

Abstract
Damage to the corticospinal and reticulospinal tract has been associated with spasticity in humans with upper motor neuron lesions. We hypothesized that these descending motor pathways distinctly contribute to the control of a spastic muscle in humans with incomplete spinal cord injury (SCI). To test this hypothesis, we examined motor-evoked potentials (MEPs) elicited by transcranial magnetic stimulation over the leg representation of the primary motor cortex, maximal voluntary contractions (MVCs), and the StartReact response (shortening in reaction time evoked by a startling stimulus) in the quadriceps femoris muscle in male and females with and without incomplete SCI. A total of 66.7% of the SCI participants showed symptoms of spasticity, whereas the other 33.3% showed no or low levels of spasticity. We found that participants with spasticity had smaller MEPs and MVCs and larger StartReact compared with participants with no or low spasticity and control subjects. These results were consistently present in spastic subjects but not in the other populations. Clinical scores of spasticity were negatively correlated with MEP-max and MVC values and positively correlated with shortening in reaction time. These findings provide evidence for lesser corticospinal and larger reticulospinal influences to spastic muscles in humans with SCI and suggest that these imbalanced contributions are important for motor recovery.
SIGNIFICANCE STATEMENT Although spasticity is one of the most common symptoms manifested in humans with spinal cord injury (SCI) to date, its mechanisms of action remain poorly understood. We provide evidence, for the first time, of imbalanced contributions of the corticospinal and reticulospinal tract to control a spastic muscle in humans with chronic incomplete SCI. We found that participants with SCI with spasticity showed small corticospinal responses and maximal voluntary contractions and larger reticulospinal gain compared with participants with no or low spasticity and control subjects. These results were consistently present in spastic subjects but not in the other populations. We showed that imbalanced corticospinal and reticulospinal tract contributions are more pronounced in participants with chronic incomplete SCI with lesser recovery.
Keywords: corticospinal pathway, muscle weakness, reticulospinal pathway, spasticity, voluntary drive
Introduction
Damage to descending motor pathways has been associated with the development of spasticity after spinal cord injury (SCI) (Frigon and Rossignol, 2006; Trompetto et al., 2014). For example, self-reported questionnaires and clinical examinations indicate that most people with incomplete SCI or residual descending voluntary drive have a high prevalence of spasticity (Little et al., 1989; Maynard et al., 1990; Sköld et al., 1999; Holtz et al., 2017). Recent electrophysiological data showed the presence of motor-evoked potentials (MEPs), likely involving the corticospinal tract, in humans with a diagnosis of clinically motor complete SCI who have spasticity but not in those individuals without spasticity (Sangari et al., 2019), suggesting that spasticity involved the presence of residual descending connections. Which descending motor pathways influence spasticity in humans with SCI, and to which extent, however, remains poorly understood.
After CNS damage, spasticity has been related to injury of the corticospinal and reticulospinal pathways (Cannon et al., 1943; Schreiner et al., 1949; Paulson et al., 1986; Nathan, 1994; Lee et al., 2016). The corticospinal and reticulospinal tracts constitute major descending motor pathways with convergent projections onto spinal motoneurons and interneurons in mammals (Jankowska and Edgley, 2006; Lemon, 2008; Baker et al., 2015). In primates, reticulospinal connections to motoneurons innervating limb muscles are strengthened after a corticospinal lesion (Zaaimi et al., 2012). In humans with stroke, spastic muscles are weaker (Pasternak-Mladzka et al., 2007) and exhibit an exaggerated response to a startle stimulus (Jankelowitz and Colebatch, 2004; Li et al., 2014; Choudhury et al., 2019), a stimulus that is thought to engage the reticulospinal tract (Brown et al., 1991; Valls-Solé et al., 1995, 1999), compared with no or less spastic muscles. Therefore, it has been proposed that the reticulospinal pathway might contribute to compensate for the loss of corticospinal axons after the injury (Pettersson et al., 2000; Schucht et al., 2002; Ballermann and Fouad, 2006; Asboth et al., 2018).
Both corticospinal (Oudega and Perez, 2012) and reticulospinal (Zaaimi et al., 2012, 2018) neurons undergo reorganization after SCI. Animal model of SCI showed that lesions of the corticospinal tract at the spinal cord level are accompanied by increased afferent sprouting at the spinal cord leading to symptoms of hyperreflexia (Murray and Goldberger, 1974). A number of individuals with SCI have weaker muscles and small corticospinal responses (Bunday et al., 2014), and those not taking a medication to reduce spasticity are unable to modulate corticospinal responses to a similar extent as control subjects (Barry et al., 2013; Bunday et al., 2014). In addition, participants with SCI showed an exaggerated response to a startle stimulus in certain conditions compared with control subjects (Jankelowitz and Colebatch, 2004; Kumru et al., 2008, 2009; Baker and Perez, 2017), suggesting a strong contribution from the reticulospinal tract after the injury. We hypothesized that individuals with incomplete SCI with spasticity show weak corticospinal and strong reticulospinal outputs compared with participants with no or low spasticity and control subjects.
To test this hypothesis, we examined MEPs elicited by transcranial magnetic stimulation (TMS) over the leg representation of the primary motor cortex, maximal voluntary contractions (MVCs), and the StartReact response (shortening in reaction time evoked by a startling stimulus) in the quadriceps femoris muscle in individuals with and without incomplete SCI.
Materials and Methods
Subjects.
Thirty individuals with SCI (mean age 51.6 ± 15.8 years, 6 female; Table 1) and 15 age-matched controls (mean age 45.5 ± 13.6 years, 4 female, p = 0.1) participated in the study. All participants gave informed consent to the experimental procedures, which were approved by the local ethics committee at the University of Miami and performed in accordance with the Declaration of Helsinki. Participants with SCI had a chronic injury (≥1 year) and were classified using the International Standards for Neurological Classification of Spinal Cord Injury examination as having a C1-L3 SCI and by the American Spinal Cord Injury Association Impairment Scale (AIS) as AIS C (n = 22) or AIS D (n = 8). AIS C and D participants show preserved voluntary anal sphincter contraction or sacral sensory sparing with motor function sparing in more than three levels below the motor level for that side of the body. AIS C is assigned if half of the key muscles below the neurological level of injury are graded as <3/5, and AIS D is assigned if half or more of the key muscles below the neurological level of injury are graded as >3/5. Sixteen SCI individuals were under anti-spastic medication (baclofen and/or gabapentin and/or tizanidine; Table 1). We tested individuals with preservation of motor output in the quadriceps femoris muscle or “motor incomplete SCI” to assess the role of residual descending motor pathways in spasticity. All subjects were asked to perform voluntary knee extension, and all of them were able to exert voluntary electromyographic (EMG) activity in the quadriceps femoris muscle. The degree of spasticity in the quadriceps was examined in all SCI participants by using the Modified Ashworth Scale (MAS).
Table 1.
Spinal cord injury participantsa
| Participant | Age (yr) | Gender | AIS | Level | Etiology | Time after injury (yr) | MAS score | Medication(s) |
|---|---|---|---|---|---|---|---|---|
| 1 | 61 | M | D | C4 | T | 13.9 | 0 | None |
| 2 | 69 | M | D | T4 | NT | 7.0 | 0 | None |
| 3 | 66 | M | C | C5 | T | 17.0 | 0 | None |
| 4 | 58 | F | C | C4 | T | 6.0 | 0 | None |
| 5 | 69 | F | C | L3 | T | 2.5 | 0 | None |
| 6 | 28 | F | C | C5 | T | 2.2 | 0 | GBP |
| 7 | 70 | M | C | T11 | T | 3.4 | 1 | BAC |
| 8 | 68 | M | D | C3 | T | 11.0 | 1 | GBP, TIZ |
| 9 | 53 | M | D | L2 | T | 31.9 | 1 | BAC |
| 10 | 61 | M | D | C4 | NT | 4.1 | 1 | GBP |
| 11 | 41 | F | C | C1 | NT | 25.0 | 2 | BAC, GBP |
| 12 | 39 | M | C | C5 | T | 12.1 | 2 | None |
| 13 | 70 | M | C | C3 | T | 7.6 | 2 | BAC |
| 14 | 21 | F | C | C4 | T | 1.9 | 2 | None |
| 15 | 34 | M | D | C2 | T | 10.3 | 3 | None |
| 16 | 61 | M | C | T5 | NT | 41.0 | 3 | None |
| 17 | 37 | M | C | C5 | T | 2.2 | 3 | BAC |
| 18 | 42 | M | C | T2 | T | 2.1 | 3 | BAC |
| 19 | 67 | M | C | C7 | T | 3.0 | 3 | None |
| 20 | 41 | M | C | C5 | T | 16.0 | 3 | None |
| 21 | 44 | M | C | C4 | T | 10.3 | 3 | None |
| 22 | 71 | M | C | T7 | NT | 9.0 | 3 | None |
| 23 | 19 | F | C | T6 | T | 6.0 | 4 | BAC |
| 24 | 61 | M | D | C5 | T | 16.0 | 4 | None |
| 25 | 59 | M | C | C5 | T | 14.3 | 4 | BAC |
| 26 | 69 | M | D | C4 | T | 46.0 | 4 | BAC |
| 27 | 34 | M | C | T12 | T | 2.3 | 4 | BAC |
| 28 | 52 | M | C | T5 | T | 1.8 | 4 | BAC |
| 29 | 43 | M | C | C4 | T | 12.1 | 4 | BAC |
| 30 | 39 | M | C | T10 | T | 1.6 | 4 | BAC |
aT, traumatic; NT, non-traumatic; BAC, baclofen; GBP, gabapentin; TIZ, tizanidine.
MAS.
This clinical scale measures resistance encountered during manual passive muscle stretching using a 6 point ordinal scale (0 = no increase in tone; 1/+1 = slight increase in tone with a catch and release or minimal resistance at the end or less than half of the range of movement, respectively; 2 = more marked increased tone through most of the range of movement but affected part easily moved; 3 = considerable increase in tone and passive movement difficult; and 4 = affected part rigid) (Bohannon and Smith, 1987). During testing, subjects were lying in a semisupine position with the trunk at an angle of 30° of flexion (Fig. 1A). This neutral position helps to avoid increases in spasticity related to the stretching of the rectus femoris or the decrease of spasticity related to less stretched and more relaxed muscle (de Azevedo et al., 2015). The same rater performed all MAS assessments. Because spasticity can be asymmetric between left and right limbs (Chen et al., 2018), both legs were tested and the leg with higher MAS score was used for all measurements to allow better comparisons between spastic and no or low spastic individuals. Participants with a MAS score of 1 and +1 were grouped together as MAS 1. Recently, we observed no differences in outcomes related to spasticity between individuals with MAS score of 0 and 1 (post hoc analysis not reported in the paper) (Sangari et al., 2019). The same result was observed here. Thus, participants were separated into those with spasticity (spastic SCI: 20 out of 30, 66.7%; MAS = 2, 3 and 4) and those with no or low spasticity (non-spastic SCI: 10 out of 30, 33.3%; MAS = 0 and 1) according to the MAS scores.
Figure 1.
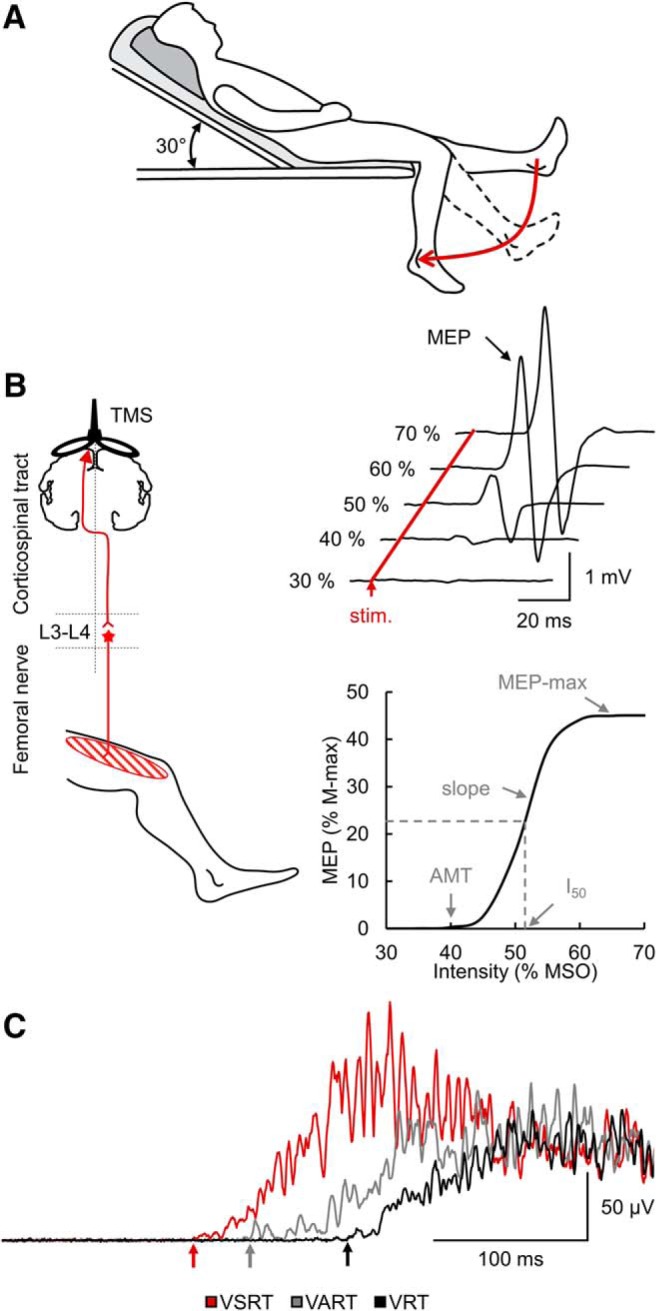
Experimental setup. A, The MAS measures resistance encountered during manual passive muscle stretching. During testing, subjects were lying in a semisupine position with the trunk at an angle of 30° of flexion. This neutral position helps to avoid increases in spasticity related to the stretching of the rectus femoris or the decrease of spasticity related to less stretched and more relaxed muscle. B, TMS was applied over the leg representation of the primary motor cortex to activate corticospinal neurons projecting directly or indirectly to quadriceps femoris motoneurons located around the third and fourth lumbar segment (L3–L4) to elicit an MEPs. MEP recruitment curves can be obtained by plotting the amplitude of the MEP against the TMS intensity and allow to define the MEP-max, I50, AMT, and the slope of the curve. C, During the StartReact response in some trials, an LED was presented with either a quiet acoustic stimulus or a startling acoustic stimulus (SAS). The StartReact response was measured between the VRT (defined as the time from cue to onset of the EMG burst in the quadriceps femoris after the LED presentation), the VART (defined as the time delay between the presentation of the quiet acoustic stimulus and the onset of the EMG response), and the VSRT (defined as the time between the SAS and the EMG onset).
EMG recordings.
EMG was recorded from the quadriceps femoris muscle of the right side in control subjects and from the leg with the higher MAS score in individuals with SCI through bipolar surface electrodes (Ag-AgCl, 10 mm diameter, 1 cm apart) secured on the skin. The signals were amplified, filtered (30–2000 Hz), and sampled at 2 kHz for off-line analysis (CED 1401 with Signal software, Cambridge Electronic Design).
Experimental paradigm.
During all testing procedures, subjects were seated comfortably in an armchair with both legs placed on a custom platform with the hip (∼120°) and knee (∼160°) flexed and the ankle restrained by straps in ∼110° of plantarflexion. During MVCs, subjects were instructed to perform an isometric knee extension, which consisted of performing three consecutive maximal efforts lasting 3 s each and separated by a rest period of 1 min. The maximal mean EMG activity measure over a period of 1 s on the rectified response generated during each MVC was analyzed, and the highest value of the three trials was used. During acquisition of MEP recruitment curves, participants were able to maintain ∼10% MVC into knee extension with the quadriceps muscle (controls = 8.2 ± 0.9% of MVC, non-spastic SCI = 8.4 ± 0.9% of MVC, non-spastic SCI = 8.9 ± 0.9% of MVC; p = 0.2).
MEP recruitment curve.
TMS was delivered over the leg representation of the primary motor cortex from a BiStim2 (Magstim) through a double-cone coil with a monophasic current waveform. The coil was positioned over the vertex and moved around this point to determine the optimal position for eliciting an MEP in the quadriceps femoris muscle during a tonic knee extension of ∼10% MVC. Participants wore a cap on which the position of the coil was marked to ensure the stability of TMS across the stimulation. The TMS intensity was ranged with 5% of the maximal stimulator output (MSO) stepwise from the active motor threshold (AMT) to the intensity producing the maximal MEP (MEP-max). The AMT was defined as the minimal stimulus intensity needed to elicit 5 out of 10 MEPs 100 μV above the EMG background. Ten stimuli (0.2 Hz) were delivered at each intensity to plot the mean peak-to-peak amplitude of the MEP from the non-rectified response against the TMS intensity in each subject (MEP recruitment curve; Fig. 1B). The experimental data were fitted with the following 3-parameter sigmoid function (Devanne et al., 1997; Carroll et al., 2001):
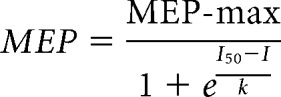 |
where MEP-max is the maximal size of MEP, I50 is the TMS intensity producing half MEP-max, and k is the Boltzmann slope parameter (Fig. 1B). Sigmoid fit was performed in each subject, individually. The estimated values of each parameter of the sigmoid (MEP-max, I50, and k) were extracted in each participant, and we calculated the mean of each parameter in each group (control, non-spastic SCI, spastic SCI). The mean estimated MEP-max, the mean estimated I50, and the mean estimated k were used to draw the mean estimated sigmoid curve in each group. MEP-max onset latencies were defined when the rectified EMG reached 2 × SD calculated over a 100 ms period of the prestimulus activity. Percutaneous electrical stimulation of the femoral nerve was delivered (1 ms rectangular electrical stimulus, DS7AH, Digitimer) through a cathode (10-mm-diameter Ag-AgCl electrode) placed in the femoral triangle and an anode (Ag-AgCl plaque) placed over the posterior aspect of the thigh. Stimulus intensities were increased up to measure the maximal motor response (M-max) at rest (0.22 Hz). The M-max was measured as peak-to-peak amplitude of the non-rectified response, and it was used to normalize MEP values in each participant.
StartReact response.
The StartReact response was tested using a previously described paradigm in humans with SCI (Baker and Perez, 2017). Here, participants were asked to observe a light-emitting diode (LED) located ∼1 m in front of the participants' head. When the LED was illuminated (20 ms), individuals were asked to perform isometric knee extension as fast as possible. We measured the visual reaction time (VRT; Fig. 1C) as the time from cue to onset of the EMG burst in the quadriceps femoris after the LED presentation. In some trials, the LED was presented with either a quiet acoustic stimulus (80 dB, 500 Hz, 50 ms) or a startling acoustic stimulus (SAS, 120 dB, 500 Hz, 50 ms) delivered through two audio speakers (T-15, Polk Audio) located right behind participant's head. The loud intensity evoked a clear startle in control subjects and in some individuals with SCI on initial presentation. The time delay between the presentation of the quiet acoustic stimulus and the onset of the EMG response was referred as the visual-auditory reaction time (VART; Fig. 1C), whereas the time between the SAS and the EMG onset was defined as the visual + startle reaction time (VSRT; Fig. 1C). A familiarization trial consisting of three repetitions of each task responding to the LED was completed at the beginning of each experiment to ensure that participants were able to complete all task and to get them familiarized with the startling cue (Fisher et al., 2013). In each task, 20 responses were recorded in each condition (VRT, VART, and VSRT) in a randomized order with an intertrial interval varying between 5 and 15 s to avoid habituation and predictability of the stimulation timing. Data were measured trial by trial. The reaction time was defined at the time point where the rectified EMG signal exceeded 2 × SD calculated over a 100 ms period of the prestimulus activity. Reaction time exceeding 700 ms were excluded. The VART and VSRT are both mediated via the cochlear nuclei, but only the high intensity sound of the VSRT activate the reticulospinal pathway (Davis et al., 1982; Brown et al., 1991; Valls-Solé et al., 1999). To estimate changes in the gain of reticulospinal output, we normalized the data as follows (Baker and Perez, 2017):
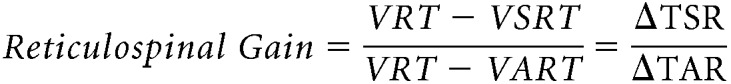 |
where ΔTSR (i.e. shortening of reaction time with a startle stimulus) is the shortening effect of a SAS on the VRT and ΔTAR (i.e. shortening of reaction time with an auditory stimulus) measures the shortening of reaction time provided by a non-startling auditory stimulus on the VRT, which presumably does not activate reticulospinal pathways.
Data analysis.
Normal distribution was tested by the Shapiro–Wilk's test and homogeneity of variances by the Levene's test of equality and Mauchly's test of sphericity. When sphericity could not be assumed, the Greenhouse–Geisser correction statistic was used. One-way ANOVA was performed to examine the effect of GROUP (controls, non-spastic SCI, spastic SCI) on MVCs, voluntary EMG level exerted during MEP recruitment curves, M-max, MEP-max, slope, AMT, I50, and ΔTSR/ΔTAR ratio. Repeated-measures ANOVAs as a mixed model were also performed to determine the effect of GROUP (controls, non-spastic SCI, spastic SCI) and CONDITION (VRT, VART, VSRT) on the reaction time and on the mean EMG activity measured over 100 ms before stimulus and from EMG burst onset. CONDITION was used a repeated-measures factor. Additional repeated ANOVA were performed on each group separately as needed. Holm–Sidak post hoc analysis was used to test for significant comparisons. Pearson correlation coefficient analysis was used as needed. If ratios were not normally distributed, a log transformation was applied. The statistical analysis was conducted using SigmaPlot (Systat Software) and the significance was set at p < 0.05. Group data are presented as mean ± SD.
Results
MAS
We found that 66.7% of all individuals with incomplete SCI tested showed spasticity (20 out of 30; MAS 2, 3 and 4), and 33.3% of them showed no or low spasticity (10 out of 30; MAS 0 and 1; Fig. 2A). The majority of SCI participants exhibited severe spasticity (MAS = 3, n = 8; and MAS = 4, n = 8) compared with a marked increase in muscle tone (MAS = 2, n = 4). However, a lesser number of SCI participants exhibited no (MAS = 0, n = 6) or low (MAS = 1, n = 4) spasticity.
Figure 2.
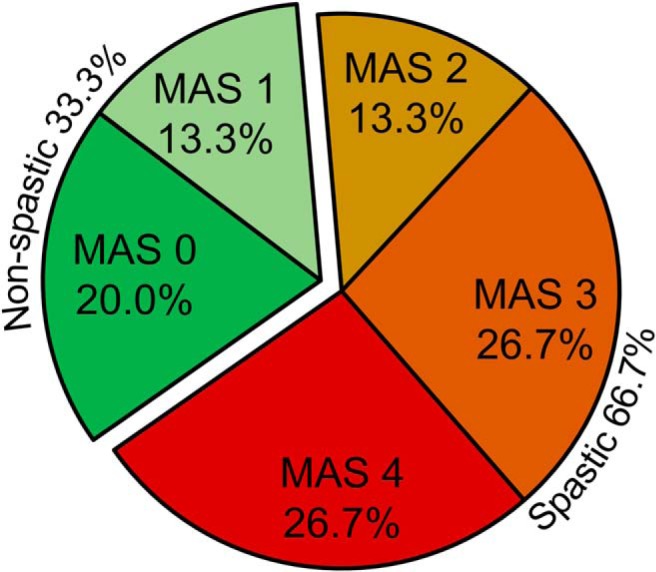
MAS score distribution. Individual MAS score distribution showed that 66.7% of all individuals with incomplete SCI showed spasticity (spastic SCI, 20 out of 30; MAS 2, 3 and 4) and 33.3% of them showed no or low spasticity (non-spastic SCI, 10 out of 30; MAS 0 and1).
MVC
Figure 3A illustrates rectified EMG activity during MVC in a control subject (blue EMG trace) and participants with SCI without (green EMG trace, MAS = 0) and with (red EMG trace, MAS = 3) spasticity. The non-spastic individual exhibited similar MVC compared with the control subject, whereas the spastic individual showed a reduced MVC compared with the other participants. One-way ANOVA showed an effect of GROUP (F(2,42) = 24.2, p < 0.001, η2 = 0.9; Fig. 3B) on MVC. Post hoc tests showed that the MVC in the quadriceps femoris muscle was reduced in spastic (0.07 ± 0.04 mV) compared with controls (0.2 ± 0.07 mV, p < 0.001) and non-spastic (0.2 ± 0.1 mV, p < 0.001) participants. No differences were found between controls and non-spastic participants (p = 0.2). In spastic (p = 0.2) and non-spastic (p = 0.7) SCI participants, MVC values remained similar between people taking and not taking anti-spastic medication.
Figure 3.
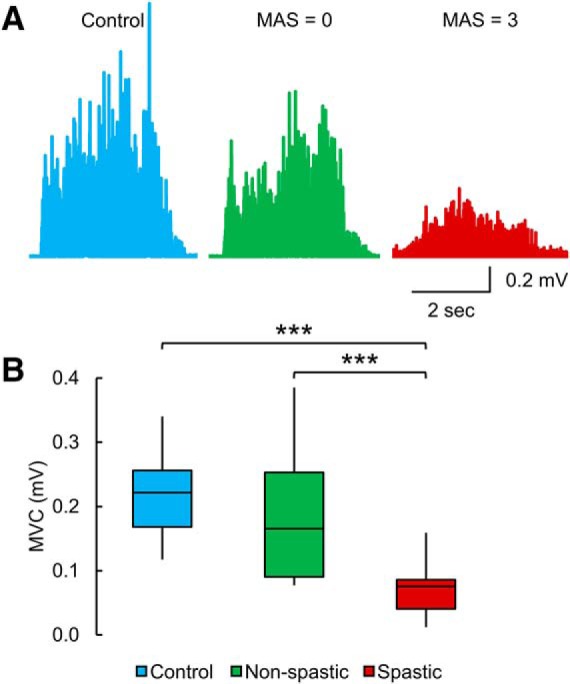
MVC. A, EMG recorded during the MVC test in a control subject and participants with SCI without (MAS = 0) and with (MAS = 3) spasticity. The non-spastic individual exhibited similar MVC compared with the control subject, whereas the spastic individual showed a reduced MVC compared with the other participants. B, Box plot charts represent the group data. The abscissa indicates the groups tested (blue bar represents controls; green bar represents non-spastic SCI; red bar represents spastic SCI), and the ordinate indicates the MVC (in millivolt). Top and bottom line of the box corresponds to the 95% CI, and the line in the box corresponds to the median. The two bars extend from the maximum and minimum value. ***p < 0.001.
MEP recruitment curve
Figure 4A illustrates the mean MEP recruitment curves in controls (blue line), non-spastic (green line), and spastic (red line) SCI groups. The graph shows in the x axis the intensity of TMS, and the y axis shows the MEP size in the quadriceps femoris muscle normalized to the M-max. The spastic group showed a reduced MEP-max and slope compared with the non-spastic group and control subjects. One-way ANOVA showed an effect of GROUP on the MEP-max (F(2,36) = 14.1, p < 0.001, η2 = 0.7; Fig. 4B) and the slope of the MEP recruitment curve (F(2,36) = 13.3, p < 0.001, η2 = 0.7; Fig. 4B). Post hoc tests showed that the MEP-max was reduced in spastic (5.0 ± 6.2% of M-max) compared with controls (35.3 ± 18.2% of M-max, p < 0.001) and non-spastic (28.5 ± 28.1% of M-max, p = 0.007) participants, whereas no differences were found between controls and non-spastic participants (p = 0.4). Post hoc tests also showed that the slope of the MEP recruitment curve was reduced in spastic (0.1 ± 0.2) compared with controls (1.1 ± 0.6, p < 0.001) and non-spastic (0.8 ± 0.9, p = 0.02) participants. No difference was found between controls and non-spastic participants (p = 0.2). We found that the M-max was reduced in SCI (6.9 ± 3.3 mV) compared with the control group (8.9 ± 2.3 mV, p = 0.04, dcohen = 0.7). However, no differences were observed between spastic (7.1 ± 3.3 mV) and non-spastic participants (6.6 ± 3.4, p = 0.7, dcohen = 0.1). In the spastic and non-spastic groups, no differences were found on the MEP-max (spastic, p = 0.8; non-spastic, p = 0.3), the slope of the MEP recruitment curve (spastic, p = 0.7; non-spastic, p = 0.4), and the M-max (spastic, p = 0.9; non-spastic, p = 0.7) between SCI participants taking or not taking anti-spastic medication.
Figure 4.
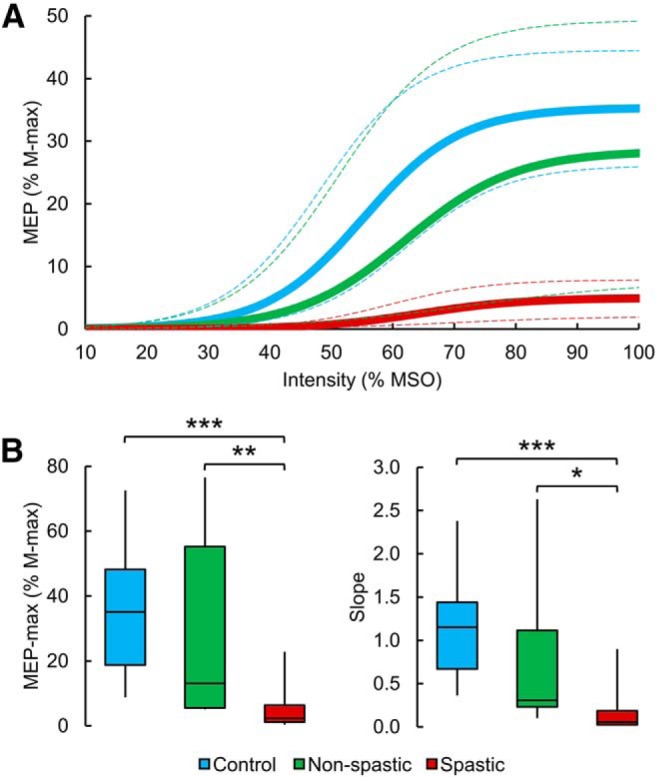
MEP recruitment curves. A, The mean MEP recruitment curves in controls (blue line), non-spastic (green line), and spastic (red line) SCI groups. The x axis indicates the intensity of TMS, and the y axis indicates the MEP size in the quadriceps femoris muscle normalized to the M-max. The 95% CIs of each parameter of the sigmoid function were used to draw the confidence bands (dashed line) around each respective recruitment curve. The spastic group showed a reduced MEP-max and slope compared with the non-spastic group and control subjects. B, Box plot charts represent the group data. The abscissa indicates the groups tested (blue bar represents controls; green bar represents non-spastic SCI; red bar represents spastic SCI), and the ordinate indicates the MEP-max (as a percentage of the M-max, left) and the slope (right). Top and bottom line of the box corresponds to the 95% CI, and the line in the box corresponds to the median. The two bars extend from the maximum and minimum value. *p < 0.05, **p < 0.01, ***p < 0.001.
Since MEPs were normalized to the M-max, we compared these responses across groups without normalization. As it was observed in normalized MEP values, raw MEP-max values (F(2,36) = 23.1, p < 0.001, η2 = 1.0) were reduced in spastic (0.3 ± 0.3 mV) compared with controls (3.1 ± 1.4 mV, p < 0.001) and non-spastic (1.7 ± 1.6 mV, p = 0.01) participants but also between controls and non-spastic participants (p = 0.02), suggesting that the normalization procedures did not influence our results. Because spastic individuals where weaker than non-spastic participants, in an additional control experiment, we asked non-spastic individuals (n = 6) to match EMG level exerted by spastic participants during acquisition of MEP recruitment curves (spastic = 0.006 ± 0.002 mV; non-spastic = 0.006 ± 0.001 mV; p = 0.9). Here, we found that the MEP-max and the slope of the recruitment curve were larger in the non-spastic (MEP-max = 27.3 ± 13.9% of M-max, p = 0.01, η2 = 1.3; slope = 0.8 ± 0.4, p = 0.02, dcohen = 1.2) compared with the spastic (MEP-max = 5.0 ± 6.2% of M-max; slope = 0.1 ± 0.2) SCI group, confirming our previous results and agreeing with evidence supporting the view that the level of voluntary contraction does not affect MEP-max value (Devanne et al., 1997).
One-way ANOVA also showed an effect of GROUP on the latency of the MEP-max (F(2,36) = 17.1, p < 0.001, η2 = 1.0) and the AMT (F(2,36) = 3.7, p = 0.03, η2 = 0.4). The MEP latency was delayed in spastic (27.9 ± 5.4 ms) compared with controls (19.9 ± 2.6 ms, p < 0.001) and non-spastic (20.3 ± 3.2 ms, p < 0.001) participants, consistent with the view that corticospinal drive is more impaired in spastic individuals. No differences were found between controls and non-spastic participants (p = 0.8). The AMT was increased in spastic (49.9 ± 11.9% of MSO) compared with controls (39.5 ± 9.4% of MSO, p = 0.03) but not with non-spastic (42.4 ± 12.3% of MSO, p = 0.6) participants. No differences were found between controls and non-spastic participants (p = 0.3). No differences were found on latency of the MEP-max (spastic, p = 0.2; non-spastic, p = 0.6) and the AMT (spastic, p = 0.8; non-spastic, p = 0.1) in both groups in SCI participants taking or not taking anti-spastic medication.
StartReact response
Repeated-measures ANOVA showed an effect of GROUP (F(2,42) = 5.5, p = 0.007, η2p = 0.2) and CONDITION (F2,84 = 442.1, p < 0.001, η2p = 0.9) and in their interaction (F4,84 = 5.9, p < 0.001, η2p = 0.2) on reaction time. Post hoc testing showed that VRTs were prolonged in spastic (288.2 ± 57.5 ms) compared with controls (232.2 ± 35.9, p = 0.002) and non-spastic (243.0 ± 39.1, p = 0.02) participants. No differences were found between controls and non-spastic participants (p = 0.6). We also found that VARTs were prolonged in spastic (236.5 ± 58.9 ms) compared with controls (183.4 ± 36.9, p = 0.003) and non-spastic (189.5 ± 36.7, p = 0.02) participants. No differences were found between controls and non-spastic participants (p = 0.7). We did not find a significant difference on the VSRT between spastic (176.9 ± 42.3 ms), controls (149.2 ± 33.7 ms, p = 0.2) and non-spastic (154.3 ± 34.3 ms, p = 0.4) participants, and between controls and non-spastic participants (p = 0.8).
Repeated-measures ANOVA showed no effect of GROUP (F(2,42) = 0.2, p = 0.8, η2p = 0.01), CONDITION (F2,84 = 0.1, p = 0.9, η2p = 0.003), nor in their interaction (F4,84 = 0.7, p = 0.6, η2p = 0.03) on mean rectified EMG activity in the quadriceps femoris muscle measured 100 ms before stimulus presentation. However, we found an effect of GROUP (F(2,42) = 13.5, p < 0.001, η2p = 0.4), CONDITION (F2, 84 = 56.4, p < 0.001, η2p = 0.6) and in their interaction (F4,84 = 8.1, p < 0.001, η2p = 0.3) on the mean rectified EMG activity in the quadriceps femoris muscle measured over 100 ms after the EMG burst onset. Here in all groups the mean EMG activity was larger during VSRT (controls = 0.1 ± 0.06 mV; spastic SCI = 0.03 ± 0.03 mV; non-spastic SCI = 0.08 ± 0.05 mV) compare with VRT (controls = 0.06 ± 0.03 mV, p < 0.001; spastic SCI = 0.022 ± 0.02 mV, p < 0.001; non-spastic SCI = 0.05 ± 0.03 mV, p < 0.001). Also, mean EMG activity was larger during VSRT compared with VART in controls (0.08 ± 0.04 mv, p < 0.001), non-spastic SCI (0.06 ± 0.03, p = 0.002) and spastic SCI (0.025 ± 0.02, p < 0.001). Mean EMG activity was larger during VART compare to VRT in controls (p < 0.001), spastic (p < 0.001) and non-spastic SCI (p = 0.006).
To ensure that any changes in reaction times were related to changes in reticulospinal gain, we compared the ΔTSR/ΔTAR ratios across groups. Figure 5A illustrates the mean EMG activity related to VRT (black), VART (gray), and VSRT (red) in a control subject and participants with SCI without (MAS = 0) and with (MAS = 3) spasticity. Reaction time was prolonged in the spastic individual in all conditions compared with the non-spastic and control participant. Notably, in the spastic participant, reaction time further decreased during VSRT (i.e., ΔTSR) but not during VART (i.e., ΔTAR) compared with VRT, in comparison of the other participants. One-way ANOVA showed an effect of GROUP (F(2,42) = 4.1, p = 0.02, η2 = 0.5; Fig. 5B) on ΔTSR/ΔTAR ratios. Post hoc tests showed that the reticulospinal gain was increased in spastic (2.3 ± 0.9) compared with controls (1.8 ± 0.4, p = 0.03) and non-spastic (1.7 ± 0.2, p = 0.02) participants. No differences were found between controls and non-spastic participants (p = 0.6). Notably, the reticulospinal gain in the spastic (p = 0.5) and non-spastic (p = 0.2) group remained similar between participants taking or not taking anti-spastic medication.
Figure 5.
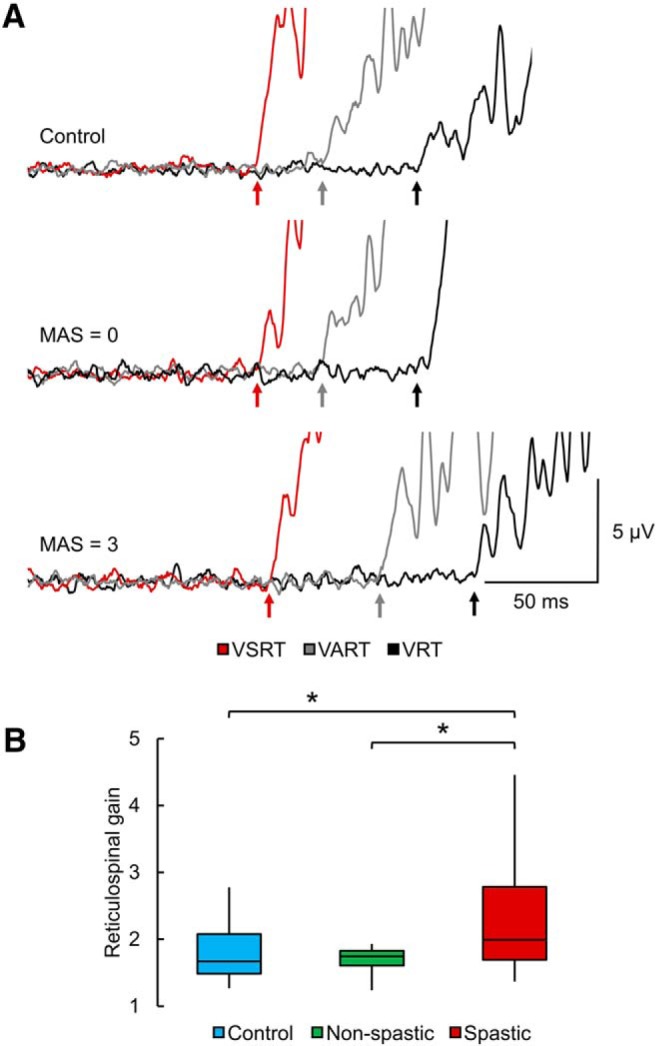
StartReact. A, The mean EMG activity related to VRT (black), VART (gray), and VSRT (red) in a control subject and participants with SCI without (MAS = 0) and with (MAS = 3) spasticity. Reaction time was prolonged in the spastic individual in all conditions compared with the non-spastic and control participant. Notably, in the spastic participant, reaction time further decreased during VSRT, but not during VART compared with VRT, in comparison of the other participants. B, Box plot charts represent the group data. The abscissa indicates the groups tested (blue bar represents controls; green bar represents non-spastic SCI; red bar represents spastic SCI), and the ordinate indicates the reticulospinal gain. Top and bottom line of the box corresponds to the 95% CI, and the line in the box corresponds to the median. The two bars extend from the maximum and minimum value. *p < 0.05.
Figure 6 shows individual data from all non-spastic (green circles) and spastic (red circles) SCI participants. The graphs show MEP-max and reticulospinal gain (Fig. 6A) and MVCs and reticulospinal gain (Fig. 6B) values expressed as a percentage of the mean value found in control subjects. In non-spastic participants, MEP-max (Fig. 6A, left), MVC (Fig. 6B, left), and reticulospinal gain values were higher, lower, or similar compared with controls. However, all spastic SCI participants showed smaller MEP-max (Fig. 6A, right) and MVC (Fig. 6B, right) and larger reticulospinal gain compared with the control group. These differences were more pronounced in weaker spastic SCI participants.
Figure 6.
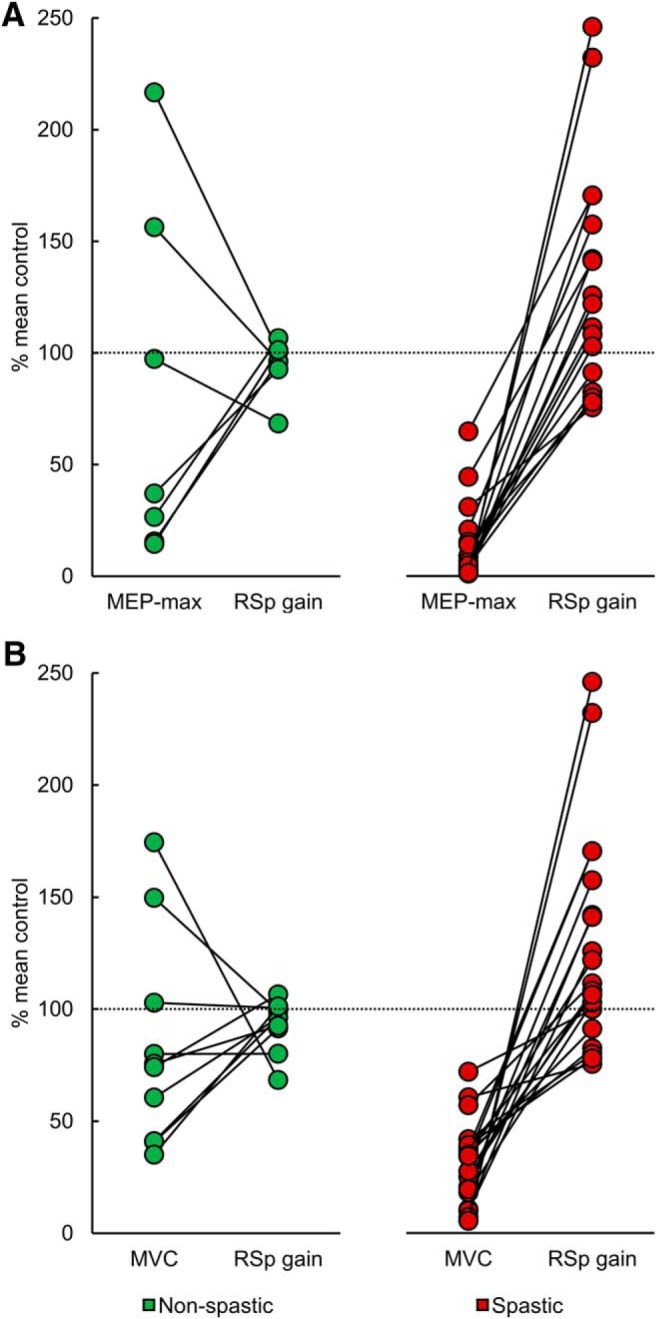
MEP-max, MVCs, and reticulospinal gain. Individual data from non-spastic (green) and spastic (red) SCI participants. The abscissa indicates the MEP-max and reticulospinal gain (RSp gain; A) and the MVCs and RSp gain (B), and the ordinate indicates values expressed as a percentage of the mean value from controls. The line connects physiological outcomes recorded in the same participant. In non-spastic SCI participants, MEP-max, MVC, and the reticulospinal gain values were higher, lower, or similar to the control group. However, all spastic SCI participants showed smaller MEP-max and MVC and larger reticulospinal gain compared with the control group.
Correlations
We found that MAS scores were negatively correlated with MEP-max (r = −0.48, p = 0.01) and MVC (r = −0.52, p = 0.003) values and positively correlated with the reticulospinal gain (r = 0.36, p = 0.03) in SCI participants. Notably, MVCs values were negatively correlated with the reticulospinal gain (r = −0.61, p < 0.01; Fig. 7A) in spastic participants. Individuals with smaller voluntary output in the quadriceps femoris muscle were those who showed larger reticulospinal gain, suggesting that imbalanced corticospinal and reticulospinal tract contributions are more pronounced in weaker spastic participants with chronic incomplete SCI, whereas in non-spastic participants (r = −0.49, p = 0.1; Fig. 7B) and control participants (r = 0.25, p = 0.4; Fig. 7C) MVCs values were not correlated with the reticulospinal gain. No correlation was also found between physiological outcomes and the time after injury (M-max, r = 0.40, p = 0.1; MEP-max, r = 0.23, p = 0.3; slope, r = 0.07, p = 0.7; AMT, r = −0.08, p = 0.7; MVC, r = 0.30, p = 0.1; reticulospinal gain, r = −0.25, p = 0.2) and medication intake (M-max, r = 0.02, p = 0.9; MEP-max, r = 0.27, p = 0.2; slope, r = 0.32, p = 0.1; AMT, r = −0.2, p = 0.4; MVC, r = −0.04, p = 0.8; reticulospinal gain, r = 0.13, p = 0.5).
Figure 7.
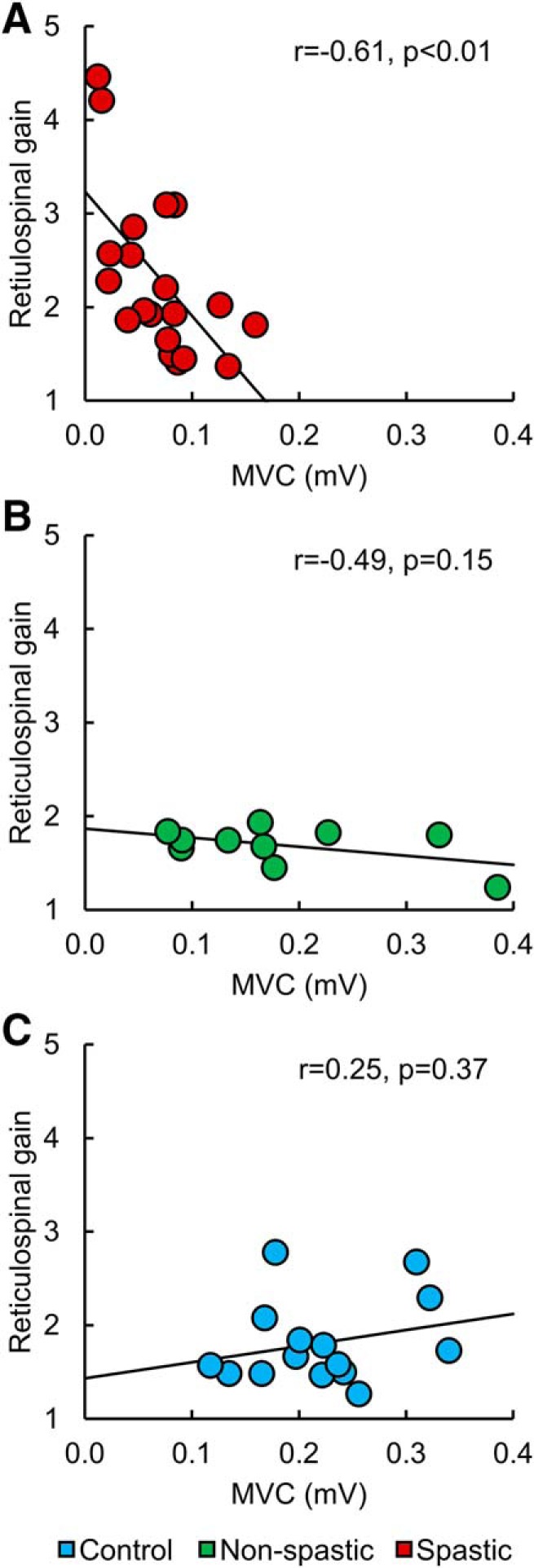
Correlations. Individual data from spastic (A, red) and non-spastic (B, green) SCI participants and control subjects (C, blue). The abscissa indicates the MVC (in millivolt), and the ordinate indicates the reticulospinal gain. MVC values were negatively correlated with the reticulospinal gain in spastic participants, but not in non-spastic and control participants.
Discussion
Our results provide evidence for imbalanced contributions of the corticospinal and reticulospinal tract to the control of a spastic muscle in humans with incomplete chronic SCI. Specifically, we found that SCI participants with spasticity had smaller MEPs and MVCs and larger StartReact response compared with those with no or low spasticity and control subjects. These results were consistently present in participants with spasticity but not in the other populations, suggesting a lesser corticospinal and larger reticulospinal influence to spastic muscles. Clinical scores of spasticity correlated negatively with MEP-max and MVC values and positively with shortening in reaction time. We hypothesize that imbalanced corticospinal and reticulospinal tract contributions are more pronounced in participants with chronic incomplete SCI with lesser recovery.
Corticospinal and reticulospinal contribution to spasticity after SCI
Two of our findings support the view that spastic muscles have reduced corticospinal drive following incomplete chronic SCI. First, we found that the size of MEPs was decreased in spastic compared with non-spastic and control participants. MEPs elicited by TMS over the primary motor cortex provide an index of corticospinal excitability (Lemon et al., 1995; Petersen et al., 2003). Because MEP recruitment curves were tested during matched levels of voluntary activity, it is less likely that changes in the excitability of spinal motoneurons (Burke and Pierrot-Deseilligny, 2010) affected our results. Animal models of SCI showed that corticospinal neurons change their pattern of synaptic connectivity after the lesion (Kim et al., 2002; Ghosh et al., 2010). Multiple studies in humans with incomplete SCI revealed that corticospinal responses have different characteristics compared with those in uninjured controls, including decreased amplitude, longer latency, and higher threshold (Ellaway et al., 2007; Perez, 2012; Jo et al., 2018). Yet, little is known about the effect of spasticity on corticospinal output following SCI. Self-reported questionnaires and clinical examinations indicate that after SCI, which typically damages the corticospinal tract, most individuals develop symptoms of spasticity (Little et al., 1989; Maynard et al., 1990; Sköld et al., 1999; Holtz et al., 2017). A few studies also reported that individuals with incomplete SCI that do not take a medication to reduce spasticity are unable to modulate corticospinal responses, as is found in individuals that do take anti-spastic medication (Barry et al., 2013; Bunday et al., 2014), which is consistent with the view that corticospinal responses are affected when spasticity is present. This also agrees with recent results in humans with motor complete SCI showing that MEPs elicited by TMS were only present in spastic but not in non-spastic participants (Sangari et al., 2019). Second, we found that the magnitude of maximal voluntary output was decreased in spastic compared with non-spastic and control subjects. This is in agreement with results demonstrating that spastic muscles are weaker in stroke patients (Pasternak-Mladzka et al., 2007). A large number of participants with SCI take baclofen (Little et al., 1989; Maynard et al., 1990), a GABAB receptor agonist commonly used to decrease spasticity (Aydin et al., 2005; Roy and Edgerton, 2012). Conflicting results have been reported about the effect of baclofen on voluntary muscle output in humans with SCI, from having limited effects (Burke, 1975; Latash et al., 1990; Domingo et al., 2012) to decreasing contractile properties of motor units (Thomas et al., 2010). The similar results in physiological outcomes found in spastic and non-spastic participants taking or not taking anti-spastic medication, suggest that it is less likely that this factor affected our results.
We also found that spastic muscles showed increased reticulospinal gain following SCI. Inferences about the contribution of the reticulospinal tract in humans can be made by using the StartReact paradigm, which measures the shortening of a voluntary reaction time when a visual cue is paired with a startle sound (Valls-Solé et al., 1995). The shortening in reaction time likely involves subcortical structures, such as the reticular formation (Davis and Gendelman, 1977; Davis et al., 1982; Valls-Solé et al., 1999; Carlsen et al., 2003, 2004, 2009; Nonnekes et al., 2014). This test is sensitive to detect changes across tasks in humans with SCI (Baker and Perez, 2017). Notably, in our study, spastic SCI participants showed reduced corticospinal drive and enhanced reticulospinal gain. This agrees with findings in stroke patients showing that individuals with lower motor function and/or severe hemiparesis have increased reticulospinal output (Owen et al., 2017; McPherson et al., 2018; Choudhury et al., 2019). An important question is whether the lesser corticospinal and larger reticulospinal influences to a spastic muscle after SCI are related. In primates, reticulospinal connections to motoneurons innervating limb muscles are strengthened after a corticospinal lesion (Zaaimi et al., 2012). A possible interpretation of these results is that the reticulospinal pathway might compensate for the loss of corticospinal axons after injury (Pettersson et al., 2000; Zaaimi et al., 2012). This might be true after SCI considering the evidence suggesting that the reticulospinal tract has a greater capacity for regeneration than the corticospinal tract (Vavrek et al., 2007; Zörner et al., 2014). However, in this study (Zaaimi et al., 2012), monkeys did not develop spasticity, suggesting that this reorganization might not contribute to spasticity. Symptoms of spasticity might be related to damage (Tower, 1940; Murray and Goldberger, 1974; Paulson et al., 1986; Nathan, 1994; Sherman et al., 2000) and/or abnormalities (Owen et al., 2017) in the reticulospinal pathway. A cat model of spasticity induced by damage to the cerebral and cerebellar suppressor system showed that a second lesion, which interrupted reticulospinal projections, abolished spasticity (Schreiner et al., 1949). In addition, abnormal recruitment of cortico-reticulospinal networks is associated with flexion synergy expression in stroke patients (McPherson et al., 2018). Reorganization in brainstem pathways after SCI has been reported in animals (Pons et al., 1991) and in humans (Jankelowitz and Colebatch, 2004; Kumru et al., 2008, 2009). The corticospinal tract is more dorsally located than the reticulospinal tract in the lateral funiculus of the human spinal cord; however, most injuries will damage both pathways. Both injured and uninjured corticospinal (Fouad et al., 2001; Bareyre et al., 2004) and reticulospinal (Filli et al., 2014; May et al., 2017) axons can form connections with nearby target neurons of other undamaged tracts. Thus, we favor the hypothesis that parallel damage to both tracts contributes to the observed symptoms. Studies suggest that damage to premotor areas leads to progressive spasticity (Kennard and Fulton, 1933; Tower, 1940). Similar results have been reported in human after extensive motor and premotor cortectomies (Laplane et al., 1977). The premotor cortex is a main source of cortico-reticular projections (Yeo et al., 2012). In chronic injuries, motoneurons are supersensitive to very small amounts of 5-HT; thus, residual 5-HT below the injury may be sufficient to endogenously activate 5-HT receptors (Harvey et al., 2006a,b). This is also consistent with evidence showing that animals with spinal cord transection showed more spasticity when residual spinal brainstem-derived monoamines are present (Schmidt and Jordan, 2000).
Functional considerations
Although >60% of people with SCI develop symptoms of spasticity (Little et al., 1989; Maynard et al., 1990; Sköld et al., 1999; Holtz et al., 2017), to date its mechanisms of action remain incompletely understood. Here, we demonstrate, for the first time, that spastic muscles of individuals with chronic incomplete SCI receive imbalanced contributions from corticospinal and reticulospinal tracts, which are more pronounced in participants with lesser voluntary control. How might these findings affect motor output of spastic muscles after SCI? The corticospinal tract is a major descending pathway, contributing to the control of voluntary movement in mammals (Lemon, 2008), but it is possible that other descending motor tracts contribute to maximal efforts. An SAS can augment the magnitude of voluntary muscle contraction in control subjects and Parkinson's patients (Anzak et al., 2011a, b). In stroke patients, the recovery of strength might involve strengthening of non-corticospinal pathways, such as the reticulospinal tract (Xu et al., 2017). We argue that this may be different in spastic muscles of SCI participants because we observed smaller MVCs in the quadriceps muscle in people with higher reticulospinal gain. Even if reticulospinal outputs contribute to the small voluntary EMG generated during maximal efforts, this might be a small contribution because these participants were the weakest in our SCI population. We favor the interpretation that, after SCI, although both tracts might have interacting actions, these interactions might be reduced. The reticulospinal tract has been related to the development of movement synergies in the lower limb (Sánchez et al., 2017). The quadriceps femoris is the primary mover for knee extension, and each of the heads of the quadriceps femoris uniquely contributes to the control of force output (Ebersole et al., 2006). Undesired synergistic actions between the heads might limit coordinated voluntary control when spasticity is present. The engagement of descending motor tracts in humans with SCI can be enhanced by neuromodulation and training strategies that reduce spasticity (Kumru et al., 2010; Benito et al., 2012). However, it is also possible that the contribution of reticulospinal inputs to spastic muscles might be augmented by training or that reticulospinal inputs contribute to motor recovery without the presence of spasticity. Thus, a better understanding of the involvement for corticospinal and reticulospinal pathways to the control of spastic and non-spastic muscles can lead to more effective interventions to induce plasticity and motor recovery following SCI.
Footnotes
M.A.P. was supported by the National Institute of Neurological Disorders and Stroke and the Veterans Administration.
The authors declare no competing financial interests.
References
- Anzak A, Tan H, Pogosyan A, Brown P (2011a) Doing better than your best: loud auditory stimulation yields improvements in maximal voluntary force. Exp Brain Res 208:237–243. 10.1007/s00221-010-2474-1 [DOI] [PubMed] [Google Scholar]
- Anzak A, Tan H, Pogosyan A, Djamshidian A, Ling H, Lees A, Brown P (2011b) Improvements in rate of development and magnitude of force with intense auditory stimuli in patients with Parkinson's disease. Eur J Neurosci 34:124–132. 10.1111/j.1460-9568.2011.07735.x [DOI] [PubMed] [Google Scholar]
- Asboth L, Friedli L, Beauparlant J, Martinez-Gonzalez C, Anil S, Rey E, Baud L, Pidpruzhnykova G, Anderson MA, Shkorbatova P, Batti L, Pagès S, Kreider J, Schneider BL, Barraud Q, Courtine G (2018) Cortico-reticulo-spinal circuit reorganization enables functional recovery after severe spinal cord contusion. Nat Neurosci 21:576–588. 10.1038/s41593-018-0093-5 [DOI] [PubMed] [Google Scholar]
- Aydin G, Tomruk S, Keles I, Demir SO, Orkun S (2005) Transcutaneous electrical nerve stimulation versus baclofen in spasticity: clinical and electrophysiologic comparison. Am J Phys Med Rehabil 84:584–592. 10.1097/01.phm.0000171173.86312.69 [DOI] [PubMed] [Google Scholar]
- Baker SN, Perez MA (2017) Reticulospinal contributions to gross hand function after human spinal cord injury. J Neurosci 37:9778–9784. 10.1523/JNEUROSCI.3368-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SN, Zaaimi B, Fisher KM, Edgley SA, Soteropoulos DS (2015) Pathways mediating functional recovery. Prog Brain Res 218:389–412. 10.1016/bs.pbr.2014.12.010 [DOI] [PubMed] [Google Scholar]
- Ballermann M, Fouad K (2006) Spontaneous locomotor recovery in spinal cord injured rats is accompanied by anatomical plasticity of reticulospinal fibers. Eur J Neurosci 23:1988–1996. 10.1111/j.1460-9568.2006.04726.x [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME (2004) The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci 7:269–277. 10.1038/nn1195 [DOI] [PubMed] [Google Scholar]
- Barry MD, Bunday KL, Chen R, Perez MA (2013) Selective effects of baclofen on use-dependent modulation of GABAB inhibition after tetraplegia. J Neurosci 33:12898–12907. 10.1523/JNEUROSCI.1552-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito J, Kumru H, Murillo N, Costa U, Medina J, Tormos JM, Pascual-Leone A, Vidal J (2012) Motor and gait improvement in patients with incomplete spinal cord injury induced by high-frequency repetitive transcranial magnetic stimulation. Top Spinal Cord Inj Rehabil 18:106–112. 10.1310/sci1802-106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohannon RW, Smith MB (1987) Interrater reliability of a modified Ashworth Scale of Muscle Spasticity. Phys Ther 67:206–207. 10.1093/ptj/67.2.206 [DOI] [PubMed] [Google Scholar]
- Brown P, Rothwell JC, Thompson PD, Britton TC, Day BL, Marsden CD (1991) New observations on the normal auditory startle reflex in man. Brain 114:1891–1902. 10.1093/brain/114.4.1891 [DOI] [PubMed] [Google Scholar]
- Bunday KL, Tazoe T, Rothwell JC, Perez MA (2014) Subcortical control of precision grip after human spinal cord injury. J Neurosci 34:7341–7350. 10.1523/JNEUROSCI.0390-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke D, Pierrot-Deseilligny E (2010) Caveats when studying motor cortex excitability and the cortical control of movement using transcranial magnetic stimulation. Clin Neurophysiol 121:121–123. 10.1016/j.clinph.2009.10.009 [DOI] [PubMed] [Google Scholar]
- Burke DJ. (1975) An approach to the treatment of spasticity. Drugs 10:112–120. 10.2165/00003495-197510020-00002 [DOI] [PubMed] [Google Scholar]
- Cannon BW, Beaton LE, Ranson SW (1943) Nature of paresis following lateral cortico-spinal section in monkeys. J Neurophysiol 6:425–429. 10.1152/jn.1943.6.5.425 [DOI] [Google Scholar]
- Carlsen AN, Hunt MA, Inglis JT, Sanderson DJ, Chua R (2003) Altered triggering of a prepared movement by a startling stimulus. J Neurophysiol 89:1857–1863. 10.1152/jn.00852.2002 [DOI] [PubMed] [Google Scholar]
- Carlsen AN, Chua R, Inglis JT, Sanderson DJ, Franks IM (2004) Can prepared responses be stored subcortically? Exp Brain Res 159:301–309. 10.1007/s00221-004-1924-z [DOI] [PubMed] [Google Scholar]
- Carlsen AN, Chua R, Inglis JT, Sanderson DJ, Franks IM (2009) Differential effects of startle on reaction time for finger and arm movements. J Neurophysiol 101:306–314. 10.1152/jn.00878.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TJ, Riek S, Carson RG (2001) Reliability of the input–output properties of the cortico-spinal pathway obtained from transcranial magnetic and electrical stimulation. J Neurosci Methods 112:193–202. 10.1016/S0165-0270(01)00468-X [DOI] [PubMed] [Google Scholar]
- Chen B, Lorentzen J, Nielsen JB, Perez MA (2018) 387.04-Bilateral asymmetries in spasticity following spinal cord injury. Society for Neuroscience, San Diego, CA: Available at https://www.abstractsonline.com/pp8/#!/4649/presentation/19319. [Google Scholar]
- Choudhury S, Shobhana A, Singh R, Sen D, Anand SS, Shubham S, Baker MR, Kumar H, Baker SN (2019) The relationship between enhanced reticulospinal outflow and upper limb function in chronic stroke patients. Neurorehabil Neural Repair 33:375–383. 10.1177/1545968319836233 [DOI] [PubMed] [Google Scholar]
- Davis M, Gendelman PM (1977) Plasticity of the acoustic startle response in the acutely decerebrate rat. J Comp Physiol Psychol 91:549–563. 10.1037/h0077345 [DOI] [PubMed] [Google Scholar]
- Davis M, Gendelman DS, Tischler MD, Gendelman PM (1982) A primary acoustic startle circuit: lesion and stimulation studies. J Neurosci 2:791–805. 10.1523/JNEUROSCI.02-06-00791.1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Azevedo ER, Maria RM, Alonso KC, Cliquet A Jr (2015) Posture influence on the pendulum test of spasticity in patients with spinal cord injury. Artif Organs 39:1033–1037. 10.1111/aor.12500 [DOI] [PubMed] [Google Scholar]
- Devanne H, Lavoie BA, Capaday C (1997) Input–output properties and gain changes in the human corticospinal pathway. Exp Brain Res 114:329–338. 10.1007/PL00005641 [DOI] [PubMed] [Google Scholar]
- Domingo A, Al-Yahya AA, Asiri Y, Eng JJ, Lam T, Spinal Cord Injury Rehabilitation Evidence Research Team (2012) A systematic review of the effects of pharmacological agents on walking function in people with spinal cord injury. J Neurotrauma 29:865–879. 10.1089/neu.2011.2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersole KT, O'Connor KM, Wier AP (2006) Mechanomyographic and electromyographic responses to repeated concentric muscle actions of the quadriceps femoris. J Electromyogr Kinesiol 16:149–157. 10.1016/j.jelekin.2005.05.005 [DOI] [PubMed] [Google Scholar]
- Ellaway PH, Catley M, Davey NJ, Kuppuswamy A, Strutton P, Frankel HL, Jamous A, Savic G (2007) Review of physiological motor outcome measures in spinal cord injury using transcranial magnetic stimulation and spinal reflexes. J Rehabil Res Dev 44:69–76. 10.1682/JRRD.2005.08.0140 [DOI] [PubMed] [Google Scholar]
- Filli L, Engmann AK, Zörner B, Weinmann O, Moraitis T, Gullo M, Kasper H, Schneider R, Schwab ME (2014) Bridging the gap: a reticulo-propriospinal detour bypassing an incomplete spinal cord injury. J Neurosci 34:13399–13410. 10.1523/JNEUROSCI.0701-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher KM, Chinnery PF, Baker SN, Baker MR (2013) Enhanced reticulospinal output in patients with (REEP1) hereditary spastic paraplegia type 31. J Neurol 260:3182–3184. 10.1007/s00415-013-7178-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouad K, Pedersen V, Schwab ME, Brösamle C (2001) Cervical sprouting of corticospinal fibers after thoracic spinal cord injury accompanies shifts in evoked motor responses. Curr Biol 11:1766–1770. 10.1016/S0960-9822(01)00535-8 [DOI] [PubMed] [Google Scholar]
- Frigon A, Rossignol S (2006) Functional plasticity following spinal cord lesions. Prog Brain Res 157:231–260. 10.1016/S0079-6123(06)57016-5 [DOI] [PubMed] [Google Scholar]
- Ghosh A, Haiss F, Sydekum E, Schneider R, Gullo M, Wyss MT, Mueggler T, Baltes C, Rudin M, Weber B, Schwab ME (2010) Rewiring of hindlimb corticospinal neurons after spinal cord injury. Nat Neurosci 13:97–104. 10.1038/nn.2448 [DOI] [PubMed] [Google Scholar]
- Harvey PJ, Li X, Li Y, Bennett DJ (2006a) Endogenous monoamine receptor activation is essential for enabling persistent sodium currents and repetitive firing in rat spinal motoneurons. J Neurophysiol 96:1171–1186. 10.1152/jn.00341.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey PJ, Li X, Li Y, Bennett DJ (2006b) 5-HT2 receptor activation facilitates a persistent sodium current and repetitive firing in spinal motoneurons of rats with and without chronic spinal cord injury. J Neurophysiol 96:1158–1170. 10.1152/jn.01088.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtz KA, Lipson R, Noonan VK, Kwon BK, Mills PB (2017) Prevalence and effect of problematic spasticity after traumatic spinal cord injury. Arch Phys Med Rehabil 98:1132–1138. 10.1016/j.apmr.2016.09.124 [DOI] [PubMed] [Google Scholar]
- Jankelowitz SK, Colebatch JG (2004) The acoustic startle reflex in ischemic stroke. Neurology 62:114–116. 10.1212/01.WNL.0000101711.48946.35 [DOI] [PubMed] [Google Scholar]
- Jankowska E, Edgley SA (2006) How can corticospinal tract neurons contribute to ipsilateral movements? A question with implications for recovery of motor functions. Neuroscientist 12:67–79. 10.1177/1073858405283392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo HJ, Di Lazzaro V, Perez MA (2018) Effect of coil orientation on motor-evoked potentials in humans with tetraplegia. J Physiol 596:4909–4921. 10.1113/JP275798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennard MA, Fulton JF (1933) The localizing significance of spasticity, reflex grasping, and the signs of Babinski and Rossolimo. Brain 56:213–225. 10.1093/brain/56.2.213 [DOI] [Google Scholar]
- Kim ES, Kim GM, Lu X, Hsu CY, Xu XM (2002) Neural circuitry of the adult rat central nervous system after spinal cord injury: a study using fast blue and the bartha strain of pseudorabies virus. J Neurotrauma 19:787–800. 10.1089/08977150260139156 [DOI] [PubMed] [Google Scholar]
- Kumru H, Vidal J, Kofler M, Benito J, Garcia A, Valls-Solé J (2008) Exaggerated auditory startle responses in patients with spinal cord injury. J Neurol 255:703–709. 10.1007/s00415-008-0780-3 [DOI] [PubMed] [Google Scholar]
- Kumru H, Kofler M, Valls-Solé J, Portell E, Vidal J (2009) Brainstem reflexes are enhanced following severe spinal cord injury and reduced by continuous intrathecal baclofen. Neurorehabil Neural Repair 23:921–927. 10.1177/1545968309335979 [DOI] [PubMed] [Google Scholar]
- Kumru H, Murillo N, Samso JV, Valls-Solé J, Edwards D, Pelayo R, Valero-Cabre A, Tormos JM, Pascual-Leone A (2010) Reduction of spasticity with repetitive transcranial magnetic stimulation in patients with spinal cord injury. Neurorehabil Neural Repair 24:435–441. 10.1177/1545968309356095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplane D, Talairach J, Meininger V, Bancaud J, Bouchareine A (1977) Motor consequences of motor area ablations in man. J Neurol Sci 31:29–49. 10.1016/0022-510X(77)90004-1 [DOI] [PubMed] [Google Scholar]
- Latash ML, Penn RD, Corcos DM, Gottlieb GL (1990) Effects of intrathecal baclofen on voluntary motor control in spastic paresis. J Neurosurg 72:388–392. 10.3171/jns.1990.72.3.0388 [DOI] [PubMed] [Google Scholar]
- Lee D, Song J, Kim JW, Ahn TB (2016) Spasticity secondary to isolated involvement of the pyramidal tract. J Neurol Sci 368:130–131. 10.1016/j.jns.2016.06.072 [DOI] [PubMed] [Google Scholar]
- Lemon RN. (2008) Descending pathways in motor control. Annu Rev Neurosci 31:195–218. 10.1146/annurev.neuro.31.060407.125547 [DOI] [PubMed] [Google Scholar]
- Lemon RN, Johansson RS, Westling G (1995) Corticospinal control during reach, grasp, and precision lift in man. J Neurosci 15:6145–6156. 10.1523/JNEUROSCI.15-09-06145.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Chang SH, Francisco GE, Verduzco-Gutierrez M (2014) Acoustic startle reflex in patients with chronic stroke at different stages of motor recovery: a pilot study. Topics Stroke Rehabil 21:358–370. 10.1310/tsr2104-358 [DOI] [PubMed] [Google Scholar]
- Little JW, Micklesen P, Umlauf R, Britell C (1989) Lower extremity manifestations of spasticity in chronic spinal cord injury. Am J Phys Med Rehabil 68:32–36. 10.1097/00002060-198902000-00009 [DOI] [PubMed] [Google Scholar]
- May Z, Fenrich KK, Dahlby J, Batty NJ, Torres-Espín A, Fouad K (2017) Following spinal cord injury transected reticulospinal tract axons develop new collateral inputs to spinal interneurons in parallel with locomotor recovery. Neural Plast 2017:1–15. 10.1155/2017/1932875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard FM, Karunas RS, Waring WP 3rd (1990) Epidemiology of spasticity following traumatic spinal cord injury. Arch Phys Med Rehabil 71:566–569. [PubMed] [Google Scholar]
- McPherson JG, Chen A, Ellis MD, Yao J, Heckman CJ, Dewald JP (2018) Progressive recruitment of contralesional cortico-reticulospinal pathways drives motor impairment post stroke. J Physiol 596:1211–1225. 10.1113/JP274968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray M, Goldberger ME (1974) Restitution of function and collateral sprouting in the cat spinal cord: the partially hemisected animal. J Comp Neurol 158:19–36. 10.1002/cne.901580103 [DOI] [PubMed] [Google Scholar]
- Nathan PW. (1994) Effects on movement of surgical incisions into the human spinal cord. Brain 117:337–346. 10.1093/brain/117.2.337 [DOI] [PubMed] [Google Scholar]
- Nonnekes J, Oude Nijhuis LB, de Niet M, de Bot ST, Pasman JW, van de Warrenburg BP, Bloem BR, Weerdesteyn V, Geurts AC (2014) StartReact restores reaction time in HSP: evidence for subcortical release of a motor program. J Neurosci 34:275–281. 10.1523/JNEUROSCI.2948-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oudega M, Perez MA (2012) Corticospinal reorganization after spinal cord injury. J Physiol 590:3647–3663. 10.1113/jphysiol.2012.233189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen M, Ingo C, Dewald JP (2017) Upper extremity motor impairments and microstructural changes in bulbospinal pathways in chronic hemiparetic stroke. Front Neurol 8:257. 10.3389/fneur.2017.00257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternak-Mladzka I, Mladzki Z, Bedzinski R, Baran B (2007) Objective measurements of muscle force in a group of after-stroke patients with hemiparesis. Acta Bioeng Biomech 9:19–23. [PubMed] [Google Scholar]
- Paulson GW, Yates AJ, Paltan-Ortiz JD (1986) Does infarction of the medullary pyramid lead to spasticity? Arch Neurol 43:93–95. 10.1001/archneur.1986.00520010087030 [DOI] [PubMed] [Google Scholar]
- Perez MA. (2012) Transcranial magnetic stimulation and spinal cord injury. In: Cortical connectivity: brain stimulation for assessing and modulating cortical connectivity and function (Chen R, Rothwell JC, eds), pp 323–336. Berlin: Springer. [Google Scholar]
- Petersen NT, Pyndt HS, Nielsen JB (2003) Investigating human motor control by transcranial magnetic stimulation. Exp Brain Res 152:1–16. 10.1007/s00221-003-1537-y [DOI] [PubMed] [Google Scholar]
- Pettersson LG, Blagovechtchenski E, Perfiliev S, Krasnochokova E, Lundberg A (2000) Recovery of food-taking in cats after lesions of the corticospinal (complete) and rubrospinal (complete and incomplete) tracts. Neurosci Res 38:109–112. 10.1016/S0168-0102(00)00143-7 [DOI] [PubMed] [Google Scholar]
- Pons TP, Garraghty PE, Ommaya AK, Kaas JH, Taub E, Mishkin M (1991) Massive cortical reorganization after sensory deafferentation in adult macaques. Science 252:1857–1860. 10.1126/science.1843843 [DOI] [PubMed] [Google Scholar]
- Roy RR, Edgerton VR (2012) Neurobiological perspective of spasticity as occurs after a spinal cord injury. Exp Neurol 235:116–122. 10.1016/j.expneurol.2012.01.017 [DOI] [PubMed] [Google Scholar]
- Sánchez N, Acosta AM, Lopez-Rosado R, Stienen AH, Dewald JP (2017) Lower extremity motor impairments in ambulatory chronic hemiparetic stroke: evidence for lower extremity weakness and abnormal muscle and joint torque coupling patterns. Neurorehabil Neural Repair 31:814–826. 10.1177/1545968317721974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangari S, Lundell H, Kirshblum S, Perez MA (2019) Residual descending motor pathways influence spasticity after spinal cord injury. Ann Neurol 86:28–41. 10.1002/ana.25505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt BJ, Jordan LM (2000) The role of serotonin in reflex modulation and locomotor rhythm production in the mammalian spinal cord. Brain Res Bull 53:689–710. 10.1016/S0361-9230(00)00402-0 [DOI] [PubMed] [Google Scholar]
- Schreiner LH, Lindsley DB, Magoun HW (1949) Role of brain stem facilitatory systems in maintenance of spasticity. J Neurophysiol 12:207–216. 10.1152/jn.1949.12.3.207 [DOI] [PubMed] [Google Scholar]
- Schucht P, Raineteau O, Schwab ME, Fouad K (2002) Anatomical correlates of locomotor recovery following dorsal and ventral lesions of the rat spinal cord. Exp Neurol 176:143–153. 10.1006/exnr.2002.7909 [DOI] [PubMed] [Google Scholar]
- Sherman SJ, Koshland GF, Laguna JF (2000) Hyper-reflexia without spasticity after unilateral infarct of the medullary pyramid. J Neurol Sci 175:145–155. 10.1016/S0022-510X(00)00299-9 [DOI] [PubMed] [Google Scholar]
- Sköld C, Levi R, Seiger A (1999) Spasticity after traumatic spinal cord injury: nature, severity, and location. Arch Phys Med Rehabil 80:1548–1557. 10.1016/S0003-9993(99)90329-5 [DOI] [PubMed] [Google Scholar]
- Thomas CK, Häger-Ross CK, Klein CS (2010) Effects of baclofen on motor units paralysed by chronic cervical spinal cord injury. Brain 133:117–125. 10.1093/brain/awp285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower SS. (1940) Pyramidal lesion in the monkey. Brain 63:36–90. 10.1093/brain/63.1.36 [DOI] [Google Scholar]
- Trompetto C, Marinelli L, Mori L, Pelosin E, Currà A, Molfetta L, Abbruzzese G (2014) Pathophysiology of spasticity: implications for neurorehabilitation. Biomed Res Int 2014:354906. 10.1155/2014/354906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valls-Solé J, Solé A, Valldeoriola F, Muñoz E, Gonzalez LE, Tolosa ES (1995) Reaction time and acoustic startle in normal human subjects. Neurosci Lett 195:97–100. 10.1016/0304-3940(94)11790-P [DOI] [PubMed] [Google Scholar]
- Valls-Solé J, Rothwell JC, Goulart F, Cossu G, Muñoz E (1999) Patterned ballistic movements triggered by a startle in healthy humans. J Physiol 516:931–938. 10.1111/j.1469-7793.1999.0931u.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vavrek R, Pearse DD, Fouad K (2007) Neuronal populations capable of regeneration following a combined treatment in rats with spinal cord transection. J Neurotrauma 24:1667–1673. 10.1089/neu.2007.0290 [DOI] [PubMed] [Google Scholar]
- Xu J, Ejaz N, Hertler B, Branscheidt M, Widmer M, Faria AV, Harran MD, Cortes JC, Kim N, Celnik PA, Kitago T, Luft AR, Krakauer JW, Diedrichsen J (2017) Separable systems for recovery of finger strength and control after stroke. J Neurophysiol 118:1151–1163. 10.1152/jn.00123.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo SS, Chang MC, Kwon YH, Jung YJ, Jang SH (2012) Corticoreticular pathway in the human brain: diffusion tensor tractography study. Neurosci Lett 508:9–12. 10.1016/j.neulet.2011.11.030 [DOI] [PubMed] [Google Scholar]
- Zaaimi B, Edgley SA, Soteropoulos DS, Baker SN (2012) Changes in descending motor pathway connectivity after corticospinal tract lesion in macaque monkey. Brain 135:2277–2289. 10.1093/brain/aws115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaaimi B, Soteropoulos DS, Fisher KM, Riddle CN, Baker SN (2018) Classification of neurons in the primate reticular formation and changes after recovery from pyramidal tract lesion. J Neurosci 38:6190–6206. 10.1523/JNEUROSCI.3371-17.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zörner B, Bachmann LC, Filli L, Kapitza S, Gullo M, Bolliger M, Starkey ML, Röthlisberger M, Gonzenbach RR, Schwab ME (2014) Chasing central nervous system plasticity: the brainstem's contribution to locomotor recovery in rats with spinal cord injury. Brain 137:1716–1732. 10.1093/brain/awu078 [DOI] [PubMed] [Google Scholar]