

Abstract
Alzheimer's disease (AD) is associated with the cleavage of the amyloid precursor protein (APP) to produce the toxic amyloid-β (Aβ) peptide. Accumulation of Aβ, together with the concomitant inflammatory response, ultimately leads to neuronal death and cognitive decline. Despite AD progression being underpinned by both neuronal and immunological components, therapeutic strategies based on dual targeting of these systems remains unexplored. Here, we report that inactivation of the p110δ isoform of phosphoinositide 3-kinase (PI3K) reduces anterograde axonal trafficking of APP in hippocampal neurons and dampens secretion of the inflammatory cytokine tumor necrosis factor-alpha by microglial cells in the familial AD APPswe/PS1ΔE9 (APP/PS1) mouse model. Moreover, APP/PS1 mice with kinase-inactive PI3Kδ (δD910A) had reduced Aβ peptides levels and plaques in the brain and an abrogated inflammatory response compared with APP/PS1 littermates. Mechanistic investigations reveal that PI3Kδ inhibition decreases the axonal transport of APP by eliciting the formation of highly elongated tubular-shaped APP-containing carriers, reducing the levels of secreted Aβ peptide. Importantly, APP/PS1/δD910A mice exhibited no spatial learning or memory deficits. Our data highlight inhibition of PI3Kδ as a new approach to protect against AD pathology due to its dual action of dampening microglial-dependent neuroinflammation and reducing plaque burden by inhibition of neuronal APP trafficking and processing.
SIGNIFICANCE STATEMENT During Alzheimer's disease (AD), the accumulation of the toxic amyloid-β (Aβ) peptide in plaques is associated with a chronic excessive inflammatory response. Uncovering new drug targets that simultaneously reduce both Aβ plaque load and neuroinflammation holds therapeutic promise. Using a combination of genetic and pharmacological approaches, we found that the p110δ isoform of phosphoinositide 3-kinase (PI3K) is involved in anterograde trafficking of the amyloid precursor protein in neurons and in the secretion of tumor necrosis factor-alpha from microglial cells. Genetic inactivation of PI3Kδ reduces Aβ plaque deposition and abrogates the inflammatory response, resulting in a complete rescue of the life span and spatial memory performance. We conclude that inhibiting PI3Kδ represents a novel therapeutic approach to ameliorate AD pathology by dampening plaque accumulation and microglial-dependent neuroinflammation.
Keywords: Alzheimer's disease, amyloid precursor protein, axon trafficking, neuroinflammation, p110delta, TNF-alpha
Introduction
Alzheimer's disease (AD) is characterized by the accumulation of toxic amyloid-β (Aβ) peptide, which leads to synaptic dysfunction, axonal degeneration, and, ultimately, neuronal death (Selkoe, 2002). Two complementary systems play essential roles in the pathogenesis and progression of AD: neuronal dysfunction and inflammation.
The neuronal aspect involves the aggregation of secreted Aβ into fibrils and its deposition in amyloid plaques, one of the main pathological hallmarks of AD (Dickson, 1997). Although the primary event that results in the abnormal accumulation of Aβ is thought to be a dysregulated proteolytic processing of the amyloid precursor protein (APP), the specific cellular location of APP cleavage and Aβ release remains unclear. Endogenous APP is present, however, in presynaptic vesicles that undergo stimulation-dependent exo-endocytic recycling (Groemer et al., 2011), and axonal tracks have been shown to be crucial compartments in the production of Aβ (Stokin et al., 2005).
The immune system also plays a significant role in AD progression. Indeed, the development of amyloid plaques is associated with glial activation (i.e., hemopoietic-derived astrocytes and microglia) and the secretion of proinflammatory cytokines by these cells (Wyss-Coray et al., 2001). Although it is thought that the inflammatory response has a beneficial impact in dampening the early stages of disease progression, chronic neuroinflammation and the associated production of proinflammatory cytokines ultimately lead to neuronal death (Mandrekar-Colucci and Landreth, 2010). One of the earliest proinflammatory cytokines released from microglia is tumor necrosis factor-alpha (TNFα). Excessive secretion of TNFα as a result of neuroinflammation causes neuronal apoptosis, which is also observed during AD (Collins et al., 2000). Furthermore, elevated TNFα levels promote Aβ production (Liao et al., 2004), thereby driving a vicious cycle of chronic neuroinflammation and disease progression. Blocking TNFα signaling therefore has the potential to decrease Aβ-associated neuroinflammation to attenuate AD progression. However, undesired secondary effects have been observed when the downstream signaling of the TNFα pathway is completely inhibited (Montgomery et al., 2013). Blocking the release of excessive pathogenic TNFα while maintaining homeostatic basal pools of this cytokine is therefore considered a more cautious and pragmatic therapeutic approach to control neuroinflammation in conditions such as AD or stroke (Low et al., 2014).
Phosphoinositide 3-kinases (PI3Ks) are modulators of cellular membrane lipids involved in signaling and trafficking events. In the brain, PI3Kδ is expressed in neurons and microglia (Eickholt et al., 2007; Low et al., 2014; Hood et al., 2019). Despite its established role in controlling functions in autoimmunity and inflammation (Okkenhaug and Vanhaesebroeck, 2003; Ali et al., 2014), the role of PI3Kδ in neurons remains largely unknown, with the effects of inhibiting this isoform in the context of neurological disorders also remaining unexplored.
In this study, we crossed the double-transgenic APP/PS1 mouse model of AD (Jankowsky et al., 2004) with the PI3K-p110δD910A mouse line (hereafter referred to as δD910A), in which the endogenous PI3Kδ is rendered kinase-dead via the germline introduction of a point mutation in its ATP-binding site (Okkenhaug et al., 2002), to generate the APP/PS1/δD910A composite strain. Using genetic and pharmacological approaches in vitro and in vivo, we then investigated the effect of inhibiting PI3Kδ on the trafficking of APP in neurons and the development of AD neuropathology in APP/PS1 and APP/PS1/δD910A mice. Our results highlight novel roles for PI3Kδ in mediating the accumulation of Aβ plaques and the concomitant chronic inflammation, opening potential therapeutic opportunities to interfere with AD progression through the targeting of PI3Kδ activity.
Materials and Methods
Animals.
APP/PS1 transgenic mice (Jax Mice Database strain B6C3 Tg (APPswe, PSEN1dE9) 85Dbo/Mmjax; stock #34829-JAX | APP/PS1) express human APP695 with Swedish mutations (K595N and M596L) and mutant human presenilin 1 carrying the exon-9-deleted variant (Jankowsky et al., 2004). Hemizygous APP/PS1 transgenic males aged 6.5 months were used, together with age-matched nontransgenic littermate controls. Only male mice were used in these studies as gender differences in plaque deposition for this transgenic line have been reported (Burgess et al., 2006). The δD910A kinase-dead mice were generated by a germline knock-in mutation in the ATP-binding site of p110δ, as described previously (Okkenhaug et al., 2002). Double transgenic mice were derived from APP/PS1 × δD910A and APP/PS1/δD910A/WT × δD910A/D910A crosses. DNA was extracted from toe samples of each mouse and appropriately digested and genotyped as previously described (Okkenhaug et al., 2002). Mice were housed in a climate-controlled room on a 12 h light/dark with access to standard chow and water ad libitum. For each genotypic cohort, animals were age-matched based on their week of birth. All experiments were approved by the Animal Ethics Committee at the University of Queensland and conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Primary neuronal cultures.
Hippocampal neurons were cultured from embryonic day 16 (E16) mice. For live-imaging, neurons were plated onto 35 mm glass-bottom dishes (Cellvis). For immunocytochemistry, neurons were plated onto 12 mm coverslips (Thermo Scientific). In both situations, the glass surfaces were coated with poly-l-lysine (Sigma-Aldrich). Primary neuronal culture was performed as described previously (Joensuu et al., 2017). Briefly, the dissected hippocampi were digested with 0.25% trypsin-EDTA and DNase I for 20 min at 37°C, followed by mechanical trituration. Neurons were then plated in “neuronal plating medium” (Neurobasal medium supplemented with penicillin/streptomycin, 1% Glutamax, and 10% FBS), and 4 h later the medium was changed to “neuronal culture medium” (Neurobasal medium supplemented with penicillin/streptomycin, 1% Glutamax, and 2% B27). The neurons were transfected after 12 d in vitro (DIV12) using the Lipofectamine 2000 (Invitrogen) reagent, with experiments being performed at DIV14–18.
Plasmids.
APP695-GFP was a generous gift from Dr. Olav Andersen (Aarhus Universitet) (Andersen et al., 2005; Schmidt et al., 2007). RFP-fused vesicular stomatitis virus membrane glycoprotein (VSVG-RFP) was a generous gift from J.L. Daniotti (Universidad Nacional de Córdoba, Ciudad Universitaria, X5000 HUA Córdoba, Argentina; Keller et al., 2001).
Reagents.
CAL-101 was purchased from Active Biochem. A methoxypyridyl substituted quinazoline analogous to Leniolisib (CDZ173) (Compound 12) was synthesized and provided by the Queensland Emory Drug Discovery Initiative (QEDDI, Australia) based on Hoegenauer et al. (2016). Both compounds were dissolved in DMSO (Sigma-Aldrich).
Live imaging of APP-GFP anterograde carriers.
Treatment and live imaging of APP-GFP anterograde carriers was performed on hippocampal neurons at DIV14–18 using a Roper iLas2 microscope (Roper Scientific) (100X). CAL-101 or DMSO (0.1%) was added to the neurons, after which they were immediately returned to the incubator for 1 h 45 min. Following incubation, the medium was replaced with prewarmed imaging medium (0.5 mm MgCl2, 2.2 mm CaCl2, 5.6 mm KCl, 145 mm NaCl, 5.6 mm d-glucose, 0.5 mm ascorbic acid, 0.1% w/v BSA and 15 mm HEPES, pH 7.4). Under the microscope, five axons from five different neurons transfected with APP-GFP were selected and each position was saved using the Multidemensional Acquisition App of MetaMorph software (Molecular Devices). This took ∼15 min. Images were then immediately acquired (2500 frames every 100 ms). Kymographs were generated for the resulting images using FIJI-ImageJ (Schindelin et al., 2012) and the plugins Multi-Kymograph and Trackmate (Tinevez et al., 2017).
Immunofluorescence staining.
For double immunofluorescence labeling of brain tissue, 40 μm thickness floating sections were blocked with 5% normal horse serum (NHS) and 0.1% Triton X-100 in phosphate buffer saline (PBS) for 1 h. To immunostain microglia and astrocytes, sections were then incubated with anti-Iba1 (1:500, Abcam) and anti-GFAP (1:500, Abcam) primary antibodies, respectively, in blocking solution overnight at 4°C. After washing, labeling was visualized by incubating the sections with an appropriate mixture of secondary antibodies (Alexa Fluor 488-conjugated donkey anti-rabbit and Alexa Fluor 647-conjugated donkey anti-goat, respectively; 1:1000; Invitrogen); cell nuclei were stained with 4′,6-diamindino-2-phenylindole dihydrochloride (DAPI, 1:5000, Sigma-Aldrich). Sections were then washed, mounted onto Superfrost Plus slides (Lomb Scientific) and coverslipped with fluorescence mounting medium (Dako).
For immunofluorescence labeling of primary cultures, neurons were fixed during 10 min with PBS containing 4% paraformaldehyde (PFA) and 4% sucrose, permeabilized for 10 min with PBS containing 0.1% Triton X-100 and blocked with PBS containing 5% NHS and 1% BSA for 1 h. Primary antibodies anti-APP (1:500, Abcam), anti MAP2 (1:1000, Synaptic Systems), anti-NF (1:2000, BioLegend), and anti-GM130 (1:500, BD Transduction Laboratories) were diluted in PBS containing 3% BSA and incubated overnight at 4°C. After washing, labeling was visualized by incubating the neurons with an appropriate mixture of secondary antibodies (Alexa Fluor 488-conjugated donkey anti-rabbit and Alexa Fluor 647-conjugated donkey anti-goat, respectively; 1:1000; Invitrogen) diluted into PBS containing 5% NHS for 1 h. Neurons were mounted with fluorescent mounting medium (Dako).
Thioflavin S staining.
For Thioflavin S staining, 40-μm-thick floating sections were mounted on slides and left to dry overnight. The following day, the slides were washed in distilled water, incubated in the dark with twice-filtered 0.1% Thioflavin S (Sigma-Aldrich) solution for 5 min and then washed with 70% ethanol followed by distilled water to remove excess stain. Finally, the slides were left to dry and then mounted in fluorescent mounting media (Dako).
Image acquisition, processing, and quantification.
The imaging for immunofluorescence microscopy of fixed primary neurons was performed on a Zeiss LSM 510 inverted confocal microscope (63x) and analyzed with FIJI-ImageJ (Schindelin et al., 2012). For the analysis, 30 to 40 fields (142.86 μm × 142.86 μm) each one containing 1 to 2 somas were selected from 2 technical replicate dishes, and 3 independent cultures were used per analysis. The morphology of the axon was derived from masks using the neurofilament channel, and the fluorescence intensity of the APP signal within the masked regions was measured using FIJI-ImageJ. The morphology of the soma was derived from masks using the MAP2 channel, and the degree of colocalization between the APP signal and GM130 signal was measured using the JACoP plug-in of FIJI-ImageJ.
Fluorescence images of labeled brain slices were acquired using an upright Axio Imager microscope equipped with an AxioCam HRc digital camera and AxioVision Microscopy software (Z1; Carl Zeiss). High-resolution mosaic images were compiled from Z-stack images obtained using a 20x objective (4 optical sections 2 μm thick) of the cortex and hippocampus. All images were transposed into a mean intensity projection using the Zeiss AxioVision Microscopy software.
To quantify Aβ plaques, the images were opened in FIJI-ImageJ and the number of plaques in the brain regions of interest (hippocampus and cortex) was counted manually. The boundary of the hippocampus was determined by anatomical landmarks delineated using a brain atlas (Franklin and Paxinos, 2013). To determine the total area covered by plaques, FIJI-ImageJ was used to set an intensity threshold to encapsulate the area of Thioflavin S staining. The number of plaques and the area covered by plaques was averaged across three representative sections per brain at the level of the hippocampus and cortex. The results are expressed as the mean and the SEM for four brains analyzed for each cohort of mice.
To analyze the brain infiltration of astrocytes and microglia, a Z stack of the hippocampus and cortex containing markers for astrocytes (GFAP) and microglia (Iba1) was rendered into 3D using Imaris 7.5 software (Bitplane Scientific Software). The total number of voxels occupied by GFAP or Iba1 staining was modeled by creating an automatic isosurface. The volume occupied by GFAP or Iba1 staining was normalized to the volume of the hippocampal or cortical region. This volume was then averaged across three representative sections per brain at the level of the hippocampus and cortex. The results are expressed as the mean and the SEM of four brains analyzed for each cohort of mice.
Tissue lysis and protein quantification.
Animals were culled by cervical dislocation. The brain was immediately dissected to isolate the cortex and the hippocampus of both hemispheres. To extract the proteins, the dissected tissue was homogenized in ice-cold lysis buffer (20 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 1% Nonidet P-40, Sigma-Aldrich) containing complete protease inhibitors (Roche). Brain tissue homogenates were then centrifuged at 15,000 rpm for 15 min at 4°C. The total protein concentration was assessed for each supernatant using the Pierce Bicinchoninic Acid (BCA) Protein Assay Kit (Thermo Scientific) before SDS-PAGE protein separation and analysis by Western blotting.
Analysis of protein expression.
Western blotting was used to assess the protein expression levels of TNFα, APP, Aβ40/42, PSD-95, and Synapsin-1 in the cortex of mice. Samples were prepared by mixing 20 μg of protein supernatant with sample buffer (4× NuPAGE® LDS sample buffer, Invitrogen) and dithiothreitol, followed by denaturing of the sample at 95°C for 10 min. Proteins were separated on a NuPAGE® 12% Bis-Tris gel (Invitrogen) for TNFα, or on a 4–12% Novex Bis-Tris gel (Life Technologies) for APP and Aβ40/42, at 140V for 90 min using the Invitrogen XCell SureLock Gel electrophoresis system with 1x NuPAGE 3-(N-morpholino) propane sulfonic acid SDS running buffer (Invitrogen). After separation, proteins were transferred onto a polyvinylidene difluoride membrane at 90 V for 90 min submerged in Tris/glycine transfer buffer. The membrane was then blocked in 5% w/v Diploma skim milk powder in PBS containing 0.1% Tween-20 (PBST) for 1 h at room temperature. Membranes were incubated on a rotator overnight at 4°C in TNFα primary antibody (1:1000, Abcam) and β-3-tubulin for a loading control (1:1000, Calbiochem); 6E10 antibody (1:2000, BioLegend) and GAPDH for a loading control (1:4000, Cell Signaling Technology); or PSD-95 antibody (1:1000, Santa Cruz Biotechnology), Synapsin-1 antibody (1:1000, Synaptic Systems) and GAPDH for a loading control (1:4000, Cell Signaling Technology). After 3 × 15 min washes in PBST, the membrane was incubated with the appropriate secondary antibodies in blocking solution (680 Infrared anti-mouse and 800 Infrared anti-rabbit, LI-COR Biosciences). After three washes, the membranes were imaged using the Odyssey Infrared Imaging System (LI-COR Biosciences). Densitometry analysis was conducted using the Odyssey Fc Imaging System and Image StudioTM (LI-COR Biosciences) and FIJI-ImageJ software was used to quantify changes in protein expression.
Measurement of Aβ secretion and metabolic rate in primary neuronal cultures.
These experiments were performed independently by QPS Austria. Primary cortical mouse neurons (E18) were isolated using standard protocols and seeded into 24-well (MSD assay; 3 × 105 neurons per well) and 96-well plates (MTT assay; 5 × 104 neurons per well) previously coated with poly-d-lysine. Neurons were cultivated for 7 d in the presence of the test compound (CAL-101) or vehicle control (DMSO). At DIV4 and DIV6, half of the medium was replaced with fresh culture medium containing CAL-101 or vehicle (culture medium/0.1% DMSO). On DIV8 (chronic), supernatants were collected and cells were lysed in RIPA buffer (150 mm NaCl, 50 mm Tris-HCl pH 7.4, 1 mm EDTA, 1% Nonidet P40, 0.25% Natriumdeoxycholat, 0.1% SDS, Protease Inhibitor Cocktail I, Calbiochem). Secreted Aβ38, 40 and 42 and intracellular Aβ38, 40 and 42 levels were measured using a commercially available immunosorbent Meso Scale Discovery (MSD) assay kit from Mesoscale Diagnostics, applying 4G8 antibody. The immune assay was performed according to the manual and plates were read on the Sector Imager 2400. Analyte levels were evaluated according to adequate Aβ peptide standards (MSD) as picograms per milliliter and as absolute signals. Furthermore, the effects of prolonged exposure to the test compound on cell viability and metabolic rate were determined using the MTT assay. MTT solution was added to each well at a final concentration of 0.5 mg/ml. After 2 h, the MTT-containing medium was aspirated. Cells were lyzed in 3% SDS and the formazan crystals were dissolved in isopropanol/HCl. The optical density (OD) was measured with a plate-reader at wavelength 570 nm. The metabolic rate was expressed as the OD. Values were calculated as a percentage of the vehicle control. These experiments were performed in five (acute) or six (chronic) technical replicates and repeated two independent times. Group differences were evaluated by one-way ANOVA followed by Bonferroni's post hoc analysis, comparing all treatment groups to the vehicle control group.
Measurement of Aβ42 from mouse brains.
Males and females mice from APP/PS1 and APP/PS1/δD910A genotypes and 5–7 months old were anesthetized and transcardially perfused with PBS. The cortex was dissected, weighed, and snap frozen with liquid nitrogen. Tissue lysates were prepared by homogenization in 10 volume (v/w) of RIPA buffer (containing Roche cOmplete protease inhibitor cocktail and PhosSTOP phosphatase inhibitor cocktail and 1 mm AEBSF) using Precellys Evolution homogenizer (Bertin Technologies). After homogenization, the lysate containing soluble Aβ42 was collected at 100,000 × g for 20 min at 4°C. To determine the level of total Aβ42 in the brain (containing fibrillary Aβ42), the homogenate was mixed with 8.2 m guanidine/82 mm Tris-HCl, pH 8.0, and then incubated for 4 h at room temperature. After diluting the homogenate with cold BSAT-DPBS buffer (0.2 g/L KCl, 0.2 g/L KH2PO4, 8.0 g/L NaCl, 1.150 g/L Na2HPO4, 5% BSA and 0.03% Tween-20, pH 7.4) containing protease inhibitor cocktails, the supernatant was collected after centrifugation at 16,000 × g for 20 min at 4°C. The level of Aβ42 in the tissue was assessed by an Amyloid β 42 Human ELISA Kit (Invitrogen) as per the manufacturer's instructions. The resulting measurements were normalized for tissue weight.
Measurement of TNFα and other cytokine secretion and metabolic rate in primary microglia cultures.
These experiments were performed independently by QPS Austria. Primary mouse microglia from newborn pups at postnatal day 1 (P1) to P4 were isolated using standard protocols and seeded into poly-d-lysine-coated T75 flasks (one pup per flask) in “microglia culture medium” (DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 2 mm l-glutamine). The medium was changed every 3–4 d and cells were cultured for 14 d before harvesting the microglia by shaking the flasks at 200 rpm at 37°C for 2 h. Microglia cells were seeded into uncoated 96-well plates at 8.2 × 104 cells/well and incubated for 2 h at 37°C. The plates were then rocked at 100 rpm for 5 min to detach the oligodendrocytes. The medium containing the floating oligodendrocytes was discarded and exchanged with fresh medium. Microglial cells were incubated for 2 d at 37°C before lipopolysaccharide (LPS, Sigma-Aldrich) treatment. For the LPS stimulation assay, the culture medium was removed completely 1 h before the addition of LPS and replaced by “microglia treatment medium” (DMEM supplemented with 5% FBS and 2 mm l-glutamine) containing three different concentrations of “compound 12” or DMSO (0.1%). One hour later, cells were stimulated with control medium or LPS (100 ng/ml). In one condition, dexamethasone (Sigma-Aldrich, 10 μm) was added in parallel to LPS stimulation as an inhibitor of cytokine release. Twenty-four hours after LPS stimulation, the cell supernatants were collected. The levels of TNFα, IFNγ, IL-6, and IL-10 were measured by an immunosorbent assay (U-PLEX Custom Mouse Cytokine, Mesoscale Discovery) according to the manufacturer's instructions and evaluated compared with calibration curves provided in the kit. Results are given as picograms per milliliters or as intensity signal arbitrary units (AUs). The experiment was performed in six technical replicates for all groups and repeated two independent times. The metabolic rate and viability after 24 h of LPS stimulation of the cultures was determined by the MTT assay as described for the neuronal cultures. Group differences were evaluated by one-way ANOVA followed by Bonferroni's post hoc test comparing all groups with the LPS control group.
Measurement of TNFα and other cytokine secretion in Bv-2 cells.
These experiments were performed in collaboration with UniQuest and the Queensland Emory Drug Discovery Initiative (Australia). The murine microglial cell line Bv-2 was thawed and cultivated in “microglia culture medium.” For the LPS stimulation assay, 5 × 104 Bv-2 cells per well were plated out in a 96-well dish. After 48 h, the medium was changed to “microglia treatment medium” supplemented with CAL-101 at one of three different concentrations or DMSO (0.1%). One hour later, the cells were stimulated by the addition of LPS (10 ng/ml) to the medium. Cells treated with DMSO and LPS served as controls. After 8 h of stimulation, cell supernatants were collected for cytokine measurement. TNFα, IFNγ, IL-6, IL-10, and IL-1β were measured using specific ELISA plates (https://elisakit.com/) according to the manufacturer's instructions. The experiment was performed in four technical replicates for all groups and repeated three independent times. Group differences were evaluated by one-way ANOVA followed by Bonferroni's post hoc test comparing all groups to the LPS control group.
Morris water maze spatial learning and memory test.
Animals were handled for two days by the experimenter before the initiation of behavioral experiments. The Morris water maze apparatus, consisting of a circular platform 28 cm high and 10 cm in diameter, was placed at a fixed location within a 100 cm diameter bath containing water (21−23°C) that was colored with nontoxic white paint to a depth of 1.5 cm over the platform. Mice were released into the bath from two alternating starting points and given four swim trials a day for 5 d, 10–15 min apart, in which they had up to 60 s to find a hidden platform and remain resting on it for 10 s before being assisted back into their home cage. Mice failing to find the platform or remain on it for <10 s during trials were assisted to do so before being returned to their cages. The animals were then subjected to a single probe test on the sixth day in which the platform was removed and their search strategy was recorded over 60 s. Latency to cross the position where the platform was located (latency to platform) and frequency of crossing the platform position were determined for each probe trial. Videos were recorded using Cyberlink Power Director V5 and analyzed using Ethovision 5 software.
Statistical analysis.
All data were analyzed using GraphPadPrism 5 for Mac OS X software (GraphPad Software, Inc). The D'Agostino and Pearson test was used to test for normality. The unpaired two-tailed Student's t test was used for comparison of two groups. For datasets comparing more than two groups, ANOVA followed by Bonferroni's post hoc test were used with corrections for multiple comparisons. Statistical comparisons in cell culture experiments were performed on a per-cell basis, with neurons collected from at least three independent experiments. The neurons in cell culture experiments were randomly selected within the dishes and blindly analyzed. Statistical comparisons of protein expression, histological data and animal behavioral experiments were performed on an animal basis, comparing different genotypes. Values are represented as the mean ± SEM. The tests used are indicated in the respective figure legends.
Results
PI3Kδ inactivation prevents APP anterograde axonal trafficking in cultured hippocampal neurons
We previously demonstrated that PI3Kδ is present in both neurons and microglia (Eickholt et al., 2007; Low et al., 2014). PI3Kδ controls the anterograde post-Golgi trafficking of TNFα in both microglia and macrophages (Low et al., 2010). Based on the notion that TNFα and APP share structural similarities and are both recognized as members of the TNF receptor superfamily (Nikolaev et al., 2009), we investigated whether APP trafficking in neurons could also be regulated by PI3Kδ. We first transfected cultured mouse hippocampal neurons at DIV12 with APP-GFP to image APP-GFP-positive carriers by time-lapse fluorescence imaging in axons at DIV14–18 (Fig. 1A and Movie 1). To manipulate PI3Kδ activity, we used a combination of genetic (δD910A neurons) and pharmacological approaches, incubating neurons with CAL-101, a selective PI3Kδ inhibitor that is approved for the treatment of B-cell malignancies (Lannutti et al., 2011). The percentage of APP-GFP-positive carriers undergoing anterograde transport decreased dramatically following either genetic or pharmacological inhibition of PI3Kδ (Fig. 1B–F and Movie 2). Treatment with CAL-101 (2 h, 1 μm) resulted in a selective reduction in the number of vesicles undergoing anterograde trafficking (Fig. 1A). A similar inhibition was found in hippocampal neurons from δD910A mice (Fig. 1A). Importantly, no additive effect was observed in hippocampal neurons from δD910A mice treated with CAL-101, ruling out off-target effects (Fig. 1C). We also tested increasing concentrations of CAL-101 in hippocampal neurons and found a dose-dependent inhibition of anterograde trafficking (Fig. 1D). The selectivity of such trafficking was tested using RFP-fused vesicular stomatitis virus membrane glycoprotein (VSVG-RFP), which is a marker for Golgi-derived plasma membrane carriers. When VSVG-RFP was coexpressed with APP-GFP in hippocampal neurons (Fig. 1E), we found that acute CAL-101 treatment also impacted the anterograde trafficking of VSVG-RFP carriers (Fig. 1F,G). These results suggest that the axonal anterograde trafficking APP and VSVG are controlled by PI3Kδ activity in hippocampal neurons.
Figure 1.

Axonal anterograde trafficking of APP in hippocampal neurons is reduced by pharmacological or genetic inhibition of PI3Kδ. A, Detail of a hippocampal neuron transfected with APP-GFP. Asterisks represent different APP-containing vesicles. The boxed area is magnified in the right panels. The panels represent images of a time series showing a vesicle containing APP-GFP (arrowhead) moving anterogradely along the axon. B, Representative kymographs of APP-GFP-containing vesicles moving along the axons of hippocampal neurons from mice of different genotypes and treated with control vehicle (DMSO) or CAL-101 (1 μm) for 2 h. Each analyzed region is shown just above the respective kymograph. C, Quantification of the percentage of anterograde axonally transported APP-positive vesicles in DMSO or CAL-101 (1 μm) treated neurons from WT or δD910A mice. D, Quantification of the percentage of anterograde axonally moving APP-positive vesicles in WT neurons treated with different concentrations of CAL-101. E, Representative images of triple fluorescence immunocytochemistry for APP-GFP (green), VSVG-RFP (red), and GM130 (blue) of WT hippocampal cultures of DIV10 neurons treated with control DMSO (top) or CAL-101 (1 μm) (bottom) for 2 h. F, Quantification of the alteration of neuronal trafficking of VSVG-RFP in neurons treated with DMSO or CAL-101 (1 μm). G, Quantification of the alteration of neuronal trafficking of APP-GFP in neurons treated with DMSO or CAL-101 (1 μm). The subcellular trafficking was evaluated as the ratio of each protein (APP or VSVG) in axonal compartments to that colocalizing with cis-Golgi GM130-positive structures in the soma. Scale bars, 10 μm in A and B and 20 μm in E,. Data are displayed as mean ± SEM. n = 25–38 neurons from 3 independent neuronal cultures in C and D and n = 11–14 neurons in F and G; one-way ANOVA followed by Bonferroni's post hoc test comparing each genetic group with its vehicle control (DMSO) and with the WT or APP/PS1 control group (DMSO) in C and D; Student's t test in F and G, *p < 0.05, **p < 0.01, ***p < 0.001.
APP-GFP vesicles moving along the axon of hippocampal neuron (DIV14). Video at 100 frames per second (fps), with each frame being acquired every 0.1 s. The arrow indicates the anterograde direction. Images acquired before 60 min treatment with CAL-101 (1 μM). Scale bar represents 10 μm.
APP-GFP vesicles moving along the axon of hippocampal neuron (DIV14). Video at 100 frames per second (fps), with each frame being acquired every 0.1 s. The arrow indicates the anterograde direction. Images acquired after 60 min treatment with CAL-101 (1 μM). Scale bar represents 10 μm.
We next performed immunostaining of endogenous APP in DIV14 hippocampal neurons from wild-type (WT), δD910A, APP/PS1, and APP/PS1/δD910A mice, either untreated or treated with CAL-101 (1 μm) for 2 h, followed by fixation and coimmunostaining for APP and the cis-Golgi marker GM130 (Fig. 2A). A significant increase in APP immunoreactivity was detected in GM130-positive structures following treatment of WT and APP/PS1 neurons with CAL-101 (Fig. 2C). Similarly, high levels of colocalization between APP and GM130 were detected in δD910A and APP/PS1/δD910A hippocampal neurons (Fig. 2A). Consistent with previous observations, CAL-101 did not induce a detectable additive effect in neurons derived from δD910A mice (Fig. 2C).
Figure 2.

Levels of axonal APP in neurons upon pharmacological or genetic inhibition of PI3Kδ. A, Representative images of double fluorescence immunocytochemistry for APP (red), GM130 (green), and MAP2 (blue) of cultures of hippocampal neurons from different genotypes treated with DMSO or CAL-101 (1 μm) for 2 h. B, Representative images of double fluorescence immunocytochemistry for neurofilament (NF, green) and APP (red) of cultures of hippocampal neurons from different genotypes and treated with DMSO or CAL-101 (1 μm) for 2 h. C, Quantification of the retention of APP in the cis-Golgi, as a percentage of the APP signal in GM130-positive structures, in neurons from different genotypes and treated with DMSO or CAL-101 (1 μm). D, Quantification of the levels of axonal APP, as the relative APP signal in NF-positive structures, in neurons from different genotypes treated with DMSO or CAL-101 (1 μm). Scale bar represents 10 μm. Data are displayed as mean ± SEM, n = 30–46 neurons from 3 independent neuronal cultures, one-way ANOVA followed by Bonferroni's post hoc test comparing each genetic group with its vehicle control (DMSO) and with the WT or APP/PS1 control group (DMSO), *p < 0.05, **p < 0.01, ***p < 0.001.
Additional analysis of endogenous APP immunoreactivity in axons (stained with neurofilament) from these neurons showed that CAL-101 reduced the density of APP staining along the axons of WT and APP/PS1 neurons. The APP axonal immunostaining signal was also lower in δD910A neurons and no further decrease in intensity was observed upon treatment with CAL-101. Similar results were obtained in APP/PS1/δD910A neurons (Fig. 2B,D).
A more detailed evaluation of axonal APP-GFP carriers from δD910A hippocampal neurons revealed the presence of highly-elongated tubular cargoes displaying back-and-forth mobility (Fig. 3A and Movie 3). Whereas the average diameter of APP-containing vesicles in WT neurons was 0.46 μm, the average length of these tubular carriers was 2.42 μm, reaching up to 15 μm for the longest ones, increasing the total area occupied by the carriers from 0.25 to 3 μm2 (Fig. 3B). These long tubular APP carriers were found in 20% of neurons from δD910A animals, whereas only slightly elongated APP carriers were found in 1% of WT neurons (Fig. 3C). Interestingly, the tubular carriers from δD910A neurons represented 53.9% of the APP-positive total population (Fig. 3B). The 2 h treatment of WT neurons with CAL-101 (1 μm) also produced elongated carriers, but only in some neurons (data not shown), suggesting that prolonged PI3Kδ inhibition is required for the formation of these structures. Kymograph analysis of the anterograde tubular carriers revealed a predominant back-and-forth mobility, with a 40% increase in the dwelling time (Fig. 3D,E). These results suggest that APP trafficking is dramatically impacted by PI3Kδ inhibition, which likely reduces the downstream processing of APP as it is uncertain whether these carriers can reach their final destination at the nerve terminals.
Figure 3.
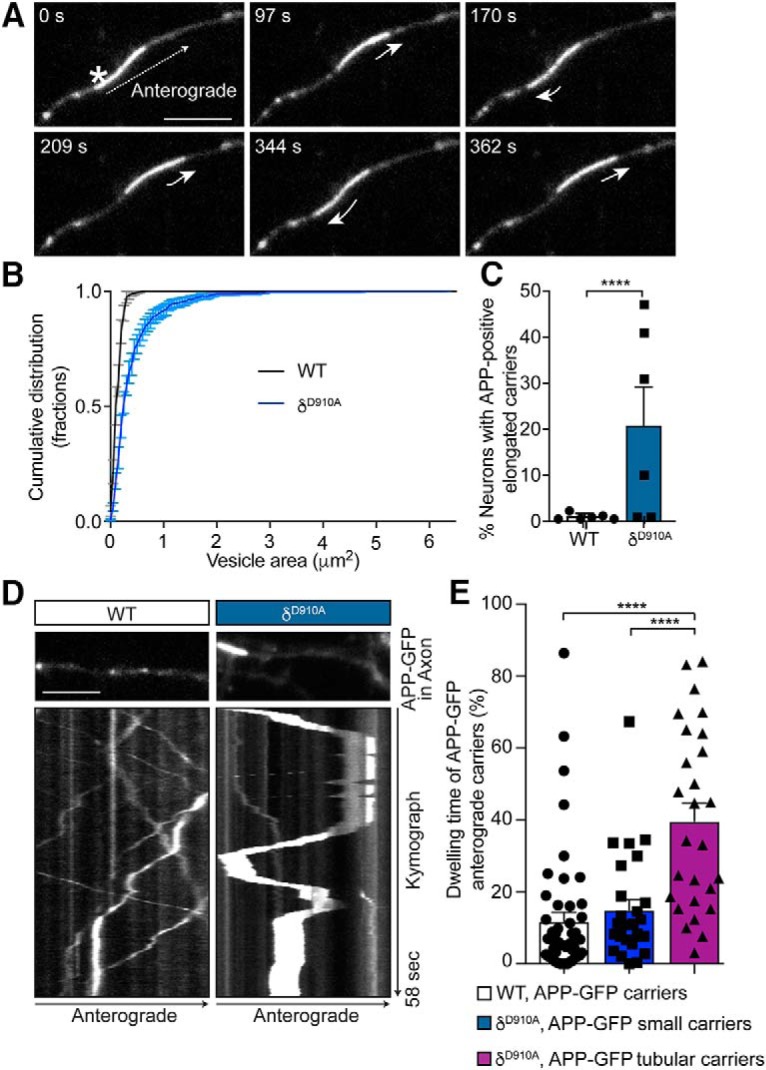
PI3Kδ inhibition leads to the formation of tubular APP-positive carriers with altered mobility parameters. A, Representative time series of hippocampal neurons from δD910A mice transfected with APP-GFP showing an elongated tubular-like carrier containing APP-GFP (asterisk) moving anterogradely along the axon. The short arrows represent changes in movement direction. B, Cumulative frequency distribution of the areas of APP-positive carriers from WT or δD910A neurons. C, Quantification of percentage of neurons containing tubular APP-GFP cargoes in neurons from WT or δD910A mice. D, Representative kymographs of APP-GFP-containing carriers moving along axons of hippocampal neurons from WT or δD910A mice. Each analyzed region is shown just above the respective kymograph. E, Quantification of the percentage of dwelling time relative to the total trafficking time in anterograde carriers from WT neurons and small anterograde carriers from δD910A neurons, and in large tubular anterograde carriers from δD910A neurons. The dwelling time is defined as the time the carriers spent with an instant speed between 0 and 0.7 μm/s. Scale bar represents 5 μm. Data are displayed as mean ± SEM; in B, n = 600–800 carriers from 12–16 neurons from 3 independent neuronal cultures; in C, n = 6 independent neuronal cultures with 10–27 neurons analyzed in each experiment; n = 27–50 carriers from 10 neurons. Student's t test in C and one-way ANOVA in E, followed by Bonferroni's post hoc test comparing all groups, ****p < 0.0001.
APP-GFP tubular vesicles moving along the axon of a hippocampal neuron (DIV18). Video at 100 frames per second (fps), with each frame being acquired every 0.1 s. The arrow indicates the anterograde direction. Scale bar represents 5 μm.
Collectively, these data suggest that PI3Kδ activity is critical for the anterograde axonal trafficking of APP from the Golgi to presynaptic nerve terminals, and that blocking PI3Kδ activity prevents this trafficking and leads to APP accumulation in the Golgi apparatus.
PI3Kδ deficiency attenuates Aβ levels and plaque burden in APP/PS1 mice
Pathological proteolysis of APP results in the accumulation of Aβ, which is secreted from neurons. To test whether altered APP trafficking affects Aβ formation, we measured the levels of secreted Aβ1–38, Aβ1–40 and Aβ1–42 in WT primary mouse cortical neurons (Fig. 4A,B). Treatment with CAL-101 (7 d) impacted on Aβ1–40 and Aβ1–42 secretion at the highest dose tested (1 μm) (Fig. 4A). However, we did not detect any difference in Aβ levels from total lysates (Fig. 4B). It is worth noting that CAL-101 treatment also slightly reduced the metabolic rate of neurons (Fig. 4C). We next analyzed the effect of PI3Kδ inactivation in vivo. Measurement of synaptic markers indicated, first of all, that PI3Kδ inactivation does not significantly affect the integrity of the synapses (Fig. 5A–C). We next measured the levels of APP, as well as soluble and insoluble Aβ1–40/42 peptides in cortical lysates from 6- to 7-month-old mice (Fig. 5D–G). This revealed slightly reduced levels of total APP in APP/PS1/δD910A mice compared with APP/PS1 mice (Fig. 5E), and a complete blockade of Aβ1–40/42 production and accumulation in PI3Kδ-deficient APP/PS1 mice (Fig. 5F,G).
Figure 4.

Impact of pharmacological PI3Kδ inactivation on Aβ levels in primary neuronal cell cultures. A, B, Primary mouse cortical neurons were treated with CAL-101 (1 μm) for 7 d. At DIV8, secreted and intracellular Aβ1–38, Aβ1–40, and Aβ1–42 were measured in supernatants (A) and lysates (B), respectively. Results are expressed as picograms per milliliter. C, Primary mouse cortical neurons were treated with vehicle (DMSO) or CAL-101 (0.1, 0.3 and 1 μm) for 7 d. On DIV8, neurons were subjected to the MTT assay of cell viability. Data are displayed as mean ± SEM, n = 5–6 in A and B, n = 6 in C. One-way ANOVA followed by Bonferroni's post hoc test comparing all groups with the vehicle control group in A–C, **p < 0.01, ***p < 0.001.
Figure 5.

Impact of genetic PI3Kδ inactivation on Aβ and APP levels in brains of APP/PS1 mice. A, Representative Western blot of PSD-95 and Synapsin-1 from brain extracts prepared from a different cohort of 12-month-old mice of indicated genotypes (3 animals per group). B, Densitometric measurements of PSD-95 normalized to GADP levels and to the average level obtained in WT mice. C, Densitometric measurements of Synapsin-1 normalized to GADP levels and to the average level obtained in WT mice. D, Representative Western blot of APP and Aβ1–40/42 from brain extracts prepared from the same 12-month-old cohort of mice. E, Densitometric measurements of APP normalized to GADP levels and to the average level obtained in APP/PS1 mice. F, Densitometric measurements of Aβ1–40/42 normalized to GADP levels. G, Quantification of the amount of total and soluble Aβ42 in cortical lysates of APP/PS1 and APP/PS1/δD910A mice by ELISA. The data of both genders of APP/PS1 were combined. No significant differences were found between males and females (p = 0.7922, data not shown). Data are displayed as mean ± SEM, n = 3 in B–D and F, n = 11 and 3 mice in G. One-way ANOVA followed by Bonferroni's post hoc test comparing all groups with the WT group in B and C. Student's t test in E–G, *p < 0.05, **p < 0.01, ****p < 0.0001.
Having established that blocking PI3Kδ activity affects both APP trafficking and Aβ secretion, we next assessed whether these changes led to a reduction in the plaque load. APP/PS1 mice show plaque deposition throughout the cortex and hippocampus as early as 4–6 months of age (Edwards et al., 2014), which was confirmed by staining 6.5-month-old brains for aggregated Aβ deposition using Thioflavin S. Specifically, hippocampal sections from APP/PS1 animals contained a significant number of Thioflavin S-positive deposits, whereas no plaques were observed in brain sections from either age-matched WT or δD910A mice (Fig. 6A). Strikingly, both the number and size of the Thioflavin S-positive plaques were significantly reduced in the hippocampus of APP/PS1/δD910A mice (Fig. 6B,C). Although we did not detect a significant reduction on plaque size in the cortex, there was a 75% reduction in plaque number in cortical sections (Fig. 6D) of APP/PS1/δD910A mice compared with APP/PS1 animals (Fig. 6E,F). The statistical power of the results is specified in the Fig. 6-1. PI3Kδ inactivation on plaque size between the cortex and hippocampus could be a reflection of the intrinsically distinct spatial patterns of Aβ deposition in different regions of the brain (Bero et al., 2011; Whitesell et al., 2019). Furthermore, amyloid plaque levels are also directly affected by Aβ degradation and clearance from the brain tissue by microglia. The PI3K/Akt pathway is specifically involved in microglia migration (Horvath and DeLeo, 2009), and PI3Kδ itself is involved in controlling the secretion of TNFα from microglia (Low et al., 2014). Thus, local alterations of Aβ clearance by microglial cells could also partially explain the brain area-specific effect in plaque size.
Figure 6.
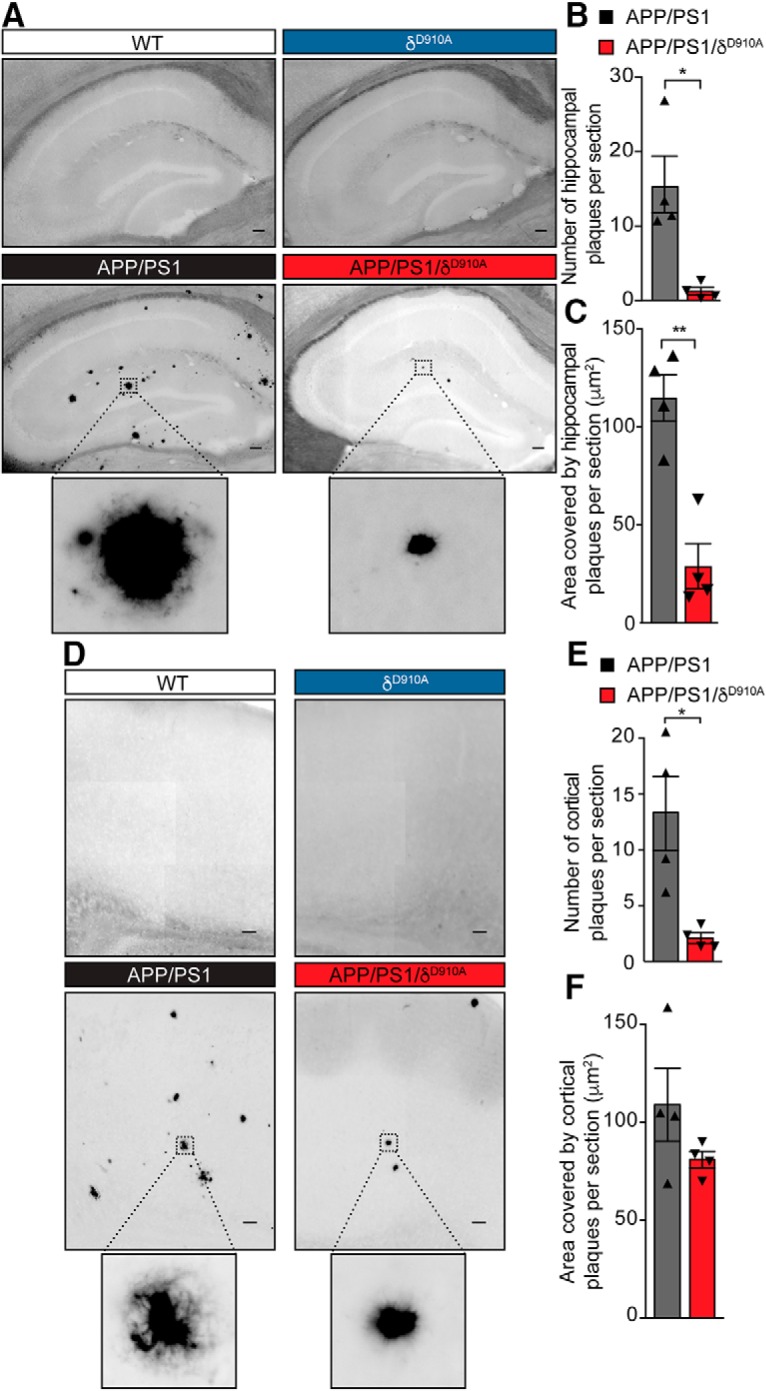
Amyloid plaque load is reduced in brain sections by genetic inactivation of PI3Kδ. A, Representative photomicrographs of hippocampal sections from mice of different genotypes stained for Aβ plaques using Thioflavin S. B, C, Quantification of plaque number (B) and size (C) in the hippocampus of APP/PS1 and APP/PS1/δD910A mice. D, Representative photomicrograph of cortical sections stained with Thioflavin S. E, F, Quantification of plaque numbers (E) and area covered by plaques (F) in the cortex of experimental mice. Scale bar represents 100 μm. Data are displayed as mean ± SEM, n = 4 animals aged 6.5-months-old per genotype and 3 replicates per animal (each replicate is a different section from the same brain region i. e. hippocampus or cortex), Student's t test, *p < 0.05, **p < 0.01. The statistical power of the results is specified in the Figure 6-1.
Extended data table related to Fig. 6. Table with statistical power calculation. Table with the values corresponding to the calculation of the effect size (Cohen's D), the post hoc statistical power and the p-value from the Student's t-test performed when analysing the results from Fig. 6. Download Figure 6-1, DOCX file (14.8KB, docx)
PI3Kδ activity controls TNFα levels and brain gliosis in APP/PS1 mice
TNFα is increased in patients with AD (Tarkowski et al., 2003). We therefore next investigated the effect of PI3Kδ on the immune response associated with AD. Pharmacological inactivation of PI3Kδ with CAL-101 dose-dependently reduced TNFα secretion by murine microglia-derived Bv2 cells (Fig. 7A), consistent with previous results in primary murine microglia (Low et al., 2014). A different potent PI3Kδ inhibitor generated by Novartis, a methoxypyridyl substituted quinazoline analogous to Leniolisib (CDZ173), hereafter named “compound 12” (Hoegenauer et al., 2016), induced similar effects to CAL-101 in primary microglia (Fig. 7B), without affecting the cells' metabolic rate (Fig. 7C). We also evaluated the effect of PI3Kδ inhibition on the release of other proinflammatory (IL-1β, IFNγ, IL-6) and anti-inflammatory (IL-6, IL-10) cytokines by Bv2 cells (Fig. 7E,G,I) and primary microglia (Fig. 7D,F,H,J). None of these cytokines was affected to the same extent as TNFα by pharmacological PI3Kδ inhibition in either of the cellular systems, suggesting some level of specificity for PI3Kδ in regulating TNFα release.
Figure 7.

Genetic and pharmacological inactivation of PI3Kδ activity ameliorate LPS-dependent release of TNFα in microglia and control TNFα levels in APP/PS1 mice. A, Relative TNFα release from Bv-2 murine-derived microglial cells, stimulated with LPS (10 ng/ml) for 8 h and treated with control vehicle (DMSO) or increasing concentrations of CAL-101 (0.1 μm, 0.3 μm, 1 or 3 μm). The data are expressed as percentage relative to LPS stimulation and DMSO treatment. B, TNFα release by mouse primary microglia stimulated for 24 h with control vehicle or LPS (100 ng/ml) and treated with DMSO or with increasing concentrations of “compound 12” (0.1 μm, 0.3 or 1 μm). C, MTT viability assay of mouse microglia after 24 h of control vehicle or LPS (100 ng/ml) stimulation and treatment with DMSO or increasing concentrations of “compound 12” (0.1 μm, 0.3 or 1 μm). D, F, H, J, Measurement of IL-1β, IFNγ, IL-6, and IL-10 release, respectively, by mouse primary microglia stimulated for 12 h with control vehicle or LPS (100 ng/ml) and treated with DMSO or with increasing concentrations of “compound 12” (0.1 μm, 0.3 or 1 μm). The data are expressed as pg/ml or as intensity signal arbitrary units (AU) of the cytokine present in the collected supernatants. E, G, I, Measurement of IFNγ, IL-6, and IL-10 release, respectively, from of Bv-2 murine-derived microglial cells, stimulated for 6 h with control vehicle or LPS (10 ng/ml) and treated with control (DMSO) or increasing concentrations of CAL-101 (0.3 or 1 μm). The data are expressed as pg/ml of the cytokine present in the collected supernatants. K, Representative Western blot of TNFα from brains of 6.5-month-old mice of different phenotypes. L, Densitometric measurements of TNFα normalized to β-tubulin levels. Data are displayed as mean ± SEM, n = 3 independent experiments with n = 4 replicates in each experiment in A, n = 5–6 in B–J, n = 3 animals per genotype in J. One-way ANOVA followed by Bonferroni's post hoc test comparing all groups with the LPS group, *p < 0.05, **p < 0.01 and ***p < 0.001 in A–J; one-way ANOVA followed by Bonferroni's post hoc test comparing all groups with the APP/PS1 group, ***p < 0.001 in L.
We next determined the impact of PI3Kδ deficiency on TNFα levels in APP/PS1 mice, measured by Western blotting of brain lysates from 6.5-month-old mice (Fig. 7K). In accordance with previous results from animal models (Jin et al., 2008), significantly higher levels of TNFα were detected in APP/PS1 mice than in WT animals (Fig. 7L). This increase was virtually abolished in APP/PS1 × δD910A mice, with comparable TNFα levels obtained in lysates from age-matched WT and δD910A mice (Fig. 7L). These data demonstrate that the high TNFα levels in the APP/PS1 AD mouse model can be normalized by PI3Kδ inactivation.
We then further explored whether the expression levels of TNFα also correlated with the level of overall inflammation in the brain. Increased numbers of microglia (Iba1-positive cells) and astrocytes (GFAP-positive cells) were found in APP/PS1 mice compared with WT, δD910A, and APP/PS1/δD910A mice, in both the hippocampus (Fig. 8A–C) and cortex (Fig. 8D–F). A 75% reduction in the relative staining volume of microglia and astrocytes was found within the hippocampus of APP/PS1/δD910A mice compared with their APP/PS1 counterparts (Fig. 8B,C). The statistical power of the results is specified in the Fig. 8-1. significant difference between the APP/PS1/δD910A mice and age-matched WT or δD910A control groups, indicating full penetrance of the phenotypic rescue in the APP/PS1/δD910A mice.
Figure 8.

Brain gliosis in APP/PS1 mice is controlled by PI3Kδ activity. A, D, Representative images of double fluorescence immunohistochemistry for microglia (Iba1, red), astrocytes (GFAP, green) and nuclei (DAPI, blue) in coronal hippocampal sections (A) and coronal cortical sections (D). B, C, E, F, Tissue volume occupied by immunopositive structures calculated as a proportion of the total hippocampal volume imaged. Percentage volume of microglia (B, E) and astrocytes (C, F) in the hippocampus (B, C) and the cortex (E, F) of animals of different genotypes. Insets represent magnifications from each boxed area, showing regions of microglia and astrocyte accumulation. Scale bars, 100 μm in A and D. Data are displayed as mean ± SEM of n = 4 mice aged 6.5 months old per genotype and 3 replicates per animal (each replicate is a different section from the same brain structure -hippocampus or cortex-), one-way ANOVA followed by Bonferroni's post hoc test comparing all groups to the APP/PS1 group, **p < 0.01, ***p < 0.001, ****p < 0.0001 in B, C, E, F. The statistical power of the results is specified in the Figure 8-1.
Extended data table related to Fig. 8. Table with statistical power calculation. Table with the values corresponding to the calculation of the effect size, the post hoc statistical power and the p-value from the ANOVA test performed when analysing the results from Fig. 8. Download Figure 8-1, DOCX file (14.8KB, docx)
Inactivation of PI3Kδ activity increases the survival of APP/PS1 mice and prevents cognitive dysfunction
Having established that PI3Kδ inhibition affects both the formation of amyloid plaques and the neuroinflammation response associated with AD progression, we next tested whether the cognitive impairment associated with this AD model was alleviated. Given that APP/PS1 mice suffer from premature death at the time at which Aβ begins to be deposited as plaques (Minkeviciene et al., 2009), we also assessed whether survival was altered in the absence of PI3Kδ activity. Interrogation of colony breeding and maintenance data revealed that 42.7% (56 of 131) of APP/PS1 mice had died at the 210 d time point. In contrast, only 3% (3 of 100) APP/PS1/δD910A mice had succumbed by this age, demonstrating a decrease in the premature mortality of the APP/PS1 mice in response to PI3Kδ inactivation (Fig. 9A).
Figure 9.

Genetic inactivation of PI3Kδ increases survival and rescues spatial learning and memory of APP/PS1 mice. A, APP/PS1 transgenic mice (n = 131) present with a pronounced premature mortality that is ameliorated by inactivating PI3Kδ (p < 0.0001). Whereas the lethality was 42.7% in the APP/PS1 mice at the 210 d time point, in APP/PS1/δD910A animals (n = 100) it was reduced to 3%. B, Representative images of swimming tracks for mice of different genotypes on days 1, 3, and 5 of the learning phase of the Morris water maze. C, Learning acquisition plot measured as the time taken for mice to reach the submerged platform (latency) during the learning phase (daya 1, 3, and day 5). D, Latency to target the platform area during the probe test. E, Number of platform crossings during the probe test. F, Schematic diagram summarizing a model for the regulation of APP and TNFα trafficking by PI3Kδ in neurons and microglia. PI3Kδ is involved in the trafficking of APP and TNFα by controlling the formation of TGN-derived cargoes in neurons and microglia, respectively. By pharmacological modulation of PI3Kδ activity using CAL-101, the release of neuronal Aβ and microglial TNFα can be attenuated, reducing the extent of Aβ plaque formation and neuroinflammation. Data are displayed as mean ± SEM of n = 10 animals aged 6.5 months old per genotype, one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.001.
We then evaluated the cognitive impairment associated with AD progression using the Morris water maze spatial navigation task. APP/PS1 mice present synaptic and memory deficits at 3–6 months of age, concomitant with an increase in amyloid deposition (Trinchese et al., 2004). Nontransgenic WT mice and δD910A controls, in line with previously published data (Eickholt et al., 2007), learned to navigate to the platform within a 5-day training period using a direct navigation strategy (Janus, 2004) (Fig. 9B), and displayed a preference for the original platform location during the probe trial (Fig. 9B). In contrast, 6.5-month-old APP/PS1 mice displayed significant deficits in performance during the acquisition phase of the Morris water maze (Fig. 9B). The strong cognitive impairment of the APP/PS1 mice was also evident from their inability to remember the position of the submerged platform during the probe trial (i.e., time spent in target area) (Fig. 9C). On average, APP/PS1 mice took three times longer to pass over the learned location of the platform compared with WT or δD910A mice (Fig. 9D); they also crossed the platform position less often than controls during the probe trial (Fig. 9E). Strikingly, the cognitive deficits observed in the APP/PS1 mice were completely absent in APP/PS1/δD910A mice. On average, APP/PS1/δD910A mice found the escape platform in a time that was equivalent to that of WT and δD910A mice during training sessions, and they performed equally well as controls in all analyzed parameters of the probe trial (Fig. 9C–E). Together, these results demonstrate that the impairment in spatial learning and memory displayed by APP/PS1 mice can be rescued by PI3Kδ inactivation. A proposed model for the regulation of APP and TNFα trafficking by PI3Kδ in neurons and microglia is illustrated in Figure 9F.
Discussion
Here, we show that inactivation of PI3Kδ acts on both neurons and microglia to prevent anterograde post-Golgi trafficking of APP and TNFα, respectively. Both APP trafficking and TNFα-mediated neuroinflammation are critically involved in neurodegenerative diseases such as AD. By simultaneously targeting these pathways, we demonstrate that, compared with APP/PS1 mice, APP/PS1/δD910A mice exhibit dramatically reduced Aβ levels, Aβ plaque number and size, as well as significant reductions in gliosis and TNFα levels, with complete protection from early cognitive impairment.
Because of its importance in immune signaling, PI3Kδ is considered an important therapeutic target in the treatment of inflammation and autoimmunity (Okkenhaug et al., 2002). Based on its high expression in leukocytes, it is also a target in hematological malignancies (Lampson and Brown, 2017), including chronic lymphocytic leukemia (Furman et al., 2014). Compared with leukocytes, PI3Kδ is also expressed at moderate levels in neurons (Eickholt et al., 2007; Low et al., 2014), and we demonstrate here that PI3Kδ inhibition blocks APP anterograde trafficking in hippocampal neurons. Interestingly, the elongated tubular shape of some axonal APP carriers observed in δD910A neurons resembles the tubular TGN-derived TNFα-containing cargoes described in macrophages in response to PI3Kδ inhibition (Low et al., 2010). These results therefore suggest that PI3Kδ might also be involved in the fission machinery required at the TGN for the exit of APP-containing cargoes in neurons. Although the basis of the commonality of action of PI3Kδ on TNFα and APP is currently unknown, it is important to note that PI3Kδ is expressed in the Golgi of macrophages (Low et al., 2010), microglia, and neurons (Low et al., 2014). Like TNFα, APP is a type I transmembrane protein that is synthesized at the endoplasmic reticulum and then undergoes complex intracellular trafficking via the TGN, going through the secretory pathway and endocytic, and recycling routes, in the neuronal soma, dendrites and axons (Muresan and Ladescu Muresan, 2015). In addition, APP and TNFα share structural similarities and APP has been shown to bind the TNF receptor superfamily member Death Receptor 6 (DR6) (Nikolaev et al., 2009). This common structural and trafficking pathway is likely to underpin the high sensitivity of both APP and TNFα trafficking to PI3Kδ inhibition.
It was not only APP axonal anterograde trafficking that was significantly reduced by PI3Kδ inhibition; the secretion of Aβ peptides was also reduced after pharmacological inhibition of PI3Kδ in WT cultured neurons, with dramatically reduced levels of Aβ1–40/42 toxic peptides and Aβ42 fibrils in vivo in the brains of 6- to 7-month-old APP/PS1/δD910A mice compared with age-matched APP/PS1 animals. Although inhibition of PI3Kδ activity is unlikely to have a direct effect on tau phosphorylation and inclusion, the protection afforded by reducing APP processing, Aβ release, and plaque formation might provide a protective environment that reduces tau hyperphosphorylation. However, such protection is unlikely to prevent from tauopathies mediated by tau mutations that are driving toxicity. Further experiments would be required to evaluate these effects. In summary, our data strongly suggest that accumulation of Aβ1–40/42 in the AD brain model used is greatly dampened by PI3Kδ inhibition.
The level of amyloid plaques is a consequence of a fine balance between the secretion of toxic Aβ peptides, their oligomerization and deposition, and their degradation and clearance from the brain by microglia (Bates et al., 2009). Microglia have a complex role in amyloid plaque formation. In the early stages of AD, microglia accumulate in the vicinity of developing amyloid plaques and exposure to extracellular fibrillary Aβ converts these immune cells into an “activated” phenotype (Ferrera et al., 2014). Aβ-activated microglia show enhanced phagocytosis and clearance of plaques (Bolmont et al., 2008). Furthermore, a reduction in the early accumulation of microglia at the site of inflammation in AD brains hastens the progression of the disease (El Khoury et al., 2007). However, microglia also have detrimental activities in AD, including the exacerbation of neurotoxic inflammatory signals (Colonna and Butovsky, 2017). The initial low, basal level of TNFα release drives microglia to clear and eradicate plaques (Viel et al., 2001) without enabling the activation of deleterious chronic inflammation. However, at later stages of disease, excess TNFα levels in the brain in response to amyloid aggregates and fibrils can inhibit the microglial clearance of Aβ (Hickman et al., 2008), inducing surrounding astrocytes into a toxic “A1” state that causes neuronal death (Liddelow et al., 2017). Consistent with previous reports of increased levels of brain TNFα in animal models of AD (Jin et al., 2008), and consistent with the reports of elevated levels of this cytokine in the blood (Fillit et al., 1991) and CSF (Tarkowski et al., 2003) of patients with AD, we found that the brain level of TNFα was increased in APP/PS1 mice. The role of microglia in neuroinflammation associated with AD is well established and several microglial receptors such as TREM2, CD33, and CR3, as well as their membrane adaptor TYROBP/DAP12 (Pottier et al., 2016), are risk genes in the development of late-onset sporadic AD (Lambert et al., 2013). We previously demonstrated that TNFα secretion after an inflammatory stimulus can be blocked by inhibiting PI3Kδ, leading to the amelioration of the in vivo toxicity associated with ischemic stroke (Low et al., 2014). Here, we show that blockade of PI3Kδ activity in primary microglia specifically affects the release of the proinflammatory cytokine TNFα; none of the other proinflammatory or anti-inflammatory cytokines that we tested (IFNγ, IL-6, IL-1β, and IL-10) was affected. Remarkably, we found that the elevation of TNFα levels observed in APP/PS1 mice was prevented in APP/PS1/δD910A animals. Our results from Bv2 cells and primary cultured microglia suggest a direct effect of PI3Kδ inhibition on TNFα trafficking and secretion. However, the reduced Aβ secretion and deposition observed in the APP/PS1/δD910A brains may also contribute to the reduction of TNFα levels (Jin et al., 2008). The genetic inactivation of PI3Kδ activity might therefore have prevented the chronic AD-associated chronic inflammatory response, as TNFα release from microglia is drastically reduced and the cytokine expression in the brain remains at basal levels, ensuring an effective clearance of newly formed Aβ in the brains of APP/PS1/δD910A mice. Our results also clearly show that PI3Kδ inhibition in a mouse model of AD strongly attenuates the accumulation of both astrocytes and microglia around plaques, suggesting attenuated proinflammatory feedback cascades and a decreased astrocytic toxic response, thereby reducing the neurotoxic microenvironment associated with amyloid plaques in the brain. Interestingly, genetic downregulation of TYROBP/DAP12 exerts a similar protective effect in the same mouse model (APP/PS1) of AD (Haure-Mirande et al., 2017). It is, however, unlikely that TYROBP/DAP12 and PI3Kδ act in the same pathway, as our results show that PI3Kδ acts on post-Golgi trafficking in both microglia (Low et al., 2014) and neurons whereas TYROBP/DAP12 is expressed in microglia and dendritic cells (Audrain et al., 2018). The dual action of PI3Kδ inhibition on neuronal APP processing and microglial TNFα secretion might ultimately be responsible for the combined beneficial effects observed in mice lacking this enzyme.
As reported previously (Serrano et al., 2010), the survival of APP/PS1 mice is reduced in comparison with that of WT animals. In addition to attenuating tissue pathology, genetic inhibition of PI3Kδ also increased the life expectancy of double mutant APP/PS1/δD910A animals. Coincident with the accumulation of Aβ plaques within the brains of APP/PS1 mice, we and others have further demonstrated that these animals exhibit cognitive decline, as measured by the Morris water maze test at 6.5 months of age (Trinchese et al., 2004; Puzzo et al., 2014). Importantly, in APP/PS1/δD910A mice, these memory deficits were fully rescued. There is a wealth of evidence to indicate that soluble Aβ oligomers can negatively affect synaptic transmission and plasticity (Klyubin et al., 2012). Aβ plaques can also physically impede neurotransmission (Xu et al., 2015), making this the most likely explanation for the impaired cognitive performance of the APP/PS1 animals. The reduced Aβ plaque load in the APP/PS1/δD910A mice is therefore likely to underpin the rescue of the Aβ-induced cognitive decline in these animals. Significant neuroinflammation has also been correlated with both the onset and severity of cognitive impairment in AD, being associated with neuronal dysfunction in animal models (Parachikova et al., 2007). TNFα is directly involved in the progression of this chronic inflammatory cascade (Bhaskar et al., 2014). Lowering TNFα and neuroinflammation reduces the progression of AD, as reported previously by others (Cheng et al., 2014) and thus the inhibition of the TNFα-driven chronic inflammatory cascade in APP/PS1/δD910A mice may contribute to the protection of these animals against cognitive impairment.
Selectively controlling chronic inflammation and Aβ plaques without affecting basal TNFα levels, as proposed here, offers significant advantages over current therapeutic strategies aimed at inhibiting TNFα action, the side-effects of which limit their potential. PI3Kδ inhibitors have been approved for the treatment of B-cell malignancies and are undergoing trials in various inflammatory conditions (Bilanges et al., 2019). Our study therefore reinforces the importance of targeting both the neuronal and immunological components in the progression of AD, and highlights the potential for pharmacologically targeting PI3Kδ in the clinic to simultaneously reduce Aβ generation and TNFα secretion. Such an approach is more specific and could be considered as a novel therapeutic for AD, although it would require the development of PI3Kδ antagonists that can effectively cross the blood–brain barrier.
Footnotes
This work was supported by the National Health and Medical Research Council (NHMRC) (Project Grants 1049236 to E.J.C., 1005964 to F.A.M., and 1060538 to M.J.R.). Work in the laboratory of B.V. is supported by the Biotechnology and, Biological Sciences Research Council (BB/I007806/1), Cancer Research UK (C23338/A25722) and the National Institute for Health Research (NIHR) University College London Hospitals Biomedical Research Centre. F.A.M. is supported by an NHMRC Senior Research Fellowship (569596). R.M.-M. is supported by the Clem and Jones Foundation, the State Government of Queensland, and the NHMRC Boosting Dementia Research Initiative. T.W. is supported by a Discovery Early Career Researcher Fellowship (DE170100546). M.J.R. is supported by a SpinalCure Australia Career Development Fellowship. We thank members of the Meunier and Coulson laboratories for technical assistance; Rumelo Amor for expert help with microscopy; Alan Ho, Pranesh Padmanabhan, and Tristan Wallis for expert help with statistical analysis; Rodrigo Medeiros and Judit Camats for providing frozen mouse brain samples; Nick Valmas for graphic design; Rowan Tweedale for critical reading and editing of the manuscript; UniQuest and the Queensland Emory Drug Discovery Initiative team for commercial insights and technical assistance; and the technical staff of the University of Queensland's Biological Resources and the Queensland Brain Institute animal facility for maintaining the various mouse lines used in this study.
B.V. is a consultant for Karus Therapeutics (Oxford, UK), iOnctura (Geneva, Switzerland), and Venthera (Palo Alto, California) and received speaker fees from Gilead (Foster City, California). The remaining authors declare no competing financial interests.
References
- Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H, Friedman L, Turner M, Okkenhaug K, Vanhaesebroeck B (2014) Inactivation of PI(3)K p110delta breaks regulatory T-cell-mediated immune tolerance to cancer. Nature 510:407–411. 10.1038/nature13444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen OM, Reiche J, Schmidt V, Gotthardt M, Spoelgen R, Behlke J, von Arnim CA, Breiderhoff T, Jansen P, Wu X, Bales KR, Cappai R, Masters CL, Gliemann J, Mufson EJ, Hyman BT, Paul SM, Nykjaer A, Willnow TE (2005) Neuronal sorting protein-related receptor sorLA/LR11 regulates processing of the amyloid precursor protein. Proc Natl Acad Sci U S A 102:13461–13466. 10.1073/pnas.0503689102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audrain M, Haure-Mirande JV, Wang M, Kim SH, Fanutza T, Chakrabarty P, Fraser P, St George-Hyslop PH, Golde TE, Blitzer RD, Schadt EE, Zhang B, Ehrlich ME, Gandy S (2018) Integrative approach to sporadic Alzheimer's disease: deficiency of TYROBP in a tauopathy mouse model reduces C1q and normalizes clinical phenotype while increasing spread and state of phosphorylation of tau. Mol Psychiatry. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates KA, Verdile G, Li QX, Ames D, Hudson P, Masters CL, Martins RN (2009) Clearance mechanisms of Alzheimer's amyloid-beta peptide: implications for therapeutic design and diagnostic tests. Mol Psychiatry 14:469–486. 10.1038/mp.2008.96 [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM (2011) Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci 14:750–756. 10.1038/nn.2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar K, Maphis N, Xu G, Varvel NH, Kokiko-Cochran ON, Weick JP, Staugaitis SM, Cardona A, Ransohoff RM, Herrup K, Lamb BT (2014) Microglial derived tumor necrosis factor-alpha drives Alzheimer's disease-related neuronal cell cycle events. Neurobiol Dis 62:273–285. 10.1016/j.nbd.2013.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilanges B, Posor Y, Vanhaesebroeck B (2019) PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. In press. [DOI] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, Calhoun ME (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 28:4283–4292. 10.1523/JNEUROSCI.4814-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess BL, McIsaac SA, Naus KE, Chan JY, Tansley GH, Yang J, Miao F, Ross CJ, van Eck M, Hayden MR, van Nostrand W, St George-Hyslop P, Westaway D, Wellington CL (2006) Elevated plasma triglyceride levels precede amyloid deposition in Alzheimer's disease mouse models with abundant A beta in plasma. Neurobiol Dis 24:114–127. 10.1016/j.nbd.2006.06.007 [DOI] [PubMed] [Google Scholar]
- Cheng X, Shen Y, Li R (2014) Targeting TNF: a therapeutic strategy for Alzheimer's disease. Drug Discov Today 19:1822–1827. 10.1016/j.drudis.2014.06.029 [DOI] [PubMed] [Google Scholar]
- Collins JS, Perry RT, Watson B Jr, Harrell LE, Acton RT, Blacker D, Albert MS, Tanzi RE, Bassett SS, McInnis MG, Campbell RD, Go RC (2000) Association of a haplotype for tumor necrosis factor in siblings with late-onset alzheimer disease: the NIMH alzheimer disease genetics initiative. Am J Med Genet 96:823–830. [DOI] [PubMed] [Google Scholar]
- Colonna M, Butovsky O (2017) Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol 35:441–468. 10.1146/annurev-immunol-051116-052358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW. (1997) The pathogenesis of senile plaques. J Neuropathol Exp Neurol 56:321–339. 10.1097/00005072-199704000-00001 [DOI] [PubMed] [Google Scholar]
- Edwards SR, Hamlin AS, Marks N, Coulson EJ, Smith MT (2014) Comparative studies using the Morris water maze to assess spatial memory deficits in two transgenic mouse models of Alzheimer's disease. Clin Exp Pharmacol Physiol 41:798–806. 10.1111/1440-1681.12277 [DOI] [PubMed] [Google Scholar]
- Eickholt BJ, Ahmed AI, Davies M, Papakonstanti EA, Pearce W, Starkey ML, Bilancio A, Need AC, Smith AJ, Hall SM, Hamers FP, Giese KP, Bradbury EJ, Vanhaesebroeck B (2007) Control of axonal growth and regeneration of sensory neurons by the p110delta PI 3-kinase. PLoS One 2:e869. 10.1371/journal.pone.0000869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD (2007) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med 13:432–438. 10.1038/nm1555 [DOI] [PubMed] [Google Scholar]
- Ferrera D, Mazzaro N, Canale C, Gasparini L (2014) Resting microglia react to Abeta42 fibrils but do not detect oligomers or oligomer-induced neuronal damage. Neurobiol Aging 35:2444–2457. 10.1016/j.neurobiolaging.2014.05.023 [DOI] [PubMed] [Google Scholar]
- Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G (1991) Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett 129:318–320. 10.1016/0304-3940(91)90490-K [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G (2013) Paxinos and Franklin's The mouse brain in stereotaxic coordinates, Ed 4 Amsterdam: Academic. [Google Scholar]
- Furman RR, Sharman JP, Coutre SE, Cheson BD, Pagel JM, Hillmen P, Barrientos JC, Zelenetz AD, Kipps TJ, Flinn I, Ghia P, Eradat H, Ervin T, Lamanna N, Coiffier B, Pettitt AR, Ma S, Stilgenbauer S, Cramer P, Aiello M, et al. (2014) Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med 370:997–1007. 10.1056/NEJMoa1315226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groemer TW, Thiel CS, Holt M, Riedel D, Hua Y, Huve J, Wilhelm BG, Klingauf J (2011) Amyloid precursor protein is trafficked and secreted via synaptic vesicles. PLoS One 6:e18754. 10.1371/journal.pone.0018754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haure-Mirande JV, Audrain M, Fanutza T, Kim SH, Klein WL, Glabe C, Readhead B, Dudley JT, Blitzer RD, Wang M, Zhang B, Schadt EE, Gandy S, Ehrlich ME (2017) Deficiency of TYROBP, an adapter protein for TREM2 and CR3 receptors, is neuroprotective in a mouse model of early Alzheimer's pathology. Acta Neuropathol 134:769–788. 10.1007/s00401-017-1737-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J (2008) Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci 28:8354–8360. 10.1523/JNEUROSCI.0616-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoegenauer K, Soldermann N, Hebach C, Hollingworth GJ, Lewis I, von Matt A, Smith AB, Wolf RM, Wilcken R, Haasen D, Burkhart C, Zécri F (2016) Discovery of novel pyrrolidineoxy-substituted heteroaromatics as potent and selective PI3K delta inhibitors with improved physicochemical properties. Bioorg Med Chem Lett 26:5657–5662. 10.1016/j.bmcl.2016.10.069 [DOI] [PubMed] [Google Scholar]
- Hood VL, Berger R, Freedman R, Law AJ (2019) Transcription of PIK3CD in human brain and schizophrenia: Regulation by pro inflammatory cytokines. Hum Mol Genet. Advance online publication. Retrieved June 18, 2019. doi: 10.1093/hmg/ddz144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath RJ, DeLeo JA (2009) Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci 29:998–1005. 10.1523/JNEUROSCI.4595-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Gonzales V, Jenkins NA, Copeland NG, Borchelt DR (2004) APP processing and amyloid deposition in mice haplo-insufficient for presenilin 1. Neurobiol Aging 25:885–892. 10.1016/j.neurobiolaging.2003.09.008 [DOI] [PubMed] [Google Scholar]
- Janus C. (2004) Search strategies used by APP transgenic mice during navigation in the morris water maze. Learn Mem 11:337–346. 10.1101/lm.70104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K (2008) Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation 5:23. 10.1186/1742-2094-5-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joensuu M, Martinez-Mármol R, Padmanabhan P, Glass NR, Durisic N, Pelekanos M, Mollazade M, Balistreri G, Amor R, Cooper-White JJ, Goodhill GJ, Meunier FA (2017) Visualizing endocytic recycling and trafficking in live neurons by subdiffractional tracking of internalized molecules. Nat Protoc 12:2590–2622. 10.1038/nprot.2017.116 [DOI] [PubMed] [Google Scholar]
- Keller P, Toomre D, Díaz E, White J, Simons K (2001) Multicolour imaging of post-Golgi sorting and trafficking in live cells. Nat Cell Biol 3:140–149. 10.1038/35055042 [DOI] [PubMed] [Google Scholar]
- Klyubin I, Cullen WK, Hu NW, Rowan MJ (2012) Alzheimer's disease abeta assemblies mediating rapid disruption of synaptic plasticity and memory. Mol Brain 5:25. 10.1186/1756-6606-5-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thorton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, et al. (2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat Genet 45:1452–1458. 10.1038/ng.2802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampson BL, Brown JR (2017) PI3Kdelta-selective and PI3Kalpha/delta-combinatorial inhibitors in clinical development for B-cell non-Hodgkin lymphoma. Expert Opin Investig Drugs 26:1267–1279. 10.1080/13543784.2017.1384815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, Byrd JC, Tyner JW, Loriaux MM, Deininger M, Druker BJ, Puri KD, Ulrich RG, Giese NA (2011) CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood 117:591–594. 10.1182/blood-2010-03-275305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS (2004) Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem 279:49523–49532. 10.1074/jbc.M402034200 [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, et al. (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487. 10.1038/nature21029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low PC, Misaki R, Schroder K, Stanley AC, Sweet MJ, Teasdale RD, Vanhaesebroeck B, Meunier FA, Taguchi T, Stow JL (2010) Phosphoinositide 3-kinase delta regulates membrane fission of Golgi carriers for selective cytokine secretion. J Cell Biol 190:1053–1065. 10.1083/jcb.201001028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low PC, Manzanero S, Mohannak N, Narayana VK, Nguyen TH, Kvaskoff D, Brennan FH, Ruitenberg MJ, Gelderblom M, Magnus T, Kim HA, Broughton BR, Sobey CG, Vanhaesebroeck B, Stow JL, Arumugam TV, Meunier FA (2014) PI3Kdelta inhibition reduces TNF secretion and neuroinflammation in a mouse cerebral stroke model. Nat Commun 5:3450. 10.1038/ncomms4450 [DOI] [PubMed] [Google Scholar]
- Mandrekar-Colucci S, Landreth GE (2010) Microglia and inflammation in Alzheimer's disease. CNS Neurol Disord Drug Targets 9:156–167. 10.2174/187152710791012071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fulop L, Penke B, Zilberter Y, Harkany T, Pitkänen A, Tanila H (2009) Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29:3453–3462. 10.1523/JNEUROSCI.5215-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SL, Narrow WC, Mastrangelo MA, Olschowka JA, O'Banion MK, Bowers WJ (2013) Chronic neuron- and age-selective down-regulation of TNF receptor expression in triple-transgenic Alzheimer disease mice leads to significant modulation of amyloid- and tau-related pathologies. Am J Pathol 182:2285–2297. 10.1016/j.ajpath.2013.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan V, Ladescu Muresan Z (2015) Amyloid-beta precursor protein: multiple fragments, numerous transport routes and mechanisms. Exp Cell Res 334:45–53. 10.1016/j.yexcr.2014.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M (2009) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457:981–989. 10.1038/nature07767 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Okkenhaug K, Vanhaesebroeck B (2003) PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol 3:317–330. 10.1038/nri1056 [DOI] [PubMed] [Google Scholar]
- Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJ, Vanhaesebroeck B (2002) Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297:1031–1034. 10.1126/science.1073560 [DOI] [PubMed] [Google Scholar]
- Parachikova A, Agadjanyan MG, Cribbs DH, Blurton-Jones M, Perreau V, Rogers J, Beach TG, Cotman CW (2007) Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging 28:1821–1833. 10.1016/j.neurobiolaging.2006.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottier C, Ravenscroft TA, Brown PH, Finch NA, Baker M, Parsons M, Asmann YW, Ren Y, Christopher E, Levitch D, van Blitterswijk M, Cruchaga C, Campion D, Nicolas G, Richard AC, Guerreiro R, Bras JT, Zuchner S, Gonzalez MA, Bu G, Younkin S, et al. (2016) TYROBP genetic variants in early-onset Alzheimer's disease. Neurobiol Aging 48:222.e9–222.e15. 10.1016/j.neurobiolaging.2016.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzzo D, Lee L, Palmeri A, Calabrese G, Arancio O (2014) Behavioral assays with mouse models of Alzheimer's disease: practical considerations and guidelines. Biochem Pharmacol 88:450–467. 10.1016/j.bcp.2014.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt V, Sporbert A, Rohe M, Reimer T, Rehm A, Andersen OM, Willnow TE (2007) SorLA/LR11 regulates processing of amyloid precursor protein via interaction with adaptors GGA and PACS-1. J Biol Chem 282:32956–32964. 10.1074/jbc.M705073200 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. (2002) Alzheimer's disease is a synaptic failure. Science 298:789–791. 10.1126/science.1074069 [DOI] [PubMed] [Google Scholar]
- Serrano J, Fernández AP, Martínez-Murillo R, Martínez A (2010) High sensitivity to carcinogens in the brain of a mouse model of Alzheimer's disease. Oncogene 29:2165–2171. 10.1038/onc.2009.503 [DOI] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307:1282–1288. 10.1126/science.1105681 [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Andreasen N, Tarkowski A, Blennow K (2003) Intrathecal inflammation precedes development of Alzheimer's disease. J Neurol Neurosurg Psychiatry 74:1200–1205. 10.1136/jnnp.74.9.1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez JY, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, Eliceiri KW (2017) TrackMate: an open and extensible platform for single-particle tracking. Methods 115:80–90. 10.1016/j.ymeth.2016.09.016 [DOI] [PubMed] [Google Scholar]
- Trinchese F, Liu S, Battaglia F, Walter S, Mathews PM, Arancio O (2004) Progressive age-related development of alzheimer-like pathology in APP/PS1 mice. Ann Neurol 55:801–814. 10.1002/ana.20101 [DOI] [PubMed] [Google Scholar]
- Viel JJ, McManus DQ, Smith SS, Brewer GJ (2001) Age- and concentration-dependent neuroprotection and toxicity by TNF in cortical neurons from beta-amyloid. J Neurosci Res 64:454–465. 10.1002/jnr.1097 [DOI] [PubMed] [Google Scholar]
- Whitesell JD, Buckley AR, Knox JE, Kuan L, Graddis N, Pelos A, Mukora A, Wakeman W, Bohn P, Ho A, Hirokawa KE, Harris JA (2019) Whole brain imaging reveals distinct spatial patterns of amyloid beta deposition in three mouse models of Alzheimer's disease. J Comp Neurol 527:2122–2145. 10.1002/cne.24555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L (2001) TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med 7:612–618. 10.1038/87945 [DOI] [PubMed] [Google Scholar]
- Xu K, Olsen O, Tzvetkova-Robev D, Tessier-Lavigne M, Nikolov DB (2015) The crystal structure of DR6 in complex with the amyloid precursor protein provides insight into death receptor activation. Genes Dev 29:785–790. 10.1101/gad.257675.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APP-GFP vesicles moving along the axon of hippocampal neuron (DIV14). Video at 100 frames per second (fps), with each frame being acquired every 0.1 s. The arrow indicates the anterograde direction. Images acquired before 60 min treatment with CAL-101 (1 μM). Scale bar represents 10 μm.
APP-GFP vesicles moving along the axon of hippocampal neuron (DIV14). Video at 100 frames per second (fps), with each frame being acquired every 0.1 s. The arrow indicates the anterograde direction. Images acquired after 60 min treatment with CAL-101 (1 μM). Scale bar represents 10 μm.
APP-GFP tubular vesicles moving along the axon of a hippocampal neuron (DIV18). Video at 100 frames per second (fps), with each frame being acquired every 0.1 s. The arrow indicates the anterograde direction. Scale bar represents 5 μm.
Extended data table related to Fig. 6. Table with statistical power calculation. Table with the values corresponding to the calculation of the effect size (Cohen's D), the post hoc statistical power and the p-value from the Student's t-test performed when analysing the results from Fig. 6. Download Figure 6-1, DOCX file (14.8KB, docx)
Extended data table related to Fig. 8. Table with statistical power calculation. Table with the values corresponding to the calculation of the effect size, the post hoc statistical power and the p-value from the ANOVA test performed when analysing the results from Fig. 8. Download Figure 8-1, DOCX file (14.8KB, docx)