

SUMMARY
Neoantigen-specific T cells are increasingly viewed as important immunotherapy effectors, but physically isolating these rare cell populations is challenging. Here, we describe a sensitive method for the enumeration and isolation of neoantigen-specific CD8+ T cells from small samples of patient tumor or blood. The method relies on magnetic nanoparticles that present neoantigen-loaded major histocompatibility complex (MHC) tetramers at high avidity by barcoded DNA linkers. The magnetic particles provide a convenient handle to isolate the desired cell populations, and the barcoded DNA enables multiplexed analysis. The method exhibits superior recovery of antigen-specific T cell populations relative to literature approaches. We applied the method to profile neoantigen-specific T cell populations in the tumor and blood of patients with metastatic melanoma over the course of anti-PD1 checkpoint inhibitor therapy. We show that the method has value for monitoring clinical responses to cancer immunotherapy and might help guide the development of personalized mutational neoantigen-specific T cell therapies and cancer vaccines.
Graphical Abstract
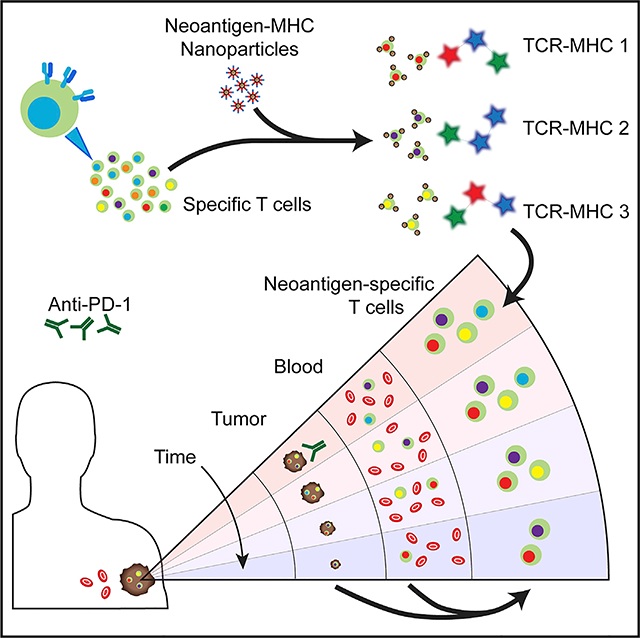
In Brief
Peng et al. report a sensitive method to detect tumor-associated neoantigen-specific T cells. Neoantigens and fluorescent DNA barcodes, presented on nanoparticle scaffolds, permit multiplex capture and analysis of specific T cell populations from blood or tumor. Neoantigen-specific T cell numbers track tumor volume in a melanoma patient responding to immunotherapy.
INTRODUCTION
Tumor neoantigens have been implicated in T cell recognition of tumors and are useful in the design of personalized cancer vaccines (Carreno et al., 2015; Gubin et al., 2014; Ott et al., 2017) and T cell receptor (TCR)-engineered adoptive cell therapies (Stroncek et al., 2012; Zacharakis et al., 2018). Neoantigens are mutation-containing peptide fragments of tumor-associated mutant proteins that can be presented by major histocompatibility complex (MHC) class I protein complexes for CD8+ T cell surveillance. These neoantigens are potentially recognized by highly specific TCRs, thus avoiding off-target interactions. The tumor specificity of neoantigens, coupled with the ability of neoantigen-specific T cells to selectively kill cancer cells (Berger and Mardis, 2018; Lu et al., 2014; Robbins et al., 2013), have made them increasingly important for cancer immunotherapy.
Putative neoantigen peptides can be predicted by analyzing the tumor exome for mutated genes that may result in the presentation of a mutational peptide to T cells (Gee et al., 2018; Lu et al., 2014; Robbins et al., 2013; van Rooij et al., 2013; Yadav et al., 2014). Candidates are typically ranked according to level of expression and the predicted peptide-MHC (pMHC) binding affinity (Fritsch et al., 2014; Nielsen et al., 2007). Experimental testing of which candidate neoantigens are actually generating an anti-tumor T cell response is challenging. For example, considering only somatic mutations, a given tumor might yield 50 or more putative neoantigens with 500 nM or lower calculated binding constant (KD) to a given HLA allele, and each patient will have 6 or so such alleles. Second, any given neoantigen-specific T cell clone is likely to exist in low abundance. Yet, harnessing neoantigen-specific T cells for therapy has yielded promising clinical results, highlighting the value of meeting these challenges. One approach involves directly expressing putative neoantigens within antigen-presenting target cells that are HLA-genotype matched with the patient, and then incubating those cells with tumor infiltrating lymphocytes (TILs) or T cells from peripheral blood mononuclear cells (PBMCs) to identify neoantigen reactive T cell populations (Linnemann et al., 2015; Robbins et al., 2013). This approach can identify such populations but cannot quantitatively enumerate them. A second approach involves the use of multi-color-labeled MHC tetramers for multiplex flow cytometry (Andersen et al., 2012). pMHC tetramers labeled for mass cytometry analysis (Fehlings et al., 2017; Newell et al., 2013), or DNA-labeled tetramers designed for sequencing analysis (Bentzen et al., 2016; Zhang et al., 2018), have also been reported. These flow cytometry methods typically require reasonably large cell populations for analysis and are often used to analyze in vitro-expanded T cells (Rizvi et al., 2015). Such expansion can significantly alter T cell population profiles. Nevertheless, these methods have been used to identify neoantigen-specific CD8+ T cell populations in high mutation burden tumors (McGranahan et al., 2016).
A further measurement challenge is to match the neoantigen-specificity of a T cell with the TCR α and b chains. The (single cell) pairSEQ technique (Howie et al., 2015) provides an elegant approach for assembling the full TCR gene sequence but does not establish the antigen specificity of that gene (Glanville et al., 2017; Han et al., 2014). In general, the dual challenge of identifying neoantigen-specific T cells and matching them with their cognate TCR genes increases in difficulty as abundance of the individual T cell populations drops.
Here, we report on the method of nanoparticle (NP)-barcoded nucleic acid cell sorting (NACS) for the sensitive enumeration and isolation of antigen-specific CD8+ T cells from small samples of patient tumor cells or blood. This method relies on pMHC tetramers coupled, by DNA linkers, to magnetic NPs. The NPs provide a convenient handle to isolate the desired cell populations and permit a high loading of pMHC tetramers on the NP surface to increase tetramer avidity. The DNA linkers can be further used as barcodes for multiplexed antigen-specific T cell population profiling. We first demonstrate that the approach exhibits significantly superior recovery of antigen-specific T cells spiked into a background of healthy human PBMCs, relative to the multi-color tetramer-based flow cytometry gold standard method (Andersen et al., 2012). We then apply the method to profile neoantigen-specific T cell populations in the tumor and blood from patients with melanoma over the course of their treatment with anti-PD1 immunotherapy. We also demonstrate TCRα/β gene sequencing of captured antigen- and neoantigen-specific T cells. These results demonstrate that NP-barcoded NACS is a sensitive tool for antigen-specific CD8+ T cell capture and analysis. They also indicate that such analysis can provide a valuable and highly specific immune profiling tool for monitoring cancer immunotherapy responses in patients.
RESULTS
Construction of NP-Barcoded NACS Reagents and Sensitivity Analysis for Antigen-Specific T Cell Capture
The basic components of a NP-barcoded NACS reagent are shown in Figure 1A. The reagent is comprised of pMHC tetramers linked to magnetic NPs by single-stranded DNA (ssDNA) oligomers, and each component is designed for one or more specific purposes. The biotin-labeled pMHC components were prepared as described in the literature (Bakker et al., 2008; Celie et al., 2009; Rodenko et al., 2006). For the tetramer scaffold, we used cysteine-modified streptavidin (SAC) (Ramachandiran et al., 2007), which is engineered for ssDNA labeling in a site-specific manner (Kwong et al., 2009) that does not interfere with biotin binding. We explored a size range of iron oxide magnetic NPs to find that a radius (r) = 500 nm was optimal. Smaller particles (r = 50 nm) require strong magnetic fields and extensive processing for cell enrichment, which reduces cell recovery. Particles of r >1 μm rapidly precipitate, making them difficult to manipulate. Each NP can present up to 300,000 ssDNA oligomers, which provide handles for hybridization with DNA-functionalized SAC molecules. This modularity of components, coupled with DNA-directed assembly, makes the preparation of a pNP library straightforward; most components can be prepared ahead of time and cryogenically stored until ready for use. In addition, as elaborated below, the sequence of each DNA oligomer can serve as a barcode for the identity of a specific peptide antigen, thus enabling multiplexed analysis. An assembled NP-barcoded NACS reagent is called a pNP. Each pNP presents >20,000 pMHC tetramers (Figure S1A; STAR Methods). To capture and enumerate a single antigen-specific T cell population using NP-barcoded NACS, a small cocktail of pNPs is prepared, each with a different fluorophore. One pNP is specific to the T cell population of interest, and the others serve as controls. That cocktail is mixed with viability-stained CD8+ T cells (Figure 1B). The pNP-bound cells and free particles are then isolated with a magnet, and the captured cells are imaged and counted on a hemocytometer chip (Figure 1B, top right). A cell is considered specific to the neoantigen of interest if it is decorated with a multiplicity (typically 5–20) of NPs of a single fluorescent color. To enumerate mixed populations of antigen-specific T cells, one can either repeat this type of analysis in a serial manner (Figure 1B, bottom right) or implement a parallel NP-barcoded NACS protocol by using the oligomer barcode, as described below.
Figure 1. Peptide-MHC (pMHC) Tetramer Functionalized Magnetic Nanoparticles (NPs) for Antigen-Specific T Cell Enumeration.

(A) Biotinylated pMHC, DNA-labeled cysteine-modified streptavidin (SAC-DNA), and an NP decorated with DNA (NP-DNA) are coupled by biotin-streptavidin interaction and DNA hybridization. This produces a pMHC tetramer NP (pNP), with each particle presenting at least 20,000 pMHC tetramers. In addition to a SAC hybridization site, the DNA on the NP also has a fluorescent oligo docking site (barcode) for hybridizing dyelabeled ssDNA.
(B) In a typical antigen-specific T cell enumeration, a specific pNP reagent (e.g., peptide 1) is mixed with around 10,000 viability-stained CD8+ T cells. For sensitivity comparison in (C), two irrelevant pNPs labeled with different oligo dyes are also added to characterize non-specific binding. After incubating the pNP reagents with the CD8+ T cells, pNP-bound cells and free particles are isolated with a magnet and spread across a hemocytometry chip for enumeration by microscopy. The optical and fluorescent micrograph series shows a typical antigen-specific T cell (top right). In the bright field image, the T cell appears black due to the pNPs that decorate its surface. In fluorescent images, the viability stain (green) confirms the presence of a live cell, and its colocalization with only peptide 1 pNP (red) confirms its specificity. Scale bars represent 10 μm. Serial NP-barcoded NACS: to analyze more than one antigen-specific population using this method, the supernatant containing the free cells is sequentially analyzed with different antigen-presenting pNPs (e.g., peptide antigen 1, 2, …n).
(C) Plots comparing the sensitivity of pNP capture (left, n = 4) and flow cytometry (right, n = 3) for the analysis of MART-1 TCR-engineered T cells spiked into 10,000 CD8+ donor PBMCs. Each plot includes a straight-line fit, the slope, percent recovery, and the R2 fitting metric.
See also Figure S1.
We first tested the sensitivity of a pNP reagent for antigen-specific CD8+ T cell capture by comparing its capture efficiency against the gold standard multi-color flow cytometry method (Andersen et al., 2012) (Figure 1C). For this comparison, we used the realistic background of healthy donor CD8+ PBMCs. Into this background we spiked a known number of anti-MART-1 (F5) TCR engineered T cells (Johnson et al., 2006). To engineer those cells, we used recently reported high-efficiency CRISPR gene editing methods to knock out the endogenous TCR and knock in the F5 TCR (Roth et al., 2018). The efficiency of that preparation, as assessed by flow cytometry analysis of CD3+ cells, was around 34% (Figure S1B). Between 8 and 128 of these engineered cells were spiked into 10,000 CD8+ donor PBMCs. The analysis preparation included viability staining as well as MART-1 and control pMHC to help differentiate spiked-in and background cells and exclude non-specific cells (STAR Methods; Figures S1C–S1F). We observed that non-specific binding of pNPs to cells is easily distinguished from specific cells (Figures S1D–S1F). Both NP-barcoded NACS and flow cytometry yielded linear performance for detecting the MART-1-specific T cells spiked into healthy donor PBMCs (Figure 1C). However, the NP-barcoded NACS method captured 94% of the expected MART-1 specific T cells, whereas the flow method detected only 52%. The increased capture efficiency of the pNPs likely arises from the increased avidity enabled by the NP scaffold. A few individual cells identified as expressing MART-1-specific TCRs by either the NP-barcoded NACS method (n = 3) or by the multicolor flow method (n = 5) were analyzed and found, in fact, to express the F5 TCR. This analysis shows that although both methods capture the targeted T cell populations, the NP-barcoded NACS method exhibits an almost 2-fold increased capture efficiency.
Multiplexed Analysis of TILs from Biopsies of Patients with Metastatic Melanoma by using NP-Barcoded NACS
We applied the NP-barcoded NACS approach to enumerate multiple antigen-specific T cell populations from biopsies of four patients with metastatic melanoma responding to anti-PD1 (pembrolizumab) cancer immunotherapy within a phase I trial (Ribas et al., 2016). To this end, we constructed libraries of putative neoantigens by analyzing the exome and transcriptome of pre-therapy resected tumor materials from three patients expressing HLA-A*02:01 allele (Table S1). The fourth patient expressed the HLA-A*03:01 allele. The analysis provided input for in silico prediction of putative neoantigens, rank-ordered by pMHC binding affinity, and filtered for gene expression. The putative neoantigen lists for the patients, along with gene expression and wild-type peptide information, are found in Table S2. For the fourth patient, we did a more limited search to demonstrate the generality of the approach, as described below.
We developed a parallel NP-barcoded NACS protocol by modifying the process flow of Figure 1B in two ways, as illustrated in Figure 2. First, we created a barcoded pNP library with each library element defined by a 3-position, 3-color barcode, to yield 27 (33) distinct DNA barcodes to be paired to a unique putative neoantigen (Figure 2A). Second, we developed a microfluidic chip to trap individual pNP-labeled (barcoded) cells (Figures 2B and S2A) for decoding. The chip design contained a series of 60 micro-chambers, each with 10 cell traps, and connected by microfluidic channels. This chip design permitted about 10% capture efficiency of barcoded cells, which is suitable to sample population trends of expanded T cell specimens. A near term goal is the design of a cell-trap microchip that permits more efficient cell capture. By comparison, the hemocytometer chip yielded near 100% cell capture.
Figure 2. Parallel NP-Barcoded NACS and Sample TIL Analysis.

(A) A barcoded pNP library is formed, with each library element containing a unique peptide and 3-position oligo docking site (barcode). Each position can be one of three unique sequences for hybridizing green, yellow, or red-labeled ssDNA, creating 27 (3 3 3 3 3) unique DNA barcodes. All elements of the barcoded pNP library are combined and mixed with CD8+ cells, and the particle-bound cells (barcoded cells) are isolated and physically immobilized in an array of microfluidic cell traps. To decode the barcode, sets of dye-labeled ssDNA are sequentially hybridized, imaged, and displaced for three rounds (one for each position). Thus, each of the 27 unique peptides is associated with a unique sequence of three colors from the fluorescent readout.
(B) Optical micrograph shows a microfluidic chamber equipped with 10 cell traps, 9 of which contain single barcoded T cells. This microchip has 60 such cell capture microchambers. Scale bar represents 50 μm.
(C) The fluorescent micrograph series are the sequential reads of the three barcode positions, with the fluorescent readouts for the 9 trapped cells provided to the right (R, red; G, green; Y, yellow). The cell at position viii does not provide a clean read, while the cell at position ix was lost during the process. Some traps contain 2 cells (for example, iv), and so reads are only done on those cells that are clearly delineated in the images. Scale bar represents 5 μm.
(D) The DNA barcode key shows each of the 27 color sequences and their corresponding neoantigen identity or, at position #8, the MART-1 tumor antigen. For example, the cell at position #3 reads “YRG” and corresponds to neoantigen 12. The Y(D) notation (red font) implies a Y for D mutation in the neoantigen sequence.
See also Figure S2.
We carried out the parallel NP-barcoded NACS protocol on patient #1 CD8+ TILs by mixing a 27-element barcoded pNP library with the specimen (Figure 2A). The barcoded cells were isolated from unbound cells by using a magnet, purified of free NPs by using a transwell membrane, and immobilized in individual traps on the microchip. To decode the barcodes, a set of dyelabeled ssDNA was hybridized to the first position and read out using fluorescence microscopy. The dye-labeled ssDNAs were removed using displacement ssDNAs, and a second set of dye-labeled ssDNAs was added to read position two and so forth, until all 3 positions were decoded for each cell (Figures 2A and 2C). Each of the 27 antigen specificities was associated with a unique barcode sequence (Figure 2D). The chemistry associated with barcode readout, along with validation data, is provided in Figures S2B–S2E. The sample sorting scheme prior to analysis is provided in Figure S2F. For the full list of the ssDNA reagents and their corresponding barcode usage, refer to Table S3.
Figures 2B–2D are representative data from the analysis of TILs from patient #1. The fluorescent micrographs (Figure 2C) are sequential reads of the 9 trapped cells seen in Figure 2B. The DNA barcode key (Figure 2D) pairs the 27 color sequences with the pNP antigen identity. For example, the cell at position iii reads “YRG,” which assigns it as a neoantigen 12-specific CD8+ T cell. Typically, we obtained either a high-fidelity read (position viii) or a nonsense read to be discarded (position ix). In some instances, a trap may capture two cells (see position iv), which can be separately barcoded by microscopic inspection. The combination of fluorescent barcoding plus cell imaging thus yields a high fidelity to the approach.
We tested the reproducibility of the parallel NP-barcoded NACS approach by separately analyzing 2 separate vials of TILs from the same tumor biopsy, using freshly prepared reagents each time, and carrying out each analysis more than 1 week apart. From these analyses, we determined that 4% of the CD8+ TILs exhibited specificity to library elements 1–27 (Figure S3A). We also recorded a 0.5% non-selective capture rate, based upon testing the library against CD4+ T cells from the same sample. Importantly, both analyses yielded the same seven neoantigen-specific T cell populations (Figure S3B). Here, we define neoantigen-specific T cell populations as detected if identified by 3 or more unambiguous reads, or identified in more than one analysis of patient #1 TILs.
The above TIL analyses revealed a larger number of neoantigen-specific T cell populations than have been reported by using flow cytometry (Carreno et al., 2015; Gubin et al., 2014; McGranahan et al., 2016; Schumacher and Schreiber, 2015). This prompted us to analyze expanded TILs from patient #1 by using the multiplexed flow cytometry method (Andersen et al., 2012). For this analysis, we prepared a 14-element tetramer library presenting a subset of 13 putative neoantigens (9 of which were detected using parallel NP-barcoded NACS) and MART-1. With a conservative gating scheme reflective of the multiplexed flow method (Andersen et al., 2012), we only identified neoantigen 12 in patient #1 TILs (Figures S3C–S3E). With a non-conservative gating scheme (Figures S3F and S3G), we identified neoantigen 12, plus signals (>7 cells) for 3 populations (5, 15, and 27). The 7-cell cutoff for the non-conservative gating scheme was selected based upon a background level of 7 cells identified as specific for the conditional (J) pMHC tetramer.
We also used the serial hemocytometer NP-barcoded NACS method (Figure 1B) to analyze patient TILs. We first established the selectivity of this method by using the full pNP library (Table S2) associated with patient 1 to analyze TILs from a different patient with metastatic melanoma on the same clinical trial, as well as PBMCs from a healthy donor. The list of neoantigens is unique to patient #1 and should not capture T cell populations from the other specimens. The results for both controls were similar. The patient #1 pNP library captured on average 2 cells per library element from a total of 10,000 CD8+ TILs, with a SD of 1.4 (Figures S3H and S3I). Thus, we set a detection threshold of 5 cells (2 SDs above the mean). For either control sample, no library element captured more than 5 cells, and there was no correlation between the controls, indicating that none of the patient #1 library elements exhibited intrinsically low selectivity. This finding was consistent with the analysis of spiked specimens. We used serial NP-barcoded NACS to analyze patient #2 TILs and observed a similar number of neoantigen-specific T cell populations, as were found for patient #1 (Figures S4A and S4B). In contrast, serial analysis of patient #3 TILs yielded fewer populations compared to patients #1 and #2 (Figures S4C–S4E).
Kinetics of Neoantigen-Specific T Cell Populations in a Patient with Metastatic Melanoma Responding to Anti-PD1 Immunotherapy
Because therapeutic responses were mediated by tumor-infiltrating CD8+ cells (Tumeh et al., 2014), we reasoned that the kinetics of neoantigen-specific T cell populations might provide insights into cancer patient responses to anti-PD1 checkpoint inhibitor treatments. We, thus, carried out a comparative analysis of neoantigen-specific TILs and PBMCs from patient #1 and explored how those detected neoantigen-specific T cell populations evolved over time and how they related to independent measures of tumor volume in that patient. The response of patient #1 to therapy is documented in the computed tomography (CT) scans of Figure 3A and in the lesion size timeline of Figure 3B.
Figure 3. Analysis of Neoantigen-Specific CD8+ T Cell Populations from Patient #1 TILs and PBMCs Over the Course of Treatment with Anti-PD1 Therapy.

(A) Representative CT scan of patient #1, who had metastatic melanoma to the chest wall, pleura, and lung, progressing after three prior lines of therapy with high doses of interleukin-2, vemurafenib, and TIL adoptive cell therapy. Upon administration of pembrolizumab, the patient had a transient progression for 3 months, with an increase in the size of the chest wall masses and pleural effusion (Dec 2012), followed by a long-lasting tumor regression.
(B) Timeline of the lesion size and neoantigen-specific T cell counts in CD8+ PBMCs. Day 0 is the start of treatment. A baseline tumor biopsy was collected for genomic and transcriptomic analysis at day –28 (star symbol). Black dots represent CT-scan measurement of lesion size (left y axis), while black squares represent total neoantigen-specific T cells counted from 104 CD8+ PBMCs (right y axis). PBMCs analyzed at day 439 yielded no detectable neoantigen-specific populations. Earlier points of analyses are represented by arrows, color-coded for the bar graphs in (C).
(C) Neoantigen-specific T cell populations detected from day 187 TILs (top graph) and PBMCs (middle graphs) over the course of the therapy, along with mutation-containing mRNA read counts for the mutant proteins (bottom graph) from the baseline RNA-seq. All plots are arranged by descending mRNA read counts, and putative neoantigens with undetectable transcripts (on the right) are controls. The horizontal dashed lines in the TIL and PBMC plots represent the signal threshold above which the identification of a T cell population is statistically significant.
(D) ELISpot assays of IFN-γ secretion from each of the neoantigen-specific T cell populations detected from day 41 PBMCs. The baseline (dashed line) was established as the average background level by using an identical number of CD8+ T cells from a healthy donor. The left y axis represents the neoantigen-specific cells detected in (C) and the right y axis represents, in an analysis of a different vial of day 41 CD8+ PBMCs, the numbers of ELISpots detected. Neoantigen numbers 1 and 10 (red font x axis label) are controls. Micrographs of representative ELISpot assays are shown for a healthy donor and for a neoantigen-specific T cell population. Scale bar represents 500 μm.
See also Figures S2–S4.
The above-described datasets from the patient #1 TIL analyses are combined in the top histogram of Figure 3C, along with a third parallel NP-barcoded NACS analysis that used a pNP library built from 23 additional putative neoantigens predicted to be more weakly binding (Figure S3B). Each bin on the histogram is associated with a specific putative neoantigen. The numerical identifier of each neoantigen indicates its rank order of calculated antigen-MHC binding strength. A lower numerical index indicates a stronger binder. In the histograms, the neoantigens are arranged, from left to right, according to the measured expression level of the associated transcript (bottom histogram of Figure 3C). The transcriptome analysis was from a tumor biopsy collected 28 days prior to the start of therapy (start of Figure 3B timeline). For the 10 rightmost putative neoantigens (numbers 3, 5, 11, etc.), zero transcripts were detected.
In addition to TILs analyzed at day 187 following the start of anti-PD1 therapy, we performed serial NP-barcoded NACS to analyze non-expanded PBMCs from patient #1 at days 41, 187, 208, and 439 (middle three histograms of Figure 3C). There are several things to note about Figure 3C. First, all but one of the neoantigen-specific T cell populations observed in the TILs were also observed in at least one PBMC analysis. Second, for the neoantigen-specific T cell populations seen in more than one analysis, the detectable numbers of T cells were strongly correlated with the relevant transcript expression level (R2 = 0.76) (Figure S3J) and only loosely correlated with the predicted pMHC binding affinity (R2 = 0.20) (Figure S3K). Of the 10 putative neoantigens predicted from rare transcripts below the RNA sequencing (RNA-seq) detection limit, only T cells specific to the strongly binding neoantigen 5 were detected. Third, the detected numbers of neoantigen-specific CD8+ TILs strongly correlated with measured lesion size (Figure 3B). By day 439, no T cell populations were detected at all (Figure S3L). In contrast, for PBMCs collected at day 41, when the tumors appeared to be growing even after the start of therapy (an effect known as pseudo progression; Brandsma et al., 2008), neoantigen-specific T cells were present in their greatest relative abundance.
We also performed serial NP-barcoded NACS analysis of PBMCs from patient #3 (Figures S4C–S4E). For this patient, two neoantigen-specific T cell populations (numbers 13 and 20) were detected in TILs. The most dominant population (#13) was also detected in PBMCs at day 25 following the start of therapy, along with four additional neoantigen-specific populations. Only T cell populations specific to neoantigens 13 and 14 were detected at the time point (day 87) closest to maximum therapeutic response. Although patient #3 was participating in the same sample trial as patient #1 and #2, patient #3 showed a very different response profile to anti-PD1 therapy and had a lower abundance of neoantigen-specific T cells.
Functional and Genetic Validation of Identified Neoantigen-Specific T Cells
To validate the neoantigen-specific populations identified by NP-barcoded NACS, we analyzed functional activity by an ELISpot cell secretion assay and, for one population, carried out single-cell TCR gene sequencing. Exposure of antigen-specific CD8+ T cells to their cognate antigen-MHC leads to functional activation, which is commonly detected by interferon gamma (IFN-γ) release. To confirm the functional activation of the neoantigen-specific CD8+ populations from patient #1 PBMCs collected at day 41, we prepared a 14-element tetramer library, stimulated the PBMCs with the individual tetramers, and measured IFN-γ secretion with an ELISpot single cell assay (Figure 3D). The baseline (dashed line) was established as the average background level by using an identical number of CD8+ T cells from a healthy donor. Of the 12 neoantigen-specific elements in the 14-element library, 11 of them exhibited ELISpot counts above background, whereas no ELISpots were detected in the negative controls (neoantigen numbers 1 and 10).
We also used NP-barcoded NACS to isolate a single neoantigen-specific cell from patient #1 PBMCs and determined both its antigen specificity and TCRα/β genes (Figure 4). For this experiment, we designed a cell capture microchip with a low-density of cell traps (STAR Methods) so that the cell could be barcoded to identify the antigen specificity (Figure 4A), and then punched out of the device for TCR sequencing. For this case, the identity of the cell was neoantigen 12. The sequenced TCR genes were cloned into a retroviral vector. Jurkat cells (a CD3+ T cell immortalized line) that were transduced to express the TCR were found to bind to the neoantigen 12 tetramer (Figure 4B), demonstrating that matched TCR-antigen pairs can be identified from single cells isolated by NP-barcoded NACS. Importantly, these two neoantigen-specific T cell characterization assays confirm that the populations detected by NP-barcoded NACS are, in fact, present in the patient-derived specimens and are functionally active.
Figure 4. Determination of T Cell Receptor (TCR) Genes for a Corresponding Neoantigen-Specific T Cell by Using NP-Barcoded NACS.

(A) The optical micrograph shows a captured barcoded T cell from patient #1, followed by the 3 sequential fluorescent readout steps to identify specificity against neoantigen 12. Scale bars represent 20 μm. The captured single T cell was punched out for RT-PCR to obtain TCRα and TCRβ gene sequences (DNA ladder: 100 bp). The TCR gene was then assembled and inserted into a retroviral vector and transduced into Jurkat T cells for analysis by flow cytometry.
(B) Flow cytometry results for untransduced Jurkats (left), Jurkats transduced with LNGFR expression reporter only (center), and Jurkats transduced with both LNGFR and the TCR specific for neoantigen 12 (right).
See also Figures S3 and S4.
Extension to HLA-A*03:01
We carried out a limited library analysis (patient #4, 5 neoantigens + MART-1; Table S2) of non-expanded PBMCs from the patient expressing the HLA-A*03:01 allele and, by using the hemocytometry approach, identified a single neoantigen-specific T cell population (Figures S4F and S4G). The neoantigen identified (SLHAHGLSYK, gene F5) had a high predicted pMHC binding strength (8.22 nM), although cell populations for putative neoantigens with similar binding strength (7.44, 16.23 nM) were not found. We subsequently expanded the neoantigen-specific cells and used tetramer staining to sort single cells of that same neoantigen-specific T cell population into individual microwells for TCR sequencing (Han et al., 2014). Of 24 wells where single cells were sorted, TCR β chains were sequenced from 10 wells (Figure S4H). One β chain was identified from 4 different wells and another b chain was identified in 3 different wells, demonstrating a polyclonal response to this neoantigen, but also demonstrating the generality of this method beyond HLA-A*02:01.
DISCUSSION
Individual populations of neoantigen-specific T cells are typically scarce in patient tumor tissues or blood, and this can make them extremely challenging to study. Nevertheless, knowledge about those populations can provide guidance for various vaccination (Gubin et al., 2014) or cell-based cancer immunotherapies (Tran et al., 2016). The NP-barcoded NACS approaches described here are simple to implement and, yet, outperform state-of-the-art multi-color flow cytometry for the detection of these low-abundance cells. The high capture efficiency of the NP-barcoded NACS approach likely arises from the increased avidity of the NP-presentation format for the pMHC tetramers, which is especially useful for small sample sizes. The parallel and serial NP-barcoded NACS approaches used similar reagents for cell capture but with distinct advantages. The serial approach has limited multiplexing but features near the unity cell capture efficiency (Figure 1C), whereas the parallel approach has excellent multiplexing (Figure 2) but lower capture efficiency. The captured cells remain viable for further functional or genetic investigations (Figure 4).
In the analysis of TILs and/or PBMCs collected from patients responding to anti-PD1 therapy, we made three major observations. First, we observed that 6%–32% of the top predicted neoantigens for HLA-A*02:01 can be paired with T cell populations (Figures 3C, S3B, S4B, and S4D). Including additional HLA alleles for each patient (Table S1), as well as including weaker binding neoantigens, would almost certainly yield additional T cell populations. The implication is that a few tens of % of the CD8+ TILs within these patient tumors may be neoantigen-specific. This number is high relative to previous reports from patient sample analyses (Carreno et al., 2015; McGranahan et al., 2016) but is consistent with a prior result in mice (Gubin et al., 2014). The neoantigen-specific T cell population numbers detected here are consistent with immune responses to tumor antigens that have been observed in healthy donors (Strønen et al., 2016). In that work, PBMCs from the healthy donor were expanded in the presence of a library of neoantigens so as to amplify those rare populations that exhibit specificity against library elements. With the additional sensitivity gained by the NP-barcoded NACS method, cell expansion and the associated distortion of T cell populations may no longer be necessary.
A second observation is that the measured spectrum of neoantigen-specific T cell populations only correlates loosely with the calculated antigen-MHC binding affinity, but there is a strong correlation with mRNA expression levels (Figures S3J and S3K), which is consistent with recent work on antigen profiling (Abelin et al., 2017). These data should provide guidance for the refinement of neoantigen prediction algorithms (Balachandran et al., 2017).
A third observation is that the same neoantigen-specific populations detected in the tumor are also found in the blood, albeit at a lower abundance relative to all CD8+ PBMCs. This is consistent with a recent observation in patients with melanoma (Gros et al., 2016). In patient #3, a neoantigen-specific T cell population was found in blood 25 days after the start of checkpoint inhibitor immunotherapy, which was also observed in TILs a month prior to start of therapy (Figure S4D). In the analysis of patient #1, neoantigen-specific populations were detected in blood a full 2 months prior to the observation of actual tumor shrinkage by CT scan (Figures 3B and 3C). In this patient, we observed a strong correlation between the kinetics of tumor shrinkage and the abundance kinetics of neoantigen-specific T cells in PBMCs. The NP-barcoded NACS method for enumeration is currently challenging to integrate with flow cytometry for single-cell isolation and sequencing of TCRs due to the NP light scattering properties. It is likely that smaller diameter NP scaffolds may be useful for this purpose (Bentzen et al., 2016).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James R. Heath (jheath@systemsbiology.org). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patients, Treatment, and Specimen Collection
Tumor biopsy and peripheral blood cell collection and analyses were approved by UCLA IRBs 11–001918 and 11–003066. Patients with metastatic melanoma were selected for the current analysis by being HLA-A*02:01 positive, having an adequate baseline biopsy as well as an on-treatment biopsy, and exhibiting an objective tumor response while participating in a phase 1 trial of pembrolizumab. Patients #1, #2 and #3 received single agent pembrolizumab intravenously at 10 mg/kg every 3 weeks (10Q3W). Tumor responses were evaluated starting at 12 weeks, confirmed 4 weeks after first response, and imaged every 12 weeks thereafter. Response was characterized by both the Response Evaluation Criteria in Solid Tumors (RECIST) and the immune-related response criteria (irRC). Tumor biopsies from the patients analyzed were obtained at baseline and on therapy and were processed with one aliquot immediately fixed in formalin followed by paraffin embedding for pathological analyses, a second aliquot snap frozen by immediate immersion in liquid nitrogen for genetic analyses, and a third aliquot minced fresh under sterile conditions followed by DNase/collagenase digestion to create single cell suspensions (s.c.s) before cryopreservation in liquid nitrogen. Peripheral blood mononuclear cells (PBMCs) were prepared from fresh whole blood by Ficoll-Paque density gradient centrifugation and cryopreserved for NP-NACS analysis. To evaluate the generality of the methods developed here, we also analyzed PBMCs of an HLA-A*03:01 positive patient (patient #4). Patient information for patients #1, #2, and #3 is detailed in Table S1.
Primary Cell Culture, Expansion, and Sorting Prior to Analysis
Unless specified otherwise, primary tumor infiltrating lymphocytes (TILs), PBMCs or gene-edited T cells expressing anti-MART-1 (F5) TCR (a gift from PACT pharma) were cultured in R10 media (RPMI 1640 containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin) in a humidified incubator at 37°C with 5% CO2. TILs were expanded from cryopreserved s.c.s by incubating in R10 media supplemented with anti-CD3 antibody (OKT3, 50 ng/mL, 48hr exposure) and IL-2 (300 IU/mL) and re-cyropreserved at 5×106 cells/mL after 2–4 weeks of expansion in the same media without anti-CD3 antibody. Prior to analysis, TILs or unexpanded PBMCs were thawed and treated with DNase for 45 min, and stained with antibodies to CD4 (BV510, BioLegend, San Diego, CA) and CD8 (BV605, BioLegend, San Diego, CA). Live (7AAD-negative) populations of CD4 and CD8 single-positive cells were sorted using a fluorescence activated cell sorter (FACS) (BD Biosciences, San Jose, CA). Some PBMC specimens were sorted using magnetically activated cell sorting (MACS) kits (Miltenyi biotec, Bergisch Gladbach, Germany). Following the manufacturer’s protocol, live cells were first purified using the dead cell removal kit, and then the CD8+ population was enriched using the CD8+ T Cell Isolation kit. Briefly, thawed cells were incubated with the dead cell removal microbeads, and the microbead-bound apoptotic cells were removed using a magnetic column, while live cells were collected in media. Subsequently, the live cells were incubated with a biotinylated antibody cocktail that binds to non-CD8+ cells in PBMCs, as well as streptavidin-coated microbeads. The microbead-bound cells were removed using a magnetic column, while live CD8+ cells were collected in media for analysis. For single-cell TCR sequencing of Patient #4 PBMCs, the cells were activated by culturing them for 2 days in expansion media (ImmunoCult-XF T Cell Expansion Medium, STEMCELL Technologies Inc.) supplemented with IL-2 (50 IU/ml) and magnetic particles coated with anti-CD3 and anti-CD28 antibodies (Dynabeads Human T-Activator CD3/CD28, ThermoFisher), and then expanded for up to 7 days in the same media without the particles.
Cell Lines
Cell lines (HEK293T/17 and Jurkat E6–1) were purchased from American Type Culture Collection (ATCC) and cultured a humidified incubator at 37°C with 5% CO2. HEK293T/17 cells were grown in D10 media (DMEM containing 10% fetal bovine serum, 100 U/mL penicillin, and 100 mg/mL streptomycin) and Jurkat T cells were grown in R10 media. The cells were split every 2–3 days to maintain < 80% confluency for HEK293T/17 or < 106 cells/mL density for Jurkat.
METHOD DETAILS
Reagents and Materials
Unless otherwise specified, reagents were purchased from Sigma-Aldrich. Deionized water with resistivity of >18 megaohms∙cm was used to prepare all aqueous solutions. Roswell Park Memorial Institute (RPMI) 1640 Medium, Dulbecco’s Modified Eagle Medium (DMEM), phosphate buffered saline (PBS), and fluorescent-labeled streptavidins were purchased from ThermoFisher Scientific (Waltham, MA). Fetal bovine serum (FBS) was purchased from ATCC (Mannassas, VA). Penicillin-Streptomycin mixture (17–602E) was purchased from Lonza (Basel, Switzerland). Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, Iowa).
Whole-Exome Sequencing (WES), Mutation Calling, and HLA-Typing
Both DNA and RNA were extracted simultaneously from snap-frozen tumor biopsies using the QIAGEN AllPrep Kit, which consists of purification using two separate spin columns. DNA from tumors and matched normal blood samples were sequenced at the UCLA Clinical Microarray Core to determine candidate neoantigens. Paired-end 2×100bp sequencing was carried out on the HiSeq 2000 platform (Illumina, San Diego, CA) following exon capture using the Nimblegen SeqCap EZ Human Exome Library v3.0 (Roche), which targets 65 Mb of genome. Each targeted base was covered by an average of 90–150 reads. Sequences were aligned to the UCSC hg19 human genome reference using the BWA-mem algorithm (v0.7.9) (Li and Durbin, 2010). Preprocessing followed the GATK Best Practices Workflow v3, including duplicate removal (Picard Tools), indel realignment, and base quality score recalibration. Somatic mutations were called with methods modified from (Shi et al., 2014), using MuTect (v1.1.7) (Cibulskis et al., 2013), Varscan2 Somatic (v2.3.6) (Koboldt et al., 2012), and the GATK-HaplotypeCaller (HC, v3.3). Only high-confidence mutations were retained, defined as those identified by at least two out of three programs. For the GATK-HC, somatic variants were determined using a one-sided Fisher’s Exact Test (p value cut-off % 0.01) between tumor/normal pairs. Variants were annotated by Oncotator (Ramos et al., 2015), with non-synonymous mutations being those classified as Nonsense, Missense, Splice_Site, or Nonstop Mutations, as well as Frame_Shift, In_Frame, or Start_Codon altering insertions/deletions. HLA-typing was performed by ATHLATES (Liu et al., 2013) from the whole-exome sequencing data.
RNA Sequencing
RNA sequencing was performed using the Illumina HiSeq 2500 platform on 100-bp paired-end libraries prepared using the Illumina-TruSeq RNA sample preparation kit per the manufacturer’s instructions, to compare mutated RNA read counts with neoantigen-specific cells detected. Reads were mapped to hg19 using TopHat2 v2.0 (Kim et al., 2012), and were quantified and normalized using Cufflinks v2.2.1 (Trapnell et al., 2012) and CuffNorm to generate normalized expression tables by library size (fragments per kilobase of exon per million fragments mapped, FPKM) using the geometric normalization method. Mutation-containing read counts from RNA data were identified from the aligned bam files using the samtools mpileup feature as implemented by the pysam 0.15.0 software library, and verified by visual inspection in the Integrated Genomics Viewer (IGV) (Robinson et al., 2011).
Peptide HLA Binding Prediction and Neoantigen Candidate Identification
Peptide binding predictions to HLA-A*02:01 and HLA-A*03:01 were generated by netMHC3.4 (Lundegaard et al., 2008) for 9-mer and 10-mer peptides in a sliding window around each non-synonymous amino acid-altering mutation. Peptide sequences were derived from Ensembl GRCh37 release 74. Candidate peptides were binned by 1) those with mutation-containing reads identified by RNA-seq, 2) those with RNA expression (FPKM >0) but no identified mutated reads, and 3) all others without detectable RNA-seq expression. Peptides were ranked and sorted by HLA binding affinity within each bin. For patient #1, peptides with predicted binding affinity less than 500 nM were selected for further analysis. For patient #2 and #3, peptides with predicted binding affinity less than 50 nM were selected for further analysis. For patient #4, 5 peptides spanning binding affinities between 0 and 250 nM were selected for further analysis.
Production of SAC-DNA Conjugates
The SAC-DNA conjugate was produced following a previously published protocol (Kwong et al., 2009). Briefly, SAC was first expressed from the pTSA-C plasmid containing the SAC gene (Addgene) (Sano and Cantor, 1990). Before conjugation to DNA, SAC (1 mg/ml) was buffer exchanged to PBS containing Tris(2-Carboxyethyl) phosphine hydrochloride (TCEP, 5 mM) using Zeba desalting columns (Pierce). Then 3-N-Maleimido-6-hydraziniumpyridine hydrochloride (MHPH, 100 mM, Solulink) in DMF was added to SAC at a molar excess of 300:1. In the meantime, succinimidyl 4-formylbenzoate (S-4FB, 100mM, Solulink) in DMF was added to 5′-amine modified ssDNA (500 μM) in a 40:1 molar ratio. After reacting at rt for 4 hours, MHPH-labeled SAC and SFB-labeled DNA were buffer exchanged to citrate buffer (50 mM sodium citrate, 150 mM NaCl, pH 6.0), and then mixed at a 20:1 ratio of DNA to SAC to react at rt overnight. SAC-DNA conjugate was purified using the Superdex 200 gel filtration column (GE health) and concentrated with 10K MWCO ultra-centrifuge filters (Millipore).
Monomeric Human MHC Class I Neoantigen pMHC Library Construction
We used the well-established method of conditional antigen exchange to enable the rapid construction of a library of antigen-MHC tetramers by the release of a photo-labile peptide for HLA-A*02:01 (KILGFVFJV) or HLA-A*03:01 (RIYRJGATR) (Bakker et al., 2008; Celie et al., 2009; Rodenko et al., 2006). The photo-labile peptide and other neoantigen peptides were synthesized with standard automated Fmoc-peptide synthesis methodology (J, (S)-3-(Fmoc-amino)-3-(2-nitrophenyl) propionic acid, is the photo-labile amino acid residue) using Liberty/Titan peptide synthesizers (CEM/Aapptec). Plasmids encoded with DNA sequence for bacterial expression of human MHC Class I HLA-A*02:01 heavy chain subunit, human MHC Class I HLA-A*03:01 heavy chain subunit, and human β-2-microglobulin subunit were kind gifts from Ton N M Schumacher. E. coli (BL21-CodonPlus (DE3)-RIPL strain) transformed with these plasmids were induced to express protein inclusion bodies for their respective MHC subunits. The inclusion bodies were then extracted, purified, and folded in the presence of a photo-labile peptide according to the previously published protocol(Garboczi et al., 1992). The folded pMHC was then biotinylated overnight with BirA biotin ligase (2.5 μg BirA enzyme per 10 nmol protein) in 10 mM ATP, 10 mM MgOAc, 50 μM d-biotin, 50 mM bicine buffer at pH 8.3. To exchange the photo-labile peptide with a target peptide, mixtures of the photo-labile peptide-loaded MHC protein (0.5 μM) and each neoantigen peptide (50 μM) were exposed to 365 nm UV light for 1 hour.
Neoantigen pNP Library Construction
Streptavidin coated NPs (500 nm radius, Invitrogen Dynabeads MyOne T1) were prepared according to the manufacturer’s recommended protocol for biotinylated nucleic acid attachment. Briefly, these NPs were mixed with biotin-ssDNA at 1:20 ratio to obtain NP-DNA in the recommended DNA-binding buffer (10 mM Tris-HCl, 1 mM EDTA, 2 M NaCl, pH 7.5). Excess DNA was removed by washing and resuspending the NPs with PBS three times on a magnetic stand, and the particles are resuspended in PBS with 2 mM MgCl2. In parallel, neoantigen pMHC monomer library was added to SAC-DNA at a 4:1 ratio, incubating at rt with rotation for 30 min, to form the pMHC tetramer-DNA. To generate pNPs, equal amounts (in terms of DNA molar ratio) of NP-DNA and pMHC tetramer-DNA were hybridized at 37°C for 30 min, and washed once with PBS with 0.1% BSA and 2 mM MgCl2. For analysis of Patient #3, we used a simpler method of pNP construction by incubating the streptavidin NP with biotinylated pMHC monomer at a ratio of 1:4 to 1:8 at rt for 30min. Excessive pMHC monomer was removed by washing the NP twice with PBS with 0.1% BSA and 2 mM MgCl2. Typically, each NP-barcoded NACS analysis of < 10,000 cells uses 2.5 μL of stock NPs (28.2 million particles total) per library element.
Determining pMHC Tetramer Valency on NP
Two equivalents of NP particle (56.4 million total) were prepared with NY-ESO pMHC (A2 NY-ESO antigen: SLLMWITQV) as described above. The pMHC tetramer on NP was dehybridized in deionized water at 98°C, and concentrated with 10K MWCO ultra-centrifuge filters. The total protein was measured using a bicinchoninic acid protein assay kit (ThermoFisher), following manufacturer’s instructions. The total volume of concentrated protein was 45.9 μL, and the assay volume used was 25 mL. The standards were prepared and analyzed in triplicates. The theoretical maximum pMHC tetramer occupancy (M) is calculated as follows (Singha et al., 2017):
where d (the hydrodynamic diameter of pMHC tetramer) is estimated to be 8 nm, and D (diameter of a NP plus DNA) is estimated to be 1053.46 nm, which is calculated by adding the NP diameter (1000 nm) plus two times the length of the final DNA linker (2 × 26.73 nm). Here, the DNA linker is formed from biotin-DNA barcode hybridized to DNA-SAC (71 plus 10 base pairs). Thus, theoretical max number of pMHC tetramer on surface (M) is 70,400.
Sensitivity Comparison of NP-Barcoded NACS and Multicolor Flow Cytometry
Varying numbers (0, 8, 16, 32, 64, and 128) of F5 engineered T cells were spiked into 10,000 CD8+ donor PBMCs. The F5 T cells were stained with Calcein green, while the background PBMCs were stained with Calcein violet. These samples were prepared and analyzed in triplicate for flow cytometry, and quadruplicate for NP-barcoded NACS. For multicolor flow analysis, cells were stained with R-Phycoerythrin (PE) MART-1 tetramer, Allophycocyanin (APC) MART-1 tetramer, BV510 NY-ESO tetramer, and PerCP/Cy5.5 anti-CD8 antibody. For the NP-barcoded NACS comparison, cells were incubated with an equal mixture of MART-1 pNP (DNA-dyed with Cy5), CMV pNP (DNA-dyed with Cy3), and NY-ESO pNP (DNA-dyed with Alexa Fluor 750) at rt for 15–30min (A2 CMV p65 antigen: NLVPMVATV, A2 MART-1 antigen: ELAGIGILTVl). The magnetically labeled T cells were enriched using a magnet, re-suspended in 10 μl PBS with 0.1% BSA, and loaded into a plastic hemocytometer chip. The hemocytometer was imaged using bright field and fluorescence microscopy. To sequence the F5 TCR, individual cells from both techniques were sorted into wells of a 96-well plate containing cell lysis buffer (10mM Tris, pH = 8, with 1U/μl RNase inhibitor, Promega). Following 1 hour of lysis at −80°C, the cell lysates were split in half to perform separate alpha and beta gene amplification. TCR α and β domain genes were cloned from split cell lysates using a OneStep RT PCR kit (QIAGEN) with one primer set for F5-TRAV (5′-CAACAGAAGGAGGTGGAGCAGAA –3′ and 5′-TGGGTTTCACAGATAACTCCGTTCC-3′) and one for F5-TRBV (5′-ATCACCCAGGCACCAACATCTCA-3′ and 5′-CTACAACTGTGAGTCTGGTGCCT-3′). cDNA products were then used as templates in a second semi-nested amplification with a set of primers with Illumina upstream binding regions underlined (alpha forward primer: 5′- CCAGGGTTTTCCCAGTCACGACACAGAAGGAGGTG GAGCAGAATT –3′, alpha reverse primer: 5′- AGCGGATAACAATTTCACACAGGAGTTTCACAGATAACTCCGTTCCCTG –3′; beta forward primer: 5′- CCAGGGTTTTCCCAGTCACGACCCCAGGCACCAACATCTCAGATC –3′, beta reverse primer: 5′- AGCGGATAACAATTTCACACAGGACAACTGTGAGTCTGGTGCCTTGT –3′). Libraries were further amplified adapters were attached (Han et al., 2014), and libraries of TCR genes were sequenced on an Illumina MiSeq machine. For HLA-A*03:01 sorting, 3 neoantigen tetramers not detected using NP-NACS (patient #4, neoantigens 3–5 in Table S2) were labeled with APC and used as negative selection during FACS sorting before single cell TCR sequencing. TCR CDR3 regions were aligned using MixCR (Bolotin et al., 2015).
Serial NP-Barcoded NACS
Live CD8+ T cells from PBMC were either sorted by FACS or MACS. CD8+ T cells were then stained with Calcein AM (a green-fluorescent live cell stain, ThermoFisher) and incubated with each individual pNP library element at rt for 15–30 min. Neoantigen-specific cells were enriched by magnet pulldown. The non-captured T cells in the supernatant were collected for further incubation with other pNP library elements. The enriched T cells were washed by PBS with 0.1% BSA once to remove any non-specific cell pulldown. Cells were then loaded into a hemocytometer chip. The whole area in the hemocytometer was counted to obtain the total pulldown cell number. Healthy donor PBMC and/or PBMC from an unrelated melanoma patient were used as control to obtain the background.
Cell-Trapping Microfluidic Device Fabrication
Devices were fabricated using standard soft lithography techniques. First, a 20 to 25-μm height master mold with cell traps (multiple traps or single traps) was prepared using the SU-8 2025 photoresist. Briefly, SU-8 was spin coated at 3000 RPM for 60 s, and the SU-8 layer was exposed to UV (Karl Suss) and developed. After trimethylchlorosilane coating to prevent adhesion on the mold, Sylgard 184 (A:B = 10:1) mixture was then poured onto the mold, degassed, and cured at 80°C for 2 hours. In the meantime, a thin layer of PDMS was spin-coated onto a glass slide at 2000 rpm for 1 min and cured at 80°C for 1 hour. The PDMS device and PDMS-coated glass were treated with O2 plasma for 1 min and bound together to yield the final cell-trapping microfluidic device.
Parallel NP-Barcoded NACS
A patient-specific pNP library was incubated with CD8+ human T cells for 15–30 min at rt. The pNP-bound T cells were enriched by magnetic pulldown and washed in PBS with 0.1% BSA and 2 mM MgCl2 to remove any pulled-down T cells not bound to magnetic pNPs. The cells were then loaded into Costar transwell polycarbonate membranes (Corning, 5 μm pore) to remove free NPs. Then, the cells were loaded into the cell-trapping microfluidic device and sequentially barcoded. First, 3 different DNA-dyes (Cy3, Cy5 and Alex 488) complementary to the first position of their respective DNA barcodes were loaded into the device to hybridize with the barcode DNA on the NP at 37°C for 15 min. After a brief washing, fluorescent images were taken to obtain the first round barcode image for each trapped cell. Displacement DNAs were added to the device at 37°C for 15 min to remove the first round DNA dyes. Similar procedures were employed to obtain the second and third round barcoding images. The three colors imaged for NPs covering each cell were matched to the corresponding peptide identity (Table S3). DNA-dyes and Displacement DNAs were prepared in PBS with 0.1% BSA and 2 mM MgCl2 at 1 μM.
ELISPOT Assay
8000 CD8+ T cells from PBMCs (Day 41 from patient #1 or heathy donor, 100 μl in growth media supplemented with 300 IU/ml IL-2) were stimulated with 0.1 μM final concentration of neo-antigen tetramers for 26 hours in a 37°C CO2 incubator. Secreted human IFN-γ was detected by an ELISpot assay (R & D) following the manufacturer’s protocols, except with less cells (8000 instead of 50,000) and with more stringent washing by increasing the repeated washing to 6 times in each washing step. The number of spot forming cells were enumerated by bright field microscopy using a 2x objective.
Multicolor Flow Cytometry for Analysis of Patient #1 Expanded TILs
In Figure 3C (top panel) of the main text we present an analysis of patient #1’s expanded TILs using the parallel NP-barcoded NACS approach. We analyzed another vial of those similarly expanded TILs using the multicolor flow cytometry method. For this analysis, we prepared a 14-element tetramer library presenting a subset of 13 putative neoantigens (9 of which were detected using parallel NP-barcoded NACS) and MART-1. In Figures S3C–S3G, we present multiple analyses of the patient #1 TILs dataset. We study CD8+ cells, as well as a highly expressing subset termed CD8++ cells, which express CD8 at the highest decade of expression. For both CD8+ and CD8++ cells, we apply a tight and loose gating strategy. The loose gate is determined by visual separation of double-stained cells from single-stained and unstained cells in the FACS plot. The tight gate was calibrated using single-stained and unstained controls, and is set to eliminate any possible leak of control cells into the gate of the double-stained cells. The top table (Figure S3C) provides the dye labels that define each neoantigen-specific barcode. For example, a T cell that is dyed for both APC and PE will be identified as specific to neoantigen 1. In Figures S3D–S3G, the bottom left table provides the numbers of cells that were detected positive for only single colors. The bottom right table provides the numbers of cells that were detected as positive for two colors. Figure S3D represents the most stringent selection process – only CD8++ cells, with a tight gating strategy, were selected for analysis. Under this gating scheme (which is reflective of the multiplexed flow method; Andersen et al., 2012), one population of cells (#12) was stained with both BV-421 and PE dyes, and 699 such cells were detected. Only a total of 4 other cells are detected as stained with just a single dye. An analogous analyses of a healthy donor PBMC control did not detect any neoantigen-specific T cell (data not shown).
Single Cell TCR Cloning
Barcoded neoantigen-specific T cells were trapped in a microfluidic device with single cell traps, which are designed to be 2 mm apart from each other. This permits the isolation of the specific trapped cell by a 1 mm PDMS punch without disturbing other trapped cells. This process removes a 1 mm wide core of PDMS including the trapped cell. Brief sonication was applied to release the cell from the punched-out PDMS into 25 μL of cell lysis buffer (10mM Tris, pH = 8, with 1U/μl RNase inhibitor, Promega). Rearranged Vα and Vβ domain genes were cloned from single cells using a OneStep RT PCR kit (QIAGEN) with multiplexed forward primers that bind TRAV and TRBV gene segments (Table S4) and reverse primers that bind the constant Cα (5′-GCCACAGCACTGTTGCTCTTGAAGTCC-3′) and Cβ (5′-CCACCAGCTCAGCTCCACGTG-3′) domain genes. cDNA products were used as templates in a second semi-nested amplification with a universal set of primers (alpha forward primer: 5′-TGGCCTGCTTTGTTTGCCGTGGTTACAGGAAGCCTCAGCA-3′, alpha reverse primer: 5′-GCCACAGCACTGTTGCTCTTGAAGTCCATAG-3′; beta forward primer: 5′-CGG GCTCCTTTGCCTACCGTGCCTGCAGGAGGGCTCGGCA-3′, beta reverse primer: 5′-CGTGCTGACCCCACTGTGCACCTCCTTCCCATTCACCCACCAGCTCAGCTCCACGTGGTC-3′). Vα and Vβ cDNA were Sanger sequenced and re-amplified using single TRAV/TRBV forward primers to correct mispriming artifacts introduced through multiplexed PCR. Retroviral vectors were constructed for functional testing through PCR assembly as described previously (Bethune et al., 2016; Bethune et al., 2018). Briefly, the Vα and Vβ domain genes were assembled with human growth hormone (HGH) signal peptides, constant regions of the TCRα and TCRβ chains, and a 2A ribosomal skipping sequence, then digested with restriction enzymes and ligated into a MSCV-based non-replicative retro-viral backbone. To produce retrovirus, HEK293T/17 cells were transfected via calcium phosphate precipitation with the TCR vector, a packaging vector encoding gag-pol, and a pseudotyping vector encoding RD114 envelope glycoprotein. D10 media was replaced 24 hours following transfection and viral supernatant was collected 48 hours following transfection. An equal volume of viral supernatant was added to Jurkat T cells in R10 media (final density: 0.5 × 106 cells/mL) and polybrene was added to a final concentration of 5 μg/mL. Cell spinfection was carried out at 1350×g for 90 minutes at 30°C, and then cells were incubated with virus overnight at 37°C, 5% CO2. Half of the media was replaced 24 hours following infection and cells were assayed for TCR specificity 48 hours following infection via flow cytometry using cognate fluorescent pMHC tetramers.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data plotting, curve fitting, and statistical analyses (i.e., determination of fitting metric/correlation, R2) were performed using GraphPad Prism (GraphPad Software, San Diego, CA) and JMP (SAS, Cary, NC). Unless otherwise stated, individual values are plotted without averaging, and the number of experimental replicates are given in the figure legends.
DATA AND CODE AVAILABILITY
Putative neoantigens, mutated gene expression, and predicted binding affinity for patient #1, #2 and #3 are available in Table S2. A limited number of putative neoantigens and predicted binding affinity for patient #4 are available in Table S2. The published article also includes neoantigen-specific T cell counts enumerated during this study for the indicated neoantigen. The whole-exome sequencing data have been deposited to the Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra) under accession number NCBI SRA: SRP067938 for a separate study (Hugo et al., 2016). The transcriptome data have been deposited (NCBI GEO: GSE78220) for a separate study (Hugo et al., 2016). The patient ID numbering in NCBI SRA: SRP67938 and NCBI GEO: GSE78220 are not the same as in this manuscript; use direct SRR mapping in Table S1 for correctness.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE anti-human CD3 Antibody | BioLegend | Cat# 317307; RRID: AB_571912 |
| Brilliant Violet 510 anti-human CD4 Antibody | BioLegend | Cat# 317443; RRID: AB_2561377 |
| APC/Cyanine7 anti-human CD4 Antibody | BioLegend | Cat# 357415; RRID: AB_2616809 |
| Brilliant Violet 605 anti-human CD8 Antibody | BioLegend | Cat# 344741; RRID: AB_2566512 |
| PerCP/Cyanine5.5 anti-human CD8 Antibody | BioLegend | Cat# 344709; RRID: AB_2044009 |
| Alexa Fluor® 488 anti-human CD8 Antibody | BioLegend | Cat# 344716; RRID: AB_1054930 |
| PE anti-human CD271 (NGFR) Antibody | BioLegend | Cat# 345105; RRID: AB_2282827 |
| Bacterial and Virus Strains | ||
| E. coli (BL21-CodonPlus (DE3)-RIPL strain) | Fisher Scientific | Cat# NC9122855 |
| Biological Samples | ||
| TILs and PBMCs from patients with metastatic melanoma, see Table S1 | This paper | N/A |
| Gene-edited T cells expressing anti-MART-1 (F5) TCR | PACT pharma | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human IL-2 Protein | STEMCELL Technologies Inc. | Cat# 78036 |
| Tris(2-Carboxyethyl) phosphine hydrochloride (TCEP) | Millipore-Sigma | CAS# 51805-45-9 |
| 3-N-Maleimido-6-hydraziniumpyridine hydrochloride (MHPH) | Solulink | Cat# S-1009 |
| succinimidyl 4-formylbenzoate (S-4FB) | Solulink | Cat# S-1004 |
| Photo-labile peptide for HLA-A*02:01 (KILGFVFJV) | Andersen et al., 2012 | N/A |
| Photo-labile peptide for HLA-A*03:01 (RIYRJGATR) | Bakker et al., 2008 | N/A |
| Putative neo-antigens for patient samples, see Table S2 | This paper | N/A |
| BirA-500: BirA biotin-protein ligase standard reaction kit | Avidity LLC | Cat# BirA500 |
| Dynabeads MyOne T1 streptavidin-coated NPs (500 nm radius) | Invitrogen | Cat# 65602 |
| Calcein AM | ThermoFisher | Cat# C3099 |
| CellTrace Calcein Violet, AM, for 405 nm excitation | ThermoFisher | Cat# C34858 |
| 7-AAD Viability Staining Solution | BioLegend | Cat# 420403 |
| Critical Commercial Assays | ||
| Pierce BCA Protein Assay Kit | ThermoFisher | Cat# 23225 |
| AllPrep DNA/RNA Mini Kit | QIAGEN | Cat# 80204 |
| OneStep RT PCR kit | QIAGEN | Cat# 210210 |
| Human IFN-gamma ELISpot Kit | R&D systems | Cat# EL285 |
| Nimblegen SeqCap EZ Human Exome Library v3.0 | Roche | Cat# 06465684001 |
| Deposited Data | ||
| Whole exome sequencing data | Hugo et al., 2016 | NCBI SRA: SRP067938 |
| Transcriptome data | Hugo et al., 2016 | NCBI GEO: GSE78220 |
| Experimental Models: Cell Lines | ||
| Jurkat T cells | ATCC | ATCC® TIB-152 |
| HEK293T/17 | ATCC | ATCC® CRL-11268 |
| Oligonucleotides | ||
| DNA for NP modification (ssDNA, 5′-biotin-) (27), see Table S3 | This paper | N/A |
| DNA for barcoding (ssDNA, 5-Cy5/Cy3/AlexaFluor488/AlexaFluor750) (9), see Table S3 | This paper | N/A |
| DNA for displacement (ssDNA) (9), Table S3 | This paper | N/A |
| DNA for streptavidin labeling (ssDNA, 5-NH2-) (1), see Table S3 | This paper | N/A |
| Single cell TCRα cloning primers (57), see Table S4 | This paper | N/A |
| Single cell TCRβ cloning primers (65), see Table S4 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid containing SAC gene | Sano and Cantor, 1990 | Addgene Plasmid #17329 |
| HLA-A*02:01 plasmid | Andersen et al., 2012 | N/A |
| HLA-A*03:01 plasmid | Andersen et al., 2012 | N/A |
| β-2-microglobulin plasmid | Andersen et al., 2012 | N/A |
| Software and Algorithms | ||
| Cellsense v1.18 | Olympus | https://www.olympus-lifescience.com/en/software/cellsens/ |
| ImageJ/FIJI | Univ. of Wisc. Madison | https://loci.wisc.edu/software/fiji |
| Cell Profiler 3.0 | Cellprofiler | https://cellprofiler.org/ |
| FlowJo v.9 | FlowJo | https://www.flowjo.com/ |
| BWA-mem algorithm (v0.7.9) | Li and Durbin., 2010 | http://bio-bwa.sourceforge.net/ |
| MuTect v1.1.7, Varscan2 Somatic v2.3.6, and GATK-HaplotypeCaller v3.3 | Broad Institute | https://software.broadinstitute.org/gatk/ |
| ATHLATES | Liu et al., 2013 | https://www.broadinstitute.org/viral-genomics/athlates |
| TopHat2 v2.0 | Kim etal., 2012 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Cufflinks v2.21 | Trapnell et al., 2012 | https://github.com/cole-trapnell-lab/cufflinks |
| Integrated Genomics Viewer | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| netMHC3.4 | Lundegaard et al., 2008 | http://www.cbs.dtu.dk/services/NetMHC-3.4/ |
| GraphPad Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
| JMP 13 | SAS | https://www.jmp.com/en_us/home.html |
Highlights.
Peptide-MHC tetramers are displayed on nanoparticles to enhance T cell capture
Fluorescent barcodes enable multiplex capture of antigen-specific T cells
Neoantigen-specific T cell numbers track tumor regression in a melanoma patient
Neoantigen-specific T cell populations in the blood mirror those in the tumor
ACKNOWLEDGMENTS
We acknowledge the following agencies and foundations for support: The Stand Up to Cancer Foundation and the Cancer Research Institute (A.R. and J.R.H.), the National Cancer Institute (1U54 CA199090 and R01-CA170689 to J.R.H. and R35 CA197633 to A.R.), the Jean Perkins Foundation (J.R.H., principal investigator [PI]), and Caltech (internal support through a CI2 grant). O.N.W. is supported by the Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research. A.H.C.N. is supported by a Banting Postdoctoral Fellowship from the Government of Canada. A.M.X. is supported by a Ruth L. Kirschstein F32 Postdoctoral Fellowship from the National Cancer Institute (1F32CA213966–01). We thank Eugene Barsov and Richard Morgan (NCI) for providing the retroviral MSGV vector.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.07.106.
DECLARATION OF INTERESTS
D.B., A.R., and J.R.H. are scientific co-founders of PACT Pharma, a company that is seeking to commercialize certain aspects of the NP-barcoded NACS technology. S.P., A.H.C.N., W.C., J.C., and J.R.H. have at least one patent related to this work.
REFERENCES
- Abelin JG, Keskin DB, Sarkizova S, Hartigan CR, Zhang W, Sidney J, Stevens J, Lane W, Zhang GL, Eisenhaure TM, et al. (2017). Mass Spectrometry Profiling of HLA-Associated Peptidomes in Mono-allelic Cells Enables More Accurate Epitope Prediction. Immunity 46, 315–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen RS, Kvistborg P, Frøsig TM, Pedersen NW, Lyngaa R, Bakker AH, Shu CJ, Straten Pt., Schumacher TN, and Hadrup SR (2012). Parallel detection of antigen-specific T cell responses by combinatorial encoding of MHC multimers. Nat. Protoc. 7, 891–902. [DOI] [PubMed] [Google Scholar]
- Bakker AH, Hoppes R, Linnemann C, Toebes M, Rodenko B, Berkers CR, Hadrup SR, van Esch WJ, Heemskerk MH, Ovaa H, and Schumacher TN (2008). Conditional MHC class I ligands and peptide exchange technology for the human MHC gene products HLA-A1, -A3, -A11, and -B7. Proc. Natl. Acad. Sci. USA 105, 3825–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, et al. ; Australian Pancreatic Cancer Genome Initiative; Garvan Institute of Medical Research; Prince of Wales Hospital; Royal North Shore Hospital; University of Glasgow; St Vincent’s Hospital; QIMR Berghofer Medical Research Institute; University of Melbourne, Centre for Cancer Research; University of Queensland, Institute for Molecular Bioscience; Bankstown Hospital; Liverpool Hospital; Royal Prince Alfred Hospital, Chris O’Brien Lifehouse; Westmead Hospital; Fremantle Hospital; St John of God Healthcare; Royal Adelaide Hospital; Flinders Medical Centre; Envoi Pathology; Princess Alexandria Hospital; Austin Hospital; Johns Hopkins Medical Institutes; ARC-Net Centre for Applied Research on Cancer (2017). Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentzen AK, Marquard AM, Lyngaa R, Saini SK, Ramskov S, Donia M, Such L, Furness AJS, McGranahan N, Rosenthal R, et al. (2016). Large-scale detection of antigen-specific T cells using peptide-MHC-I multimers labeled with DNA barcodes. Nat. Biotechnol. 34, 1037–1045. [DOI] [PubMed] [Google Scholar]
- Berger MF, and Mardis ER (2018). The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 15, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethune MT, Gee MH, Bunse M, Lee MS, Gschweng EH, Pagadala MS, Zhou J, Cheng D, Heath JR, Kohn DB, et al. (2016). Domain-swapped T cell receptors improve the safety of TCR gene therapy. eLife 5, e19095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bethune MT, Li X-H, Yu J, McLaughlin J, Cheng D, Mathis C, Moreno BH, Woods K, Knights AJ, Garcia-Diaz A, et al. (2018). Isolation and characterization of NY-ESO-1–specific T cell receptors restricted on various MHC molecules. Proc. Natl. Acad. Sci. USA 115, E10702–E10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381. [DOI] [PubMed] [Google Scholar]
- Brandsma D, Stalpers L, Taal W, Sminia P, and van den Bent MJ (2008). Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol. 9, 453–461. [DOI] [PubMed] [Google Scholar]
- Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, Ly A, Lie WR, Hildebrand WH, Mardis ER, and Linette GP (2015). Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celie PH, Toebes M, Rodenko B, Ovaa H, Perrakis A, and Schumacher TN (2009). UV-induced ligand exchange in MHC class I protein crystals. J. Am. Chem. Soc. 131, 12298–12304. [DOI] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehlings M, Simoni Y, Penny HL, Becht E, Loh CY, Gubin MM, Ward JP, Wong SC, Schreiber RD, and Newell EW (2017). Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8+ T cells. Nat. Commun. 8, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsch EF, Rajasagi M, Ott PA, Brusic V, Hacohen N, and Wu CJ (2014). HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol. Res. 2, 522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garboczi DN, Hung DT, and Wiley DC (1992). HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc. Natl. Acad. Sci. USA 89, 3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee MH, Han A, Lofgren SM, Beausang JF, Mendoza JL, Birnbaum ME, Bethune MT, Fischer S, Yang X, Gomez-Eerland R, et al. (2018). Antigen Identification for Orphan T Cell Receptors Expressed on Tumor-Infiltrating Lymphocytes. Cell 172, 549–563.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanville J, Huang H, Nau A, Hatton O, Wagar LE, Rubelt F, Ji X, Han A, Krams SM, Pettus C, et al. (2017). Identifying specificity groups in the T cell receptor repertoire. Nature 547, 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros A, Parkhurst MR, Tran E, Pasetto A, Robbins PF, Ilyas S, Prickett TD, Gartner JJ, Crystal JS, Roberts IM, et al. (2016). Prospective identification of neoantigen-specific lymphocytes in the peripheral blood of melanoma patients. Nat. Med. 22, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber W-J, et al. (2014). Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han A, Glanville J, Hansmann L, and Davis MM (2014). Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol. 32, 684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW, Kirsch I, Vignali M, Rieder MJ, Carlson CS, et al. (2015). Highthroughput pairing of T cell receptor α and β sequences. Sci. Transl. Med. 7, 301ra131. [DOI] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Heemskerk B, Powell DJ Jr., Cohen CJ, Morgan RA, Dudley ME, Robbins PF, and Rosenberg SA (2006). Gene transfer of tumor-reactive TCR confers both high avidity and tumor reactivity to nonreactive peripheral blood mononuclear cells and tumor-infiltrating lymphocytes. J. Immunol. 177, 6548–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-M, Bhonsle L, Besgen P, Nickel J, Backes A, Held K, Vollmer S, Dornmair K, and Prinz JC (2012). Analysis of the paired TCR α- and β-chains of single human T cells. PLoS One 7, e37338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong GA, Radu CG, Hwang K, Shu CJY, Ma C, Koya RC, Comin-Anduix B, Hadrup SR, Bailey RC, Witte ON, et al. (2009). Modular nucleic acid assembled p/MHC microarrays for multiplexed sorting of antigen-specific T cells. J. Am. Chem. Soc. 131, 9695–9703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, Behjati S, Velds A, Hilkmann H, Atmioui DE, et al. (2015). High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 21, 81–85. [DOI] [PubMed] [Google Scholar]
- Liu C, Yang X, Duffy B, Mohanakumar T, Mitra RD, Zody MC, and Pfeifer JD (2013). ATHLATES: accurate typing of human leukocyte antigen through exome sequencing. Nucleic Acids Res. 41, e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y-C, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y, et al. (2014). Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin. Cancer Res. 20, 3401–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, and Nielsen M (2008). NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 36, W509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. (2016). Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell EW, Sigal N, Nair N, Kidd BA, Greenberg HB, and Davis MM (2013). Combinatorial tetramer staining and mass cytometry analysis facilitate T-cell epitope mapping and characterization. Nat. Biotechnol. 31, 623–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M, Lundegaard C, Blicher T, Lamberth K, Harndahl M, Justesen S, Røder G, Peters B, Sette A, Lund O, and Buus S (2007). NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PLoS One 2, e796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. (2017). An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandiran V, Grigoriev V, Lan L, Ravkov E, Mertens SA, and Altman JD (2007). A robust method for production of MHC tetramers with small molecule fluorophores. J. Immunol. Methods 319, 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, and Getz G (2015). Oncotator: cancer variant annotation tool. Hum. Mutat. 36, E2423–E2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, Joshua AM, Patnaik A, Hwu WJ, Weber JS, et al. (2016). Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 315, 1600–1609. [DOI] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PF, Lu Y-C, El-Gamil M, Li YF, Gross C, Gartner J, Lin JC, Teer JK, Cliften P, Tycksen E, et al. (2013). Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 19, 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenko B, Toebes M, Hadrup SR, van Esch WJE, Molenaar AM, Schumacher TNM, and Ovaa H (2006). Generation of peptide-MHC class I complexes through UV-mediated ligand exchange. Nat. Protoc. 1, 1120–1132. [DOI] [PubMed] [Google Scholar]
- Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, Hiatt J, Saco J, Krystofinski P, Li H, et al. (2018). Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano T, and Cantor CR (1990). Expression of a cloned streptavidin gene in Escherichia coli. Proc. Natl. Acad. Sci. USA 87, 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN, and Schreiber RD (2015). Neoantigens in cancer immunotherapy. Science 348, 69–74. [DOI] [PubMed] [Google Scholar]
- Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, Chodon T, Guo R, Johnson DB, Dahlman KB, et al. (2014). Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 4, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singha S, Shao K, Yang Y, Clemente-Casares X, Solé P, Clemente A, Blanco J, Dai Q, Song F, Liu SW, et al. (2017). Peptide-MHC-based nanomedicines for autoimmunity function as T-cell receptor microclustering devices. Nat. Nanotechnol. 12, 701–710. [DOI] [PubMed] [Google Scholar]
- Stroncek DF, Berger C, Cheever MA, Childs RW, Dudley ME, Flynn P, Gattinoni L, Heath JR, Kalos M, Marincola FM, et al. (2012). New directions in cellular therapy of cancer: a summary of the summit on cellular therapy for cancer. J. Transl. Med. 10, 48–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strønen E, Toebes M, Kelderman S, van Buuren MM, Yang W, van Rooij N, Donia M, Böschen M-L, Lund-Johansen F, Olweus J, and Schumacher TN (2016). Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 352, 1337–1341. [DOI] [PubMed] [Google Scholar]
- Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, Pasetto A, Zheng Z, Ray S, Groh EM, et al. (2016). T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N. Engl. J. Med. 375, 2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJM, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, van Dijk LJA, Behjati S, Hilkmann H, El Atmioui D, et al. (2013). Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 31, e439–e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576. [DOI] [PubMed] [Google Scholar]
- Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. (2018). Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat. Med. 24, 724–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S-Q, Ma K-Y, Schonnesen AA, Zhang M, He C, Sun E, Williams CM, Jia W, and Jiang N (2018). High-throughput determination of the antigen specificities of T cell receptors in single cells. Nat. Biotechnol. 36, 1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Putative neoantigens, mutated gene expression, and predicted binding affinity for patient #1, #2 and #3 are available in Table S2. A limited number of putative neoantigens and predicted binding affinity for patient #4 are available in Table S2. The published article also includes neoantigen-specific T cell counts enumerated during this study for the indicated neoantigen. The whole-exome sequencing data have been deposited to the Sequence Read Archive (SRA, https://www.ncbi.nlm.nih.gov/sra) under accession number NCBI SRA: SRP067938 for a separate study (Hugo et al., 2016). The transcriptome data have been deposited (NCBI GEO: GSE78220) for a separate study (Hugo et al., 2016). The patient ID numbering in NCBI SRA: SRP67938 and NCBI GEO: GSE78220 are not the same as in this manuscript; use direct SRR mapping in Table S1 for correctness.