

Abstract
Lymphoma is one of the most curable types of cancer. However, drug resistance is the main challenge faced in lymphoma treatment. Peroxisomal acyl-CoA oxidase 1 (ACOX1) is the rate-limiting enzyme in fatty acid β-oxidation. Deregulation of ACOX1 has been linked to peroxisomal disorders and carcinogenesis in the liver. Currently, there is no information about the function of ACOX1 in lymphoma. In this study, we found that upregulation of ACOX1 promoted proliferation in lymphoma cells, while downregulation of ACOX1 inhibited proliferation and induced apoptosis. Additionally, over-expression of ACOX1 increased resistance to doxorubicin, while suppression of ACOX1 expression markedly potentiated doxorubicin-induced apoptosis. Interestingly, downregulation of ACOX1 promoted mitochondrial location of Bad, reduced mitochondrial membrane potential and provoked apoptosis by activating caspase-9 and caspase-3 related apoptotic pathway. Overexpression of ACOX1 alleviated doxorubicin-induced activation of caspase-9 and caspase-3 and decrease of mitochondrial membrane potential. Importantly, downregulation of ACOX1 increased p73, but not p53, expression. p73 expression was critical for apoptosis induction induced by ACOX1 downregulation. Also, overexpression of ACOX1 significantly reduced stability of p73 protein thereby reducing p73 expression. Thus, our study indicated that suppression of ACOX1 could be a novel and effective approach for treatment of lymphoma.
Keywords: ACOX1, Apoptosis, Doxorubicin, Drug resistance, Lymphoma, p73, Protein degradation
INTRODUCTION
Malignant lymphoma is one of the most common hematologic malignancies and comprises 3.37% of all malignancies globally (1). Incidence rate of all lymphoma, Non-Hodgkin or Hodgkin lymphoma is approximately 20.3%, 17.37% or 2.92% (2), respectively. Incidence of malignant lymphoma globally increased at a rate of approximately 3–4% per year (1). Overall five-year survival rate for people with Non-Hodgkin or Hodgkin lymphoma is 53.7% or 78.9% (2), respectively. Standard treatments for lymphoma are anthracycline-based combinatorial chemotherapy regimens. Although patients achieve complete response after initial treatment, a substantial proportion of patients relapse. Relapsed lymphomas are refractory to subsequent treatments and exhibit cross-resistance to a variety of anti-cancer drugs (3, 4). Emergence of acquired chemoresistance poses a challenge in successful treatment of lymphoma. Thus, understanding the mechanism of drug resistance is crucial for successful treatment of lymphoma.
Acyl-CoA oxidase 1 (ACOX1) is a flavoenzyme that catalyzes the first and rate-limiting reaction of desaturation of very-long-chain acyl-CoAs to 2-trans-enoyl-CoAs and transfers electrons to molecular oxygen to generate hydrogen peroxide (5). ACOX1 deficiency usually causes peroxisomal disorders and has been linked to human diseases, such as pseudoneonatal adrenoleukodystrophy (6). Recent studies indicate that ACOX1 may involve in cancer development. ACOX1 knockout mice display growth retardation, infertility, excess very-long-chain fatty acids in the blood, microvesicular steatohepatitis, apoptosis, liver regeneration, and oxidative stress (7, 8). ACOX1 knockout mice eventually develop hepatocellular carcinoma (9). Findings suggest that peroxisomal ACOX1 dysfunction contributes to development of chronic liver disease and hepatocarcinogenesis. Currently, little is known about the role of ACOX1 in other cancers, including lymphoma, and the mechanism behind it remains to be elucidated.
p73, a member of the p53 family of tumor suppressors, shares a remarkable homology in DNA sequence and protein structure with p53 (10). p73 displays a certain degree of functional overlap with p53 (10). p53 is usually inactivated in human cancer by point mutations, but the p73 gene is rarely mutated in human cancers (10). Currently, p73 was found to be suppressed through various mechanisms including epigenetic silencing and post-translational modifications (11). In malignant lymphoma, mechanisms of epigenetic silencing or deletion are commonly responsible for inactivation of the p73 gene (12). However, how post-translational modifications regulate p73 protein stability has not been fully elucidated in malignant lymphoma.
In this study, we examined the role of ACOX1 in lymphoma cells. We found that ACOX1 was essential for proliferation of lymphoma cells. Overexpression of ACOX1 reduced the sensitivity of lymphoma cells to doxorubicin. While down-regulation of ACOX1 significantly enhanced doxorubicin-induced apoptosis. Additionally, ACOX1 participated in regulation of apoptosis by regulating activation of caspase-9 and caspase-3, and mitochondrial membrane potential. Importantly, p73, but not p53, was critical for mediating ACOX1 regulated apoptosis response. ACOX1 reduced p73 expression by destabilizing p73 protein. Our data indicated that ACOX1 could be a novel target for increasing drug sensitivity and improving treatment of lymphoma.
RESULTS
ACOX1 regulates proliferation and apoptosis
To evaluate the role of ACOX1 in lymphoma, ACOX1-Flag or ACOX1 shRNA were stably expressed via lentivirus-mediated gene transfer in lymphoma cells. As shown in Fig. 1A and Supplementary Fig. 1A, ACOX1-Flag was overexpressed in lymphoma cells. Overexpression of ACOX1-Flag significantly promoted proliferation in lymphoma cells (Fig. 1B, Supplementary Fig. 1B). While knockdown of ACOX1 expression (Fig. 1C, Supplementary Fig. 1C) markedly suppressed proliferation of lymphoma cells (Fig. 1D, Supplementary Fig. 1D). These data indicated that ACOX1 was essential for regulating lymphoma cell proliferation. We further examined if ACOX1 may participate in regulation of apoptosis. As shown in Fig. 1E, F and Supplementary Fig. 1E, F, overexpression of ACOX1-Flag did not cause apoptosis. While downregulation of ACOX1 slightly induced apoptosis as compared with negative control (NC) group (Fig. 1G, H, and Supplementary Fig. 1G, H). To further confirm the effect of ACOX1 on apoptosis, TUNEL assay was performed. Consistently, upregulation of ACOX1 did not induce apoptosis (Fig. 1I, J), while downregulation of ACOX1 induced apoptosis (Fig. 1K, L). These data implied that ACOX1 may participate in regulation of apoptosis in lymphoma cells.
Fig. 1.
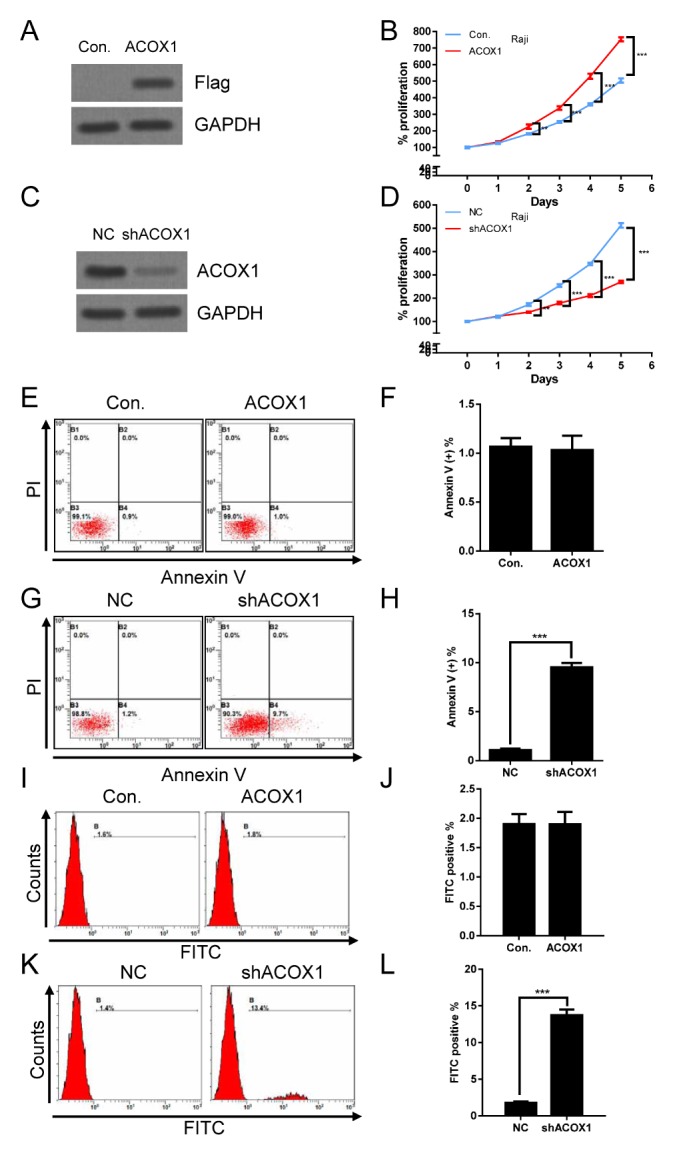
ACOX1 regulates proliferation and apoptosis. (A) ACOX1 was stably expressed in Raji lymphoma cells via lentivirus-mediated gene transfer. Cells were subjected to WB analysis. (B) Cells (1 × 105) stably expressed ACOX1 were culture for an indicated time. Then MTT assay was used to evaluate proliferation. (C) ACOX1 shRNA was stably expressed in Raji lymphoma cells via lentivirus-mediated gene transfer. Cells were subjected to WB analysis. (B) Cells (1 × 105) stably expressed ACOX1 shRNA were culture for an indicated time. Then MTT assay was used to evaluate proliferation. (E, F) Cells (1 × 106) stably expressed ACOX1 were subjected to apoptosis analysis (E). Statistical results are shown (F). (G, H) Cells (1 × 106) stably expressed ACOX1 were subjected to apoptosis analysis (G). Statistical results are shown (H). (I, J) Treatment was the same as (E) and (F). Cells were subjected to TUNEL assay analysis (I). Statistical results are shown (J). (K, L) Treatment was the same as (G) and (H). Cells were subjected to TUNEL assay analysis (K). Statistical results are shown (L). The bar represents mean ± SD of three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001, The ANOVA test, followed by Least Significant Difference test, were used to make statistical comparisons).
ACOX1 modulates doxorubicin-induced cytotoxic effect
Anthracycline-based combinatorial chemotherapy regimens are the most commonly used and efficacious regimens in lymphoma. Due to ACOX1 participation in regulation of apoptosis (Fig. 1G, H, and Supplementary Fig. 1G, H), we next evaluated if ACOX1 may affect cytotoxic effects of doxorubicin. As shown in Fig. 2A, B and Supplementary Fig. 2A, B, 0.6 μM doxorubicin treatment alone induced significant apoptosis in lymphoma cells. However, overexpression of ACOX1 markedly reduced doxorubicin caused apoptosis (Fig. 2A, B and Supplementary Fig. 2A, B). Also, our data showed that the combination of ACOX1 downregulation and doxorubicin treatment resulted in greater apoptosis in lymphoma cells than was achieved with either ACOX1 downregulation or doxorubicin alone (0.1 μM) (Fig. 2C, D and Supplementary Fig. 2C, D). To further confirm the above results, the TUNEL assay was performed. Consistently, upregulation of ACOX1 reduced doxorubicin-induced apoptosis (Fig. 2E, F), while downregulation of ACOX1 enhanced doxorubicin-induced apoptosis (Fig. 2G, H). These data indicated that ACOX1 could be a potential target to enhance cytotoxic effects or prevent drug resistance of doxorubicin.
Fig. 2.
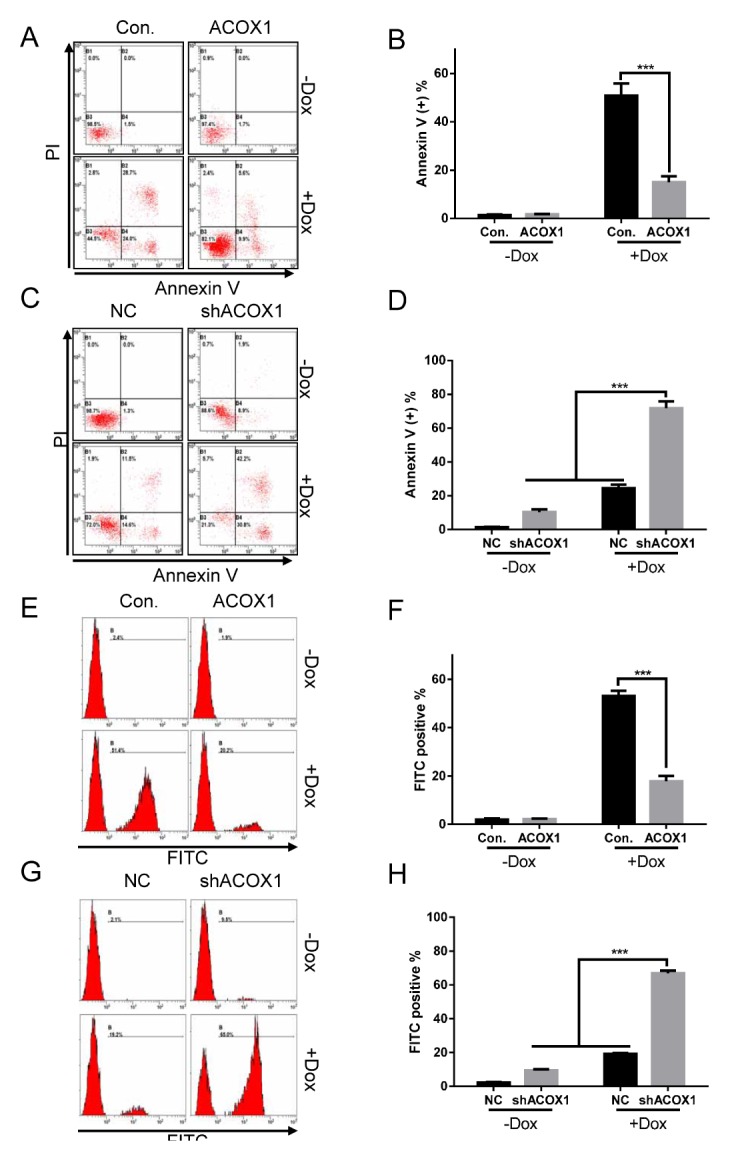
ACOX1 modulates doxorubicin-induced cytotoxic effect. (A, B) ACOX1 expressed Raji cells and control cells (Con.) were treated with doxorubicin (0.6 μM) or control solvent for 48 hours. Cells were subjected to apoptosis analysis (A). Statistical results are shown (B). (C, D) shACOX1 expressed Raji cells and control cells (NC) were treated with doxorubicin (0.1 μM) or control solvent for 48 hours. Cells were subjected to apoptosis analysis (C). Statistical results are shown (D). (E, F) Treatment was the same as (A) and (B). Cells were subjected to TUNEL assay analysis (E). Statistical results are shown (F). (G, H) Treatment was the same as (C) and (D). Cells were subjected to TUNEL assay analysis (G). Statistical results are shown (H). The bar represents mean ± SD of three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001, The ANOVA test, followed by Least Significant Difference test, were used to make statistical comparisons).
Downregulation of ACOX1 induced apoptosis through the intrinsic pathway
We next examined the mechanism of ACOX1 regulated apoptosis. As shown in Fig. 3A and Supplementary Fig. 3A, downregulation of ACOX1 significantly increased activities of caspase-9 and caspase-3, correlating with increased PARP cleavage. Cleavage of procaspase-8 was not significantly altered after downregulation of ACOX1 (Fig. 3A and Supplementary Fig. 3A). We confirm this by using different caspase inhibitors, including LEHD (inhibition of caspase-9 or caspase-3), DEVD (inhibition of caspase-3) and IETD (inhibition of caspase-8). As shown in Fig. 3B, LEHD or DEVD effectively reversed shACOX1 induced apoptosis. While IETD did not reverse shACOX1 induced apoptosis (Fig. 3B). These data imply that ACOX1 mainly regulated apoptosis through the intrinsic apoptotic pathway. Consistently, the combination of ACOX1 downregulation and doxorubicin treatment induced a more significant increase of activated caspase-9 and caspase-3, and PARP cleavage as compared with either ACOX1 downregulation or doxorubicin alone (Fig. 3A and Supplementary Fig. 3A). Bad translocation induces perme-abilization of the outer mitochondrial membrane and leads to reduction of mitochondrial membrane potential (MMP) and cytochrome C releasing (13). Indeed, we observed that downregulation of ACOX1 induced mitochondrial translocation of Bad and pBad (Ser112) (Fig. 3C and Supplementary Fig. 3B), cytoplasmic translocation of cytochrome C (Fig. 3C and Supplementary Fig. 3B), and reduction of MMP (Fig. 3D and Supplementary Fig. 3C). We further evaluated if overexpression of ACOX1 affected the above effects. As shown in Fig. 3E and Supplementary Fig. 3D, overexpression of ACOX1 reversed doxorubicin-induced activation of caspase-9 and caspase-3 and PARP cleavage. Consistently, overexpression of ACOX1 effectively rescued doxorubicin-induced reduction of MMP (Fig. 3F and Supplementary Fig. 3E).
Fig. 3.
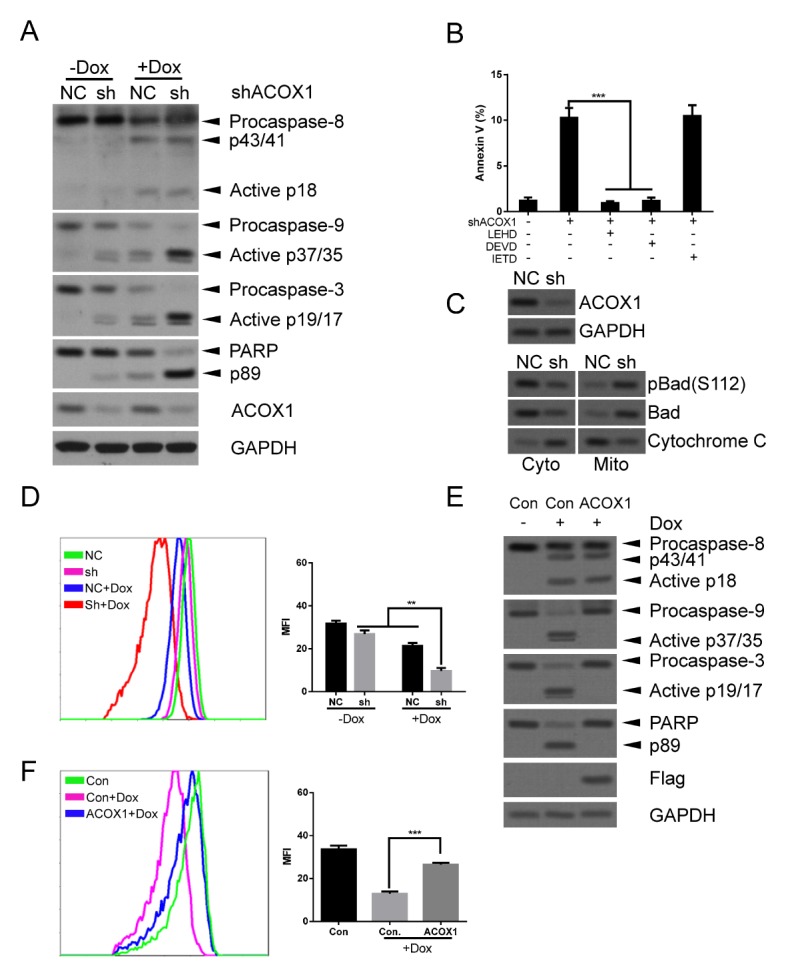
Downregulation of ACOX1 induced apoptosis through the intrinsic pathway. (A) shACOX1 expressed Raji cells (sh) and control cells (NC) were treated with doxorubicin (0.1 μM) or control solvent for 48 hours. Cells were subjected to WB analysis. (B) shACOX1 expressed Raji cells (sh) and control cells (NC) were treated with LEHD, DEVD, IETD or control solvent for 24 hours. Cells were subjected to apoptosis analysis. Statistical results are shown. (C) shACOX1 expressed Raji cells (sh) and control cells (NC) were subjected to total protein extraction or cytoplasmic/mitochondria protein extraction. These proteins were subjected to WB analysis. (D) Treatment was similar with (A). Cells were subjected to mitochondrial membrane potential analysis. (E) ACOX1 expressed Raji cells and control cells (Con.) were treated with doxorubicin (0.6 μM) or control solvent for 48 hours. Cells were subjected to WB analysis. (F) The treatment was similar with (E). Cells were subjected to mitochondrial membrane potential analysis. The bar represents mean ± SD of three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001, The ANOVA test, followed by Least Significant Difference test, were used to make statistical comparisons).
ACOX1 destabilizes p73 to reduce apoptosis
We further evaluated the downstream target of ACOX1. We found that p53 expression was not induced by downregulating ACOX1 (Fig. 4A and Supplementary Fig. 4A), while p73 expression was enhanced by downregulating ACOX1 (Fig. 4A and Supplementary Fig. 4A). We also examined the expression of two p53 phosphorylated sites (Ser15 and Ser20) required for p53-dependent apoptosis (14). Our data showed that phosphorylation of these sites did not change when ACOX1 was downregulated (Supplementary Fig. 4B). These data indicate that p73 was critical for ACOX1 regulated apoptosis. We inhibited p73 expression under downregulation of ACOX1 to evaluate apoptosis. Our data shows that downregulation of ACOX1 failed to induce apoptosis when the p73 expression was inhibited (Fig. 4B, C and Supplementary Fig. 4C, D). We further confirmed regulation of p73 expression by ACOX1. As shown in Fig. 4D and Supplementary Fig. 4E, doxorubicin treatment activated expression of p53 and p73. Overexpression of ACOX1 abolished doxorubicin-induced p73 expression but not p53 expression (Fig. 4D and Supplementary Fig. 4E). We also examined downstream targets of p73. As shown in Fig. 4E and Supplementary Fig. 4F, downregulation of ACOX1 promoted expression of p73 downstream targets, including p21, Bax, and GADD45 (15). We also found that ACOX1 did not regulate p73 expression at transcription level (Fig. 4E) or translation level (Supplementary Fig. 4G). We found that MG132 treatment abolished ACOX1 induced reduction of p73 expression (Fig. 4F), suggesting that ACOX1 may regulate p73 expression at the post-translational level. Interestingly, our data shows that overexpression of ACOX1 significantly reduced protein stability of p73, while downregulation of ACOX1 enhanced protein stability of p73 (Fig. 4G, H, and Supplementary Fig. 4H, I). To confirm this, a pulse-chase assay was performed. Consistently, p73 stability was reduced by overexpressing ACOX1 (Supplementary Fig. 4J, K). We also found that ACOX1 interacted with p73 protein (Supplementary Fig. 4L), indicating that ACOX1 may regulate p73 stability via protein interaction. Together, our data suggests that ACOX1 regulated apoptosis by modulating protein stability of p73. A previous study showed that PPARα was critical for ACOX1 regulated effects. Indeed, downregulation of ACOX1 upregulated PPARα expression (Supplementary Fig. 5A). We found that increasing of PPARα expression reduced doxorubicin-induced apoptosis (Supplementary Fig. 5B, C), suggesting that ACOX1 induced PPARα expression may counteract the apoptotic effect.
Fig. 4.

ACOX1 destabilizes p73 to reduce apoptosis. (A) shACOX1 expressed Raji cells (sh) and control cells (NC) were subjected to WB analysis. (B, C) shACOX1 expressed Raji cells (sh) and control cells (NC) were transfected with p73 siRNA for 48 hours. Cells were subjected to WB analysis (B) or apoptosis analysis (C). (D) ACOX1 expressed Raji cells and control cells (Con.) were treated with doxorubicin (0.6 μM) or control solvent for 48 hours. Cells were subjected to WB analysis. (E) shACOX1 expressed Raji cells (sh) and control cells (NC) were subjected to RNA extraction and PCR analysis. (F) Cells were treated with MG132 (10 μM). Cells were subjected to Western blot analysis. (G, H) ACOX1 expressed, shACOX1 expressed Raji cells and control cells were treated with CHX for the indicated time. WB analysis of p73, ACOX1, and GAPDH was performed (G). The densitometric quantification of p73 normalized to GAPDH was plotted against various time points to determine its half-life (H).
DISCUSSION
In this study, we elucidated the oncogenic role of ACOX1 in lymphoma. Our data shows ACOX1 was essential for proliferation of lymphoma cells. Additionally, overexpression of ACOX1 alleviated doxorubicin-induced apoptosis, while downregulation of ACOX1 aggravated doxorubicin-induced apoptosis. Also, ACOX1 participated in regulation of apoptosis by modulating mitochondrial membrane potential and activation of caspase-9 and caspase-3. Importantly, ACOX1 destabilizes p73 to suppress activation of the intrinsic apoptosis pathway.
Peroxisomes play a critical role in metabolism of reactive oxygen species, bile acids, very-long-chain, and branched-chain fatty acids (16). Cancer cells usually reprogram metabolism to support their malignant phenotype. Dysfunction of peroxisomes may lead to metabolic alterations and contribute to cancer development (17). Current study suggests that deregulation of peroxisome function may contribute to drug resistance in lymphoma cells (18). Peroxisomal ACOX1 is commonly expressed in most tissues and is critical for desaturation of very-long-chain acyl-CoAs to 2-trans-enoyl-CoAs. ACOX1 deficiency caused endoplasmic reticulum stress, accumulation of reactive oxygen species, VLCFAs and enlarged peroxisomes and is connected with human disease (19, 20). In this study, we show that ACOX1 functioned as an oncogene in lymphoma cells to promote proliferation and counteract doxorubicin-induced apoptosis. Our data implies that suppression of ACOX1 could be a novel and effective approach for treatment of lymphoma. Deficiency of ACOX1 results in activation of PPARα and induces progressive endoplasmic reticulum stress contributing to hepatocarcinogenesis (9). The lack of ACOX1 and PPARα causes absence of spontaneous peroxisome proliferation in aged mice (21), indicating that PPARα is critical for ACOX1 regulated effects. In our study, upregulation of PPARα was also observed when ACOX1 expression was downregulated. In this situation, upregulation of PPARα may be a protective mechanism for lymphoma cells to escape apoptosis. This phenomenon implies that the combination of ACOX1 and PPARα suppression could achieve more significant anti-cancer effects.
Expression of p73 is maintained at low levels in mammalian cells and is usually controlled through mechanisms including epigenetic silencing and post-translational modifications (11). In this study, our data suggests that p73 was destabilized via a proteasome related-degradation mechanism during ACOX1 overexpression in malignant lymphoma cells. The mechanism of p73 degradation has not been fully elucidated. A previous study showed that Itch, a Nedd4-like HETC-E3 ubiquitin ligase, selectively binds and ubiquitinates p73 to induce proteasome-dependent degradation of p73 (22). Also, NEDL2 bind to p73, p73 is ubiquitinated and stabilized in a NEDL2-dependent manner (23). Additionally, NQO1 physically interacts with p73 in an NADH-dependent manner and protects it from 20S proteasomal degradation (24). Additionally, DNA damage can induce phosphorylation of p73 by c-Abl, leading to stabilization of p73 protein (25). Our data provides a novel ACOX1-related mechanism to demonstrate deregulation of p73 expression in lymphoma. p73 is induced in response to DNA damage. A previous study showed that deficiency of ACOX1 results in increased intracellular H2O2 levels (9), usually causing DNA damage and stress. We speculated that destabilization of p73 may be caused by downregulating ACOX1 induced DNA damage and stress response. Besides, our data indicates that ACOX1 interacted with p73, implying that interaction between ACOX1 and p73 may interfere with degradation of p73 protein via proteasome related-mechanism. However, more data about the physical interaction between ACOX1 and p73 is needed to elucidate the mechanism.
In conclusion, our study uncovers a previously unknown oncogenic function of ACOX1 in lymphoma. We anticipate that our findings will provide a new target for overcoming drug resistance of traditional chemotherapy in lymphoma.
MATERIALS AND METHODS
Detailed information is provided in the Supplementary Information.
Reagents and cell culture
Doxorubicin, Cycloheximide (CHX), MG132, WY-14643, and Annexin V-FITC Apoptosis Detection Kit were purchased from Sigma-Aldrich (MA, USA). Antibodies against Flag, Caspase-3, Caspase-8, Caspase-9, PARP, Bax, phospho-Bad (Ser112), Cytochrome C, p53, p53(Ser15), p53(Ser20), p73, and GAPDH were purchased from Cell Signaling Technology (Boston, USA). Antibodies against ACOX1 was purchased from Abcam (Cambridge, U.K.). Antibody against puromycine was purchased from Merk (Darmstadt, Germany). LEHD, DEVD, and IETD were purchased from R&D Systems (MN, USA). Raji and Daudi cells were obtained from the ATCC (Manassas, VA, USA) and were cultured in RPMI-1640 (Thermo Fisher Scientific, USA) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, USA). Cells were maintained at 37°C in a humidified 5% CO2 atmosphere.
Western blot (WB) analysis
Cells were lysed in RIPA buffer containing protease inhibitors. Equal amounts of protein were analyzed by electrophoresis in SDS-PAGE gels. Proteins were transferred to nitrocellulose membrane. Nitrocellulose membranes were blocked by 3% BSA in TBST at RT and then were incubated with indicated antibodies at 4°C overnight. Nitrocellulose membranes were incubated for one hour at room temperature with secondary antibodies. Antibody binding was evaluated with an enhanced chemiluminescence kit (Pierce, USA).
Lentivirus preparation and transfection
Lentivirus preparation and transfection were described previously (26). 5 × 106 293T cells were transfected with 12 μg lentiviral vector, 9 μg psPAX2 (Addgene, MA, USA) and 3 μg pMD2.G (Addgene, MA, USA). Supernatants were collected every 24 hours after 12 hours transfection and concentrated via ultracentrifugation. Viral titer was determined by serial dilutions. Multiplicity of infection for transfection was 10.
Statistical analysis
Statistical analyses were performed using SPSS software, version 16.0 (SPSS Inc.). The ANOVA test, followed by Least Significant Difference test, was used when performing multiple comparisons (27). Level of significance was set at P < 0.05.
Supplementary Information
ACKNOWLEDGEMENTS
This study was supported by the National Natural Science Foundation of China (No. 81602585 to Fei-Meng Zheng), Science and Technology Planning Project of Guangdong Province (No. 2017A030313522 to Fei-Meng Zheng), Fundamental Research Funds for the Central Universities (No. 18ykpy13 to Fei-Meng Zheng), Kelin Nova Program of The First Affiliated Hospital of Sun Yat-sen University (Fei-Meng Zheng) and the National Natural Science Foundation of China (No. 81401905 to Wang-Bing Chen). We thank the members of Quentin Liu’s and Zi-Jie Long’s laboratory for their critical comments and technical support.
Footnotes
CONFLICTS OF INTEREST
The authors have no conflicting interests.
REFERENCES
- 1.Huh J. Epidemiologic overview of malignant lymphoma. Korean J Hematol. 2012;47:92–104. doi: 10.5045/kjh.2012.47.2.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith A, Crouch S, Lax S, et al. Lymphoma incidence, survival and prevalence 2004–2014 sub-type analyses from the UK’s Haematological Malignancy Research Network. Br J Cancer. 2015;112:1575–1584. doi: 10.1038/bjc.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shanbhag S, Ambinder RF. Hodgkin lymphoma: A review and update on recent progress. CA Cancer J Clin. 2018;68:116–132. doi: 10.3322/caac.21438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Armitage JO, Gascoyne RD, Lunning MA, Cavalli F. Non-Hodgkin lymphoma. Lancet. 2017;390:298–310. doi: 10.1016/S0140-6736(16)32407-2. [DOI] [PubMed] [Google Scholar]
- 5.Zeng J, Li D. Expression and purification of his-tagged rat peroxisomal acyl-CoA oxidase I wild-type and E421 mutant proteins. Protein Expr Purif. 2004;38:153–160. doi: 10.1016/j.pep.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 6.Fournier B, Saudubray JM, Benichou B, et al. Large deletion of the peroxisomal acyl-CoA oxidase gene in pseudoneonatal adrenoleukodystrophy. J Clin Invest. 1994;94:526–531. doi: 10.1172/JCI117365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan CY, Pan J, Chu R, et al. Hepatocellular and hepatic peroxisomal alterations in mice with a disrupted peroxisomal fatty acyl-coenzyme A oxidase gene. J Biol Chem. 1996;271:24698–24710. doi: 10.1074/jbc.271.40.24698. [DOI] [PubMed] [Google Scholar]
- 8.Fan CY, Pan J, Usuda N, Yeldandi AV, Rao MS, Reddy JK. Steatohepatitis, spontaneous peroxisome proliferation and liver tumors in mice lacking peroxisomal fatty acyl-CoA oxidase. Implications for peroxisome proliferator-activated receptor alpha natural ligand metabolism. J Biol Chem. 1998;273:15639–15645. doi: 10.1074/jbc.273.25.15639. [DOI] [PubMed] [Google Scholar]
- 9.Huang J, Viswakarma N, Yu S, et al. Progressive endoplasmic reticulum stress contributes to hepatocarcinogenesis in fatty acyl-CoA oxidase 1-deficient mice. Am J Pathol. 2011;179:703–713. doi: 10.1016/j.ajpath.2011.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melino G, De Laurenzi V, Vousden KH. p73: Friend or foe in tumorigenesis. Nat Rev Cancer. 2002;2:605–615. doi: 10.1038/nrc861. [DOI] [PubMed] [Google Scholar]
- 11.Oberst A, Rossi M, Salomoni P, et al. Regulation of the p73 protein stability and degradation. Biochem Biophys Res Commun. 2005;331:707–712. doi: 10.1016/j.bbrc.2005.03.158. [DOI] [PubMed] [Google Scholar]
- 12.Nemajerova A, Petrenko O, Trumper L, Palacios G, Moll UM. Loss of p73 promotes dissemination of Myc-induced B cell lymphomas in mice. J Clin Invest. 2010;120:2070–2080. doi: 10.1172/JCI40331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zorova LD, Popkov VA, Plotnikov EY, et al. Mitochondrial membrane potential. Anal Biochem. 2018;552:50–59. doi: 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chao C, Herr D, Chun J, Xu Y. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 2006;25:2615–2622. doi: 10.1038/sj.emboj.7601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones EV, Dickman MJ, Whitmarsh AJ. Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochem J. 2007;405:617–623. doi: 10.1042/BJ20061778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antonenkov VD, Grunau S, Ohlmeier S, Hiltunen JK. Peroxisomes are oxidative organelles. Antioxid Redox Signal. 2010;13:525–537. doi: 10.1089/ars.2009.2996. [DOI] [PubMed] [Google Scholar]
- 17.Dahabieh MS, Di Pietro E, Jangal M, et al. Peroxisomes and cancer: The role of a metabolic specialist in a disease of aberrant metabolism. Biochim Biophys Acta Rev Cancer. 20181870:103–121. doi: 10.1016/j.bbcan.2018.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Dahabieh MS, Ha Z, Di Pietro E, et al. Peroxisomes protect lymphoma cells from HDAC inhibitor-mediated apoptosis. Cell Death Differ. 2017;24:1912–1924. doi: 10.1038/cdd.2017.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferdinandusse S, Denis S, Hogenhout EM, et al. Clinical, biochemical, and mutational spectrum of peroxisomal acyl-coenzyme A oxidase deficiency. Hum Mutat. 2007;28:904–912. doi: 10.1002/humu.20535. [DOI] [PubMed] [Google Scholar]
- 20.Yu S, Rao S, Reddy JK. Peroxisome proliferator-activated receptors, fatty acid oxidation, steatohepatitis and hepatocarcinogenesis. Curr Mol Med. 2003;3:561–572. doi: 10.2174/1566524033479537. [DOI] [PubMed] [Google Scholar]
- 21.Hashimoto T, Fujita T, Usuda N, et al. Peroxisomal and mitochondrial fatty acid beta-oxidation in mice nullizygous for both peroxisome proliferator-activated receptor alpha and peroxisomal fatty acyl-CoA oxidase. Genotype correlation with fatty liver phenotype. J Biol Chem. 1999;274:19228–19236. doi: 10.1074/jbc.274.27.19228. [DOI] [PubMed] [Google Scholar]
- 22.Rossi M, De Laurenzi V, Munarriz E, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836–848. doi: 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miyazaki K, Ozaki T, Kato C, et al. A novel HECT-type E3 ubiquitin ligase, NEDL2, stabilizes p73 and enhances its transcriptional activity. Biochem Biophys Res Commun. 2003;308:106–113. doi: 10.1016/S0006-291X(03)01347-0. [DOI] [PubMed] [Google Scholar]
- 24.Asher G, Tsvetkov P, Kahana C, Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005;19:316–321. doi: 10.1101/gad.319905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73alpha and their collaboration to induce apoptosis. Nature. 1999;399:809–813. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- 26.Zheng F, Yue C, Li G, et al. Nuclear AURKA acquires kinase-independent transactivating function to enhance breast cancer stem cell phenotype. Nat Commun. 2016;7:10180. doi: 10.1038/ncomms10180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zheng FM, Long ZJ, Hou ZJ, et al. A novel small molecule aurora kinase inhibitor attenuates breast tumor-initiating cells and overcomes drug resistance. Mol Cancer Ther. 2014;13:1991–2003. doi: 10.1158/1535-7163.MCT-13-1029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.