

Abstract
Nitric oxide (NO) is involved in many biological functions, including blood pressure regulation, the immune response, and neurotransmission. However, excess production of NO can lead to the generation of reactive nitrogen species and nitrosative stress and has been linked to several neurodegenerative diseases and cardiovascular disorders. Because NO is short-lived and generally difficult to detect, its primary stable degradation product, nitrite, is frequently monitored in its place. In this paper, an improved method using microchip electrophoresis with electrochemical detection (ME-EC) was developed for the separation and detection of nitrite in cell lysates. A separation of nitrite from several electroactive cell constituents and interferences was optimized, and the effect of sample and buffer conductivity on peak efficiency was explored. It was found that the addition of 10 mM NaCl to the run buffer caused stacking of the nitrite peak and improved limits of detection. A platinum black working electrode was also evaluated for the detection of nitrite and other electroactive cellular species after electrophoretic separation. The use of a modified platinum working electrode resulted in 2.5-, 1.7-, and 7.2-fold signal enhancement for nitrite, ascorbic acid, and hydrogen peroxide, respectively, and increased the sensitivity of the method for nitrite twofold. The optimized ME-EC method was used to compare nitrite production by native and lipopolysaccharide-stimulated RAW 264.7 macrophage cells.
Introduction
Nitric oxide (NO) is an important molecule involved in cellular signaling and platelet regulation. It is generated in vivo from arginine via the enzyme nitric oxide synthase (NOS).1 A specific form of NOS, inducible nitric oxide synthase (iNOS), is activated to produce nitric oxide as part of the immune response.1,2 The resulting NO is capable of reacting with cellular components and can disrupt biological function.3 Under conditions of inflammation, NO can also react with superoxide to form peroxynitrite.1,2 Peroxynitrite can then cause nitration of proteins, peroxidation of lipids, and cleavage of the phosphate backbone of DNA.2–4 These biomolecular modifications have been implicated in the pathology of several neurodegenerative and cardiovascular diseases.2,5 It is therefore important to have robust analytical methods that are capable of monitoring reactive nitrogen and oxygen species (RNOS) in biological systems.
Currently, several methods exist to detect NO, including fluorescence, chemiluminescence, electron paramagnetic resonance spectroscopy, amperometry, and voltammetry.6–18 Fluorescence detection of RNOS in cells requires the use of a derivatization reagent that can enter cells and generate a product that is highly specific for the analyte of interest, as well as being stable and nontoxic to the biological system.6,10,19 Diaminofluorescein (DAF) dyes have been commonly used to monitor NO production in cells.20–22 However, DAF-FM has been shown to react with dehydroascorbic acid (DHA) and ascorbic acid, producing side products that must be resolved from the product of interest.19,20,23 While some approaches for correcting these issues have been developed, it is still of interest to explore other more direct detection methods for RNOS that do not require derivatization.
Electrochemical detection has been used for the direct detection of nitric oxide.10,24 In most cases, the working electrode is modified for the specific detection of NO at the expense of the simultaneous detection of other analytes.14,25–27 In addition, the very short half-life of NO in vivo makes it impossible to detect in cell samples, tissue homogenates, and microdialysate samples because it is degraded prior to analysis. Therefore, its stable primary degradation product, nitrite, is often used as an indicator of nitric oxide production.7,11
A microchip electrophoresis (ME) method for the separation of nitrosative stress markers in macrophage cells was recently described by our group.28 While ME systems are more complicated to assemble and operate than capillary electrophoresis (CE) instruments, ME has the advantage of being well-suited for further adaptation into a single cell analysis device capable of cell lysis and separation and detection of intercellular components on-chip.20 In these studies, amperometric detection (EC) was used as the detection method for nitrosative stress markers as they are electrochemically active. In addition, PDMS/glass hybrid chips were employed because they enable easier incorporation and alignment of the working electrode compared to all-glass devices.29,30 EC typically suffers from lower sensitivity and higher limits of detection than laser-induced fluorescence (LIF)-based detection for ME. While the previously developed ME-EC method was capable of detecting nitrite in the bulk cell lysates of macrophage cells, it was not sensitive enough to detect the small changes in RNOS expected in a microchip single cell analysis system.
Platinum black electrodes have been extensively employed for the improved detection of NO. Platinization, or Pt black deposition, on a variety of electrode materials, including gold, carbon, and platinum, has been shown to improve electron transfer kinetics as well as increase the active surface area of the electrode.31 Enhanced signals for NO and related compounds, including nitrite, peroxynitrite, and hydrogen peroxide have been reported.25,26,31–37 These Pt black-modified electrodes have been used for a variety of applications, including imaging of NO generated on microdisks by scanning electrochemical microscopy,38 microarrays for the detection of NO released from endothelial cells,26 amperometric sensors to measure NO generation from kidney slices,39,40 3D printed microfluidic devices for the detection NO solutions,25 and the detection of RNOS released by single cells.9 Platinized carbon electrodes have also been inserted through the cell membrane35,41 and incorporated into biosensors for pond snail homogenates33 to monitor RNOS production.
While many groups have employed platinized electrodes to obtain better sensitivity for a particular analyte, only a few have extended this to the simultaneous measurement of multiple analytes. Most sensors employing Pt black are designed specifically for NO detection and employ selective membranes such as Nafion to exclude nitrite and other electroactive anions.25,26 Alternatively, sensors that have been used for the determination of multiple analytes are based on using voltammetry to distinguishing between species.33,39,41–44 This requires precise knowledge of the electrochemical behavior of all possible interfering species under the given conditions in order to quantify the analyte of interest.
There are few reports of integrating a platinum black electrode with capillary or microchip electrophoresis. One group has incorporated Pt black into a glucose-selective biosensor as a detector for CE.45 In a separate report, a platinized decoupler and reference electrodes were used in a ME device.46 However, to our knowledge, there have been no attempts to integrate a Pt black working electrode with microchip electrophoresis for the separation and detection of multiple RNOS. In this paper, microchip electrophoresis with electrochemical detection using a Pt black-modified working electrode is evaluated for the detection of nitrite, a marker of NO production, and hydrogen peroxide, a marker of superoxide. Ascorbic acid and azide were also included as potential interfering species within the biological system. In addition, the effect of sample conductivity on the separation efficiency and overall limits of detection was investigated. The optimized method was then evaluated for the detection of nitrite in native and lipopolysaccharide (LPS)-stimulated RAW 264.7 macrophage cells.
Experimental
Reagents and materials
The following materials and chemicals were used as received: boric acid, sodium chloride, tetradecyltrimethylammonium chloride (TTAC), sodium nitrite, ascorbic acid, lead (II) acetate trihydrate, and hydrogen hexachloroplatinate (IV) solution (Sigma-Aldrich, St. Louis, MO, USA); sodium hydroxide (NaOH), acetone, 2-propanol (IPA), phosphate-buffered saline (PBS), 30% hydrogen peroxide (H2O2), lipopolysaccharide (LPS), and ethanol (Fisher Scientific, Pittsburgh, PA, USA); SU-8 photoresist, SU-8 developer, and MIF 300 developer (Micro-Chem, Newton, MA, USA); glass substrates (4” × 4” × 0.090”) coated with chrome and AZ1518 photoresist (Nanofilm, Westlake, CA, USA); buffered oxide etchant (JT Baker, Austin, TX, USA); chrome etchant (Cyantek Corp., Freemont, CA, USA); Ti and Pt targets (Kurt J. Lesker Co., Jefferson Hills, PA, USA); polydimethylsiloxane (PDMS) and curing agent (Sylgard 184 elastomer kit, Ellsworth Adhesives, Germantown, WI, USA); 4” diameter silicon wafers (Silicon, Inc., Boise, ID, USA); epoxy and Cu wire (Westlake Hardware, Lawrence, KS, USA); silver colloidal (Ted Pella, Inc., Redding, CA, USA); RAW 264.7 cells, Delbecco’s Modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin-streptomycin solution (ATCC, Manassas, VA, USA), 25 ml polystyrene culture flasks (Fisher Scientific, Pittsburgh, PA, USA), C-Chip disposable hemocytometers (Bulldog Bio, Inc., Portsmouth, NH, USA), Trypan blue exclusion assay (Fisher Scientific, Pittsburgh, PA, USA), 3 kDa molecular weight cut-off filters (VWR International, West Chester, PA, USA). All water was ultrapure (18.2 MΩ) and generated from a Milli-Q Synthesis A10 system (Millipore, Burlington, MA, USA).
PDMS microchip fabrication
The fabrication of PDMS microchips has been previously described.47 Briefly, a 4” diameter silicon wafer was coated with SU-8 10 negative photoresist to a thickness of 15 μm with a Cee 100 spincoater (Brewer Science Inc., Rolla, MO, USA). The wafer then underwent a soft bake at 65 °C for 2 min and then 95 °C for 5 min on a programmable hotplate (Thermo Scientific, Waltham, MA, USA). The designs for the microchip were drawn with AutoCad (Autodesk, San Rafael, CA, USA) and printed onto a transparency (Infinite Graphics, Minneapolis, MN, USA). The coated wafer was then covered with the negative transparency and exposed at 344 mJ cm−2 with a UV flood source (ABC Inc., San Jose, CA, USA). Next, the wafer was transferred to a programmable hotplate again for a postbake at 65 °C for 1 min and then 95 °C for 2 min. After the postbake, the wafer was developed in SU-8 developer, rinsed with IPA, and dried with nitrogen. Lastly, the wafer underwent a hard bake at 200 °C for 2 h. The final silicon master contained 15 μm thick and 40 μm wide microchannels, which were measured with an Alpha Step-200 surface profiler (KLA-Tencor Instruments, Milpitas, CA, USA). The microchip used for these studies consisted of a simple-T design with a 5 cm separation channel and 0.75 cm side arms. A PDMS microchip was made by pouring a degassed 10:1 mixture of PDMS and curing agent, respectively, over the silicon master and curing the mixture overnight at 70 °C. The PDMS microchip was then peeled from the master and reservoirs were punched into the PDMS with a 4 mm biopsy punch (Harris Uni-Core, Ted Pella, Inc., Redding, CA, USA).
Electrode fabrication
The method of fabricating in-house Pt electrodes has been previously described.29 Briefly, electrode designs are drawn with AutoCad and printed onto a transparency with a resolution of 50,000 dpi. A borofloat glass substrate (4” × 4” × 0.090”) pre-coated with a layer of chrome and then AZ1518 positive photoresist is covered by the transparency and exposed at 344 mJ cm−2 with a UV flood source for 4 s. Then the glass substrate is developed in MIF 300 developer for about 15 s and baked at 100 °C for 10 min on a programmable hotplate. At this point, the electrode pattern is imprinted in the photoresist, thereby exposing the chrome layer below. The exposed chrome is removed with chrome etchant. Trenches in the pattern of the electrodes are then created with the use of buffered oxide etchant to a depth of about 300 nm. After thoroughly washing the substrate with CaCO3 solution and water, the trench depth is measured with an Alpha Step-200 surface profiler. Next, the glass substrate is exposed to an oxygen plasma for 1 min (March Plasmod, Concord, CA, USA) and immediately placed into an AXXIS DC magnetron sputtering system (Kurt J. Lesker Co., Jefferson Hills, PA, USA). After allowing the sputterer chamber to reach a pressure of 1.0 × 10−6 Torr, Ti is deposited onto the substrate for 40 s at 220 V and 5.0 × 10−3 Torr to produce a 20 nm Ti layer. Then Pt is deposited for 15 min at 200 V and 5.0 × 10−3 Torr. Next, the substrate is washed with acetone and then chrome etchant. The final width and height of the fabricated Pt electrodes are measured with an Alpha Step-200 surface profiler.
Electrophoresis procedure
Microchips were constructed by reversibly bonding a layer of PDMS containing the simple-T design to the glass substrate containing the embedded Pt electrode. The Pt electrode was aligned at the intersection of the separation channel and the waste reservoir. Pt leads were inserted into each reservoir. Two Spellman CZE 1000R high voltage power supplies (Spellman, Hauppauge, NY, USA) controlled with a LabView (National Instruments, Austin, TX, USA) program written in-house were used for all electrophoresis procedures. Samples were injected using a gated injection protocol. A gate was established by applying –2400 V and –2200 V to the buffer and sample reservoirs, respectively, while both the buffer waste and sample waste reservoirs were grounded. Sample injection occurred by floating the voltage in the buffer reservoir for 1 s. The run buffer used in these experiments was 10 mM borate with TTAC and NaCl. TTAC was varied between 3 mM and 9 mM, and NaCl was varied between 0 mM and 15 mM. The pH was adjusted to 10 with NaOH.
Electrochemical detection and data analysis
Electrochemical detection was accomplished with a two-channel wireless isolated potentiostat (Pinnacle Technology, Inc., Lawrence, KS, USA). The potentiostat used had a sampling rate of 6.5 to 13 Hz (gain = 5,000,000 V/A, resolution = 47 fA). Sirenia software (Pinnacle Technology) was used for all data acquisition. Electrochemical measurements were taken at a working electrode potential of +1.100 V vs. Ag/AgCl reference electrode (BASi, West Layfayette, IN, USA). All data analysis was performed with Origin 8.6 software (OriginLab, Northhampton, MA, USA).
Pt black deposition
A 10 mM lead (II) acetate trihydrate stock solution was prepared. The Pt black deposition solution was then prepared by adding 1 mL of 8% w/v hydrogen hexachloroplatinate solution and 0.42 mL of 10 mM lead (II) acetate trihydrate stock solution to 5.94 mL of PBS. Pt black deposition was achieved by running a cyclic voltammogram on the Pt working electrode in the presence of the deposition solution, from +0.6 V to –0.35 V vs. Ag/AgCl reference at a scan rate of 0.02 V/s. The potential was controlled using a CHI electrochemical analyzer (CH Instruments, Inc., Austin, TX, USA) controlled by CHI software. After deposition, the area around the electrode was thoroughly washed with water to remove salt crystals formed during the deposition. Next, the modified electrode area was activated while in 10 mM borate at pH 10 by applying alternating pulses of +0.2 V and –0.5 V for 1 s each for 30 cycles.
Cell culture and sample preparation
RAW 264.7 cells were cultured, stimulated, and tested for viability as previously reported.28 Briefly, cells were cultured in DMEM containing 10% (v/v) fetal bovine serum, L-glutamine (2 mM), penicillin (50 IU/mL), and streptomycin (50 μg/mL). Cells were maintained in 25 mL polystyrene culture flasks in a humidified environment at 37 °C and 5% CO2. To prevent overgrowth, cells were passaged every 2–3 days. Cell viability was determined via the Trypan blue exclusion assay and a C-Chip disposable hemocytometer. When stimulation of NO production was desired, a flask of healthy macrophage cells was incubated for 24 h with 100 ng mL−1 purified lipopolysaccharide from the Escherichia coli line 0111:B4 (Figure 1). For such experiments, a control (native) flask of unstimulated RAW macrophage cells from the population was incubated under the same conditions.
figure 1.
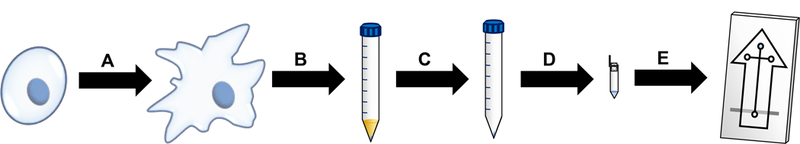
protocol for the analysis of lps-stimulated macrophage cells. (a) native cells are incubated with lps for 24 h to induce stimulation. (b) cells are harvested and centrifuged to produce a cell pellet. (c) the cell pellet is washed once with pbs to remove excess medium and once with water to remove any remaining salt. (d) the cell pellet is resuspended in borate buffer at ph 10 with 5.5 mm ttac to lyse the cells, and the lysate is centrifuged through a molecular weight cutoff filter to remove any proteins and large insoluble species. (e) filtered cell lysate is placed in the sample well of a pdms/glass microchip for me-ec analysis.
To prepare the cell lysate samples, cells grown to approximately 80% confluence (around 5 million cells per flask) were counted, harvested with a scraper, and then centrifuged at 3500 rpm for 3 min. The supernatant medium was removed, leaving behind a cell pellet. This pellet was washed once with cold 10 mM PBS and once with sterile water. The cell pellet was then lysed in 150 μL of buffer consisting of 10 mM borate at pH 10 and 5.5 mM TTAC. After lysis, the lysate was centrifuged through a 3 kDa molecular weight cut-off filter for 8 min to remove large compounds, such as membranes and proteins. The filtered lysate was run on the ME-EC device for analysis.
Results and discussion
Several strategies were investigated to improve the limits of detection for nitrite in cell lysate samples. A separation was first optimized to resolve nitrite and several other electroactive species in cells. Then, the effect of sample conductivity on the separation efficiency was evaluated. A Pt black-modified working electrode was then employed with the goal of detecting nitrite in macrophage cells lysates.
Separation optimization and stacking of nitrite
For the analysis of cell lysate samples by ME-EC, the initial experiments employed a background electrolyte consisting of 10 mM borate with 2 mM TTAC at pH 10. However, when cell lysate samples were analyzed, it was found that nitrite comigrated with an interferent peak (azide) that was present on the filters used for sample preparation. In an attempt to resolve these two compounds, the concentration of TTAC in the BGE was varied from 2 mM to 9 mM. As shown in Figure 2, the optimal TTAC concentration was determined to be 5.5 mM. These separation conditions were also capable of separating two other analytes of interest to the redox balance of the cell: ascorbic acid, an antioxidant, and hydrogen peroxide, a reactive oxygen species and product of superoxide. These four species were baseline resolved in less than 30 s.
figure 2.
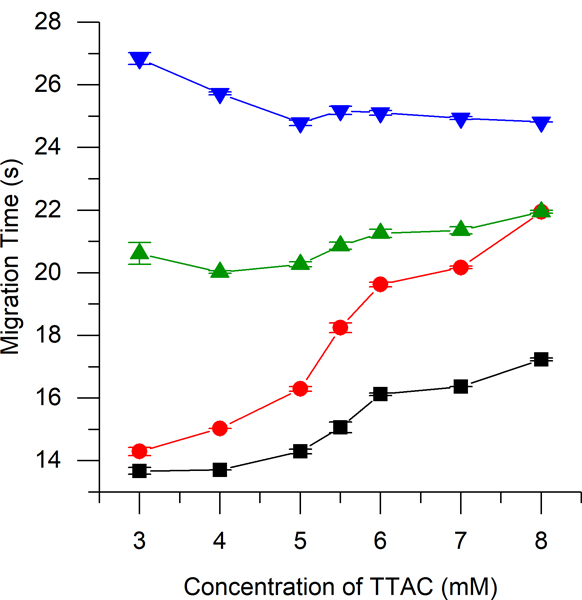
migration times of nitrite (-■-), azide (-●-), ascorbic acid (-▲-), and hydrogen peroxide (-▼-) by me-ec with a bge consisting of 10 mm borate at ph 10 with varying concentrations of ttac.
Our group has previously reported severe destacking of the nitrite peak during the analysis of cell lysate samples due to the high conductivity of the sample matrix. In those studies, cells were washed with PBS, which contains a high amount of NaCl (154 mM), prior to the final lysing with the run buffer. In this previous report, it was found that the addition of 7.5 or 10 mM NaCl to the BGE restored the peak height to that observed for low conductivity standard samples.28 However, this phenomenon was not optimized or explored further. Figure 3 shows the effects of both sample and buffer conductivities on the separation efficiency of the nitrite peak. Electropherogram 3a was obtained using standards dissolved in run buffer. If the sample contains 10 mM NaCl and is analyzed using a low conductivity buffer, the peak efficiency for nitrite decreases 6-fold compared to that of a low conductivity sample as can be seen in 3b. This loss in efficiency can be restored by adding 10 mM NaCl to the BGE, which matches the conductivity of the sample as shown in electropherogram 3c. Most importantly, it was found that if the conductivity of the BGE is significantly increased relative to the sample (such that there is 10 mM NaCl in the run buffer and no NaCl in the sample), a nearly tenfold increase in peak efficiency is observed (3d). This last set of parameters results in a greatly enhanced nitrite peak due to transient isotachophoretic stacking without affecting the peak heights or resolution of the other analytes. In order to ensure low conductivity cell samples, the sample preparation protocol was modified to include a second wash of the cell pellet with water to remove excess saline prior to lysis with run buffer.
figure 3.
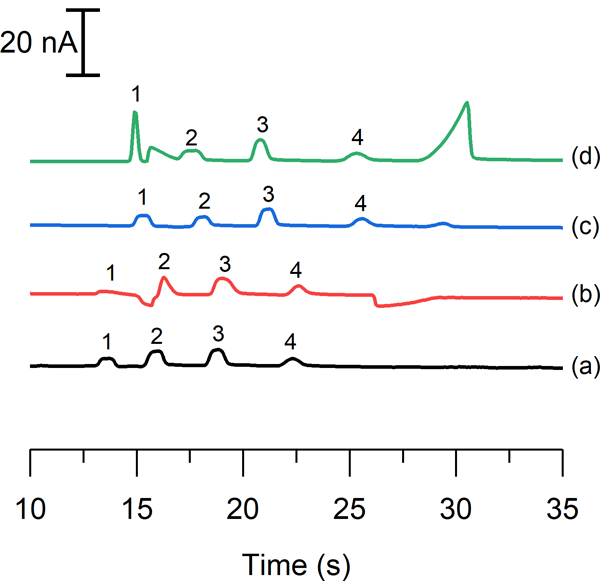
electropherograms illustrating a standard solution of 50 μm nitrite (1), 20 μm azide (2), 40 μm ascorbic acid (3), and 100 μm hydrogen peroxide (4) using (a) a run buffer and sample buffer consisting of 10 mm borate and 5.5 mm ttac at ph 10, and the effect of adding 10 mm nacl (b) to the sample buffer, (c) to both the run buffer and sample buffer, and (d) to only the run buffer.
It is hypothesized that the increase in peak height and plate numbers for nitrite is due to transient isotachophoresis (tITP) occurring prior to the separation. In tITP, the zone of the analyte of interest is stacked between a leading and a tailing electrolyte that have faster and slower electrophoretic mobilities, respectively, than the analyte of interest. In this system, the leading electrolyte would be the chloride ion because its electrophoretic mobility is slightly higher than that of nitrite under these separation conditions.48 Chloride ions have been shown to be responsible for stacking effects observed in capillary electrophoresis analysis of biological samples.49 It is postulated that the borate ion is the tailing electrolyte under these conditions. Additional evidence of this phenomenon is that the concentration of NaCl must be in excess compared to the nitrite sample concentration in order for stacking to occur.50 Figure 4A shows the effect of increasing NaCl concentration in the run buffer on the height of the nitrite peak. As the concentration of NaCl is increased from 2.5 mM to 15 mM, the nitrite peak height increases. This stacking is also reflected in the significant increase in peak efficiency for nitrite compared to those of all other analytes, which remained constant as can be seen in Figure 4B. While the addition of 15 mM NaCl also yielded higher peak heights and efficiencies compared to 10 mM NaCl, the higher salt concentration resulted in a greatly increased separation current and generated larger system peaks due to the increased buffer conductivity. Also when the separation current rises above 40 μA, bubbles can form within the PDMS channel and limit the lifetime of the device, negatively impacting the analysis. Therefore, a concentration of 10 mM NaCl was chosen for this study as it provided greatly improved peak efficiency while minimizing the negative effects of a highly conductive buffer. Using the combination of tITP and a bare Pt electrode, the LOD for nitrite was reduced to 0.50 μM from a value of 2.6 μM that was previously reported by our group.51
figure 4.
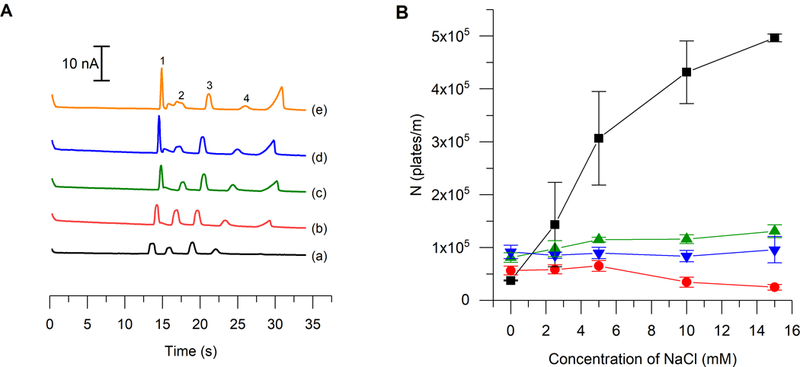
(a) electropherograms depicting the change in nitrite stacking due to transient isotachophoresis as a result of adding nacl to the run buffer at a concentration of (a) 0 mm, (b) 2.5 mm, (c) 5 mm, (d) 10 mm, and (e) 15 mm. (b) effect of increasing nacl concentration on peak efficiency of each analyte (n = 3). legend: nitrite (1, -■-), azide (2, -●-), ascorbic acid (3, -▲-), hydrogen peroxide (4, -▼-).
Deposition optimization
A Pt black working electrode was evaluated to see if it would further improve the limits of detection of the device for nitrite. Two different electrochemical deposition methods have been reported, application of a constant current density and cyclic voltammetry (CV). In both methods, the electrochemical program is applied while the electrode to be modified is in contact with a deposition solution consisting of chloroplatinic acid to generate the platinization and lead acetate to control the morphology of the deposition.
In the initial experiments, a 4-mm diameter PDMS well was aligned on top of the glass substrate such that the bare Pt electrode bisected the well. The well was filled with deposition solution, and Pt black was generated on the 4-mm length exposed to the solution using cyclic voltammetry. During these experiments, the electrode turned black, indicating that the modification was successful. However, this deposition process prevented subsequent sealing of the PDMS microchip to the glass substrate due to the height of the electrode that was produced. A typical electrode produced by sputtering is less than 200 nm in height, which is low enough that a reversible seal between the PDMS and the glass substrate can be achieved. However, this electrochemical deposition procedure resulted in an electrode with a height greater than 200 nm.
To overcome this problem and make it possible to seal the PDMS separation channel to the electrode containing the glass chip, in situ generation was evaluated. The layer of PDMS containing the separation channel microchip was first aligned with the Pt working electrode on the glass substrate. The two layers were then sealed together prior to the deposition process. Then the deposition solution was added to the bottom waste reservoir and pulled through the separation channel with an aspirator. Therefore, in this configuration, only the 40-μm wide electrode region within the channel was exposed to the solution. Solution flow was stopped during the cyclic voltammetry deposition process to allow the deposition to occur directly over the electrode. This resulted in a well-defined deposition area. It also prevented the deposition from occurring underneath the PDMS substrate and kept the seal intact. A black color was again observed on the working electrode. ME-EC experiments could immediately be performed with this device without removing the PDMS channel layer. As shown in the scanning electron micrograph in Figure 5A, this deposition procedure resulted in a greatly increased electrode surface area.
figure 5.

sem images of a (a) non-activated and (b) activated pt black-modified working electrode.
Unfortunately, it was found that depositing the Pt black onto the surface of the working electrode using only cyclic voltammetry resulted in an unstable modification. When a voltage was applied to the separation channel, a large drop in the background current at the working electrode was observed, which implied that the Pt black was detaching from the working electrode. Therefore, a combination of CV and differential pulse voltammetry (DPV) was evaluated. DPV has been reported by others to increase the stability of the Pt black modification.34 SEM images of activated and non-activated modified electrodes show a drastic difference in the physical properties of the two electrodes due to the activation procedure (Figure 5A and B).
The deposition process resulted in the formation of salt crystals in both the buffer waste reservoir and the separation channel, so the chip had to be washed with water after this process and inspected to ensure that no crystals were present. The presence of crystals can lead to irreproducible signal measurements due to irregular flows throughout the microchip channels.
Signal enhancement
Once the electrode modification procedure was optimized, the response of the Pt black electrode was compared to that obtained by a bare Pt electrode. Peak heights for nitrite, AA, and hydrogen peroxide were measured before and after the deposition with the same microchip and electrode alignment. This was important because variations in electrode alignment can cause changes in both the amplitude of the noise and signal. While using the same microchip for these enhancement measurements, the channels of the microchip were thoroughly washed with water to ensure that no run buffer remained in the channel during deposition. Figure 6A shows electropherograms of a mixture of nitrite, AA, and hydrogen peroxide standards before and after modification of a Pt working electrode using the optimized tITP separation conditions. The modified electrode generated a significant increase in signal, but also exhibited a higher background current. This increase in background current was expected since the geometric area of the electrode was increased. Average signal enhancements of 2.5 ± 0.2, 1.7 ± 0.2, and 7.2 ± 0.2 were observed for nitrite, AA, and hydrogen peroxide, respectively with the Pt black-modified electrode compared to a bare platinum electrode.
figure 6.
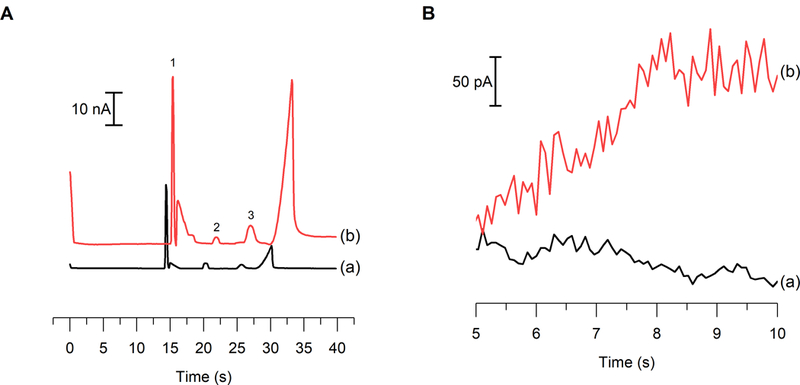
(a) electropherograms of 100 μm nitrite (1), 40 μm aa (2), and 100 μm hydrogen peroxide (3) standards (a) before and (b) after pt black modification. (b) comparison of the noise at (a) a bare pt electrode vs (b) a pt black electrode during a me separation.
An external calibration curve was generated to determine the effect of the Pt black deposition and activation on the sensitivity for nitrite (SI Figure 1). The sensitivities for the detection of nitrite with a bare Pt and Pt black-modified working electrode were 0.245 ± 0.001 and 0.580 ± 0.008 nA μM−1, respectively. The modification of the working electrode resulted in a 2.36 ± 0.03-fold enhancement in the sensitivity toward nitrite. These results are comparable with those reported by Shim et al., where a threefold sensitivity enhancement for NO was observed when a Pt working electrode underwent platinization. This increase was believed to be due to the increase in the active surface area of the electrode.38 Unfortunately, this increase in the geometric area of the electrode also resulted in an increase in noise (Figure 6B), leading to an unchanged S/N ratio and, therefore, the LOD of the system did not change. Amatore’s group has reported limits of detection for nitrite approximately 10-fold lower when using a Pt black electrode embedded in a microchannel with hydrodynamic flow. The differences in LODs of our work and these values can be attributed to both the presence of high voltage separation field and the difficulty of controlling the geometric area of the Pt black modification within the separation channel.
Nitrite in biological samples
Cell lysate samples with high conductivities have shown to suppress the nitrite peak due to destacking effects.28 Previously, our group has remedied this destacking effect by increasing the conductivity of the buffer to match that of the sample matrix. However, we observed that the nitrite peak efficiency could be greatly improved by using a buffer with a higher conductivity than the sample. Therefore, in the present study great care was taken to minimize the conductivity of the cell samples. The cell pellet was washed first with PBS, as previously reported, and then with water to eliminate the high salt content of the cell sample prior to lysing with run buffer. The addition of the water wash step in the cell preparation resulted in samples that had much lower conductivities (similar to normal standard solutions in run buffer). The lower conductivity cell lysates allowed stacking of the nitrite peak with the tITP system, which provided additional signal enhancement and peak consistency when compared to the previously published method.
It is well established that LPS-stimulated RAW 264.7 macrophages produce high amounts of NO.52 LPS is an endotoxin found on the outer membrane of gram-negative bacteria that causes the activation of iNOS in macrophages and overproduction of NO. In this study, cells were stimulated with LPS to generate higher NO production and, therefore, an increased nitrite signal. The effect of the new separation conditions and signal enhancement obtained with the Pt black modified electrodes on the determination of intracellular nitrite was investigated in both native and LPS-stimulated bulk cell lysate samples. Figure 7 shows electropherograms obtained for a LPS-stimulated cell lysate sample with the same electrode and microchip before and after the working electrode had been modified with Pt black. The identity of the nitrite peak was confirmed through spiking. The electrode modification resulted in an average 4.31 ± 0.33-fold enhancement in the nitrite signal, which was averaged over both native and stimulated (n = 3 for both sets) cell lysates. Due to the differing number of cells in each sample, the results were corrected based on their respective cell counts. When considered in combination with the tITP separation conditions facilitated by the improved washing procedure, this modification results in an increase between 10- and 20-fold in nitrite peak height in cell lysates. This improvement could prove useful in future studies to quantify nitrite electrochemically in single macrophage cells, where a significantly lower LOD may be necessary.
figure 7.
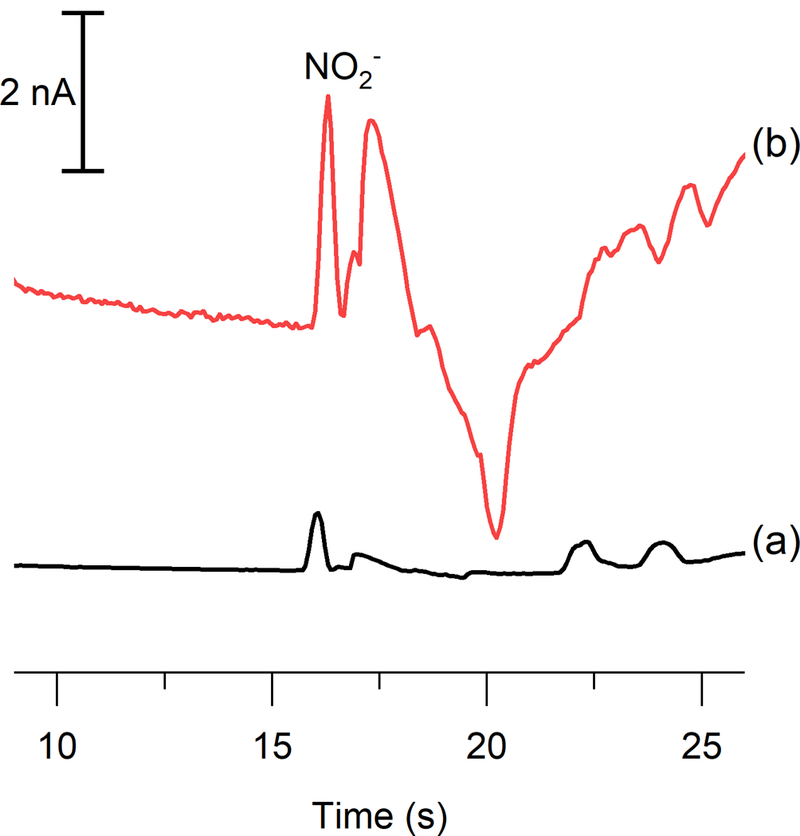
electropherograms of an lps-stimulated raw 264.7 macrophage cell lysate sample obtained using (a) bare pt and (b) pt black.
This increase in nitrite due to the presence of LPS was also calculated to ensure that the same result was achieved with both an unmodified and a modified working electrode. Upon stimulation, a 3.9 ± 0.9 times increase in nitrite was observed with the bare Pt working electrode and a 4.0 ± 0.6 times increase with the Pt black working electrode. A t-test was performed to compare these data, and it was determined that at 95% confidence there is no statistically significant difference between the two sets of data. This increase in nitrite production after exposure to LPS agrees with previous results reported by Gunasekara et al.28 Therefore, it confirms that the Pt black modification does not impact the use of the method for monitoring changes in nitrite concentration with varying cell conditions. Furthermore, these results are comparable with measurements of nitric oxide in Jurkat cells taken using laser-induced fluorescence detection, in which the indirect NO probe DAF-FM was used to observe a twofold increase in NO upon LPS stimulation.20 This suggests that electrochemical detection of nitrite is capable of providing results similar to those of indirect fluorescence detection of nitric oxide when studying cellular nitrosative stress.
Conclusions
A ME-EC method that uses transient isotachophoresis and a Pt black-modified working electrode to enhance the signal and detect nitrite as well as other electrochemically active species was reported. First, a stacking method to enhance the signal of nitrite through the addition of NaCl to the run buffer was developed that provided a 5.2-fold decrease in the LOD for nitrite. Then, a procedure to modify a Pt working electrode with Pt black for the detection of nitrite was optimized. This electrode modification was then coupled with a ME-EC method and a signal enhancement for nitrite, hydrogen peroxide, and ascorbic acid was obtained. The use of a Pt black-modified working electrode resulted in an increased sensitivity for nitrite. This method was then used to detect nitrite as an indicator of NO production in a bulk cell lysate sample. The incorporation of the tITP system and Pt black-modified working electrode improved the method previously reported by our group by increasing the sensitivity and decreasing the LOD. In the future, this method will be used to study the production of nitric oxide in microglia and other immune cells. The incorporation of this method into a single cell analysis device will make it possible to monitor short-lived species, such as NO and peroxynitrite, directly in order to investigate the conditions which lead to neurodegeneration.
Supplementary Material
Acknowledgements
This research was funded by the National Science Foundation (CHE-1411993) and National Institutes of Health (COBRE P20GM103638). J.M.S. thanks the Madison and Lila Self Graduate Fellowship for support. K.M.S. would like to acknowledge the NIH Graduate Training Program in Dynamic Aspects of Chemical Biology Grant (T32 GM008545) from the National Institutes of General Medicinal Sciences for support. G.C. acknowledges the support of an American Heart Association-Midwest Affiliate Postdoctoral Research Fellowship (NFP0075515). The authors would like to thank Nancy Harmony for editorial support, Scott Martin for assistance with the Pt black deposition procedure, Heather Shinogle and the Microscopy and Analytical Imaging Laboratory at the University of Kansas for help with obtaining SEM images, and Pinnacle Technologies, Inc. (Lawrence, KS) for loaning the lab isolated potentiostats as well as assisting in potentiostat troubleshooting.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.MacMicking J, Xie Q.-w. and Nathan C, Annu. Rev. Immunol, 1997, 15, 323–350. [DOI] [PubMed] [Google Scholar]
- 2.Pacher P, Beckman JS and Liaudet L, Physiol. Rev, 2007, 87, 315–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kröncke K-D, Fehsel K and Kolb-Bachofen V, Nitric Oxide, 1997, 1, 107–120. [DOI] [PubMed] [Google Scholar]
- 4.Calabrese V, Cornelius C, Rizzarelli E, Owen JB, Dinkova-Kostova AT and Butterfield DA, Antioxid.Redox Signal, 2009, 11, 2717–2739. [DOI] [PubMed] [Google Scholar]
- 5.Minghetti L and Levi G, Prog. Neurobiol, 1998, 54, 99–125. [DOI] [PubMed] [Google Scholar]
- 6.Gomes A, Fernandes E and Lima JL, J fluoresc, 2006, 16, 119–139. [DOI] [PubMed] [Google Scholar]
- 7.Tsikas D, J. Chromatogr. B, 2007, 851, 51–70. [DOI] [PubMed] [Google Scholar]
- 8.Tsikas D, Anal. Biochem, 2008, 379, 139–163. [DOI] [PubMed] [Google Scholar]
- 9.Amatore C, Arbault S. p. and Koh AC, Anal. Chem, 2010, 82, 1411–1419. [DOI] [PubMed] [Google Scholar]
- 10.Bedioui F and Griveau S, Electroanalysis, 2013, 25, 587–600. [Google Scholar]
- 11.Hetrick EM and Schoenfisch MH, Annu. Rev. Anal. Chem, 2009, 2, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trouillon R, Biol. Chem, 2013, 394, 17–33. [DOI] [PubMed] [Google Scholar]
- 13.Ye X, Rubakhin SS and Sweedler JV, Analyst, 2008, 133, 423–433. [DOI] [PubMed] [Google Scholar]
- 14.Hunter RA, Privett BJ, Henley WH, Breed ER, Liang Z, Mittal R, Yoseph BP, McDunn JE, Burd EM and Coopersmith CM, Anal. Chem, 2013, 85, 6066–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gunasekara DB, Hulvey MK, Lunte SM and da Silva JAF, Anal. Bioanal. Chem, 2012, 403, 2377–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bryan NS and Grisham MB, Free Radical Biol. Med, 2007, 43, 645–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Csonka C, Pali T, Bencsik P, Goerbe A, Ferdinandy P and Csont T, Br. J. Pharmacol, 2015, 172, 1620–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H-X, Chen J-B, Guo X-F, Wang H and Zhang H-S, Anal. Chem, 2014, 86, 3115–3123. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Kim W-S, Hatcher N, Potgieter K, Moroz LL, Gillette R and Sweedler JV, J. Biol. Chem, 2002, 277, 48472–48478. [DOI] [PubMed] [Google Scholar]
- 20.Metto EC, Evans K, Barney P, Culbertson AH, Gunasekara DB, Caruso G, Hulvey MK, Fracassi da Silva JA, Lunte SM and Culbertson CT, Anal. Chem, 2013, 85, 10188–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogel PA, Halpin ST, Martin RS and Spence DM, Anal. Chem, 2011, 83, 4296–4301. [DOI] [PubMed] [Google Scholar]
- 22.Letourneau S, Hernandez L, Faris AN and Spence DM, Anal. Bioanal. Chem, 2010, 397, 3369–3375. [DOI] [PubMed] [Google Scholar]
- 23.Kim W-S, Ye X, Rubakhin SS and Sweedler JV, Anal. Chem, 2006, 78, 1859–1865. [DOI] [PubMed] [Google Scholar]
- 24.Amatore C, Arbault S, Guille M and Lemaître F, Chem. Rev, 2008, 108, 2585–2621. [DOI] [PubMed] [Google Scholar]
- 25.Erkal JL, Selimovic A, Gross BC, Lockwood SY, Walton EL, McNamara S, Martin RS and Spence DM, Lab Chip, 2014, 14, 2023–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selimovic A and Martin RS, Electrophoresis, 2013, 34, 2092–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griveau S and Bedioui F, Anal. Bioanal. Chem, 2013, 405, 3475–3488. [DOI] [PubMed] [Google Scholar]
- 28.Gunasekara DB, Siegel JM, Caruso G, Hulvey MK and Lunte SM, Analyst, 2014, 139, 3265–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scott DE, Grigsby RJ and Lunte SM, Chemphyschem, 2013, 14, 2288–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saylor RA, Reid EA and Lunte SM, Electrophoresis, 2015, 36, 1912–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou L, Cheng Y and Amrein M, J. Power Sources, 2008, 177, 50–55. [Google Scholar]
- 32.Li Y, Meunier A, Fulcrand R, Sella C, Amatore C, Thouin L, Lemaître F and Guille‐Collignon M, Electroanalysis, 2016. [Google Scholar]
- 33.Fagan-Murphy A, Hachoumi L, Yeoman M and Patel B, Mech. Ageing Dev, 2016, 160, 28–31. [DOI] [PubMed] [Google Scholar]
- 34.Li Y, Sella C, Lemaître F, Guille-Collignon M, Thouin L and Amatore C, Electrochim. Acta, 2014, 144, 111–118. [Google Scholar]
- 35.Zhang XW, Qiu QF, Jiang H, Zhang FL, Liu YL, Amatore C and Huang WH, Angew. Chem, 2017, 129, 13177–13180. [DOI] [PubMed] [Google Scholar]
- 36.Lee Y, Oh BK and Meyerhoff ME, Anal. Chem, 2004, 76, 536–544. [DOI] [PubMed] [Google Scholar]
- 37.Casella IG and Salvi AM, Electroanalysis, 1997, 9, 596–601. [Google Scholar]
- 38.Shim JH and Lee Y, Anal. Chem, 2009, 81, 8571–8576. [DOI] [PubMed] [Google Scholar]
- 39.Lee Y and Kim J, Anal. Chem, 2007, 79, 7669–7675. [DOI] [PubMed] [Google Scholar]
- 40.Lee Y, Yang J, Rudich SM, Schreiner RJ and Meyerhoff ME, Anal. Chem, 2004, 76, 545–551. [DOI] [PubMed] [Google Scholar]
- 41.Li Y, Hu K, Yu Y, Rotenberg SA, Amatore C and Mirkin MV, J. Am. Chem. Soc, 2017, 139, 13055–13062. [DOI] [PubMed] [Google Scholar]
- 42.Li Y, Sella C, Lemaître F, Guille Collignon M, Amatore C and Thouin L, Anal. Chem, 2018. [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Sella C, Lemaitre F, Guille-Collignon M, Thouin L and Amatore C, Electrochim. Acta, 2014, 144, 111–118. [Google Scholar]
- 44.Park SS, Tatum CE and Lee Y, Electrochem. Commun, 2009, 11, 2040–2043. [Google Scholar]
- 45.Wang X, Zhang Y, Cheng C, Dong R and Hao J, Analyst, 2011, 136, 1753–1759. [DOI] [PubMed] [Google Scholar]
- 46.Wu C-C, Wu R-G, Huang J-G, Lin Y-C and Chang H-C, Anal. Chem, 2003, 75, 947–952. [DOI] [PubMed] [Google Scholar]
- 47.Duffy DC, McDonald JC, Schueller OJA and Whitesides GM, Anal. Chem, 1998, 70, 4974–4984. [DOI] [PubMed] [Google Scholar]
- 48.Timerbaev AR and Hirokawa T, Electrophoresis, 2006, 27, 323–340. [DOI] [PubMed] [Google Scholar]
- 49.Krivankova L, Pantuckova P, Gebauer P, Bocek P, Caslavska J and Thormann W, Electrophoresis, 2003, 24, 505–517. [DOI] [PubMed] [Google Scholar]
- 50.Szökő É, Tábi T, Halász AS, Pálfi M and Magyar K, J. Chromatogr, 2004, 1051, 177–183. [DOI] [PubMed] [Google Scholar]
- 51.Gunasekara DB, Hulvey MK and Lunte SM, Electrophoresis, 2011, 32, 832–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lorsbach RB, Murphy WJ, Lowenstein CJ, Snyder SH and Russell SW, J. Biol. Chem, 1993, 268, 1908–1913. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.