, and potentially risky interventions, providing timely and appropriate medical care.
Footnotes
Source of support: Self financing
Conflicts of interest
None.
References
- 1.Karmen A, Wroblewski F, Ladue JS. Transaminase activity in human blood. J Clin Invest. 1955;34:126–31. doi: 10.1172/JCI103055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nomenclature and criteria for diagnosis of ischemic heart disease. Report of the Joint International Society and Federation of Cardiology/World Health Organization task force on standardization of clinical nomenclature. Circulation. 1979;59:607–9. doi: 10.1161/01.cir.59.3.607. [DOI] [PubMed] [Google Scholar]
- 3.Amsterdam EA, Wenger NK, Brindis RG, et al. 2014 AHA/ACC Guideline for the Management of Patients With Non–ST-Elevation Acute Coronary Syndromes. J Am Coll Cardiol. 2014;64(24):e139–228. doi: 10.1016/j.jacc.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 4.Thygesen K, Alpert JS, Jaffe AS, et al. Executive Group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth Universal Definition of Myocardial Infarction (2018) J Am Coll Cardiol. 2018;72(18):2231–64. doi: 10.1016/j.jacc.2018.08.1038. [DOI] [PubMed] [Google Scholar]
- 5.Newby LK, Jesse RL, Babb JD, et al. ACCF 2012 expert consensus document on practical clinical considerations in the interpretation of troponin elevations: A report of the American College of Cardiology Foundation task force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2012;60:2427–63. doi: 10.1016/j.jacc.2012.08.969. [DOI] [PubMed] [Google Scholar]
- 6.Babuin L, Jaffe AS. Troponin: The biomarker of choice for the detection of cardiac injury. CMAJ. 2005;173:1191–202. doi: 10.1503/cmaj.050141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park KC, Gaze DC, Collinson PO, Marber MS. Cardiac troponins: From myocardial infarction to chronic disease. Cardiovasc Res. 2017;113(14):1708–18. doi: 10.1093/cvr/cvx183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoff J, Wehner W, Nambi V. Troponin in cardiovascular disease prevention: Updates and future direction. Curr Atheroscler Rep. 2016;18(3):12. doi: 10.1007/s11883-016-0566-5. [DOI] [PubMed] [Google Scholar]
- 9.Lewandrowski K, Chen A, Januzzi J. Cardiac markers for myocardial infarction. A brief review. Am J Clin Pathol. 2002;118(Suppl):S93–99. doi: 10.1309/3EK7-YVV9-228C-E1XT. [DOI] [PubMed] [Google Scholar]
- 10.Parmacek MS, Solaro RJ. Biology of the troponin complex in cardiac myocytes. Prog Cardiovasc Dis. 2004;47:159–76. doi: 10.1016/j.pcad.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 11.Wilkinson JM, Grand RJ. Comparison of amino acid sequence of troponin I from different striated muscles. Nature. 1978;271:31–35. doi: 10.1038/271031a0. [DOI] [PubMed] [Google Scholar]
- 12.Melanson SE, Conrad MJ, Mosammaparast N, Jarolim P. Implementation of a highly sensitive cardiac troponin I assay: Test volumes, positivity rates and interpretation of results. Clin Chim Acta. 2008;395:57–61. doi: 10.1016/j.cca.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Apple FS, Wu AH, Jaffe AS, et al. National Academy of Clinical Biochemistry and IFCC Committee for Standardization of Markers of Cardiac Damage Laboratory Medicine practice guidelines: Analytical issues for biomarkers of heart failure. Circulation. 2007;116:e95–98. doi: 10.1161/CIRCULATIONAHA.107.185266. [DOI] [PubMed] [Google Scholar]
- 14.Halim SA, Newby LK, Ohman EM. Biomarkers in cardiovascular clinical trials: Past, present, future. Clin Chem. 2012;58:45–53. doi: 10.1373/clinchem.2011.165787. [DOI] [PubMed] [Google Scholar]
- 15.Sandoval Y, Jaffe AS. Type 2 myocardial infarction: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;73(14):1846–60. doi: 10.1016/j.jacc.2019.02.018. [DOI] [PubMed] [Google Scholar]
- 16.Morrow DA, Cannon CP, Jesse RL, et al. National Academy of Clinical Biochemistry Laboratory Medicine Practice Guidelines: Clinical characteristics and utilization of biochemical markers in acute coronary syndromes. Circulation. 2007;115:e356–75. doi: 10.1161/CIRCULATIONAHA.107.182882. [DOI] [PubMed] [Google Scholar]
- 17.Kajioka S, Takahashi-Yanaga F, Shahab N, et al. Endogenous cardiac troponin T modulates Ca2+-mediated smooth muscle contraction. Sci Rep. 2012;2:979. doi: 10.1038/srep00979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bleier J, Vorderwinkler KP, Falkensammer J, et al. Different intracellular compartmentations of cardiac troponins and myosin heavy chains: A causal connection to their different early release after myocardial damage. Clin Chem. 1998;44:1912–18. [PubMed] [Google Scholar]
- 19.Katus HA, Remppis A, Neumann FJ, et al. Diagnostic efficiency of troponin T measurements in acute myocardial infarction. Circulation. 1991;83:902–12. doi: 10.1161/01.cir.83.3.902. [DOI] [PubMed] [Google Scholar]
- 20.Gerhardt W, Katus H, Ravkilde J, et al. S-troponin T in suspected ischemic myocardial injury compared with mass and catalytic concentrations of S-creatine kinase isoenzyme MB. Clin Chem. 1991;37:1405–11. [PubMed] [Google Scholar]
- 21.Consolini AE, Ragone MI, Bonazzola P, Colareda GA. Mitochondrial bioenergetics during ischemia and reperfusion. Adv Exp Med Biol. 2017;982:141–67. doi: 10.1007/978-3-319-55330-6_8. [DOI] [PubMed] [Google Scholar]
- 22.McDougal AD, Dewey CF., Jr Modeling oxygen requirements in ischemic cardiomyocytes. J Biol Chem. 2017;292(28):11760–76. doi: 10.1074/jbc.M116.751826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eggers KM, Lindahl B. Application of cardiac troponin in cardiovascular diseases other than acute coronary syndrome. Clin Chem. 2017;63(1):223–35. doi: 10.1373/clinchem.2016.261495. [DOI] [PubMed] [Google Scholar]
- 24.White HD. Pathobiology of troponin elevations: Do elevations occur with myocardial ischemia as well as necrosis? J Am Coll Cardiol. 2011;57:2406–8. doi: 10.1016/j.jacc.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 25.Yang CW, Li H, Thomas L, et al. Retrospective cause analysis of troponin I elevation in non-CAD patients: Special emphasis on sepsis. Medicine (Baltimore) 2017;96(37):e8027. doi: 10.1097/MD.0000000000008027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hessel MH, Atsma DE, van der Valk EJ, et al. Release of cardiac troponin I from viable cardiomyocytes is mediated by integrin stimulation. Pflugers Arch. 2008;455:979–86. doi: 10.1007/s00424-007-0354-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammarsten O, Mair J, Möckel M, et al. Possible mechanisms behind cardiac troponin elevations. Biomarkers. 2018;23(8):725–34. doi: 10.1080/1354750X.2018.1490969. [DOI] [PubMed] [Google Scholar]
- 28.Mittal M, Siddiqui MR, Tran K, et al. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20(7):1126–67. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Forrester SJ, Kikuchi DS, Hernandes MS, et al. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122(6):877–902. doi: 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pasini E, Corsetti G, Aquilani R, et al. Protein-amino acid metabolism disarrangements: The hidden enemy of chronic age-related conditions. Nutrients. 2018;10(4) doi: 10.3390/nu10040391. pii: E391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flati V, Pasini E, D’Antona G, et al. Intracellular mechanisms of metabolism regulation: The role of signaling via the mammalian target of rapamycin pathway and other routes. Am J Cardiol. 2008;101(11A):16E–21E. doi: 10.1016/j.amjcard.2008.02.075. [DOI] [PubMed] [Google Scholar]
- 32.Scarabelli TM, Gottlieb RA. Functional and clinical repercussions of myocyte apoptosis in the multifaceted damage by ischemia/reperfusion injury: Old and new concepts after 10 years of contributions. Cell Death and Differ. 2004;11(Suppl 2):S144–52. doi: 10.1038/sj.cdd.4401544. [DOI] [PubMed] [Google Scholar]
- 33.Dellinger RP, Levy MM, Rhodes A, et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2012. Crit Care Med. 2013;41:580–637. doi: 10.1097/CCM.0b013e31827e83af. [DOI] [PubMed] [Google Scholar]
- 34.Sato R, Nasu M. A Review of sepsis-induced cardiomyopathy. J Intensive Care. 2015;3:48. doi: 10.1186/s40560-015-0112-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antonucci E, Fiaccadori E, Donadello K, et al. Myocardial depression in sepsis: From pathogenesis to clinical manifestations and treatment. J Crit Care. 2014;29(4):500–11. doi: 10.1016/j.jcrc.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 36.Celes MR, Torres-Dueñas D, Malvestio LM, et al. Disruption of sarcolemmal dystrophin and beta-dystroglycan may be a potential mechanism for myocardial dysfunction in severe sepsis. Lab Invest. 2010;90:531–42. doi: 10.1038/labinvest.2010.3. [DOI] [PubMed] [Google Scholar]
- 37.Kim JS, Kim M, Kim YJ, et al. Troponin testing for assessing sepsis-induced myocardial dysfunction in patients with septic shock. J Clin Med. 2019;8(2) doi: 10.3390/jcm8020239. pii: E239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vallabhajosyula S, Sakhuja A, Geske JB, et al. Role of admission troponin-T and serial troponin-T testing in predicting outcomes in severe sepsis and septic shock. J Am Heart Assoc. 2017;6(9) doi: 10.1161/JAHA.117.005930. pii: e005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bessiere F, Khenifer S, Dubourg J, et al. Prognostic value of troponins in sepsis: A meta-analysis. Intensive Care Med. 2013;39(7):1181–89. doi: 10.1007/s00134-013-2902-3. [DOI] [PubMed] [Google Scholar]
- 40.Zhang L, Wang Z, Qi S. Cardiac troponin elevation and outcome after subarachnoid hemorrhage: A systematic review and meta-analysis. J Stroke Cerebrovasc Dis. 2015;24(10):2375–84. doi: 10.1016/j.jstrokecerebrovasdis.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 41.Krishnamoorthy V, Mackensen GB, Gibbons EF, Vavilala MS. Cardiac dysfunction after neurologic injury: What do we know and where are we going? Chest. 2016;149(5):1325–31. doi: 10.1016/j.chest.2015.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang HM, Okwuosa TM, Scarabelli T, et al. Best practices in cardio-oncology: Part 2. JACC. 2017;70:2552–65. doi: 10.1016/j.jacc.2017.09.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen-Scarabelli C, McRee C, et al. Comprehensive review on cardio-oncology: Role of multimodality imaging. J Nucl Cardiol. 2017;24(3):906–35. doi: 10.1007/s12350-016-0535-y. [DOI] [PubMed] [Google Scholar]
- 44.Cardinale D, Sandri MT, Colombo A, et al. Prognostic value of troponin I in cardiac risk stratification of cancer patients undergoing high-dose chemotherapy. Circulation. 2004;109(22):2749–54. doi: 10.1161/01.CIR.0000130926.51766.CC. [DOI] [PubMed] [Google Scholar]
- 45.Zhang S, Liu X, Bawa-Khalfe T, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18(11):1639–42. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 46.Ichikawa Y, Ghanefar M, Bayeva M, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014;124(2):617–30. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang HM, Moudgil R, Scarabelli T, et al. Best practices in cardio-oncology: Part 1. JACC. 2017;70:2536–51. doi: 10.1016/j.jacc.2017.09.1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cardinale D, Sandri MT, Martinoni A, et al. Myocardial injury revealed by plasma troponin I in breast cancer treated with high-dose chemotherapy. Ann Oncol. 2002;13(5):710–15. doi: 10.1093/annonc/mdf170. [DOI] [PubMed] [Google Scholar]
- 49.Ewer MS, Ewer SM. Troponin I provides insight into cardiotoxicity and the anthracycline-trastuzumab interaction. J Clin Oncol. 2010;28(25):3910–19. doi: 10.1200/JCO.2010.30.6274. [DOI] [PubMed] [Google Scholar]
- 50.Cardinale D, Colombo A, Torrisi R, et al. Trastuzumab-induced cardiotoxicity: Clinical and prognostic implications of troponin I evaluation. J Clin Oncol. 2010;28(25):3910–16. doi: 10.1200/JCO.2009.27.3615. [DOI] [PubMed] [Google Scholar]
- 51.Seidman A, Hudis C, Pierri MK, et al. Cardiac dysfunction in the trastuzumab clinical trials experience. J Clin Oncol. 2002;20:1215–21. doi: 10.1200/JCO.2002.20.5.1215. [DOI] [PubMed] [Google Scholar]
- 52.Mackam AM, Nguyen OK. Use of cardiac biomarker testing in the Emergency Department. JAMA Intern Med. 2015;175(1):67–75. doi: 10.1001/jamainternmed.2014.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fihn SD, Gardin JM, Abrams J, et al. 2012 ACCF/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the Diagnosis and Management of Patients with Stable Ischemic Heart Disease: Executive Summary: A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, and the American College of Physicians, American Association for Thoracic Surgery, Preventive Cardiovascular Nurses Association, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2012;60(24):2564–603. doi: 10.1016/j.jacc.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 54.Newby LK, Jesse RL, Babb JD, et al. ACCF 2012 expert consensus document on practical clinical considerations in the interpretation of troponin elevations: A report of the American College of Cardiology Foundation task force on Clinical Expert Consensus Documents. J Am Coll Cardiol. 2012;60(23):2427–63. doi: 10.1016/j.jacc.2012.08.969. [DOI] [PubMed] [Google Scholar]
- 55.Penumetsa SC, Mallidi J, Friderici JL, et al. Outcomes of patients admitted for observation of chest pain. Arch Intern Med. 2012;172:873–77. doi: 10.1001/archinternmed.2012.940. [DOI] [PubMed] [Google Scholar]
- 56.Tamis-Holland JE, Jneid H, Reynolds HR, et al. Contemporary diagnosis and management of patients with myocardial infarction in the absence of obstructive coronary artery disease. Circulation. 2019;139:e891–908. doi: 10.1161/CIR.0000000000000670. [DOI] [PubMed] [Google Scholar]
- 57.Fanaroff AC, Rymer JA, Goldstein SA, et al. Does this patient with chest pain have acute coronary syndrome?: The rational clinical examination systematic review. JAMA. 2015;314:1955–65. doi: 10.1001/jama.2015.12735. [DOI] [PubMed] [Google Scholar]
- 58.Gallagher S, Jones DA, Anand V, Mohiddin S. Diagnosis and management of patients with acute cardiac symptoms, troponin elevation and culprit-free angiograms. Heart. 2012;98(13):974–81. doi: 10.1136/heartjnl-2011-301121. [DOI] [PubMed] [Google Scholar]
- 59.Ahmed AN, Blonde K, Hackam D, et al. Prognostic significance of elevated troponin in non-cardiac hospitalized patients: A systematic review and meta-analysis. Ann Med. 2014;46(8):653–63. doi: 10.3109/07853890.2014.959558. [DOI] [PubMed] [Google Scholar]
- 60.Sara JD, Holmes DR, Jr, Jaffe AS. Fundamental concepts of effective troponin use: important principles for internists. Am J Med. 2015;128(2):111–19. doi: 10.1016/j.amjmed.2014.08.030. [DOI] [PubMed] [Google Scholar]