

Abstract
The clinical onset of type 1 diabetes is characterized by the destruction of the insulin-producing β cells of the pancreas, and is caused by autoantigen-induced inflammation (insulitis) of the islets of Langerhans. The current standard of care for type 1 diabetes mellitus (T1DM) patients allows for management of the disease with exogenous insulin, but patients eventually succumb to many chronic complications such as limb amputation, blindness and kidney failure. New therapeutic approaches now on the horizon are looking beyond glycemic management and are evaluating new strategies from protecting and regenerating endogenous islets to treating the underlying autoimmunity through selective modulation of key immune cell populations. Currently, there are no effective treatments for the autoimmunity that causes the disease, and strategies that aim to delay or prevent the onset of the disease will play an important role in the future of diabetes research. In this review, we summarize many of the key efforts underway that utilize molecular approaches to selectively modulate this disease and look at new therapeutic paradigms that can transform clinical treatment.
Graphical Abstract
INTRODUCTION
Type 1 diabetes mellitus (T1DM) is a global epidemic affecting over 30 million people, and is one of the most common endocrine and metabolic conditions occurring in childhood.1 The incidence of T1DM has increased 5.3% annually and the economic cost is estimated between $14.4–14.9 billion in the US alone.2–4 T1DM is characterized by the autoimmune destruction of the insulin secreting β cells of the pancreatic islets of Langherhans, leading to insulin deficiency and unregulated blood glucose levels. The current standard of care entails a rigorous routine of blood glucose monitoring coupled to daily exogenous insulin injections. Despite advances in insulin therapies, these individuals still suffer chronic diabetic complications including cardiovascular disease, retinopathy, nephropathy, ketoacidosis, nonketotic hyperosmolar coma, or death.5 Whole organ pancreas transplantation has been explored, however it requires patients to receive systemic immunosuppressants and after 5 years 90% of patients are once again dependent on exogenous insulin.6 Polymeric encapsulation of donor insulin-producing tissue to overcome the need for systemic immunosuppression has gained momentum with the recent development of new materials and formulations.7–10 This therapeutic approach to tissue replacement promises to restore glycemic control for fully symptomatic patients with little to no remaining β cells. To complement this strategy, there is growing interest in interventional strategies that aim to tackle the underlying autoimmunity of the disease and preserve as much endogenous β cells as possible. Currently there are no clinically-approved interventional therapies to treat the underlying autoimmunity, but new therapeutic agents are being clinically tested and numerous new approaches are on the horizon.
Pathogenesis.
Development of an interventional therapy for T1DM has proven challenging owing to its polygenic and heterogeneous nature. There are a plethora of purported environmental triggers whose role in pathogenic processes are poorly understood, while genetic, and phenotypic characteristics show marked variation.1 Over 40 loci play a role in T1DM susceptibility, with the major histocompatibility (MHC) class II HLA-DR and HLA-DQ genotypes providing an estimated half of the genetic susceptibility.11,12 While these genetic risk factors are necessary for T1DM development, they are not sufficient. Recent attention has turned to a variety of environmental factors including infant diet, vitamin D and the vitamin D pathway constituents, enteroviruses, the hygiene hypothesis, and the gut microbiome.1,13 However, no evident influence on pathogenesis has been identified and the exact triggering mechanism remains unknown.
The thymus plays a paramount role in eliminating self-reactive T cell populations through positive and negative selection, termed central tolerance.14 The transcription factor autoimmune regulator AIRE promotes the expression of self-antigens on the surface of medullary thymic epithelial cells (mTECs). The self-antigens are presented through MHC complexes to allow for targeted removal of potentially autoreactive T cell clones from the repertoire.15 Such regulation fails in T1DM, leading to escape of autoreactive T cell populations to the periphery. Diabetic MHC class II proteins presenting peptides recognized by these autoreactive T cells form a trimolecular complex with the T cell receptor (TCR) that leads to T cell activation and expansion. This is followed by pancreatic infiltration by T cells, macrophages, B lymphocytes and plasma cells, and subsequent autoimmune destruction of insulin secreting β cells.16 Symptoms and diagnosis typically occur well after two-thirds of β cells are lost (Figure 1).
Figure 1.

Progression of β cell loss and primary cells involved in the pathogenesis of T1DM. Predisposition from bone marrow, thymus, and immune populations followed by a precipitating event lead to β cell mass loss prior to clinical diagnosis and therapeutic intervention.
Interventional Treatments under Clinical Evaluation.
Several clinical trials evaluating immunomodulatory agents in the past 40 years are discussed and summarized in Table 1. These trials include the systemic immunosuppressants cyclosporine, azathioprine, and mofetil, and immune interfering antibodies against CD20, cytotoxic T lymphocyte antigen-4 (CTLA-4), Interleukin 2 (IL-2), and CD3.1 The ladder case involving anti-CD3 monoclonal antibodies (mAb) suggested a reversal of hyperglycemia in preclinical studies and phase I trials through inactivation of effector T cells (Teff) and an expansion of the regulatory CD4+CD25+ T cell (Treg) populations.17 However, two different anti-CD3 mAb, Otelixizumab and Teplizumab, showed disappointing results in maintaining C-peptide levels in phase III clinical trials.18,19 Likewise, all other interventional trials have failed to meet phase III endpoints. This highlights the dire need for both new targets and methods for selectively modulating the immune system, and for mechanistic biomarkers to aid in selecting both appropriate treatments and therapeutic windows.
Table 1.
T1DM and Immunoregulatory Clinical Trials
| Therapy Group | Therapy | Phase | ID | Comments |
|---|---|---|---|---|
| Cytokine Blockade | Anti-IL-1β (Canakinumab) | II | No C-peptide level difference.26 | |
| IL-1R antagonist (Anakinra) | II | No C-peptide level difference.25 | ||
| Soluble TNF-α receptor (Etanercept) | I/II | Significant reduction of HA1c and increase in C-peptide levels.31 | ||
| Inducing Antigenic Tolerance | Human recombinant GAD-alum | II | Decreased Teff, increased Tregs, but no C-peptide difference.45 | |
| HSP60 (DiaPep277) | II | N/A | Delayed decrease in C-peptide levels.49 | |
| HSP60 (DiaPep277) | II | N/A | 30 children, no C-peptide, insulin dose, or HA1c level difference.48 | |
| Altered peptide ligand of B9–23 epitope (NBI-6024) | I | N/A | T cell population shift from TH1 to TH2.52 | |
| Altered peptide ligand of B9–23 epitope (NBI-6024) | I | Repeated administration shows no efficacy.53 | ||
| Multiple Islet peptide (MultPepT1De) | Ib | Recruiting.54 | ||
| Lymphocyte Modulation: T cell | Anti-CD3 (Otelixizumab) | III | No C-peptide level difference.18 | |
| Anti-CD3 (Teplizumab) | II/III | Primary outcome not met, 5% of patients were not taking insulin 1 year after treatment.19 | ||
| Anti-CD3 | II | Reduced insulin dosage requirements.63 | ||
| Anti-CD3 (Teplizumab) | II | No C-peptide level difference. Reduced insulin requirements.64 | ||
| IL-2 (Aldesleukin) | I & II | Selectively activates Tregs. No efficacy endpoints.82 | ||
| IL-2/Rapamycin combination | I | Treg, NK and Teff populations increased. B cell function impaired.84 | ||
| IL-2 (Aldesleukin) | I & II | Dosage testing. No results reported.85 | ||
| Ex vivo expanded human autologous polyclonal Tregs | I | Safe. Subset were long-lived. C-peptide levels stabilized through 2 years.73 | ||
| Ex vivo expanded CD4+CD25+CD127-Tregs | II | ISRCTN06128462 | Safe. Increased C-peptide levels. Reduced insulin requirements.87 | |
| Ex vivo expanded human Tregs | II | Follow up study. No results reported.77 | ||
| Anti-CTLA4 (Abatacept) | II | Slowed β cell reduction for 24 months, but no improved function.97 | ||
| Lymphocyte Modulation: APC | Methyldopa | Ib | Reduced inflammatory T cell response to insulin.120 | |
| Methyldopa | II | Recruiting. | ||
| Anti-CD40 ligand for Lupus | I | N/A | Terminated due to risk of life-threating thrombotic events.130 | |
| Lymphocyte Modulation: B cell | Anti-CD20 (Rituximab) | II | Inhibits T cell activation and further insulitis over a 1 year period. Follow-up study showed increase of asymptomatic viremia.146,147 | |
| Immuno-regulatory | Anti-PD1 for various cancers | N/A | N/A | Various different trials report development of insulin-dependent diabetes following anti-PD1 therapy.159 |
| Islet Protection | Tyrosine Kinase Inhibitor (Imatinib) | II | Preserved β cell function, reduced insulin requirements up to 1 year (unpublished, presented at American Diabetes Association Scientific Sessions 2017). | |
| β Cell Imaging | GLP-1R targeted probe 111In-DTPA-Exendin | N/A | Found difference in pancreatic uptake between healthy and T1D patients.262 | |
| VMAT2 targeted probe [11C]-DTBZ | N/A | Did not correlate β cell function to mass.266 |
APPROACHES FOR IMMUNOMODULATION
Here, we highlight emerging methods for targeting the underlying mechanisms of T1DM through the view of recent advances in immunomodulatory therapies, protective and regenerative strategies, and new targeting modalities. A brief perspective is offered into the future direction of treatment for T1DM and how next generation therapies can achieve improved clinical outcomes.
Cytokine Blockade.
T1DM is characterized by an imbalance in pro-inflammatory and anti-inflammatory cytokines which manifest as the disease progresses.20,21 Elevated serum levels of pro-inflammatory cytokines, such as IL-1β, tumor necrosis factor α (TNF-α), IL-6, and IL-7 have spurred clinical investigation into the therapeutic utility of blocking these signaling pathways. IL-1 is an inflammatory cytokine involved in the differentiation of T helper 1 (TH1) and TH17 T cells that has profound pro-apoptotic effects on β cells.22,23 In non-obese diabetic (NOD) mice, knockout (KO) of the IL-1 receptor (IL-1R) slowed the progression of T1DM, but did not prevent disease onset.24 Initial clinical trials with the IL-1 antagonist anakinra showed promising but mixed results with treated participants having similar hemoglobin A1c (HbA1c) levels requiring significantly less insulin than placebo cohort after 1 month of treatment.25 Separate phase IIa clinical trials failed to find efficacy with two different IL-1 antagonists, canakinumab and anakinra.26 Both treatments were found to be safe, but failed to find any significant difference in C-peptide levels. In T1DM patients, monocyte expression of IL-1β is high at time of diagnosis but normalizes within 1 month.27 This suggests anti-IL1 treatment may only be effective at early time points in disease progression.
TNF-α is a cytokine involved in regulating the maturation of dendritic cells (DCs) and has been affiliated with the activation of islet-specific T cells in pancreatic lymph nodes (PLNs).28 T1DM patients have significantly higher levels of plasma TNF-α compared to healthy subjects.29 In NOD mice, the systemic administration of TNF-α led to a 5 week earlier onset of disease and a higher incidence rate compared to control mice,30 indicating the therapeutic value in modulating this target. However, efficacy in treatments targeting TNF-α may depend strongly on time of administration during disease progression. Administration of anti-TNF-α mAb in 2-week-old NOD mice protected against T1DM development compared to control mice, whereas 8-week-old treated mice showed only 50% incidence of the disease.30 A small clinical trial had promising results with treated patients having lower HbA1c levels and increased C-peptide levels, but there have been no subsequent studies.31
IL-7 is a pro-inflammatory cytokine that impacts proliferation and survival of naïve T cells.32,33 It is implicated in multiple autoimmune diseases and represses the suppressive functions of Tregs.34,35 Two studies showed blocking the IL-7R in NOD mice resulted in a significant reduction of T1DM incidence and even remission of established T1DM.36,37 In addition, both studies found that IL-7 suppression resulted in increased expression of the Programmed Death-1 (PD-1) receptor on the surface of Teff cells. Strikingly, NOD mice that received anti-IL-7 treatment followed by a PD-1 antagonist show increased incidence of T1DM, suggesting that PD-1 expression is required for anti-T1DM effects (discussed further below).36 Additionally, blocking IL-7R also resulted in an increase in Tregs without affecting their suppressive activity.37
IL-6 is a cytokine involved in host defense against pathogens, but is also involved in autoimmune diseases with increased TH17 cell development and inhibited Treg development.38 In patients with T1DM, IL-6 sensitivity correlated with increased expression of genes involved in T cell migration and inflammatory responses, including CD4+ cells.39 These results suggest IL-6 may be a target for modulating T1DM and other autoimmune diseases. NOD/Wehi mice treated with an anti-IL-6 antibody resulted in a three-fold reduction in T1DM incidence compared to treatment with a control antibody.40 Inhibition of IL-6 can be a promising therapeutic strategy and further validation and studies elucidating the mechanism of action should prove insightful.
Inducing Antigenic Tolerance.
T1DM pathogenesis is characterized by a loss of central tolerance and the production of autoreactive T cells that have escaped thymic negative selection. Antigen-specific immunotherapy seeks to provide a targeted approach towards inducing peripheral immune tolerance without leading to systemic immune inhibition and associated complications that are typically seen in current non-antigenic therapies.41 Antigenic therapies seek to achieve tolerance through induction of Tregs (active tolerance) that inhibit TH1/TH17 populations or removal of autoreactive cell populations (passive tolerance) by eliminating autoantigen expression or the cell populations that propagate autoantigen sensitivity. Here we focus on promising targets involving antigenic responses that may lead to re-establishing immune tolerance.
The T1DM autoantigen GAD65, encoded by the GAD2 gene, is responsible for production of the inhibitory neurotransmitter γ-aminobutyric acid (GABA)42,43 which impairs insulin secretion.44 Administration of human recombinant GAD65 formulated with aluminum hydroxide (GAD65-alum) induces a favorable immune response, with decreased GAD-specific CD4+ and CD8+ Teff cells and increased GAD65-specific Tregs. However, after 4 years of treatment there was no detectable change in T cell populations over placebo.45 Thus, while antigenic treatment with GAD65 does activate immune tolerance pathways, a combinatorial approach may be necessary for long-term efficacy.
Heat-shock protein 60 (HSP60) is an implicated autoantigen in T1DM, yet is involved in preventing stress-induced damage to proteins and functioning as a chaperone protein. This duality has been explored by administration of a peptide derived from HSP60 (p277), which protected NOD mice from both induced and spontaneous diabetes.46 Additionally, a stable version of p277 (DiaPep277) promotes anti-inflammatory effects, cell adhesion, and inhibits migration though interaction with Toll-like receptor 2 (TLR2).45,47 DiaPep277 in a phase II T1DM trial ultimately failed to meet endpoints despite previous encouraging results of a shift in T cell populations to a TH2 phenotype.48–50 Combining DiaPep277 with therapies that inhibit inflammation may be needed to enable stronger Treg induction.51
Peptide immunotherapy seeks to achieve immune tolerance by expanding Tregs and forcing pathogenic T cells through anergy and/or deletion. Recent clinical peptide immunotherapy efforts have involved administration of either an altered peptide ligand of Insulin B:9–23 or natural peptide sequences from proinsulin.52–54 The former failed to show efficacy and a single peptide sequence of proinsulin trial did not look at efficacy. One promising preclinical peptide therapy involves generating “navacims” through coating peptide-MHC complexes onto nanoparticles followed by administration to induce immune tolerance. Using this method, several autoimmune-disease-relevant peptide-MHC class II complexes triggered expansion of Type 1 regulatory-like CD4+ T cells and restored normoglycemia in the NOD mouse model.55 Importantly, these peptide-MHC coated nanoparticles did not compromise systemic immunity, which has been a major challenge in antigen-specific peptide immunotherapy.
Taken together, antigenic induced immune tolerance shows efficacy in diabetic mouse models but have so far failed to replicate in human populations when administered as monotherapies. Inadequate dosages and the inherent challenges in attempting to reverse symptomatic T1DM rather than delay the onset of the disease may be contributors to these results. T1DM prevention studies in humans also lack robust early biomarkers. Preclinical studies have suggested that combinatorial administration of antigenic therapies with immune modulators may be required for efficacy in T1DM patients.56–58 Future clinical studies with this approach are needed.
Lymphocyte Modulation.
T cells.
The persistence and proliferation of autoreactive T cells are at the root of the autoimmune reaction in T1DM. CD3 is a component of the TCR complex and is required for T cell activation, and multiple anti-CD3 therapies have emerged and are comprehensively reviewed.59–61 Briefly, blockade of CD3 in NOD mice resulted in prevention of T1DM incidence and remission of established T1DM in 60–80% of mice.62 In the first clinical study of anti-CD3 treatment, 9 of 12 treated patients maintained or reduced their insulin requirements compared to 2 of 12 in the control cohort.63 These results were promising and sparked extensive clinical trials of anti-CD3 therapies, with mixed outcomes. In a phase III clinical trial with teplizumab, effects were only seen at the highest dosing regimens and the trial failed to reach primary outcomes of patients using less than 0.5 U/kg of insulin per day and HbA1C reduction of less than 6.5% after 1 year.18 A subsequent trial identifying drug responders and nonresponders showed no significant difference in C-peptide levels between drug responders and nonresponders, however they had significant differences in their baseline insulin requirements and lower levels of CD8+ effector memory cells.64
There is rapidly growing interest in targeting T cell metabolism in autoimmune diseases. Mitochondrial metabolic activity plays a key role in modulating T cell activation, proliferation, and programmed cell dealth.65 Additionally, upon antigen recognition the T cell mitochondria modulates processes important for IL-2 production.66–68 A recent study shows that T1DM patients have an altered mitochondrial and cytokine response following TCR stimulation linked to persistent T cell mitochondrial inner membrane hyperpolarization.69 Of particular interest are metabolic pathways that inhibit proliferation and effector functions of both CD4+ and CD8+ T cells through mechanisms such as mTOR inhibition and selective reduction of glucose uptake. Blockage of glucose metabolism in the pre-diabetic NOD mouse model using 2-deoxy-D-glucose (2DG), a competitive inhibitor of phosphoglucose isomerase, reduces insulitis and the frequency of a subset of diabetogenic CD8+ T cells.70 While promising, a key challenge for metabolic therapies is identifying the optimal period for inhibition, as currently there are no predictive biomarkers for autoreactive T cell activation.
A major suppressor of autoimmunity are Tregs, which make up less than 10% of CD4+ T cells in peripheral circulation.71 Tregs play a primary role for maintaining immune tolerance by downregulating the induction of other immune cells including CD4+ and CD8+ T cells, B cells, NK cells, macrophages, and DCs.72,73 T1DM patients have been found to possess an imbalance between Teff and Treg,51 leading to an inability to properly inhibit Teff responses.72,74,75 In NOD mice, Treg populations initially increase in the inflamed pancreas but decrease as insulitis progresses.76 Numerous studies have attempted to restore Treg populations and are reviewed elsewhere in detail,71,77 with more recent progress mentioned here.
Perturbation in IL-2Rα (CD25) expression in Tregs has shown T1DM susceptibility by a reduction in STAT5 phosphorylation.78 Additionally, IL-2 deprivation in T1DM patients results in low levels of anti-apoptotic protein Bcl-2, leaving Tregs prone to apoptosis.79–81 Administration of a low dose IL-2 therapy overcomes IL-2 signaling defects in patients and was effective at increasing Treg populations.82 However, other CD25 expressing populations are also activated with low dose IL-2 administration and when combined with rapamycin an increase in both CD56high NK cells and CD4+ memory cell populations are observed and negatively impacts β cell function.83,84 A clinical trial addressing dosing has completed, however no results are reported.85
These results from IL-2 therapies led to interest in using Tregs themselves as a therapeutic approach. Indeed, adoptive transfer of Tregs have been investigated in two clinical trials and are safe, long-lived, and stable.73,86,87 Follow-up phase II studies and an interesting study of Tregs in combination with low-dose IL-2 are still ongoing.77 Antigen-specific Tregs may be more effective at preventing insulitis,88 and chimeric antigen receptor (CAR) engineered Tregs show promising results in various pre-clinical models of autoimmunity.89–92 Other avenues to expand Tregs are being explored by targeting memory T cells.93 Of note are Teff-depleting agents teplizumab and alefacept, which show promise for sustaining remission and tolerance.64,94 Alternatively, polarizing TH17 pro-inflammatory cells towards a Treg phenotype has been achieved using small molecule glutamate receptor enhancers.95
A major mechanism of maintaining tolerance is through T cell anergy, the inactivation of autoreactive T cells. T cell anergy can manifest through MHC-peptide-TCR complex recognition in the absence of co-stimulation through CD28 (T cell) and B7 (APC) where the T cell cannot produce IL-2, thus preventing proliferation.96 Similar effects happen with CTLA-4 (related to CD28) which blocks the CD28-dependent T cell activation. Abatacept, a CTLA4-Ig approved for rheumatoid arthritis, prevents disease progression in T1DM animal models. However, T1DM clinical trials involving CTLA-4 have failed to meet endpoints.97 Encouragingly, recent work with a selective CD28 antagonist in combination with rapamycin (mTOR pathway inhibition) inhibits T cell activation and migration into the pancreas in NOD mice.98 Indeed, tolerogenic APCs display low numbers of co-stimulatory molecules like CD80, CD86, and CD40, and anergy can be achieved using rapamycin, corticosteroids, IL-10, and transforming growth factor β 1 (TGFβ−1).99,100 These studies provide additional options to rapamycin in combination trials with CTLA4-Ig.
Tregs can also play a similar role in T cell anergy through ligation of CTLA4 to CD80 and CD86, which induces APCs to express Indoleamine 2,3-dioxygenase (IDO) resulting in abolished T cell activation.101–103 Additionally, Tregs can inhibit APC maturation resulting in T cell anergy.104 Increased expression and binding of lymphocyte activation gene 3 (LAG-3) to MHC II induces the ITAM-mediated inhibitory signaling pathway, resulting in suppression of DC maturation and immunostimulatory capacity. Additionally, CTLA4-Ig therapies may be beneficial to modifying DCs present in inflammatory microenvironments through CTLA4-Ig binding to B7 while simultaneously inducing DC expression of IDO which leads to Treg expansion.105 Subsequently, Tregs are reported to downregulate CD80/86 expression on APCs and DCs, which modifies them towards a tolerogenic state.106,107
A complementary approach to T cell anergy in restoring immune tolerance is inducting apoptosis of autoreactive cell populations. Activated periphery T cells express death receptors from the TNF family (Fas/FasL), making them susceptible to activated-induced cell death (AICD).108,109 Specifically, TH1 cells are most susceptible to AICD while TH2/Treg cells are considered protective against AICD.110,111 A potential therapy that induces Fas would have to be administered pre-insulitis due to inflammation induced upregulated expression of Fas in β cells leading to Fas-mediated β cell apoptosis during insulitis.112,113 The therapeutic implications of inhibiting the Fas/FasL interaction alone after early insulitis have been debated elsewhere and is thought to have low efficacy due to redundant pro-inflammatory mechanisms.114 However, recently the combination of engineered microgels displaying FasL with a short course of rapamycin achieved localized immunomodulation of islet allografts for over 200 days.115 Since there are still no predictive early biomarkers for T1DM, overactivation of the Fas/Fas-ligand pathway must be avoided since it is associated with cancer and can lead to liver toxicity.109,116
Antigen Presenting Cells.
An ideal therapy for targeting APCs will mask the presentation of self-antigens known to activate T cells. One of the major autoantigens is proinsulin, with 90% of CD4+ insulin-reactive T cell clones from prediabetic NOD mice targeting the insulin B:9–23 peptide.117,118 Preventing autoreactive T cells from recognizing insulin bound to MHC class II could delay or attenuate T1DM autoimmunity. The therapeutic potential of this approach was explored by Kappler and coworkers through generation of mAb287 that specifically recognizes NOD I-Ag7 with insulin B:9–23 bond in register 3.119 Administration of mAb287 blocked CD4+ TCR interaction with the complex resulting in a delayed disease onset and the majority of 30-week-old mice had intact islets with only mild pre-insulitis. This work established that blocking TCR recognition of the MHC-insulin complex gives a beneficial physiological response in NOD mice, and human validation is a next step for mAb287.
Recently, inhibiting autoantigen presentation was expanded to using small molecules in humans. In silico screening methods of FDA approved drugs against DQ8 identified methyldopa, and treatment in NOD mice showed a reduction in primary antigen-specific T cell responses to insulin.120 Additionally, a phase Ib dose-escalation study with methyldopa in patients with recent-onset T1DM showed reduced inflammatory T cell responses to insulin. These results further highlight the potential of this approach and that small molecules, not just antibodies, can disrupt autoantigen presentation. However, because methyldopa is a clinically approved anti-hypertension drug,121 repurposing methyldopa for immunomodulation may have its own challenges.
CD40 is a member of the TNF receptor family and is expressed by professional APCs, B cells, and T cells.122–124 The natural ligand for CD40 is CD154, in either soluble or membrane-bound form, and the CD40-CD154 interaction is important for the initiation of insulitis.124–129 Initial therapeutic attempts to block this interaction in lupus patients using anti-CD154 resulted in risk of life-threatening thrombotic events.130 Current efforts to avoid these antibody-induced thrombotic events utilize a small peptide, KGYY15, which uses the primary CD154 domain that interacts with CD40.131 In NOD mice, KGYY15 administration was shown to prevent diabetes onset, and human validation is a next step.
B cells.
In recent years, it has become clear that B lymphocytes contribute to autoimmunity and secrete autoantibodies prior to insulitis.132–139 In NOD mice B cells support the expansion of pathogenic T cell responses, and these pathogenic populations contribute to T1DM progression in humans.140–142 Abnormalities in the thymus B cell compartment prior to T1DM presentation have also been found in the NOD mouse.143 This suggests thymic B cells in T1DM have a potentially harmful impact on the effectiveness of negative selection on autoreactive T cell precursors, thus making thymus B cells a potential target for T1DM therapies.
The generation of pathogenic B cell populations is thought to take place during affinity maturation through aberrant selection leading to expansion of autoreactive B lymphocytes.144,145 Clinical trials targeting B cells with a depleting P-specific therapy, rituximab, failed to preserve C-peptide production past two years and a follow-up study showed an increase of asymptomatic viremia.146,147 This highlights the challenge that systemic non-antigenic therapies face. Promising preclinical approaches have targeted B cells undergoing affinity maturation by disrupting the activated-induced cytidine deaminase protein (AID), a protein involved in the dsDNA break repair mechanism mediated by the RAD51 complex.148 Inhibition of RAD51 showed subsequent inhibition of T1DM development in NOD mice partially through expansion of CD73+ B lymphocyte populations that regulate pathogenic T cell responses. Interestingly, these results were mirrored through CRISPR-Cas9 directed ablation of the gene encoding AID.148
Immunoregulatory.
In recent years, therapies targeting critical junction points controlling immune inhibition/activation has revolutionized how cancer is treated. Recognition that many types of cancer modulate the immune system led to the development of several immune checkpoint inhibitors (CPIs) that has drastically improved patient outcomes in several cancers.149 Drawing from this success, approaches aimed at attenuating autoimmunity through checkpoint modulation are now actively being explored.
IDO is inducible in APCs and is associated with inhibition of T cell proliferation, prevention of memory T cell formation, and induction of Treg differentiation.150 Although the precise mechanism of activation and IDO’s role is still being explored, that IDO could have a therapeutic role in diabetes is becoming more apparent.151 Notably, treatment with IDO inhibitor 1-methyl-D-tryptopan (1MT) accelerated T1DM progression in NOD mice.152 Conversely, increased IDO expression from bystander fibroblasts can inhibit macrophage and infiltration of CD3+ cells into islet xenogeneic grafts, thus impairing pro-inflammatory responses.153 Recently, IDO expression from encapsulated Sertoli cells attenuated T1DM in experimental autoimmune encephalomyelitis (EAE) induced T1DM in C57BL/6 mice.154 However, no clinical trials have commenced for increasing IDO in T1DM.
The programmed death ligand 1 (PD-L1) is a ligand for PD-1, an inhibitory receptor expressed on activated lymphocytes that serves as an immune checkpoint to regulate tolerance.155,156 Malignant cancers hijack this checkpoint to locally suppress anti-tumor immunity, and clinically successful CPIs have been developed to inhibit formation of the PD-1/PD-L1 complex.157 Interestingly, both CD274 encoding (PD-L1) and PDCD1LG2 (encoding PD-L2) are up-regulated in newly onset T1DM,158 suggesting that PD-L1 might have a protective effect on T1DM. Additionally, cancer patients on anti-PD-1 therapy can develop T1DM.159 The NOD mouse model recapitulates these results, with anti-PD-L1 therapies accelerating disease progression.160
Overexpression of PD-L1 may prove more useful for T1DM treatment. PD-L1 expressing autologous hematopoietic stem and progenitor cells (HSPCs) transplanted into NOD revert diabetes in recent onset mice.161 Migration of these PD-L1.Tg HSPCs to the pancreas appears to be driven by the chemokine profile of the inflamed islets through high expression of C-X-C chemokine receptor type 4 (CXCR4) in these HSPCs. PD-L1 also plays a significant role in HSPC immunobiology and a there is a strong link between PD-L1 defects and T1DM.160,162,163 Taken together, HSPC expression of PD-L1 plays an important role in maintenance of immunocompetence and a cell based therapy using HSPCs to correct PD-L1 expression deficiency in T1DM is a promising approach.
Targeting Innate Immunity.
The innate immune system is involved in the detection and removal of pathogens.164 Innate immune receptors, including pattern recognition receptors, such as TLRs, have been shown to play a role in the development of T1DM.165 Modulation of TLRs and other proteins involved in their signaling pathways have been explored for modulating the incidence of T1DM. The role of innate immunity in T1DM has been reviewed elsewhere,166,167 and here we highlight some of the important molecular findings from recent studies.
MyD88.
The adapter protein myeloid differentiation primary response gene 88 (MyD88) is involved in signaling pathways for multiple TLRs and IL-1R.168 NOD MyD88 KO mice are protected from T1DM when housed in normal specific pathogen-free (SPF) conditions, but had high incidence of T1DM when housed in germ-free conditions.169 Interestingly, MyD88 KO mice showed tolerization of T cells in the PLNs under SPF conditions, whereas T cells in the spleen and mesenteric lymph nodes were still autoreactive to diabetes-associated peptides. Additionally, MyD88 KO mice in SPF conditions have changes in gut microbiota composition and further studies on the interplay between the innate immune system and gut microbiome have yielded promising results.169 In a related study, MyD88 inhibition utilizing a small molecule probe, TJ-M2010–6, in NOD mice had two-fold lower T1DM incidence, an increase in Tregs, and a reduction in CD4+/CD8+ T cell proliferation.170 More work is underway to confirm the role of the microbiome in T1DM and targeted approaches to altering it.
TRIF.
The TIR-domain-containing adapter-inducing interferon-β (TRIF) is an adapter molecule involved in TLR3 and TLR4 signaling. NOD TRIF KO mice were protected from T1DM when housed with other TRIF−/− mice, but not when cohoused with WT mice.171 Similar to MyD88 KO mice, TRIF KO lead to a different microbiota compared to WT mice, along with DCs that had reduced levels of inflammatory cytokines, such as TNF-α, and increased TGF-β.172 In addition, TRIF KO mice APCs had impaired antigen presentation and ultimately reduced T cell activation and proliferation.
TLRs.
Patients with T1DM have increased expression levels of TLR2 and TLR4 in monocytes.173 Studies with TLR4 KO mouse models have yielded mixed results from inflammation reduction, no effect, to an increase in T1DM incidence.169,174,175 Antibody inhibition of the TLR4/MD-2 interaction, mediated by both MyD88 and TRIF, resulted in a significant reduction of T1DM incidence accompanied by a significant increase in Fox3p+ Tregs.176 A decrease in APC-mediated T cell proliferation is one potential mechanism, as NOD scid mice treated with TLR4-Ab followed by addition of NOD CD4+ and CD8+ T cells were protected from T1DM incidence. Underscoring its clinical potential, treatment in NOD mice with established T1DM led to a permanent reversal in 71% of treated mice.176
NETs.
Neutrophils are the most abundant leukocyte in mammals, play important roles in both immunosurveillance and inflammation.177 To fight bacterial infection, neutrophils eject nuclear chromatin and bactericidal proteins to form structures called neutrophil extracellular traps (NETs).177 Patients with T1DM have been shown to have increased levels of neutrophil elastase and proteinase 3, two neutrophil serine proteases, and corresponding higher levels of NET formation.178 In NOD mice, neutrophil infiltration and NET formation appears as early as 3 weeks of age. Upon further investigation, depleting NOD mice of neutrophils resulted in a three-fold decrease in T1DM incidence.179 In a separate study, Lactococcus lactis, a food grade microorganism, was used to deliver staphylococcal nuclease (SNase) to disrupt NETs. NOD mice treated with L. lactis expressing SNase had a two-fold lower T1DM incidence, enhanced glucose tolerance, and a three-fold reduction of severe neutrophil infiltration of the islets,180 indicating clinical potential.
Islet Protection.
A complementary therapeutic approach to immunomodulation is to inhibit pathways involved in cytokine-mediated cell death of β cells. Cytokine-induced stress is known to induce autoimmune-mediated destruction of islets cells through endoplasmic reticulum (ER) dysfunction, altering cellular metabolism, and deregulating calcium homeostasis. Here we discuss progress made in modulating these pathways to preserve β cell mass and function.
Cellular Stress.
ER stress and the affiliated unfolded protein response (UPR) has an adverse effect in many autoimmune diseases, including T1DM.181 During ER stress, the ER transmembrane kinase/endoribonuclease (RNase), IRE1α, degrades mRNA to promote apoptosis. Treatment of NOD mice with the tyrosine kinase inhibitor Imatinib led to T1DM remission in 80% of mice in an ABL protein kinase-dependent manner, leading to inactivation of IRE1α.182,183 Accordingly, direct inhibition of IRE1α with the small molecule KIRA8 also led to 90% reversal of T1DM in NOD mice.184 Imatinib is currently being investigated in a phase II trial for treatment of T1DM (Table 1), highlighting that inhibition of proteins affiliated with ER stress and the UPR are promising targets to preserve β cell mass.
T1DM patients display islet cells with hyperexpression of HLA Class I proteins along with elevated expression of signal transducer and activator of transcription 1 (STAT1).185 A recent analysis indicated JAK-STAT and IFN signaling being highly enriched in multiple autoimmune diseases.186 Suppression of STAT1 in transgenic NOD mice can also reduce incidence of T1DM.187 Inhibition of JAK1/JAK2, enzymes that phosphorylate and activate STAT1, with the small molecule inhibitor AZD1480 in NOD mice reduced MHC Class I expression on β cells and results in three-fold lower incidence of T1DM.188 Directly inhibiting STAT1 to reduce MHC Class I expression is a promising therapeutic strategy, but an inhibitor of STAT1 has yet to be developed.
Cellular Metabolism.
Sphingolipid metabolism and sphingosine 1-phosphate (S1P) play an important role in the development of several inflammatory and autoimmune disorders, and their role has been thoroughly reviewed elsewhere.189 Briefly, S1P is a bioactive signaling molecule that is degraded by the ER protein S1P lyase (SPL) and is involved in the activation of innate and adaptive immune pathways, including the TH1/Treg balance.190 Treatment with fingolimod, an antagonist for the S1P receptor, increases survival and reduces insulitis scoring in NOD mice.191,192 In INS-1 cells, overexpression of SPL protects against cytokine-related cell death and caspase-3 activation, likely through prevention of Ca2+ leakage from the ER into the cytosol.193 Disruption of Ca2+ homeostasis in the β cell from cytokine exposure has been linked to ER stress and contributes to β cell death.194,195 Modulating Ca2+ homeostasis in INS-1 cells with the small molecules dantrolene and sitagliptin displayed less cytokine-related cell death and lower expression of thioredoxin-interacting protein (TXNIP). Knockdown of TXNIP resulted in a similar protection from cytokine-mediated cell death,196 suggesting that TXNIP is a mediator in Ca2+ homeostasis and a potential target for reducing β cell death.
Oxidative stress also contributes to T1DM pathogenesis.197 Of note, reactive oxygen species (ROS) act on the innate immune system to induce the production of inflammatory cytokines, such as TNF-α and IL-1β that ultimately led to the activation of CD4+ and CD8+ T cells.197,198 The high levels of ROS in T1DM has prompted studies utilizing antioxidants for their ability to prevent β cell destruction in T1DM. The antioxidant quercetin preserves β cell mass compared in streptozotocin (STZ) treated mice.199 Quercetin treatment of INS-1 cells protects against oxidative death from H2O2 and increased glucose-induced insulin secretion.200 Unfortunately, clinical investigations utilizing antioxidants in children with recently onset T1DM have failed to find any effects on C-peptide levels.201
Modulating specific pathways involved in ROS production also affect T1DM development. The enzyme 12/15-Lipoxygenase (12/15-LOX) catalyzes the oxygenation of arachidonic acid to form 12- and 15-hydroxyeicosatetraenoic acid (HETE), a pro-inflammatory mediator of insulitis.202,203 12-HETE also decreases insulin secretion and increases β cell death in human islets.203,204 In both STZ and NOD models, 12/15-LOX KO mice had a lower incidence of T1DM.205,206 Inhibition of the 12-LOX pathway protects mouse and human islets against cytokine-mediated cell death/disregulation.207–210 A follow-up study in STZ-induced T1DM mice showed treatment with the 12/15-LOX inhibitor ML351 displayed similar glucose tolerance and identical β cell mass to control C57BL/6 mice,211 making the 12-LOX pathway a promising target for β cells preservation.
IL-35 is a newly discovered cytokine believed to have immunosuppressive effects and an important role in autoimmunity.212,213 Systemic administration of recombinant IL-35 reverses established T1DM in a NOD mouse model through tolerization by Tregs.214 Transgenic expression and viral transfection of IL-35 in the pancreatic β cells of NOD mice displayed a four-fold lower T1DM incidence compared to control NOD mice and had reduced insulitis.215 While these preliminary results are promising, more work is needed to confirm the β cell protective effects of IL-35.
Epigenetic histone modifications have been shown to play a role in T1DM.216,217 Histone deacetylases (HDACs) are enzymes that catalyze the removal of acetyl groups from the lysine residues of histone proteins, generally reducing gene expression and play a role in transcriptional regulation.218 HDAC inhibitors have anti-inflammatory properties and are actively being explored as therapeutics for cancer, rheumatoid arthritis, and T1DM.219 The HDAC inhibitor ITF2357 protected islet cells from STZ-mediated destruction and INS-1 cells from cytokine-mediated cell death through inhibition of HDAC3.220–222 Furthermore, NOD mice treated with the specific HDAC3 inhibitor BRD3308 showed a significantly lower incidence in T1DM and significantly less mononuclear islet infilltration.223 Thus far, β cell protective strategies have produced promising results in animal models and may be candidates for combination therapies with immunomodulatory agents.
Regeneration.
β Cell Protection and Proliferation.
The restoration of lost or at-risk β cells is a major therapeutic focus in both T1DM and T2DM. These efforts focus on either selectively expanding or protecting β cells and reprograming non-β cells into functional β cells. Pancreas protection and regeneration studies were initially spurred by signs of β cell regeneration in adolescence and during times of stress such as pregnancy and obesity.224 However, adult endocrine islets have limited regenerative capacity. Recent studies have shown that α cells (glucagon producing) and δ cells (somatostatin producing) can undergo transdifferentiation to functional β cells or β-like cells.225,226 Although the precise mechanism and protection of β cells is unclear, potential in vivo therapeutic targets have been identified.
Inducing β cells to undergo mitosis through increases in relevant cyclins, cyclin-dependent kinases (CDKs), and with reductions in cell cycle inhibitors can protect against losses in β cell mass.227 A high-throughput small molecule screen identified harmine as a cell mitogenic compound and two relevant protein targets: tyrosine-regulated kinase-1a (DYRK1A) and the Nuclear Factors of activated T cells (NFAT).227 Harmine inhibits the ATP binding pocket of DYRK1A, inducing c-MYC activation, and driving β cell proliferation. Harmine also causes translocation of NFAT from the cytoplasm to the nucleus and increases expression of the transcription factors involved in β cell differentiation (NKx6.1, PDX1, and MAFA).227 Ultimately, harmine induced human β cell proliferation to levels comparable in early adolescence. This work established DYRK1A and NFAT as attractive clinical targets, but harmine has limited therapeutic utility due to off-target effects against other DYRK, CDC-like kinases (CLK), and inhibition of monoamine oxidases (MAOs).
Physiological glycemic regulation is modulated by the thyroid hormones triiodothyronine (T3) and the T3 precursor, levothyroxine (T4). T3 supplementation attenuates hyperglycemia in STZ-treated mice,228 while T4 supplementation in wild-type C57BL/6 mice enhanced glucose clearance and simultaneously increased the proliferation and apoptosis of pancreatic β cells, while increasing insulin production.229 T4 supplementation was also shown to reduce the onset of T1DM in the RIP-B7.1 model of early diabetes and increase survival of STZ-treated mice through increased expression of Akt and glucokinase. Clinical application will need to avoid the detrimental effects of high circulating levels of these hormones (thyrotoxicosis).230,231
Maintaining a stable rate of β cell turnover would be beneficial, yet an ideal therapy would also keep β cell mass within a normal range and prevent future insulitis. Substance P (SP) is a neuropeptide that acts as a pain-sensing neurotransmitter in the central nervous system and as an immune modulator.232 SP directly induces proliferation of β cells through the PI3K/Akt pathway, affects the preservation of β cells at early time points after STZ treatment, and inhibits further insulitis.233 This suggests early intervention of T1DM can benefit from both β cell and neural components in the pancreatic islets.
Novel agents that can shift a pro-inflammatory autoimmune islet environment towards an anti-inflammatory milieu are also desirable for islet protection and regeneration therapies. Recently, liver receptor homologue-1 (LRH-1) was shown to modulate the expression of genes involved in glucose homeostasis and is protective against both inflammation and ER stress.234–236 Small molecule BL001 activates LRH-1 in several T1DM mouse models resulting in a shift towards the anti-inflammatory macrophage phenotype (M2), secretion of IL-10, Treg expansion, and a reduction in TH1 cells with increasing TH2 phenotype.237 This resulted in reduced insulitis and LRH-1 stimulated regeneration of β cells through a non-proliferation/apoptosis manner, suggesting LRH-1 activation may promote α to β cell transdifferentiation.
α to β Cell Transdifferentiation.
β cell regeneration can be achieved through transdifferentiation of developmentally related pancreatic cell types into functional β cells. Notable work in elucidating the mechanism of either α or δ to β cell transdifferentiation highlights the activation of transcription factors pdx1 and pax4, the downregulation of arx and foxo1, and the influence of epigenetic modulation.225,226,238–241 Direct inhibitors to transcription factors and epigenetic modulators has traditionally been considered “undruggable”, however recent progress has been made and more specific inhibitors have been developed.242 Here we focus on extracellular and extranuclear targets that stimulate islet cell transdifferentiation to β cells which act, in part, through transcription factor modulation.
The insulin-like growth factor (IGF) signaling pathway plays a role in cell proliferation and differentiation.243 Overexpression of the IGF-binding protein 1 (IGFBP1) in zebrafish results in inhibition of IGF signaling and promotes β cell regeneration through α to β transdifferentiation.244 Importantly, IGF receptor inhibitor picropodophyllin replicates this result and increases α cell expression of PDX1, an important marker for α to β transdifferentiation. The role of IGFBP1 was further explored in transgenic mice overexpressing the protein and found to restore normoglycemia when administered exogenous IGF-I.245 Enhancing IGFBP1’s potential role in human diabetes, high serum levels of IGFBP1 was associated with a reduced risk of T2DM development.246,247
Aristaless related homeobox (ARX) is a master regulator of α cell fate and is required for maintaining glucagon production.248 Targeting ARX can modulate the pancreatic ratio of α, β, δ, and pancreatic polypeptide cells. Direct inactivation of ARX alone is insufficient to induce α to β cell conversion,241,249 and inhibition of DNA methyltransferase 1 (DNM1) is also required for conversion of α cells to functional β-like insulin secreting cells.250 Heterogeneity of expressed β cell regulators was observed, showing incomplete conversion to a full β cell morphology, but the observed response to glucose is encouraging. Indirect functional repression of ARX also shows therapeutic promise. The anti-malarial drug Artemisinin induces ARX translocation to the cytoplasm and stimulates GABAA receptor signaling, resulting in increased β cell mass in zebrafish and rodent models.251 Interestingly, GABA signaling is known to modulate the immune system and promote β cell proliferation.252,253
Taken together, β cell regeneration either through protection, proliferation, or transdifferentiation offers hope as a successful regeneration therapy and need only restore normoglycemia to greatly impact treatment for T1DM patients.
ISLET TARGETING
In addition to modulating specific pathogenic pathways, targeted technologies for therapeutic and diagnostic purposes are under investigation for T1DM. Targeted β cell/islet probes by themselves do not have therapeutic effects, but are used to image and monitor disease progression, serve as a biomarker for disease incidence, or play a role in targeting drug delivery systems. Indeed, drug-loaded targeted constructs are now being investigated to better localize existing drugs/therapies to the islet microenvironment and improve treatment outcomes. Here we discuss advances in β cell imaging probes and islet delivery technologies.
β Cell Imaging.
Current clinical tests for T1DM diagnosis, such as insulin/C-peptide measurements after a glucose challenge, serve mainly as indicators of β cell function, but not β cell mass. In addition, changes in these markers are typically only detected when nearly 80% of β cells have already lost function. Therefore, development of probes to measure/monitor β cells is urgently needed to accurately monitor T1DM progression.
β cells are scattered in the pancreas in the islets of Langerhans and constitute only 1–3% of total pancreas mass, thus imaging requires a probe with high specificity and high signal-to-noise ratio. While there are a number of imaging probes used to monitor β cell mass, only a small number of probes have been tested clinically, including exendin-4/exenatide, tetrabenazine, 5-hydroxy-tryptophan (HTP), sulfonylurea, and (+)-4-propyl-9-hydroxynaphthoxazine (PHNO) based probes.254–257
Exendin-4, also known as exenatide, is a 39 amino acid peptide isolated from Heloderma horridum that shares 50% homology with endogenous glucagon-like peptide 1 (GLP-1), but has much slower clearance and high stability in vivo.258 Clinically, exendin-4 is typically prescribed with metformin for treatment of T2DM to increase insulin secretion and reduce body weight.259 GLP-1R expression is restricted to β cells in the pancreas, with low expression in exocrine tissue. Exendin-4 has been extensively utilized as a targeting ligand for β cell imaging probes.260 An exendin-4 based probe conjugated to diethylenetriaminepentaacetic acid (DTPA) chelated with Indium-111 was successfully used to image transplanted islets in humans.261 Additional clinical studies with a 111In-DTPA-Exendin probe found a linear relationship between β cell mass and pancreatic uptake of the probe, but failed to find any statistical significance in uptake between healthy and T1DM human subjects.262
Vesicular monoamine transporter 2 (VMAT-2) is involved in the storage and release of amines, such as dopamine and serotonin, and is expressed both in neuroendocrine cells and β cells.263,264 Tetrabenazine and its various analogs, such as dihydrotetrabenazine (DTBZ) and fluoropropyl-dihydrotetrabenazine (FP-DTBZ), are the most widely used VMAT2 antagonists.265,266 The analog [18F]-FP-DTBZ successfully correlates β cell function and mass, after accounting for pancreatic volume,267 with T1DM subjects showing an average 59% loss in specific binding of the probe compared to healthy subjects. Currently, there is some disagreement about using VMAT2 as a biomarker for β cell mass, as binding in the pancreas may be mainly nonspecific,268 and further studies are needed.
Although they come from different germinal layers, pancreatic β cells and neurons have shared functions, including serotonin production.269 In many mammals, including humans, serotonin production is exclusive to the islets with no expression in the exocrine pancreas.270 [11C]5-hydroxy-tryptophan ([11C]5-HTP) is a probe used to visualize serotonin biosynthesis in disease and is used to monitor neuroendocrine tumors.271,272 Initial studies with [11C]5-HTP in exocrine PANC-1 cells and endocrine human insulinoma cell lines (CM) suggested signal may not be representative of β cell mass, but PANC-1 cells rapidly lost signal whereas CM cells maintained signal.273 [11C]5-HTP was further studied in non-human primates, where it had two-fold higher pancreatic uptake compared to the liver. STZ-induced diabetic mice showed 66% lower signal compared to wild-type mice, suggesting a correlation to functional β cell mass.274 Clinical studies show a correlation between plasma C-peptide levels and the pancreatic uptake of [11C]5-HTP, suggesting that the probe may be used as a marker for β cell mass.275
Sulfonylureas, such as glibenclamide, bind to the sulfonylurea receptor 1 (SUR1) on β cells and are used clinically as insulin secretagogues for treatment of T2DM.276 Glibenclamide analogs for β cell imaging have been synthesized with high affinity to SUR1 and variable lipophilicities.276,277 A promising glipizide analog conjugated to 99mTc-DTPA was developed and displays a thirty-fold higher pancreatic uptake with reduced kidney and liver uptake.278 To compliment this effort, multivalency using glibenclamide-polyamidoamine (PAMAM) probes have been evaluated. PAMAM dendrimers with 15 glibenclamide molecules/dendrimer have higher affinity for SUR1 than the unmodified glibenclamide and successfully labeled human islets in vitro and murine islets ex vivo.279
The dopamine receptors, D2 and D3, also have overlap between neural and pancreatic cells. D2/D3 receptors colocalize with insulin-containing granules suggesting that probes used to image D2/D3 receptors in the brain can also image the receptors on β cells.280 Iodobenzamide, a molecule that is used clinically to image D2 receptors in neurodegenerative diseases, was recently shown to be effective for imaging islet grafts. Unfortunately, [123I]-Iodobenzamide also had high uptake in spleen, liver, and kidney, where grafts are often placed.281 Fallypride, another D2/D3 antagonist used for clinically imaging D2 receptors in the brain, has been investigated for imaging transplanted islet grafts. [18F]Fallypride had 0.05% injected dose uptake in pancreas, but encouragingly successfully visualized transplanted islets.282 A later study confirmed that [18F]fallypride uptake was 77% lower in STZ-treated mice compared to control, indicating potential utility in monitoring β cell mass.283 A currently used compound for positron emission tomography (PET) brain radioligands, [11C](1)-4-propyl-9-hydroxynaphthoxazine (PHNO), was recently identified as a D2/D3 antagonist and used to differentiate between a healthy, T1DM C-peptide positive, and a T1DM C-peptide negative subject based on PET imaging.284
A number of other probes have been developed and tested in multiple animal models, but have failed to garner any data in humans. Of note is the G-coupled protein receptor 44 (GPR44), which binds Prostaglandin D2 (PGD2) and was identified as a β cell-specific biomarker through a proteomic screen.285 Initial investigations in vitro with human islets and EndoC-βH1 cells confirmed the surface expression of GPR44 and identified the antagonist AZD3825 as having nanomolar affinity and high endocrine-exocrine binding.286 Biodistribution studies in non-human primates and pigs with a related GPR44 antagonist indicated pancreatic binding, but the probe also accumulated in the small intestines and spleen.287
Islet Delivery.
Several targeting ligands are under investigation for their ability to selectively deliver therapeutics to the islet microenvironment. Using phage display and a library of small cyclic peptides, the peptide CHVLWSTRC was found to specifically home to islets where it binds to the EphrinA4 receptor on islet capillary cells.288 This cyclic targeting ligand has been utilized twice in attempts to deliver therapeutics specifically to the islet microenvironment. Conjugation of this peptide to a PLGA-PEG polymer and nanoformulation with the anti-inflammatory drug genistein yielded nanoparticles targeted to the islet microenvironment. In rat islet capillary endothelial cells that endogenously express EphA4, the targeted nanoparticles had three-fold higher uptake compared to untargeted/scrambled nanoparticles.289 This peptide targeting ligand was also used to deliver plasmid DNA encoding retinoic acid early inducible gene-1 (RAE-1) to islet capillary cells. The targeted polyplex displayed 2.5-fold higher uptake compared to untargeted control in MS-1 cells that express EphA4.290 Targeted constructs utilizing the CHVLWSTRC peptide have shown promising results in vitro, but will need to be validated in animal models for their ability to bind islet vasculature.
Several antibodies have been developed to specifically home to the islet microenvironment. An in vitro phage display experiment with INS-1 cells was used to identify antibody SCA B5 binding specifically to β cells with low exocrine tissue affinity.291 In vivo, 5.12% of injected dose of 125I-labeled SCA B5 localized to the pancreas with low (<0.70%) accumulation in other tissues. The putative molecular target of SCA B5 is currently unknown.291 As previously mentioned, GAD-65 is an autoantigen affiliated with T1DM, so it may be used as a unique feature in the pancreatic microenvironment to target. An anti-GAD antibody fragment (Fab) has been developed as a β cell specific nucleic acid delivery system and showed a ten-fold higher transfection in GAD-expressing MIN-6 cells compared to an untargeted construct with low transfection in GAD-negative HEK-293 cells.292 These antibodies had promising in vitro results, but need to be validated in vivo.
β cell selective imaging probes are of vital need to monitor loss of β cell mass as the disease progresses. These probes could enable β cell mass to be a new biomarker for disease incidence. Finally, these targeting ligands have potential for islet-specific delivery of highly potent therapeutics that otherwise may give detrimental effects when administered systemically.
FUTURE PERSPECTIVE
Significant progress has been made developing new therapeutic modalities that attack T1DM on multiple fronts: from immunomodulatory agents, β cell protective and regenerative strategies, to new diagnostic probes. All of these approaches take advantage of key molecular mechanisms and targets in T1DM, and while pre-clinical results have been promising, clinical translation of these approaches has remained elusive. Many of these therapeutic modalities are more effective when administered in pre-symptomatic stages of the disease, which is currently challenging in humans due to a lack of predictive and reliable diagnostic readouts. Clinical evaluation of some of these therapies have revealed adverse effects through the distribution and systemic action of these agents at unrelated tissue sites. While many monotherapies are effective in pre-clinical models, the added complexity of human biology may require combinatorial approaches to achieve clinical efficacy.
The next generation of therapeutic modalities (Figure 3) will benefit from current efforts at developing predictive biomarkers and probes in T1DM-susceptible individuals. Improved diagnostics will enable many of the therapies discussed here to be interventional and expand the temporal window for many immunomodulatory and β cell protection/regeneration therapies. Equally important will be the emergence of drug delivery systems that can bias the systemic distribution of these agents to relevant tissue sites, enabling localized therapeutic modulation and avoiding adverse effects in healthy tissues. T1DM is a complex disease where our understanding of the pathogenesis is evolving. The diversity and richness of the approaches reviewed here reveal the direction of future treatment paradigms for this disease that could dramatically improve clinical outcomes and the quality of life for these patients.
Figure 3.

Future therapeutic paradigm for localized early delivery of therapeutics for T1DM. In combination with genetic screening and early biomarker T1DM indication, formulated nanoparticles with encapsulated therapeutic agent can be conjugated to islet targeting agents that populate the nanoparticle surface. These localized delivery agents will likely need to be used in combination therapies.
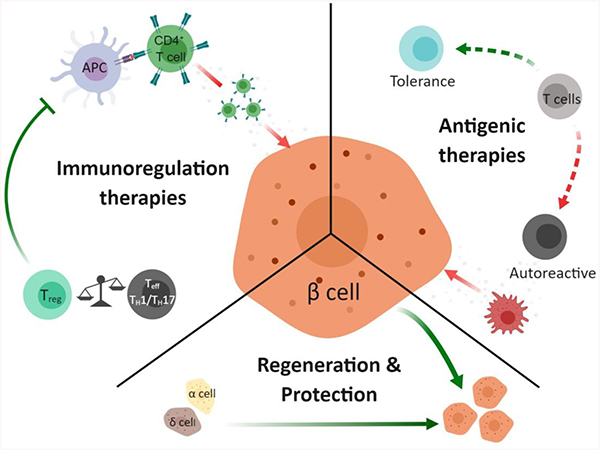
Figure 2.

Selective T1DM pre-clinical and clinical therapeutic intervention strategies through immunoregulation, β cell protection and regeneration, and antigenic therapy. Solid arrows denote a causal event. Dashed arrows denote cell population shift. Red arrows indicate the pathway leads to T1DM autoimmunity and green arrows indicated protective and immune tolerance pathways. Green text indicates increase secretion or strengthening of indicated molecular interaction. Red text indicates suppression of indicated molecular interaction. Purple text indicates additional therapies discussed herein.
ACKNOWLEDGMENT
All figures were created in Biorender (https://biorender.io).
Funding Sources
This work was supported by a National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grant (NIH NIDDK RFA-DK-15–030).
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Katsarou A, Gudbjörnsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, Jacobsen LM, Schatz DA, and Lernmark A (2017) Type 1 diabetes mellitus. Nat. Rev. Dis. Prim 3, 17016. [DOI] [PubMed] [Google Scholar]
- (2).Diaz-Valencia PA, Bougnères P, and Valleron A-J (2015) Global epidemiology of type 1 diabetes in young adults and adults: a systematic review. BMC Public Health 15, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Dall TM, Mann SE, Zhang Y, Quick WW, Seifert RF, Martin J, Huang EA, and Zhang S (2009) Distinguishing the economic costs associated with type 1 and type 2 diabetes. Popul. Health Manag 12, 103–10. [DOI] [PubMed] [Google Scholar]
- (4).Tao B, Pietropaolo M, Atkinson M, Schatz D, and Taylor D (2010) Estimating the cost of type 1 diabetes in the U.S.: a propensity score matching method. PLoS One 5, e11501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Van Belle TL, Coppieters KT, and Von Herrath MG (2011) Type 1 Diabetes: Etiology, Immunology, and Therapeutic Strategies. Physiol. Rev 91, 79–118. [DOI] [PubMed] [Google Scholar]
- (6).Vrochides D, Paraskevas S, and Papanikolaou V (2009) Transplantation for type 1 diabetes mellitus. Whole organ or islets? Hippokratia 13, 6–8. [PMC free article] [PubMed] [Google Scholar]
- (7).Vegas AJ, Veiseh O, Gürtler M, Millman JR, Pagliuca FW, Bader AR, Doloff JC, Li J, Chen M, Olejnik K, Tam HH, Jhunjhunwala S, Langan E, Aresta-Dasilva S, Gandham S, McGarrigle JJ, Bochenek MA, Hollister-Lock J, Oberholzer J, Greiner DL, Weir GC, Melton DA, Langer R, and Anderson DG (2016) Long-term glycemic control using polymer-encapsulated human stem cell-derived beta cells in immune-competent mice. Nat. Med 22, 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Veiseh O, Doloff JC, Ma M, Vegas AJ, Tam HH, Bader AR, Li J, Langan E, Wyckoff J, Loo WS, Jhunjhunwala S, Chiu A, Siebert S, Tang K, Hollister-Lock J, Aresta-Dasilva S, Bochenek M, Mendoza-Elias J, Wang Y, Qi M, Lavin DM, Chen M, Dholakia N, Thakrar R, Lacík I, Weir GC, Oberholzer J, Greiner DL, Langer R, and Anderson DG (2015) Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nat. Mater 14, 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Vegas AJ, Veiseh O, Doloff JC, Ma M, Tam HH, Bratlie K, Li J, Bader AR, Langan E, Olejnik K, Fenton P, Kang JW, Hollister-Locke J, Bochenek MA, Chiu A, Siebert S, Tang K, Jhunjhunwala S, Aresta-Dasilva S, Dholakia N, Thakrar R, Vietti T, Chen M, Cohen J, Siniakowicz K, Qi M, McGarrigle J, Lyle S, Harlan DM, Greiner DL, Oberholzer J, Weir GC, Langer R, and Anderson DG (2016) Combinatorial hydrogel library enables identification of materials that mitigate the foreign body response in primates. Nat Biotech 34, 345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Desai T, and Shea LD (2017) Advances in islet encapsulation technologies. Nat. Rev. Drug Discov 16, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Redondo MJ, and Eisenbarth GS (2002) Genetic control of autoimmunity in Type I diabetes and associated disorders. Diabetologia 45, 605–622. [DOI] [PubMed] [Google Scholar]
- (12).Erlich H, Valdes AM, Noble J, Carlson JA, Varney M, Concannon P, Mychaleckyj JC, Todd JA, Bonella P, Fear AL, Lavant E, Louey A, Moonsamy P, and Consortium, T. 1 D. G. (2008) HLA DR-DQ Haplotypes and Genotypes and Type 1 Diabetes Risk. Diabetes 57, 1084–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Burg Ashley R., Das Shaonli, Padgett Lindsey E., K. ZE and T. HM (2018) Superoxide Production by NADPH Oxidase Intensifies Macrophage Antiviral Responses during Diabetogenic Coxsackievirus Infection. J. Immunol 200, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kappler JW, Roehm N, and Marrack P (1987) T cell tolerance by clonal elimination in the thymus. Cell 49, 273–80. [DOI] [PubMed] [Google Scholar]
- (15).Abramson J, and Husebye ES (2016) Autoimmune regulator and self-tolerance - molecular and clinical aspects. Immunol. Rev 271, 127–140. [DOI] [PubMed] [Google Scholar]
- (16).Willcox A, Richardson SJ, Bone AJ, Foulis AK, and Morgan NG (2009) Analysis of islet inflammation in human type 1 diabetes. Clin. Exp. Immunol 155, 173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kuhn C, Rezende RM, M’Hamdi H, da Cunha AP, and Weiner HL (2017) IL-6 Inhibits Upregulation of Membrane-Bound TGF-β 1 on CD4 + T Cells and Blocking IL-6 Enhances Oral Tolerance. J. Immunol 198, 1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ, Bode B, Aronoff S, Holland C, Carlin D, King KL, Wilder RL, Pillemer S, Bonvini E, Johnson S, Stein KE, Koenig S, Herold KC, and Daifotis AG (2011) Teplizumab for treatment of type 1 diabetes (Protégé study): 1-year results from a randomised, placebo-controlled trial. Lancet 378, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Aronson R, Gottlieb PA, Christiansen JS, Donner TW, Bosi E, Bode BW, and Pozzilli P (2014) Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: Results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care 37, 2746–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chatzigeorgiou A, Harokopos V, Mylona-Karagianni C, Tsouvalas E, Aidinis V, and Kamper E (2010) The pattern of inflammatory/anti-inflammatory cytokines and chemokines in type 1 diabetic patients over time. Ann. Med 42, 426–438. [DOI] [PubMed] [Google Scholar]
- (21).Rabinovitch A, and Suarez-Pinzon WL (2007) Roles of cytokines in the pathogenesis and therapy of type 1 diabetes. Cell Biochem. Biophys 48, 159–163. [DOI] [PubMed] [Google Scholar]
- (22).Dinarello CA, and van der Meer JWM (2013) Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol 25, 469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mandrup-Poulsen T, Pickersgill L, and Donath MY (2010) Blockade of interleukin 1 in type 1 diabetes mellitus. Nat. Rev. Endocrinol 6, 158–166. [DOI] [PubMed] [Google Scholar]
- (24).Thomas HE, Irawaty W, Darwiche R, Brodnicki TC, Santamaria P, Allison J, and Kay TWH (2004) IL-1 Receptor Deficiency Slows Progression to Diabetes in the NOD Mouse. Diabetes 53, 113–121. [DOI] [PubMed] [Google Scholar]
- (25).Sumpter KM, Adhikari S, Grishman EK, and White PC (2011) Preliminary studies related to anti-interleukin-1β therapy in children with newly diagnosed type 1 diabetes. Pediatr. Diabetes 12, 656–667. [DOI] [PubMed] [Google Scholar]
- (26).Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Greenbaum CJ, Herold KC, Marks JB, Raskin P, Sanda S, Schatz D, Wherrett DK, Wilson DM, Krischer JP, Skyler JS, Pickersgill L, De Koning E, Ziegler AG, Böehm B, Badenhoop K, Schloot N, Bak JF, Pozzilli P, Mauricio D, Donath MY, Castaño L, Wägner A, Lervang HH, Perrild H, and Mandrup-Poulsen T (2013) Interleukin-1 antagonism in type 1 diabetes of recent onset: Two multicentre, randomised, double-blind, placebo-controlled trials. Lancet 381, 1905–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kaizer EC, Glaser CL, Chaussabel D, Banchereau J, Pascual V, and White PC (2007) Gene Expression in Peripheral Blood Mononuclear Cells from Children with Diabetes. J. Clin. Endocrinol. Metab 92, 3705–3711. [DOI] [PubMed] [Google Scholar]
- (28).Lee L-F, Xu B, Michie SA, Beilhack GF, Warganich T, Turley S, and McDevitt HO (2005) The role of TNF-alpha in the pathogenesis of type 1 diabetes in the nonobese diabetic mouse: analysis of dendritic cell maturation. Proc. Natl. Acad. Sci. U. S. A 102, 15995–6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lechleitner M, Koch T, Herold M, Dzien A, and Hoppichler F (2000) Tumour necrosis factor-alpha plasma level in patients with type 1 diabetes mellitus and its association with glycaemic control and cardiovascular risk factors. J. Internal. Medicine 248, 67–76. [DOI] [PubMed] [Google Scholar]
- (30).Yang XD, Tisch R, Singer SM, Cao ZA, Liblau RS, Schreiber RD, and McDevitt HO (1994) Effect of tumor necrosis facto α on insulin-dependent diabetes mellitus in NOD mice. I. The early development of autoimmunity and the diabetogenic process. J. Exp. Med 180, 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lucy M, Jihnhee Y, Torsten B, John B, Christine A, Shannon F, and Teresa Q (2009) Etanercept Treatment in Children With New-Onset Type 1 Diabetes. Diabetes Care 32, 1244–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, and Surh CD (2001) IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc. Natl. Acad. Sci. U. S. A 98, 8732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Monti P, and Bonifacio E (2014) Interleukin-7 and type 1 diabetes. Curr. Diab. Rep 14, 1–7. [DOI] [PubMed] [Google Scholar]
- (34).Heninger A-K, Theil A, Wilhelm C, Petzold C, Huebel N, Kretschmer K, Bonifacio E, and Monti P (2012) IL-7 Abrogates Suppressive Activity of Human CD4+CD25+FOXP3+ Regulatory T Cells and Allows Expansion of Alloreactive and Autoreactive T Cells. J. Immunol 189, 5649–5658. [DOI] [PubMed] [Google Scholar]
- (35).Calzascia T, Pellegrini M, Lin A, Garza KM, Elford AR, Shahinian A, Ohashi PS, and Mak TW (2008) CD4 T cells, lymphopenia, and IL-7 in a multistep pathway to autoimmunity. Proc. Natl. Acad. Sci. U. S. A 105, 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lee L, Logronio K, Tu GH, Zhai W, Ni I, Mei L, Dilley J, Yu J, Rajpal A, Brown C, Appah C, Mi S, Han B, Affolter T, Lin JC, Lee L, Logronio K, Huan G, Zhai W, Ni I, Mei L, Dilley J, Yu J, Chin SM, Han B, Affolter T, and Lin JC (2012) Correction for Lee et al. , Anti-IL-7 receptor-reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T-cell function. Proc. Natl. Acad. Sci 109, 16393–16393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Penaranda C, Kuswanto W, Hofmann J, Kenefeck R, Narendran P, Walker LSK, Bluestone JA, Abbas AK, and Dooms H (2012) IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc. Natl. Acad. Sci 109, 12668–12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Tanaka T, Narazaki M, and Kishimoto T (2012) Therapeutic Targeting of the Interleukin-6 Receptor. Annu. Rev. Pharmacol. Toxicol 52, 199–219. [DOI] [PubMed] [Google Scholar]
- (39).Hundhausen C, Roth A, Whalen E, Chen J, Schneider A, Long SA, Wei S, Rawlings R, Kinsman M, Evanko SP, Wight TN, Greenbaum CJ, Cerosaletti K, and Buckner JH (2016) Enhanced T cell responses to IL-6 in type 1 diabetes are associated with early clinical disease and increased IL-6 receptor expression. Sci. Transl. Med 8, 356ra119–356ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Campbell IL, Oxbrow L, and Harrison LC (1991) Essential role for interferon-gamma and interleukin-6 in autoimmune insulin-dependent diabetes in NOD/Wehi mice. 87, 739–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Tian J, and Kaufman DL (2009) Antigen-based therapy for the treatment of type 1 diabetes. Diabetes 58, 1939–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Erlander MG, Tillakaratne NJ, Feldblum S, Patel N, and Tobin AJ (1991) Two genes encode distinct glutamate decarboxylases. Neuron 7, 91–100. [DOI] [PubMed] [Google Scholar]
- (43).Hagopian WA, Michelsen B, Karlsen AE, Larsen F, Moody A, Grubin CE, Rowe R, Petersen J, McEvoy R, and Lernmark A (1993) Autoantibodies in IDDM primarily recognize the 65,000-M(r) rather than the 67,000-M(r) isoform of glutamic acid decarboxylase. Diabetes 42, 631–6. [DOI] [PubMed] [Google Scholar]
- (44).Shi Y, Kanaani J, Menard-Rose V, Ma YH, Chang PY, Hanahan D, Tobin A, Grodsky G, and Baekkeskov S (2000) Increased expression of GAD65 and GABA in pancreatic beta-cells impairs first-phase insulin secretion. Am. J. Physiol. Endocrinol. Metab 279, E684–94. [DOI] [PubMed] [Google Scholar]
- (45).Rydén AKE, Wesley JD, Coppieters KT, and Von Herrath MG (2014) Non-antigenic and antigenic interventions in type 1 diabetes. Hum. Vaccin. Immunother 10, 838–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Elias D, Reshef T, Birk OS, van der Zee R, Walker MD, and Cohen IR (1991) Vaccination against autoimmune mouse diabetes with a T-cell epitope of the human 65-kDa heat shock protein. Proc. Natl. Acad. Sci 88, 3088–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Raz I, Elias D, Avron A, Tamir M, Metzger M, and Cohen IR (2001) Beta-cell function in new-onset type 1 diabetes and immunomodulation with a heat-shock protein peptide (DiaPep277): a randomised, double-blind, phase II trial. Lancet (London, England) 358, 1749–53. [DOI] [PubMed] [Google Scholar]
- (48).Lazar L, Ofan R, Weintrob N, Avron A, Tamir M, Elias D, Phillip M, and Josefsberg Z (2007) Heat-shock protein peptide DiaPep277 treatment in children with newly diagnosed type 1 diabetes: a randomised, double-blind phase II study. Diabetes. Metab. Res. Rev 23, 286–91. [DOI] [PubMed] [Google Scholar]
- (49).Huurman VAL, Decochez K, Mathieu C, Cohen IR, and Roep BO (2007) Therapy with the hsp60 peptide DiaPep277 in C-peptide positive type 1 diabetes patients. Diabetes. Metab. Res. Rev 23, 269–275. [DOI] [PubMed] [Google Scholar]
- (50).Schloot NC, Meierhoff G, Lengyel C, Vándorfi G, Takács J, Pánczél P, Barkai L, Madácsy L, Oroszlán T, Kovács P, Sütö G, Battelino T, Hosszufalusi N, and Jermendy G (2007) Effect of heat shock protein peptide DiaPep277 on beta-cell function in paediatric and adult patients with recent-onset diabetes mellitus type 1: two prospective, randomized, double-blind phase II trials. Diabetes. Metab. Res. Rev 23, 276–85. [DOI] [PubMed] [Google Scholar]
- (51).Barbera Betancourt A, Lyu Q, Broere F, Sijts A, Rutten VPMG, and van Eden W (2017) T Cell-Mediated Chronic Inflammatory Diseases Are Candidates for Therapeutic Tolerance Induction with Heat Shock Proteins. Front. Immunol 8, 1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Alleva DG, Maki RA, Putnam AL, Robinson JM, Kipnes MS, Dandona P, Marks JB, Simmons DL, Greenbaum CJ, Jimenez RG, Conlon PJ, and Gottlieb PA (2006) Immunomodulation in Type 1 Diabetes by NBI-6024, an Altered Peptide Ligand of the Insulin B(9–23) Epitope. Scand. J. Immunol 63, 59–69. [DOI] [PubMed] [Google Scholar]
- (53).Walter M, Philotheou A, Bonnici F, Ziegler A-G, Jimenez R, and NBI-6024 Study Group. (2009) No effect of the altered peptide ligand NBI-6024 on beta-cell residual function and insulin needs in new-onset type 1 diabetes. Diabetes Care 32, 2036–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Emma Smith, Lp M (2018) Peptide immunotherapy for Type 1 Diabetes—Clinical Advances. Front. Immunol 9, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, Tsai S, Wang J, Garabatos N, Izquierdo C, Agrawal S, Keough MB, Yong VW, James E, Moore A, Yang Y, Stratmann T, Serra P, and Santamaria P (2016) Expanding antigen-specific regulatory networks to treat autoimmunity. Nature 530, 434–440. [DOI] [PubMed] [Google Scholar]
- (56).Hu C, Ding H, Zhang X, Wong FS, and Wen L (2013) Combination treatment with anti-CD20 and oral anti-CD3 prevents and reverses autoimmune diabetes. Diabetes 62, 2849–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Skelley JW, Elmore LK, and Kyle JA (2012) Teplizumab for treatment of type 1 diabetes mellitus. Ann. Pharmacother 46, 1405–12. [DOI] [PubMed] [Google Scholar]
- (58).Bresson D, Togher L, Rodrigo E, Chen Y, Bluestone JA, Herold KC, and von Herrath M (2006) Anti-CD3 and nasal proinsulin combination therapy enhances remission from recent-onset autoimmune diabetes by inducing Tregs. J. Clin. Invest 116, 1371–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Chatenoud L (2010) Immune therapy for type 1 diabetes mellituswhat is unique about anti-CD3 antibodies? Nat. Rev. Endocrinol 6, 149–157. [DOI] [PubMed] [Google Scholar]
- (60).Daifotis AG, Koenig S, Chatenoud L, and Herold KC (2013) Anti-CD3 clinical trials in type 1 diabetes mellitus. Clin. Immunol 149, 268–278. [DOI] [PubMed] [Google Scholar]
- (61).Kuhn C, and Weiner HL (2016) Therapeutic anti-CD3 monoclonal antibodies: From bench to bedside. Immunotherapy 8, 889–906. [DOI] [PubMed] [Google Scholar]
- (62).Chatenoud L, Thervet E, Primo J, and Bach JF (1994) Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc. Natl. Acad. Sci 91, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, and Bluestone JA (2002) Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 346, 1692–1698. [DOI] [PubMed] [Google Scholar]
- (64).Herold KC, Gitelman SE, Ehlers MR, Gottlieb PA, Greenbaum CJ, Hagopian W, Boyle KD, Keyes-Elstein L, Aggarwal S, Phippard D, Sayre PH, Mcnamara J, Bluestone J. a, AbATE Study Team, and Team, S. (2013) Teplizumab (Anti-CD3 mAb) Treatment Preserves C-Peptide Responses in Patients With New-Onset Type 1 Diabetes in a Randomized Controlled Trial. Diabetes 62, 3766–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Chhabra A (2010) Mitochondria-centric activation induced cell death of cytolytic T lymphocytes and its implications for cancer immunotherapy. Vaccine 28, 4566–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Baixauli F, Martín-Cófreces NB, Morlino G, Carrasco YR, Calabia-Linares C, Veiga E, Serrador JM, and Sánchez-Madrid F (2011) The mitochondrial fission factor dynamin-related protein 1 modulates T-cell receptor signalling at the immune synapse. EMBO J. 30, 1238–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Kamiński MM, Sauer SW, Kamiński M, Opp S, Ruppert T, Grigaravičius P, Grudnik P, Gröne H-J, Krammer PH, and Gülow K (2012) T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2, 1300–15. [DOI] [PubMed] [Google Scholar]
- (68).Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang C-R, Schumacker PT, Licht JD, Perlman H, Bryce PJ, and Chandel NS (2013) Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Chen J, Chernatynskaya AV, Li J-W, Kimbrell MR, Cassidy RJ, Perry DJ, Muir AB, Atkinson MA, Brusko TM, and Mathews CE (2017) T cells display mitochondria hyperpolarization in human type 1 diabetes. Sci. Rep 7, 10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Garyu JW, Uduman M, Stewart A, Rui J, Deng S, Shenson J, Staron MM, Kaech SM, Kleinstein SH, and Herold KC (2016) Characterization of Diabetogenic CD8+ T Cells. J. Biol. Chem 291, 11230–11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Gitelman SE, and Bluestone JA (2016) Regulatory T cell therapy for type 1 diabetes: May the force be with you. J. Autoimmun 71, 78–87. [DOI] [PubMed] [Google Scholar]
- (72).Tang Q, and Bluestone JA (2008) The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nat. Immunol 9, 239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, Liu W, Long SA, Masiello LM, Nguyen V, Putnam AL, Rieck M, Sayre PH, and Tang Q (2015) Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci. Transl. Med 7, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, Finkelstein D, Forbes K, Workman CJ, Brown SA, Rehg JE, Jones ML, Ni H-T, Artis D, Turk MJ, and Vignali DAA (2010) IL-35-mediated induction of a potent regulatory T cell population. Nat. Immunol 11, 1093–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Munn DH, and Mellor AL (2013) Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 34, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Tang Q, Adams JY, Penaranda C, Melli K, Piaggio E, Sgouroudis E, Piccirillo CA, Salomon BL, and Bluestone JA (2008) Central role of defective interleukin-2 production in the triggering of islet autoimmune destruction. Immunity 28, 687–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Spence A, and Tang Q (2016) Restoring Regulatory T Cells in Type 1 Diabetes. Curr. Diab. Rep 16, 110. [DOI] [PubMed] [Google Scholar]
- (78).Long SA, Cerosaletti K, Bollyky PL, Tatum M, Shilling H, Zhang S, Zhang Z-Y, Pihoker C, Sanda S, Greenbaum C, and Buckner JH (2010) Defects in IL-2R signaling contribute to diminished maintenance of FOXP3 expression in CD4(+)CD25(+) regulatory T-cells of type 1 diabetic subjects. Diabetes 59, 407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Glisic S, Klinker M, Waukau J, Jailwala P, Jana S, Basken J, Wang T, Alemzadeh R, Hagopian W, and Ghosh S (2009) Genetic association of HLA DQB1 with CD4+CD25+(high) T-cell apoptosis in type 1 diabetes. Genes Immun. 10, 334–40. [DOI] [PubMed] [Google Scholar]
- (80).Glisic-Milosavljevic S, Waukau J, Jailwala P, Jana S, Khoo HJ, Albertz H, Woodliff J, Koppen M, Alemzadeh R, Hagopian W, and Ghosh S (2007) At-risk and recent-onset type 1 diabetic subjects have increased apoptosis in the CD4+CD25+high T-cell fraction. PLoS One (Sollid, L., Ed.) 2, e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Jailwala P, Waukau J, Glisic S, Jana S, Ehlenbach S, Hessner M, Alemzadeh R, Matsuyama S, Laud P, Wang X, and Ghosh S (2009) Apoptosis of CD4+CD25high T cells in type 1 diabetes may be partially mediated by IL-2 deprivation. PLoS One (Tan, P., Ed.) 4, e6527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Yu A, Snowhite I, Vendrame F, Rosenzwajg M, Klatzmann D, Pugliese A, and Malek TR (2015) Selective IL-2 responsiveness of regulatory T cells through multiple intrinsic mechanisms supports the use of low-dose IL-2 therapy in type 1 diabetes. Diabetes 64, 2172–83. [DOI] [PubMed] [Google Scholar]
- (83).Van Gool F, Molofsky AB, Morar MM, Rosenzwajg M, Liang H-E, Klatzmann D, Locksley RM, and Bluestone JA (2014) Interleukin-5-producing group 2 innate lymphoid cells control eosinophilia induced by interleukin-2 therapy. Blood 124, 3572–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Long SA, Rieck M, Sanda S, Bollyky JB, Samuels PL, Goland R, Ahmann A, Rabinovitch A, Aggarwal S, Phippard D, Turka LA, Ehlers MR, Bianchine PJ, Boyle KD, Adah SA, Bluestone JA, Buckner JH, Greenbaum CJ, and Diabetes TrialNet and the Immune Tolerance Network. (2012) Rapamycin/IL-2 combination therapy in patients with type 1 diabetes augments Tregs yet transiently impairs β-cell function. Diabetes 61, 2340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Truman LA, Pekalski ML, Kareclas P, Evangelou M, Walker NM, Howlett J, Mander AP, Kennet J, Wicker LS, Bond S, Todd JA, and Waldron-Lynch F (2015) Protocol of the adaptive study of IL-2 dose frequency on regulatory T cells in type 1 diabetes (DILfrequency): a mechanistic, non-randomised, repeat dose, open-label, response-adaptive study. BMJ Open 5, e009799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Techmanska I, Juscinska J, Wujtewicz MA, Witkowski P, Mlynarski W, Balcerska A, Mysliwska J, and Trzonkowski P (2012) Administration of CD4+CD25highCD127-regulatory T cells preserves β-cell function in type 1 diabetes in children. Diabetes Care 35, 1817–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, Owczuk R, Szadkowska A, Witkowski P, Młynarski W, Jarosz-Chobot P, Bossowski A, Siebert J, and Trzonkowski P (2014) Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin. Immunol 153, 23–30. [DOI] [PubMed] [Google Scholar]
- (88).Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, and Bluestone JA (2004) In Vitro–expanded Antigen-specific Regulatory T Cells Suppress Autoimmune Diabetes. J. Exp. Med 199, 1455–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Wright GP, Notley CA, Xue S-A, Bendle GM, Holler A, Schumacher TN, Ehrenstein MR, and Stauss HJ (2009) Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc. Natl. Acad. Sci. U. S. A 106, 19078–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Elinav E, Adam N, Waks T, and Eshhar Z (2009) Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology 136, 1721–31. [DOI] [PubMed] [Google Scholar]
- (91).Elinav E, Waks T, and Eshhar Z (2008) Redirection of regulatory T cells with predetermined specificity for the treatment of experimental colitis in mice. Gastroenterology 134, 2014–24. [DOI] [PubMed] [Google Scholar]
- (92).Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, Harris RA, Magnusson PU, Brittebo E, and Loskog ASI (2012) CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J. Neuroinflammation 9, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Ehlers MR, and Rigby MR (2015) Targeting memory T cells in type 1 diabetes. Curr. Diab. Rep 15, 84. [DOI] [PubMed] [Google Scholar]
- (94).Rigby MR, DiMeglio LA, Rendell MS, Felner EI, Dostou JM, Gitelman SE, Patel CM, Griffin KJ, Tsalikian E, Gottlieb PA, Greenbaum CJ, Sherry NA, Moore WV, Monzavi R, Willi SM, Raskin P, Moran A, Russell WE, Pinckney A, Keyes-Elstein L, Howell M, Aggarwal S, Lim N, Phippard D, Nepom GT, McNamara J, and Ehlers MR (2013) Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol. 1, 284–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Gammon JM, Tostanoski LH, Adapa AR, Chiu Y-C, and Jewell CM (2015) Controlled delivery of a metabolic modulator promotes regulatory T cells and restrains autoimmunity. J. Control. Release 210, 169–178. [DOI] [PubMed] [Google Scholar]
- (96).Fathman CG, and Lineberry NB (2007) Molecular mechanisms of CD4+ T-cell anergy. Nat. Rev. Immunol 7, 599–609. [DOI] [PubMed] [Google Scholar]
- (97).Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, Gottlieb PA, Greenbaum CJ, Marks JB, Monzavi R, Moran A, Raskin P, Rodriguez H, Russell WE, Schatz D, Wherrett D, Wilson DM, Krischer JP, and Skyler JS (2011) Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: A randomised, double-blind, placebo-controlled trial. Lancet 378, 412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Besançon A, Goncalves T, Valette F, Mary C, Vanhove B, Chatenoud L, and You S (2018) A selective CD28 antagonist and rapamycin synergise to protect against spontaneous autoimmune diabetes in NOD mice. Diabetologia 61, 1811–1816. [DOI] [PubMed] [Google Scholar]
- (99).Gallucci S, Lolkema M, and Matzinger P (1999) Natural adjuvants: endogenous activators of dendritic cells. Nat. Med 5, 1249–55. [DOI] [PubMed] [Google Scholar]
- (100).Morelli AE, and Thomson AW (2007) Tolerogenic dendritic cells and the quest for transplant tolerance. Nat. Rev. Immunol 7, 610–21. [DOI] [PubMed] [Google Scholar]
- (101).Oderup C, Cederbom L, Makowska A, Cilio CM, and Ivars F (2006) Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology 118, 240–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Grohmann U, Orabona C, Fallarino F, Vacca C, Calcinaro F, Falorni A, Candeloro P, Belladonna ML, Bianchi R, Fioretti MC, and Puccetti P (2002) CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat. Immunol 3, 1097–101. [DOI] [PubMed] [Google Scholar]
- (103).Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, Belladonna ML, Fioretti MC, Alegre M-L, and Puccetti P (2003) Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol 4, 1206–12. [DOI] [PubMed] [Google Scholar]
- (104).Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, Flores M, Li N, Schweighoffer E, Greenberg S, Tybulewicz V, Vignali D, and Clynes R (2008) Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J. Immunol 180, 5916–26. [DOI] [PubMed] [Google Scholar]
- (105).Ko H-J, Cho M-L, Lee S-Y, Oh H-J, Heo Y-J, Moon Y-M, Kang C-M, Kwok S-K, Ju JH, Park S-H, Park K-S, and Kim H-Y (2010) CTLA4-Ig modifies dendritic cells from mice with collagen-induced arthritis to increase the CD4+CD25+Foxp3+ regulatory T cell population. J. Autoimmun 34, 111–120. [DOI] [PubMed] [Google Scholar]
- (106).Cederbom L, Hall H, and Ivars F (2000) CD4+CD25+ regulatory T cells down-regulate co-stimulatory molecules on antigen-presenting cells. Eur. J. Immunol 30, 1538–1543. [DOI] [PubMed] [Google Scholar]
- (107).Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, and Sakaguchi S (2008) CTLA-4 control over Foxp3+ regulatory T cell function. Science (5889). 322, 271–275. [DOI] [PubMed] [Google Scholar]
- (108).Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol 3, 745–56. [DOI] [PubMed] [Google Scholar]
- (109).Bohana-Kashtan O, and Civin CI (2004) Fas Ligand as a Tool for Immunosuppression and Generation of Immune Tolerance. Stem Cells 22, 908–924. [DOI] [PubMed] [Google Scholar]
- (110).Zhang X, Brunner T, Carter L, Dutton RW, Rogers P, Bradley L, Sato T, Reed JC, Green D, and Swain SL (1997) Unequal Death in T Helper Cell (Th)1 and Th2 Effectors: Th1, but not Th2, Effectors Undergo Rapid Fas/FasL-mediated Apoptosis. J. Exp. Med 185, 1837–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Varadhachary AS, Perdow SN, Hu C, Ramanarayanan M, and Salgame P (1997) Differential ability of T cell subsets to undergo activation-induced cell death. Proc. Natl. Acad. Sci. U. S. A 94, 5778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Suarez-Pinzon W, Sorensen O, Bleackley RC, Elliott JF, Rajotte RV, and Rabinovitch A (1999) Beta-cell destruction in NOD mice correlates with Fas (CD95) expression on beta-cells and proinflammatory cytokine expression in islets. Diabetes 48, 21–8. [DOI] [PubMed] [Google Scholar]
- (113).Amrani A, Verdaguer J, Thiessen S, Bou S, and Santamaria P (2000) IL-1alpha, IL-1beta, and IFN-gamma mark beta cells for Fas-dependent destruction by diabetogenic CD4(+) T lymphocytes. J. Clin. Invest 105, 459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Yolcu ES, Shirwan H, and Askenasy N (2017) Fas/fas-ligand interaction as a mechanism of immune homeostasis and β-cell cytotoxicity: Enforcement rather than neutralization for treatment of type 1 diabetes. Front. Immunol. Frontiers Media SA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Headen DM, Woodward KB, Coronel MM, Shrestha P, Weaver JD, Zhao H, Tan M, Hunckler MD, Bowen WS, Johnson CT, Shea L, Yolcu ES, García AJ, and Shirwan H (2018) Local immunomodulation with Fas ligand-engineered biomaterials achieves allogeneic islet graft acceptance. Nat. Mater 17, 732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Curtin JF, and Cotter TG (2003) Live and let die: regulatory mechanisms in Fas-mediated apoptosis. Cell. Signal 15, 983–92. [DOI] [PubMed] [Google Scholar]
- (117).Daniel D, Gill RG, Schloot N, and Wegmann D (1995) Epitope specificity, cytokine production profile and diabetogenic activity of insulin-specific T cell clones isolated from NOD mice. Eur. J. Immunol 25, 1056–62. [DOI] [PubMed] [Google Scholar]
- (118).Wegmann DR, Norbury-Glaseru M, and Danielf D (1994) Insulin-specific T cells are a predominant component of islet infiltrates in pre-diabetic NOD mice. Eur. J. Immunol 24, 1853–1857. [DOI] [PubMed] [Google Scholar]
- (119).Zhang L, Crawford F, Yu L, Michels A, Nakayama M, Davidson HW, Kappler JW, and Eisenbarth GS (2014) Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proc. Natl. Acad. Sci 111, 2656–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Ostrov DA, Alkanani A, McDaniel KA, Case S, Baschal EE, Pyle L, Ellis S, Pöllinger B, Seidl KJ, Shah VN, Garg SK, Atkinson MA, Gottlieb PA, and Michels AW (2018) Methyldopa blocks MHC class II binding to disease-specific antigens in autoimmune diabetes. J. Clin. Invest 128, 1888–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Mah GT, Tejani AM, and Musini VM (2009) Methyldopa for primary hypertension. Cochrane Database Syst. Rev CD003893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Wagner DH (2009) The co-evolution of our understanding of CD40 and inflammation. Diabetologia 52, 997–999. [DOI] [PubMed] [Google Scholar]
- (123).Wagner DH, Newell E, Sanderson RJ, Freed JH, and Newell MK (1999) Increased expression of CD40 on thymocytes and peripheral T cells in autoimmunity: a mechanism for acquiring changes in the peripheral T cell receptor repertoire. Int. J. Mol. Med 4, 231–42. [DOI] [PubMed] [Google Scholar]
- (124).Wagner DH, Vaitaitis G, Sanderson R, Poulin M, Dobbs C, and Haskins K (2002) Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc. Natl. Acad. Sci 99, 3782–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Armitage RJ, Fanslow WC, Strockbine L, Sato TA, Clifford KN, Macduff BM, Anderson DM, Gimpel SD, Davis-Smith T, Maliszewski CR, Clark EA, Smith CA, Grabstein KH, Cosman D, and Spriggs MK (1992) Molecular and biological characterization of a murine ligand for CD40. Nature 357, 80–82. [DOI] [PubMed] [Google Scholar]
- (126).Baker RL, Mallevaey T, Gapin L, and Haskins K (2012) T cells interact with T cells via CD40-CD154 to promote autoimmunity in type 1 diabetes. Eur. J. Immunol 42, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Baker RL, Wagner DH, and Haskins K (2008) CD40 on NOD CD4 T cells contributes to their activation and pathogenicity. J. Autoimmun 31, 385–392. [DOI] [PubMed] [Google Scholar]
- (128).Wagner DH (2017) Overlooked Mechanisms in Type 1 Diabetes Etiology: How Unique Costimulatory Molecules Contribute to Diabetogenesis. Front. Endocrinol. (Lausanne). 8, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Vaitaitis GM, Waid DM, Yussman MG, and Wagner DH (2017) CD40-mediated signalling influences trafficking, T-cell receptor expression, and T-cell pathogenesis, in the NOD model of type 1 diabetes. Immunology 152, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Sidiropoulos PI, and Boumpas DT (2004) Lessons learned from anti-CD40L treatment in systemic lupus erythematosus patients. Lupus 13, 391–7. [DOI] [PubMed] [Google Scholar]
- (131).Vaitaitis GM, Olmstead MH, Waid DM, Carter JR, and Wagner DH (2014) A CD40-targeted peptide controls and reverses type 1 diabetes in NOD mice. Diabetologia 57, 2366–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, Fleming SA, Leiter EH, and Shultz LD (1996) B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new "speed congenic" stock of NOD.Ig mu null mice. J. Exp. Med 184, 2049–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Akashi T, Nagafuchi S, Anzai K, Kondo S, Kitamura D, Wakana S, Ono J, Kikuchi M, Niho Y, and Watanabe T (1997) Direct evidence for the contribution of B cells to the progression of insulitis and the development of diabetes in non-obese diabetic mice. Int. Immunol 9, 1159–64. [DOI] [PubMed] [Google Scholar]
- (134).Wong FS, Visintin I, Wen L, Granata J, Flavell R, and Janeway CA (1998) The role of lymphocyte subsets in accelerated diabetes in nonobese diabetic-rat insulin promoter-B7–1 (NOD-RIP-B7–1) mice. J. Exp. Med 187, 1985–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Noorchashm H, Noorchashm N, Kern J, Rostami SY, Barker CF, and Naji A (1997) B-cells are required for the initiation of insulitis and sialitis in nonobese diabetic mice. Diabetes 46, 941–6. [DOI] [PubMed] [Google Scholar]
- (136).Bouaziz J-D, Yanaba K, Venturi GM, Wang Y, Tisch RM, Poe JC, and Tedder TF (2007) Therapeutic B cell depletion impairs adaptive and autoreactive CD4+ T cell activation in mice. Proc. Natl. Acad. Sci. U. S. A 104, 20878–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Fiorina P, Vergani A, Dada S, Jurewicz M, Wong M, Law K, Wu E, Tian Z, Abdi R, Guleria I, Rodig S, Dunussi-Joannopoulos K, Bluestone J, and Sayegh MH (2008) Targeting CD22 reprograms B-cells and reverses autoimmune diabetes. Diabetes 57, 3013–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Hu C, Rodriguez-Pinto D, Du W, Ahuja A, Henegariu O, Wong FS, Shlomchik MJ, and Wen L (2007) Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J. Clin. Invest 117, 3857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Xiu Y, Wong CP, Bouaziz J-D, Hamaguchi Y, Wang Y, Pop SM, Tisch RM, and Tedder TF (2008) B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J. Immunol 180, 2863–75. [DOI] [PubMed] [Google Scholar]
- (140).Hulbert C, Riseili B, Rojas M, and Thomas JW (2001) B cell specificity contributes to the outcome of diabetes in nonobese diabetic mice. J. Immunol 167, 5535–8. [DOI] [PubMed] [Google Scholar]
- (141).Mariño E, Tan B, Binge L, Mackay CR, and Grey ST (2012) B-cell cross-presentation of autologous antigen precipitates diabetes. Diabetes 61, 2893–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Bonifacio E, and Ziegler AG (2010) Advances in the Prediction and Natural History of Type 1 Diabetes. Endocrinol. Metab. Clin. North Am 39, 513–525. [DOI] [PubMed] [Google Scholar]
- (143).Zhao Y, Zhang L, Allison Green E, Pinto AI, Smith J, Kissack MR, and Hogg KG (2018) Thymic B cell-Mediated attack of Thymic stroma Precedes Type 1 Diabetes Development. Front. Immunol 9, 12813389–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Victora GD, and Nussenzweig MC (2012) Germinal centers. Annu. Rev. Immunol 30, 429–57. [DOI] [PubMed] [Google Scholar]
- (145).Murphy K, Travers P, Walport M, Janeway C (2008) Janeway’s Immunobiology. Garland Science, New York. [Google Scholar]
- (146).Kroll JL, Beam C, Li S, Viscidi R, Dighero B, Cho A, Boulware D, Pescovitz M, Weinberg A, and Type 1 Diabetes TrialNet Anti CD-20 Study Group. (2013) Reactivation of latent viruses in individuals receiving rituximab for new onset type 1 diabetes. J. Clin. Virol 57, 115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, Gottlieb PA, Marks JB, McGee PF, Moran AM, Raskin P, Rodriguez H, Schatz DA, Wherrett D, Wilson DM, Lachin JM, and Skyler JS (2009) Rituximab, B-Lymphocyte Depletion, and Preservation of Beta-Cell Function. N. Engl. J. Med 361, 2143–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Ratiu JJ, Racine JJ, Hasham MG, Wang Q, Branca JA, Chapman HD, Zhu J, Donghia N, Philip V, Schott WH, Wasserfall C, Atkinson MA, Mills KD, Leeth CM, and Serreze DV (2017) Genetic and Small Molecule Disruption of the AID/RAD51 Axis Similarly Protects Nonobese Diabetic Mice from Type 1 Diabetes through Expansion of Regulatory B Lymphocytes. J. Immunol 198, 4255–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).La-Beck NM, Jean GW, Huynh C, Alzghari SK, and Lowe DB (2015) Immune Checkpoint Inhibitors: New Insights and Current Place in Cancer Therapy. Pharmacother. J. Hum. Pharmacol. Drug Ther 35, 963–976. [DOI] [PubMed] [Google Scholar]
- (150).Sharma MD, Baban B, Chandler P, Hou D-Y, Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL, and Munn DH (2007) Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J. Clin. Invest 117, 2570–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Baban B, Penberthy WT, and Mozaffari MS (2010) The potential role of indoleamine 2,3 dioxygenase (IDO) as a predictive and therapeutic target for diabetes treatment: a mythical truth. EPMA J. 1, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Saxena V, Ondr JK, Magnusen AF, Munn DH, and Katz JD (2007) The countervailing actions of myeloid and plasmacytoid dendritic cells control autoimmune diabetes in the nonobese diabetic mouse. J. Immunol 179, 5041–53. [DOI] [PubMed] [Google Scholar]
- (153).Poormasjedi-Meibod M-S, Jalili RB, Hosseini-Tabatabaei A, Hartwell R, and Ghahary A (2013) Immuno-Regulatory Function of Indoleamine 2,3 Dioxygenase through Modulation of Innate Immune Responses. PLoS One 8, e71044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Fallarino F, Volpi C, Zelante T, Vacca C, Calvitti M, Fioretti MC, Puccetti P, Romani L, and Grohmann U (2009) IDO mediates TLR9-driven protection from experimental autoimmune diabetes. J. Immunol 183, 6303–12. [DOI] [PubMed] [Google Scholar]
- (155).Francisco LM, Sage PT, and Sharpe AH (2010) The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev 236, 219–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Zhang X, Schwartz J-CD, Guo X, Bhatia S, Cao E, Lorenz M, Cammer M, Chen L, Zhang Z-Y, Edidin MA, Nathenson SG, and Almo SC (2004) Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 20, 337–47. [DOI] [PubMed] [Google Scholar]
- (157).Chen L, and Han X (2015) Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J. Clin. Invest 125, 3384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Fang C, Huang Y, Pei Y, Zhang H, Chen X, Guo H, Li S, Ji X, and Hu J (2017) Genome-wide gene expression profiling reveals that CD274 is up-regulated new-onset type 1 diabetes mellitus. Acta Diabetol. 1–11. [DOI] [PubMed] [Google Scholar]
- (159).Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, Gettinger S, Sznol M, Young A, Rushakoff R, Lee J, Bluestone JA, Anderson M, and Herold KC (2018) Collateral Damage: Insulin-Dependent Diabetes Induced With Checkpoint Inhibitors. Diabetes 67, 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Ansari MJI, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, Yamazaki T, Azuma M, Iwai H, Khoury SJ, Auchincloss H, and Sayegh MH (2003) The Programmed Death-1 (PD-1) Pathway Regulates Autoimmune Diabetes in Nonobese Diabetic (NOD) Mice. J. Exp. Med 198, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Ben Nasr M, Tezza S, D’Addio F, Mameli C, Usuelli V, Maestroni A, Corradi D, Belletti S, Albarello L, Becchi G, Fadini P, Schuetz C, Markmann J, Wasserfall C, Zon L, Zuccotti GV, and Fiorina P (2017) PD-L1 genetic overexpression or pharmacological restoration in hematopoietic stem and progenitor cells reverses autoimmune diabetes. Sci. Transl. Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Fiorina P, Jurewicz M, Vergani A, Petrelli A, Carvello M, D’Addio F, Godwin JG, Law K, Wu E, Tian Z, Thoma G, Kovarik J, La Rosa S, Capella C, Rodig S, Zerwes H-G, Sayegh MH, and Abdi R (2011) Targeting the CXCR4-CXCL12 axis mobilizes autologous hematopoietic stem cells and prolongs islet allograft survival via programmed death ligand 1. J. Immunol 186, 121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Guleria I, Gubbels Bupp M, Dada S, Fife B, Tang Q, Ansari MJ, Trikudanathan S, Vadivel N, Fiorina P, Yagita H, Azuma M, Atkinson M, Bluestone JA, and Sayegh MH (2007) Mechanisms of PDL1-mediated regulation of autoimmune diabetes. Clin. Immunol 125, 16–25. [DOI] [PubMed] [Google Scholar]
- (164).Brubaker SW, Bonham KS, Zanoni I, and Kagan JC (2015) Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol 33, 257–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Tai N, Wong FS, and Wen L (2016) The role of the innate immune system in destruction of pancreatic beta cells in NOD mice and humans with type I diabetes. J. Autoimmun 71, 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Needell JC, and Zipris D (2014) Targeting innate immunity for treatment of type 1 diabetes. Curr. Diab. Rep 17, 113. [DOI] [PubMed] [Google Scholar]
- (167).Itoh A, and Ridgway WM (2017) Targeting innate immunity to downmodulate adaptive immunity and reverse type 1 diabetes. ImmunoTargets Ther. 6, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Warner N, and Nunez G (2013) Correction: MyD88: A critical adaptor protein in innate immunity signal transduction. J. Immunol 190, 3824–3824. [DOI] [PubMed] [Google Scholar]
- (169).Wen L, Ley RE, Volchkov PY, Stranges PB, Avanesyan L, Stonebraker AC, Hu C, Wong FS, Szot GL, Bluestone JA, Gordon JI, and Chervonsky AV (2008) Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 455, 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Zhang X, Xing S, Li M, Zhang L, Xie L, He W, Liu J, Chang S, Jiang F, and Zhou P (2016) Beyond knockout: A novel homodimerization-targeting MyD88 inhibitor prevents and cures type 1 diabetes in NOD mice. Metabolism. 65, 1267–1277. [DOI] [PubMed] [Google Scholar]
- (171).Burrows MP, Volchkov P, Kobayashi KS, and Chervonsky AV (2015) Microbiota regulates type 1 diabetes through Toll-like receptors. Proc. Natl. Acad. Sci 112, 9973–9977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Gülden E, Chao C, Tai N, Pearson JA, Peng J, Majewska-Szczepanik M, Zhou Z, Wong FS, and Wen L (2018) TRIF deficiency protects non-obese diabetic mice from type 1 diabetes by modulating the gut microbiota and dendritic cells. J. Autoimmun 93, 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Devaraj S, Dasu MR, Rockwood J, Winter W, Griffen SC, and Jialal I (2008) Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: Further evidence of a proinflammatory state. J. Clin. Endocrinol. Metab 93, 578–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Gülden E, Ihira M, Ohashi A, Reinbeck AL, Freudenberg MA, Kolb H, and Burkart V (2013) Toll-Like Receptor 4 Deficiency Accelerates the Development of Insulin-Deficient Diabetes in Non-Obese Diabetic Mice. PLoS One 8, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Devaraj S, Tobias P, Kasinath BS, Ramsamooj R, Afify A, and Jialal I (2011) Knockout of toll-like receptor-4 attenuates the pro-inflammatory state of diabetes. Cytokine 31, 1796–1804. [DOI] [PubMed] [Google Scholar]
- (176).Bednar KJ, Tsukamoto H, Kachapati K, Ohta S, Wu Y, Katz JD, Ascherman DP, and Ridgway WM (2015) Reversal of new-onset type 1 diabetes with an agonistic TLR4/MD-2 monoclonal antibody. Diabetes 64, 3614–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Kaplan MJ, and Radic M (2012) Neutrophil Extracellular Traps: Double-Edged Swords of Innate Immunity. J. Immunol 189, 2689–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Wang Y, Xiao Y, Zhong L, Ye D, Zhang J, Tu Y, Bornstein SR, Zhou Z, Lam KSL, and Xu A (2014) Increased neutrophil elastase and proteinase 3 and augmented NETosis are closely associated with β-cell autoimmunity in patients with type 1 diabetes. Diabetes 63, 4239–4248. [DOI] [PubMed] [Google Scholar]
- (179).Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, and Lehuen A (2013) Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat. Med 19, 65–73. [DOI] [PubMed] [Google Scholar]
- (180).Lang J, Wang X, Liu K, He D, Niu P, Cao R, Jin L, and Wu J (2017) Oral delivery of staphylococcal nuclease by Lactococcus lactis prevents type 1 diabetes mellitus in NOD mice. Appl. Microbiol. Biotechnol 101, 7653–7662. [DOI] [PubMed] [Google Scholar]
- (181).Barrera M-J, Aguilera S, Castro I, González S, Carvajal P, Molina C, Hermoso MA, and González M-J (2018) Endoplasmic reticulum stress in autoimmune diseases: Can altered protein quality control and/or unfolded protein response contribute to autoimmunity? A critical review on Sjögren’s syndrome. Autoimmun. Rev 17, 796–808. [DOI] [PubMed] [Google Scholar]
- (182).Louvet C, Szot GL, Lang J, Lee MR, Martinier N, Bollag G, Zhu S, Weiss A, and Bluestone JA (2008) Tyrosine kinase inhibitors reverse type 1 diabetes in nonobese diabetic mice. Proc. Natl. Acad. Sci. U. S. A 105, 18895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Ito Y, Pandey P, Mishra NC, Kumar S, Narula N, Kharbanda S, Saxena SP, and Kufe D (2001) Targeting of the c-Abl tyrosine kinase to mitochondria in endoplasmic reticulum stress-induced apoptosis. Mol. Cell. Biol 21, 6233–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Morita S, Villalta SA, Feldman HC, Maly DJ, Bluestone JA, Papa FR, Morita S, Villalta SA, Feldman HC, Register AC, and Rosenthal W (2017) Targeting ABL-IRE1 a Signaling Spares ER-Stressed Pancreatic b Cells to Reverse Autoimmune Diabetes Article Targeting ABL-IRE1 a Signaling Spares ER-Stressed Pancreatic b Cells to Reverse Autoimmune Diabetes. Cell Metab. 25, 883–897.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Richardson SJ, Rodriguez-Calvo T, Gerling IC, Mathews CE, Kaddis JS, Russell MA, Zeissler M, Leete P, Krogvold L, Dahl-Jørgensen K, von Herrath M, Pugliese A, Atkinson MA, and Morgan NG (2016) Islet cell hyperexpression of HLA class I antigens: a defining feature in type 1 diabetes. Diabetologia 59, 2448–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (186).Li YR, Li J, Zhao SD, Bradfield JP, Mentch FD, Maggadottir SM, Hou C, Abrams DJ, Chang D, Gao F, Guo Y, Wei Z, Connolly JJ, Cardinale CJ, Bakay M, Glessner JT, Li D, Kao C, Thomas KA, Qiu H, Chiavacci RM, Kim CE, Wang F, Snyder J, Richie MD, Flatø B, Førre øystein, Denson LA, Thompson SD, Becker ML, Guthery SL, Latiano A, Perez E, Resnick E, Russell RK, Wilson DC, Silverberg MS, Annese V, Lie BA, Punaro M, Dubinsky MC, Monos DS, Strisciuglio C, Staiano A, Miele E, Kugathasan S, Ellis JA, Munro JE, Sullivan KE, Wise CA, Chapel H, Cunningham-Rundles C, Grant SFA, Orange JS, Sleiman PMA, Behrens EM, Griffiths AM, Satsangi J, Finkel TH, Keinan A, Prak ETL, Polychronakos C, Baldassano RN, Li H, Keating BJ, and Hakonarson H (2015) Meta-analysis of shared genetic architecture across ten pediatric autoimmune diseases. Nat. Med 21, 1018–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Flodström-Tullberg M, Yadav D, Hägerkvist R, Tsai D, Secrest P, Stotland A, and Sarvetnick N (2003) Target Cell Expression of Suppressor of Cytokine Signaling-1 Prevents Diabetes in the NOD Mouse. Diabetes 52, 2696–2700. [DOI] [PubMed] [Google Scholar]
- (188).Trivedi PM, Graham KL, Scott NA, Jenkins MR, Majaw S, Sutherland RM, Fynch S, Lew AM, Burns CJ, Krishnamurthy B, Brodnicki TC, Mannering SI, Kay TW, and Thomas HE (2017) Repurposed JAK1/JAK2 inhibitor reverses established autoimmune insulitis in NOD mice. Diabetes 66, 1650–1660. [DOI] [PubMed] [Google Scholar]
- (189).Boslem E, Meikle PJ, and Biden TJ (2012) Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets 4, 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Tsai HC, and Han MH (2016) Sphingosine-1-Phosphate (S1P) and S1P Signaling Pathway: Therapeutic Targets in Autoimmunity and Inflammation. Drugs 76, 1067–1079. [DOI] [PubMed] [Google Scholar]
- (191).Tsuji T, Yoshida Y, Fujita T, and Kohno T (2012) Oral therapy for type1 diabetes mellitus using a novel immunomodulator, FTY720 (fingolimod), in combination with sitagliptin, a dipeptidyl peptidase-4 inhibitor, examined in non-obese diabetic mice. J. Diabetes Investig 3, 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Tsuji T, Inoue M, Yoshida Y, Fujita T, Kaino Y, and Kohno T (2012) Therapeutic approach for type 1 diabetes mellitus using the novel immunomodulator FTY720 (fingolimod) in combination with once-daily injection of insulin glargine in non-obese diabetic mice. J. Diabetes Investig 3, 132–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Hahn C, Tyka K, Saba JD, Lenzen S, and Gurgul-Convey E (2017) Overexpression of sphingosine-1-phosphate lyase protects insulin-secreting cells against cytokine toxicity. J. Biol. Chem 292, 20292–20304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Cardozo AK, Ortis F, Storling J, Feng Y, Rasschaert J, Tonnesen M, Eylen V, Mandrup-poulsen T, Herchuelz A, and Eizirik DL (2005) Cytokines Downregulate the Sarcoendoplasmic Reticulum Pump Ca2ⴙ ATPase 2b and Deplete Endoplasmic Reticulum Ca2+, Leading to Induction of Endoplasmic Reticulum Stress in Pancreatic beta-Cells. Diabetes 54, 452–461. [DOI] [PubMed] [Google Scholar]
- (195).Ramadan JW, Steiner SR, O’Neill CM, and Nunemaker CS (2011) The central role of calcium in the effects of cytokines on beta-cell function: Implications for type 1 and type 2 diabetes. Cell Calcium 50, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Clark AL, Kanekura K, Lavagnino Z, Spears LD, Abreu D, Mahadevan J, Yagi T, Semenkovich CF, Piston DW, and Urano F (2017) Targeting Cellular Calcium Homeostasis to Prevent Cytokine-Mediated Beta Cell Death. Sci. Rep 7, 5611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Padgett LE, Broniowska KA, Hansen PA, Corbett JA, and Tse HM (2013) The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis. Ann. N. Y. Acad. Sci 1281, 16–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, and Mescher MF (1999) Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J. Immunol 162, 3256–62. [PubMed] [Google Scholar]
- (199).Coskun O, Kanter M, Korkmaz A, and Oter S (2005) Quercetin, a flavonoid antioxidant, prevents and protects streptozotocin-induced oxidative stress and β-cell damage in rat pancreas. Pharmacol. Res 51, 117–123. [DOI] [PubMed] [Google Scholar]
- (200).Youl E, Bardy G, Magous R, Cros G, Sejalon F, Virsolvy A, Richard S, Quignard JF, Gross R, Petit P, Bataille D, and Oiry C (2010) Quercetin potentiates insulin secretion and protects INS-1 pancreatic-cells against oxidative damage via the ERK1/2 pathway. Br. J. Pharmacol 161, 799–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Ludvigsson J, Samuelsson U, Johansson C, and Stenhammar L (2001) Treatment with antioxidants at onset of type 1 diabetes in children: a randomized, double-blind placebo-controlled study. Diabetes Metab Res Rev 17, 131–136. [DOI] [PubMed] [Google Scholar]
- (202).Haeggström JZ, and Funk CD (2011) Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev 111, 5866–5896. [DOI] [PubMed] [Google Scholar]
- (203).Tersey SA, Bolanis E, Holman TR, Maloney DJ, Nadler JL, and Mirmira RG (2015) Minireview: 12-Lipoxygenase and Islet β-Cell Dysfunction in Diabetes. Mol. Endocrinol 29, 791–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Ma K, Nunemaker CS, Wu R, Chakrabarti SK, Taylor-Fishwick DA, and Nadler JL (2010) 12-Lipoxygenase Products Reduce Insulin Secretion and B-Cell Viability in Human Islets. J. Clin. Endocrinol. Metab 95, 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Bleich D, Chen S, Zipser B, Sun D, Funk CD, and Nadler JL (1999) Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J Clin Invest 103, 1431–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Mcduffie M, Maybee NA, Keller SR, Stevens BK, Garmey JC, Morris MA, Kropf E, Rival C, Ma K, Carter JD, Tersey SA, Nunemaker CS, and Nadler JL (2008) Nonobese Diabetic (NOD) Mice Congenic for a Targeted Deletion of 12/15-Lipoxygenase Are Protected From Autoimmune Diabetes. Diabetes 57, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Rai G, Joshi N, Jung JE, Liu Y, Schultz L, Yasgar A, Perry S, Diaz G, Zhang Q, Kenyon V, Jadhav A, Simeonov A, Lo EH, Van Leyen K, Maloney DJ, and Holman TR (2014) Potent and selective inhibitors of human reticulocyte 12/15-lipoxygenase as anti-stroke therapies. J. Med. Chem 57, 4035–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Luci DK, Jameson JB, Yasgar A, Diaz G, Joshi N, Kantz A, Markham K, Perry S, Kuhn N, Yeung J, Kerns EH, Schultz L, Holinstat M, Nadler JL, Taylor-Fishwick DA, Jadhav A, Simeonov A, Holman TR, and Maloney DJ (2014) Synthesis and structure-activity relationship studies of 4-((2-hydroxy-3-methoxybenzyl)amino)benzenesulfonamide derivatives as potent and selective inhibitors of 12-lipoxygenase. J. Med. Chem 57, 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Kenyon V, Rai G, Jadhav A, Schultz L, Armstrong M, Jameson JB, Perry S, Joshi N, Bougie JM, Leister W, Taylor-Fishwick DA, Nadler JL, Holinstat M, Simeonov A, Maloney DJ, and Holman TR (2011) Discovery of potent and selective inhibitors of human platelet-type 12-lipoxygenase. J. Med. Chem 54, 5485–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Taylor-Fishwick DA, Weaver J, Glenn L, Kuhn N, Rai G, Jadhav A, Simeonov A, Dudda A, Schmoll D, Holman TR, Maloney DJ, and Nadler JL (2014) Selective inhibition of 12-lipoxygenase protects islets and beta cells from inflammatory cytokine-mediated beta cell dysfunction. Diabetologia 58, 549–557. [DOI] [PubMed] [Google Scholar]
- (211).Hernandez-Perez M, Chopra G, Fine J, Conteh AM, Anderson RM, Linnemann AK, Benjamin C, Nelson JB, Benninger KS, Nadler JL, Maloney DJ, Tersey SA, and Mirmira RG (2017) Inhibition of 12/15-Lipoxygenase Protects Against β-Cell Oxidative Stress and Glycemic Deterioration in Mouse Models of Type 1 Diabetes. Diabetes 66, 2875–2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Manzoor F, Johnson MC, Li C, Samulski RJ, Wang B, and Tisch R (2017) β-cell-specific IL-35 therapy suppresses ongoing autoimmune diabetes in NOD mice. Eur. J. Immunol 47, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Su LC, Liu XY, Huang AF, and Xu WD (2018) Emerging role of IL-35 in inflammatory autoimmune diseases. Autoimmun. Rev 17, 665–673. [DOI] [PubMed] [Google Scholar]
- (214).Singh K, Kadesjö E, Lindroos J, Hjort M, Lundberg M, Espes D, Carlsson PO, Sandler S, and Thorvaldson L (2015) Interleukin-35 administration counteracts established murine type 1 diabetes - possible involvement of regulatory T cells. Sci. Rep 5, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Bettini M, Castellaw AH, Lennon GP, Burton AR, and Vignali DAA (2012) Prevention of autoimmune diabetes by ectopic pancreatic β-cell expression of interleukin-35. Diabetes 61, 1519–1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Zullo A, Sommese L, Nicoletti G, Donatelli F, Mancini FP, and Napoli C (2017) Epigenetics and type 1 diabetes: mechanisms and translational applications. Transl. Res 185, 85–93. [DOI] [PubMed] [Google Scholar]
- (217).Jerram ST, Dang MN, Leslie RD, and Leslie RD (2017) The Role of Epigenetics in Type 1 Diabetes. Curr. Diab. Rep 17, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Khan O, and La. Thangue NB (2011) HDAC inhibitors in cancer biology : emerging mechanisms and clinical applications. Immunol. Cell Biol 90, 85–94. [DOI] [PubMed] [Google Scholar]
- (219).Dinarello CA, Fossati G, and Mascagni P (2011) Histone Deacetylase Inhibitors for Treating a Spectrum of Diseases Not Related to Cancer. Mol. Med 7, 333–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Lewis EC, Blaabjerg L, Størling J, Ronn SG, Mascagni P, and Dinarello CA (2011) The Oral Histone Deacetylase Inhibitor ITF2357 Reduces Cytokines and Protects Islet β Cells In Vivo and In Vitro. Mol. Med 17, 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (221).Wagner FF, Lundh M, Kaya T, Mccarren P, Zhang Y, Chattopadhyay S, Gale JP, Galbo T, Fisher SL, Meier BC, Vetere A, Richardson S, Morgan NG, Christensen DP, Gilbert TJ, Hooker JM, Walpita D, Mandrup-poulsen T, Wagner BK, and Holson EB (2016) An Isochemogenic Set of Inhibitors To De fi ne the Therapeutic Potential of Histone Deacetylases in β ‑ Cell Protection. ACS Chem. Biol 11, 363–374. [DOI] [PubMed] [Google Scholar]
- (222).Chou DHC, Holson EB, Wagner FF, Tang AJ, Maglathlin RL, Lewis TA, Schreiber SL, and Wagner BK (2012) Inhibition of histone deacetylase 3 protects beta cells from cytokine-induced apoptosis. Chem. Biol 19, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Dirice E, Ng RWS, Martinez R, Hu J, Wagner FF, Holson EB, Wagner BK, and Kulkarni RN (2017) Isoform-selective inhibitor of histone deacetylase 3 (HDAC3) limits pancreatic islet infiltration and protects female nonobese diabetic mice from diabetes. J. Biol. Chem 292, 17598–17608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Zhou Q, and Melton DA (2018) Pancreas regeneration. Nature 557, 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, and Herrera PL (2010) Conversion of adult pancreatic α-cells to β-cells after extreme β-cell loss. Nature 464, 1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, Furuyama K, Thorel F, Gribble FM, Reimann F, and Herrera PL (2014) Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature 514, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Wang P, Alvarez-Perez J-C, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocaña A, and Stewart AF (2015) Induction of human pancreatic beta cell replication by inhibitors of dual specificity tyrosine regulated kinase. Nat Med 21, 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Lin Y, and Sun Z (2011) Thyroid hormone potentiates insulin signaling and attenuates hyperglycemia and insulin resistance in a mouse model of type 2 diabetes. Br. J. Pharmacol 162, 597–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).López-Noriega L, Cobo-Vuilleumier N, Narbona-Pérez ÁJ, Araujo-Garrido JL, Lorenzo PI, Mellado-Gil JM, Moreno JC, Gauthier BR, and Martín-Montalvo A (2017) Levothyroxine enhances glucose clearance and blunts the onset of experimental type 1 diabetes mellitus in mice. Br. J. Pharmacol 174, 3795–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Bahn RS, Burch HB, Cooper DS, Garber JR, Greenlee MC, Klein I, Laurberg P, McDougall IR, Montori VM, Rivkees SA, Ross DS, Sosa JA, Stan MN, American Thyroid Association, and American Association of Clinical Endocrinologists. (2011) Hyperthyroidism and Other Causes of Thyrotoxicosis: Management Guidelines of the American Thyroid Association and American Association of Clinical Endocrinologists. Thyroid 21, 593–646. [DOI] [PubMed] [Google Scholar]
- (231).Garmendia Madariaga A, Santos Palacios S, Guillén-Grima F, and Galofré JC (2014) The Incidence and Prevalence of Thyroid Dysfunction in Europe: A Meta-Analysis. J. Clin. Endocrinol. Metab 99, 923–931. [DOI] [PubMed] [Google Scholar]
- (232).Hong HS, Lee J, Lee E, Kwon YS, Lee E, Ahn W, Jiang MH, Kim JC, and Son Y (2009) A new role of substance P as an injury-inducible messenger for mobilization of CD29+ stromal-like cells. Nat. Med 15, 425–435. [DOI] [PubMed] [Google Scholar]
- (233).Jung N, Um J, Kim DY, Dubon MJ, Byeon Y, Kim D, Son Y, and Park K-S (2017) Substance P preserves pancreatic β-cells in streptozotocin-induced type 1 diabetic mice. Biochem. Biophys. Res. Commun 491, 958–965. [DOI] [PubMed] [Google Scholar]
- (234).Lee Y-K, and Moore DD (2008) Liver receptor homolog-1, an emerging metabolic modulator. Front. Biosci 13, 5950–8. [DOI] [PubMed] [Google Scholar]
- (235).Venteclef N, Jakobsson T, Steffensen KR, and Treuter E (2011) Metabolic nuclear receptor signaling and the inflammatory acute phase response. Trends Endocrinol. Metab 22, 333–343. [DOI] [PubMed] [Google Scholar]
- (236).Mamrosh JL, Lee JM, Wagner M, Stambrook PJ, Whitby RJ, Sifers RN, Wu S-P, Tsai M-J, DeMayo FJ, and Moore DD (2014) Nuclear receptor LRH-1/NR5A2 is required and targetable for liver endoplasmic reticulum stress resolution. Elife 3, e01694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Cobo-Vuilleumier N, Lorenzo PI, Rodríguez NG, Herrera Gómez IDG, Fuente-Martin E, López-Noriega L, Mellado-Gil JM, Romero-Zerbo SY, Baquié M, Lachaud CC, Stifter K, Perdomo G, Bugliani M, De Tata V, Bosco D, Parnaud G, Pozo D, Hmadcha A, Florido JP, Toscano MG, De Haan P, Schoonjans K, Sánchez Palazón L, Marchetti P, Schirmbeck R, Martín-Montalvo A, Meda P, Soria B, Bermúdez-Silva FJ, St-Onge L, and Gauthier BR (2018) LRH-1 agonism favours an immune-islet dialogue which protects against diabetes mellitus. Nat. Commun 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, Madsen OD, Serup P, Heimberg H, and Mansouri A (2009) The Ectopic Expression of Pax4 in the Mouse Pancreas Converts Progenitor Cells into α and Subsequently β Cells. Cell 138, 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Yang Y-P, Thorel F, Boyer DF, Herrera PL, and Wright CVE (2011) Context-specific α-to-β-cell reprogramming by forced Pdx1 expression. Genes Dev. 25, 1680–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Al-Hasani K, Pfeifer A, Courtney M, Ben-Othman N, Gjernes E, Vieira A, Druelle N, Avolio F, Ravassard P, Leuckx G, Lacas-Gervais S, Ambrosetti D, Benizri E, Hecksher-Sorensen J, Gounon P, Ferrer J, Gradwohl G, Heimberg H, Mansouri A, and Collombat P (2013) Adult duct-lining cells can reprogram into β-like cells able to counter repeated cycles of toxin-induced diabetes. Dev. Cell 26, 86–100. [DOI] [PubMed] [Google Scholar]
- (241).Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben-Othman N, Pfeifer A, Avolio F, Leuckx G, Lacas-Gervais S, Burel-Vandenbos F, Ambrosetti D, Hecksher-Sorensen J, Ravassard P, Heimberg H, Mansouri A, and Collombat P (2013) The Inactivation of Arx in Pancreatic α-Cells Triggers Their Neogenesis and Conversion into Functional β-Like Cells. PLoS Genet. (Habener, J., Ed.) 9, e1003934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Koehler AN (2010) A complex task? Direct modulation of transcription factors with small molecules. Curr. Opin. Chem. Biol 14, 331–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Mathews LS, Norstedt G, and Palmiter RD (1986) Regulation of insulin-like growth factor I gene expression by growth hormone. Proc. Natl. Acad. Sci. U. S. A 83, 9343–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Lu J, Liu K-C, Schulz N, Karampelias C, Charbord J, Hilding A, Rautio L, Bertolino P, Östenson C-G, Brismar K, and Andersson O (2016) IGFBP1 increases b-cell regeneration by promoting a-to b-cell transdifferentiation. EMBO J. 35, 2026–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Crossey PA, Jones JS, and Miell JP (2000) Dysregulation of the insulin/IGF binding protein-1 axis in transgenic mice is associated with hyperinsulinemia and glucose intolerance. Diabetes 49, 457–65. [DOI] [PubMed] [Google Scholar]
- (246).Lewitt MS, Hilding A, Östenson C-G, Efendic S, Brismar K, and Hall K (2008) Insulin-like growth factor-binding protein-1 in the prediction and development of type 2 diabetes in middle-aged Swedish men. Diabetologia 51, 1135–1145. [DOI] [PubMed] [Google Scholar]
- (247).Lewitt MS, Hilding A, Brismar K, Efendic S, Ostenson C-G, and Hall K (2010) IGF-binding protein 1 and abdominal obesity in the development of type 2 diabetes in women. Eur. J. Endocrinol 163, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Collombat P, Mansouri A, Hecksher-Sorensen J, Serup P, Krull J, Gradwohl G, and Gruss P (2003) Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 17, 2591–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Wilcox CL, Terry NA, Walp ER, Lee RA, and May CL (2013) Pancreatic α-Cell Specific Deletion of Mouse Arx Leads to α-Cell Identity Loss. PLoS One (Maedler, K., Ed.) 8, e66214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Chakravarthy H, Gu X, Enge M, Dai X, Wang Y, Damond N, Downie C, Liu K, Wang J, Xing Y, Chera S, Thorel F, Quake S, Oberholzer J, MacDonald PE, Herrera PL, and Kim SK (2017) Converting Adult Pancreatic Islet α Cells into β Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 25, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Berishvili E, Harkany T, Meyer D, Collombat P, Klughammer J, Farlik M, Sdelci S, Májek P, Pauler FM, Penz T, Stukalov A, Gridling M, Parapatics K, Colinge J, Bennett KL, Bock C, Superti-Furga G, and Kubicek S (2017) Artemisinins Target GABAAReceptor Signaling and Impair α Cell Identity. Cell 168, 86–100.e15.27916275 [Google Scholar]
- (252).Purwana I, Zheng J, Li X, Deurloo M, Son DO, Zhang Z, Liang C, Shen E, Tadkase A, Feng Z-P, Li Y, Hasilo C, Paraskevas S, Bortell R, Greiner DL, Atkinson M, Prud’homme GJ, and Wang Q (2014) GABA Promotes Human-Cell Proliferation and Modulates Glucose Homeostasis. Diabetes 63, 4197–4205. [DOI] [PubMed] [Google Scholar]
- (253).Soltani N, Qiu H, Aleksic M, Glinka Y, Zhao F, Liu R, Li Y, Zhang N, Chakrabarti R, Ng T, Jin T, Zhang H, Lu W-Y, Feng Z-P, Prud’homme GJ, and Wang Q (2011) GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc. Natl. Acad. Sci 108, 11692–11697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Malaisse WJ, Louchami K, and Sener A (2009) Noninvasive imaging of pancreatic β cells. Nat. Rev. Endocrinol 5, 394–400. [DOI] [PubMed] [Google Scholar]
- (255).Jodal A, Schibli R, and Béhé M (2017) Targets and probes for non-invasive imaging of β-cells. Eur. J. Nucl. Med. Mol. Imaging 44, 712–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Eriksson O, Laughlin M, Brom M, Nuutila P, Roden M, Hwa A, Bonadonna R, and Gotthardt M (2016) In vivo imaging of beta cells with radiotracers: state of the art, prospects and recommendations for development and use. Diabetologia 59, 1340–1349. [DOI] [PubMed] [Google Scholar]
- (257).Wu Z, and Kandeel F (2010) Radionuclide probes for molecular imaging of pancreatic beta-cells. Adv. Drug Deliv. Rev 62, 1125–1138. [DOI] [PubMed] [Google Scholar]
- (258).Raufman JP (1996) Bioactive peptides from lizard venoms. Regul. Pept 61, 1–18. [DOI] [PubMed] [Google Scholar]
- (259).Bunck M, Diamante M, Corner A, Eliasson B, Malloy J, Shaginian R, Deng W, Kendall D, Taskinen M, Smith U, Yki-Jarvinen H, and Heine R (2009) One year treatment with exenatide Improves β-cell Function, compared with insulin glargine, in metformin treated type 2 diabetic patients. Diabetes Care 32, 762–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Hubalewska-Dydejczyk A, Sowa-Staszczak A, Tomaszuk M, and Stefańska A (2015) GLP-1 and Exendin-4 for imaging endocrine pancreas. A review: Labelled glucagon-like peptide-1 analogues: Past, present and future. Q. J. Nucl. Med. Mol. Imaging 59, 152–160. [PubMed] [Google Scholar]
- (261).Pattou F, Kerr-Conte J, and Wild D (2010) GLP-1–Receptor Scanning for Imaging of Human Beta Cells Transplanted in Muscle. N. Engl. J. Med 363, 1289–1290. [DOI] [PubMed] [Google Scholar]
- (262).Brom M, Woliner-Van Der Weg W, Joosten L, Frielink C, Bouckenooghe T, Rijken P, Andralojc K, Göke BJ, De Jong M, Eizirik DL, Béhé M, Lahoutte T, Oyen WJG, Tack CJ, Janssen M, Boerman OC, and Gotthardt M (2014) Non-invasive quantification of the beta cell mass by SPECT with 111In-labelled exendin. Diabetologia 57, 950–959. [DOI] [PubMed] [Google Scholar]
- (263).Weihe E, Schäfer MKH, Erickson JD, and Eiden LE (1994) Localization of vesicular monoamine transporter isoforms (VMAT1 and VMAT2) to endocrine cells and neurons in rat. J. Mol. Neurosci 5, 149–164. [DOI] [PubMed] [Google Scholar]
- (264).Anlauf M, Eissele R, Schäfer MKH, Eiden LE, Arnold R, Pauser U, Klöppel G, and Weihe E (2003) Expression of the two isoforms of the vesicular monoamine transporter (VMAT1 and VMAT2) in the endocrine pancreas and pancreatic endocrine tumors. J. Histochem. Cytochem 51, 1027–1040. [DOI] [PubMed] [Google Scholar]
- (265).Martin WRW, Wieler M, Stoessl a J., and Schulzer M (2008) Dihydrotetrabenazine positron emission tomography imaging in early, untreated Parkinson’s disease. Ann. Neurol 63, 388–94. [DOI] [PubMed] [Google Scholar]
- (266).Goland R, Freeby M, Parsey R, Saisho Y, Kumar D, Simpson N, Hirsch J, Prince M, Maffei A, Mann JJ, Butler PC, Van Heertum R, Leibel RL, Ichise M, and Harris PE (2009) 11C-Dihydrotetrabenazine PET of the Pancreas in Subjects with Long-Standing Type 1 Diabetes and in Healthy Controls. J. Nucl. Med 50, 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Normandin MD, Petersen KF, Ding Y-S, Lin S-F, Naik S, Fowles K, Skovronsky DM, Herold KC, McCarthy TJ, Calle RA, Carson RE, Treadway JL, and Cline GW (2012) In Vivo Imaging of Endogenous Pancreatic-Cell Mass in Healthy and Type 1 Diabetic Subjects Using 18F-Fluoropropyl-Dihydrotetrabenazine and PET. J. Nucl. Med 53, 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Fagerholm V, Mikkola KK, Ishizu T, Arponen E, Kauhanen S, Nagren K, Solin O, Nuutila P, and Haaparanta M (2010) Assessment of Islet Specificity of Dihydrotetrabenazine Radiotracer Binding in Rat Pancreas and Human Pancreas. J. Nucl. Med 51, 1439–1446. [DOI] [PubMed] [Google Scholar]
- (269).Ohta Y, Kosaka Y, Kishimoto N, Wang J, Smith SB, Honig G, Kim H, Gasa RM, Neubauer N, Liou A, Tecott LH, Deneris ES, and German MS (2011) Convergence of the Insulin and Serotonin Programs in the Pancreatic-Cell. Diabetes 60, 3208–3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Ding WG, Fujimura M, Tooyama I, and Kimura H (1991) Phylogenetic study of serotonin-immunoreactive structures in the pancreas of various vertebrates. Cell Tissue Res. 263, 237–243. [DOI] [PubMed] [Google Scholar]
- (271).Eriksson B, Orlefors H, Sundin A, Skogseid B, Langstrom B, Bergstrom M, and Oberg K (1999) Positron emission tomography in neuroendocrine tumours. Ital. J. Gastroenterol. Hepatol 31 Suppl 2, S167–71. [PubMed] [Google Scholar]
- (272).Mladenovic A, Perovic M, Tanic N, Petanceska S, Ruzdijic S, and Kanazir S (2007) Validation Studies on the 5-Hydroxy-L-[b-11C]-Tryptophan/PET Method for Probing the Decarboxylase Step in Serotonin Synthesis. Synapse 61, 790–794.17568432 [Google Scholar]
- (273).Di Gialleonardo V, Signore A, Scheerstra EA, Visser AKD, van Waarde A, Dierckx RAJO, and de Vries EFJ (2012) 11C-Hydroxytryptophan Uptake and Metabolism in Endocrine and Exocrine Pancreas. J. Nucl. Med 53, 1755–1763. [DOI] [PubMed] [Google Scholar]
- (274).Eriksson O, Selvaraju RK, Johansson L, Eriksson JW, Sundin A, Antoni G, Sorensen J, Eriksson B, and Korsgren O (2014) Quantitative Imaging of Serotonergic Biosynthesis and Degradation in the Endocrine Pancreas. J. Nucl. Med 55, 460–465. [DOI] [PubMed] [Google Scholar]
- (275).Eriksson O, Espes D, Selvaraju RK, Jansson E, Antoni G, Sorensen J, Lubberink M, Biglarnia A-R, Eriksson JW, Sundin A, Ahlstrom H, Eriksson B, Johansson L, Carlsson P-O, and Korsgren O (2014) Positron Emission Tomography Ligand [11C]5-Hydroxy-Tryptophan Can Be Used as a Surrogate Marker for the Human Endocrine Pancreas. Diabetes 63, 3428–3437. [DOI] [PubMed] [Google Scholar]
- (276).Schneider S, Feilen PJ, Schreckenberger M, Schwanstecher M, Schwanstecher C, Buchholz HG, Thews O, Oberholzer K, Korobeynikov A, Bauman A, Comagic S, Piel M, Schirrmacher E, Shiue CY, Alavi AA, Bartenstein P, Rosch F, Weber MM, Klein HH, and Schirrmacher R (2005) In vitro and in vivo evaluation of novel glibenclamide derivatives as imaging agents for the non-invasive assessment of the pancreatic islet cell mass in animals and humans. Exp. Clin. Endocrinol. Diabetes 113, 388–395. [DOI] [PubMed] [Google Scholar]
- (277).Schmitz A, Shiue CY, Feng Q, Shiue GG, Deng S, Pourdehnad MT, Schirrmacher R, Vatamaniuk M, Doliba N, Matschinsky F, Wolf B, Rösch F, Naji A, and Alavi AA (2004) Synthesis and evaluation of fluorine-18 labeled glyburide analogs as β-cell imaging agents. Nucl. Med. Biol 31, 483–491. [DOI] [PubMed] [Google Scholar]
- (278).Oh C-S, Kohanim S, Kong F-L, Song H-C, Huynh N, Mendez R, Chanda M, Edmund Kim E, and Yang DJ (2012) Sulfonylurea receptor as a target for molecular imaging of pancreas beta cells with 99mTc-DTPA-glipizide. Ann. Nucl. Med 26, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Babič A, Lamprianou S, Vinet L, Stransky-Heilkron N, Xayaphoummine C, Campo MA, Glombik H, Schulte A, Juretschke HP, Montet X, Meda P, and Lange N (2016) Multivalent glibenclamide to generate islet specific imaging probes. Biomaterials 75, 1–12. [DOI] [PubMed] [Google Scholar]
- (280).Rubí B, Ljubicic S, Pournourmohammadi S, Carobbio S, Armanet M, Bartley C, and Maechler P (2005) Dopamine D2-like Receptors Are Expressed in Pancreatic Beta Cells and Mediate Inhibition of Insulin Secretion. J. Biol. Chem 280, 36824–36832. [DOI] [PubMed] [Google Scholar]
- (281).Willekens SMA, Van Der Kroon I, Joosten L, Frielink C, Boerman OC, Van Den Broek SAMW, Brom M, and Gotthardt M (2016) SPECT of Transplanted Islets of Langerhans by Dopamine 2 Receptor Targeting in a Rat Model. Mol. Pharm 13, 85–91. [DOI] [PubMed] [Google Scholar]
- (282).Garcia A, Mirbolooki MR, Constantinescu C, Pan M-L, Sevrioukov E, Milne N, Wang PH, Lakey J, Chandy KG, and Mukherjee J (2011) 18F-Fallypride PET of pancreatic islets: in vitro and in vivo rodent studies. J. Nucl. Med 52, 1125–1132. [DOI] [PubMed] [Google Scholar]
- (283).Garcia A, Venugopal A, Pan M-L, and Mukherjee J (2014) Imaging Pancreas in Healthy and Diabetic Rodent Model Using [18 F]Fallypride Positron Emission Tomography/Computed Tomography. Diabetes Technol. Ther 16, 640–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Bini J, Naganawa M, Nabulsi NB, Huang YH, Ropchan J, Lim K, Najafzadeh S, Herold KC, Cline GW, and Carson RE (2018) Evaluation of PET Brain Radioligands for Imaging Pancreatic β-Cell Mass: Potential Utility of 11 C-PHNO. J. Nucl. Med 59, 1249–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Lindskog C, Korsgren O, Pontén F, Eriksson JW, Johansson L, and Danielsson A (2012) Novel pancreatic beta cell-specific proteins: Antibody-based proteomics for identification of new biomarker candidates. J. Proteomics 75, 2611–2620. [DOI] [PubMed] [Google Scholar]
- (286).Hellström-Lindahl E, Danielsson A, Ponten F, Czernichow P, Korsgren O, Johansson L, and Eriksson O (2016) GPR44 is a pancreatic protein restricted to the human beta cell. Acta Diabetol. 53, 413–421. [DOI] [PubMed] [Google Scholar]
- (287).Eriksson O, Johnström P, Cselenyi Z, Jahan M, Selvaraju RK, Jensen-Waern M, Takano A, Sörhede Winzell M, Halldin C, Skrtic S, and Korsgren O (2018) In Vivo Visualization of β-Cells by Targeting of GPR44. Diabetes 67, 182–192. [DOI] [PubMed] [Google Scholar]
- (288).Yao VJ, Ozawa MG, Trepel M, Arap W, McDonald DM, and Pasqualini R (2005) Targeting Pancreatic Islets with Phage Display Assisted by Laser Pressure Catapult Microdissection. Am. J. Pathol 166, 625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Ghosh K, Kanapathipillai M, Korin N, McCarthy JR, and Ingber DE (2012) Polymeric nanomaterials for islet targeting and immunotherapeutic delivery. Nano Lett. 12, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Blevins KS, Jeong JH, Ou M, Brumbach JH, and Kim SW (2012) EphA2 targeting peptide tethered bioreducible poly(cystamine bisacrylamide-diamino hexane) for the delivery of therapeutic pCMV-RAE-1γ to pancreatic islets. J. Control. Release 158, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Ueberberg S, Ziegler D, Schechinger W, Dietrich JW, Akinturk S, Klein HH, and Schneider S (2010) In vitro phage display in a rat beta cell line: A simple approach for the generation of a single-chain antibody targeting a novel beta cell-specific epitope. Diabetologia 53, 1384–1394. [DOI] [PubMed] [Google Scholar]
- (292).Ji HJ, Lee M, Won JK, Yockman JW, Tae GP, Yong HK, and Sung WK (2005) Anti-GAD antibody targeted non-viral gene delivery to islet beta cells. J. Control. Release 107, 562–570. [DOI] [PubMed] [Google Scholar]