

SUMMARY
Background:
The MGTX trial demonstrated that thymectomy combined with prednisone was superior to prednisone alone in improving clinical status measured by the Quantitative MG (QMG) score in patients with non-thymomatous myasthenia gravis at 3 years. We investigated the long-term effects of thymectomy up to 5 years on clinical status, medication requirements, and adverse events.
Methods:
A multicentre, rater-blinded 2-year extension study was conducted across 36 centres in 15 countries for patients who completed the MGTX randomised, controlled trial and were willing to participate. MGTX trial patients were aged 18 to 65 years at enrollment, had generalised non-thymomatous myasthenia gravis (MG) with disease duration less than 5 years and elevated (≥1.00 nmol/l; 0.50–0.99 nmol/l allowed if confirmed by positive edrophonium or electrophysiologic testing) acetylcholine receptor antibody titers.. All patients received oral prednisone at doses titrated up to 100 mg on alternate days until they achieved minimal manifestation status. The primary endpoints were the time-weighted average of both the QMG and alternate-day prednisone dose from month 0 to month 60. Analyses were by intention-to-treat. The trial was registered on clinicaltrials.gov, number NCT00294658.
Findings:
Of 111 subjects who completed the 3-year MGTX trial, 68 (61%) entered the extension study between September 1, 2009 and August 26, 2015 (33 prednisone alone; 35 prednisone plus thymectomy). Of the 68, 50 (74%) completed the 60-month assessment (24 prednisone alone; 26 prednisone plus thymectomy). At 5 years, patients randomised to thymectomy plus prednisone continued to demonstrate improved clinical status compared to patients in the prednisone alone group based on time-weighted average QMG (5.47±3.87 vs. 9.34±5.08; 95% CI for the difference 0.71–7.04; p=0.0007) and lower average alternate-day prednisone requirements (24 mg±21 mg vs. 48±29 mg; 95% CI for the difference 12–36 mg; p=0.0002). The proportion of patients requiring hospitalisation for MG exacerbation (6% vs. 30%; 95% CI for the difference 7.1–42.1%; p=0.0105) was lower in the thymectomy group. Other MEDRA-coded adverse events were infrequent, occurring at a rate of ≤6% in both groups and did not differ significantly between them. There were no treatment-related deaths.
Interpretation:
After 5 years, thymectomy continues to confer benefits in generalised non-thymomatous MG. Although caution in predicting benefit for all such patients is appropriate since the extension study included only half of MGTX trial subjects, results available through month 60 provide further evidence to support thymectomy in this large group of patients with generalised MG.
INTRODUCTION
The Thymectomy Trial in Non-Thymomatous Myasthenia Gravis Patients Receiving Prednisone (MGTX), an international, multicentre, randomised, controlled study, recently demonstrated that extended transsternal thymectomy (ETTX) combined with a standardised prednisone protocol was superior to prednisone alone after three years in improving myasthenic weakness and lowering corticosteroid requirements in acetylcholine receptor antibody-positive generalised patients.1 Furthermore, MGTX showed that thymic resection resulted in a significantly lower requirement for azathioprine, intravenous immunoglobulin, and hospitalisations for MG exacerbation; all were reduced by more than 50% in the thymectomy group.
Results from MGTX countered doubts that had persisted for 75 years since Blalock first reported improvements in some non-thymomatous MG patients following thymectomy.2 Whether or not thymectomy offered definitive benefits in this MG population remained a heated topic, and a practice parameter in 2000 that analysed available data could only recommend thymectomy as a treatment option.3 Authors of the practice parameter and others who performed systematic literature reviews3, 4 argued for a prospective, randomised, medication-controlled trial with blinded assessments, a call that was met by MGTX.
A 3-year time point for the MGTX primary outcome was chosen based on studies reporting benefit after thymectomy in the first two to four years following the procedure, but suggesting that after that time period surgically and medically managed patients improved at comparable rates, with no additional benefit derived from thymectomy, itself.5, 6 In anticipation of an extended recruitment phase for the trial that would last several years, MGTX subjects who reached the 36-month visit were given an option to enroll in an extension study for two more years. The combined 60-month data from the MGTX Extension Study aimed to investigate the durability of treatment response related to thymectomy in this MG population and whether benefits accrue past three years.
METHODS
Study design
The extension study of the Thymectomy Trial in Non-Thymomatous Myasthenia Gravis Patients Receiving Prednisone (MGTX) was a multicentre, international, rater-blinded study performed across 36 academic medical centres in 15 countries. The extension study was open to patients who completed the 36-month MGTX trial and were willing to participate. Local institutional review board or ethics committee approvals and written informed consent were required before sites enrolled subjects in the extension study.
Participants
Participants were recruited locally; each centre was asked to screen all MG patients encountered for possible entry into the trial. Inclusion criteria for MGTX were MG duration <5 years, age between 18 and 65 years, serum acetylcholine receptor (AChR) antibody level ≥1.00 nmol/l (elevated levels of 0.50–0.99 nmol/l were accepted if diagnosis was confirmed by positive edrophonium test, abnormal repetitive nerve stimulation, or abnormal single fiber electromyography), and MG Foundation of America Clinical Classification7 Class II to IV (excluding Class I which indicates weakness only in ocular muscles, and Class V which represents crisis requiring intubation). Subjects were on optimal anticholinesterase therapy with or without oral corticosteroids. Exclusion criteria included thymoma on chest imaging, previous thymectomy, immunotherapy other than prednisone, pregnancy or lactation, unwillingness to avoid pregnancy, contraindications to corticosteroids, and significant medical illness that would prevent participation. Additional exclusion criteria for the extension study were subject’s desire to pursue thymectomy after completing the 36-month visit or enrollment into another experimental clinical trial.
Randomisation and masking
MGTX subjects were originally randomised in a 1:1 ratio to extended transsternal thymectomy (ETTX) plus prednisone or the same prednisone protocol alone. ETTX was performed within 30 days of randomisation, set as month 0. Beginning at month 4 and continuing to month 60, a blinded rater assessed the subjects who wore black, high-collared shirts to conceal transsternal incisions. There was no randomisation related to the extension study.
Procedures
The extension study followed the same prednisone protocol used for MGTX, as follows. When first enrolled into MGTX, patients not already taking prednisone had received an alternate-day oral prednisone dose starting at 10 mg, increased in 10 mg steps every other day to 100 mg on alternate days or 1.5 mg/kg, whichever was lower. For subjects already on prednisone, the dose could be increased up to 120 mg if they failed to reach Minimal Manifestation Status (MMS)7 by month 4. MMS, defined as no symptoms or functional limitations from MG although minor weakness may be present on examination, represents an accepted goal of therapy in the disease.8 The prednisone dose was maintained until MMS was achieved and the Quantitative Myasthenia Gravis (QMG) score9 [13 items, total range 0–39, with higher scores indicating more severe weakness] was <14 and also had fallen at least one point below baseline, as determined by the blinded rater. The prednisone dose was then reduced by 10 mg on alternate days every two weeks until 40 mg every other day was reached, with subsequent slowing of the taper to 5 mg every month, as long as MMS was maintained. In cases where MMS was lost, prednisone was increased by 10 mg on alternate days every two weeks until it was regained. Tapering could resume 4 weeks after MMS was restored.
Once prednisone tapering commenced, total pyridostigmine dose could not exceed 240 mg/day. In both the MGTX trial and extension study, plasmapheresis or intravenous immunoglobulin was permitted to stabilise patients at the discretion of the unblinded neurologist but not to maintain MMS. Patients who failed to achieve MMS at 12 months or who experienced intolerable side effects of prednisone could be placed on azathioprine 2.5 mg/kg/day or a substitute immunosuppressant if they could not tolerate azathioprine. Laboratory monitoring in the extension study was left to the discretion of site investigators.
From month 36 to month 60, rater-blinded QMG scores and prednisone requirements were recorded at study visits every 3 months. Prednisone intake was determined by pill counts using blister packs containing 10 mg tablets, with separate sheets provided for each dose. The alternate-day dosing was recorded in a patient diary, allowing comparison with pill counts derived from the blister packs that were checked at each visit. Pill cutters were provided for 5 mg dosing, and unused half pills were returned to the pouches.
Outcomes
The primary outcome was a staged assessment of the time-weighted average QMG score and time-weighted average required dose of prednisone through month 60. This approach examined for a potential effect of thymectomy on clinical status in addition to how thymic resection might influence long-term prednisone requirements. Rationale for a two-stage primary outcome was that an improved clinical status could be secondary to higher prednisone dosing and a poorer one to lower dosing. The first stage of the analysis compared the clinical outcomes between the two groups using the time-weighted QMG. On the basis of results of this between-group comparison of clinical outcomes (improvement, worsening, or unchanged), the difference in total prednisone requirements was analysed. QMG values and prednisone doses were collected locally, but data was centrally assessed at University of Alabama at Birmingham (UAB), Alabama, USA.
Other secondary outcomes measured from month 0 through month 60 were the MG Activities of Daily Living10 (MG-ADL; range 0–24, higher scores indicate more severe disease), proportion of subjects reaching MMS, and usage of non-steroid immunosuppressants, plasma exchange and intravenous immunoglobulin. A novel MG Quality-of-Life questionnaire11 (MG-QOL15, range 0–60, higher scores indicate more severe disease) that was developed after MGTX commenced was assessed in exploratory manner at months 39, 48 and 60. For another secondary outcome, we repeated the primary dosing analysis using a pre-specified penalty if azathioprine was added to prednisone. The two penalty methods were: (1) taking the maximum dose of prednisone before azathioprine was added, and (2) taking the prednisone dose at the time azathioprine commenced. These doses were maintained through month 60 or to the time of study withdrawal.
Other secondary outcomes assessed from months 0 to 60 focused on safety and adverse events including days of hospitalisation and surveys adapted from the cardiac transplant literature12 to assess 36 treatment-associated complications from corticosteroids.
A Data Safety Monitoring Board was assembled by NINDS that oversaw the MGTX trial and extension study until the very last study assessment was completed. The trial was registered on clinicaltrials.gov, number NCT00294658.
Statistical analysis
Data management was conducted at UAB via a web-based system. Notification of adverse events and visit tracking were performed electronically. Intention-to-treat was used for all analyses. The protocol pre-specified analysis of 3 subgroups (prior corticosteroid use, gender, and disease onset below and above 40 years). There were no planned adjustments for multiple secondary outcomes. For the main MGTX trial, sample size calculations were based on a reduction of the time-weighted average prednisone dose of ≥30% in favor of one treatment. This reduction was deemed the minimum to be clinically valuable by a consensus of international MG specialists participating in the trial. The sample size calculation assumed a two-group comparison of the treatment means, with the distribution of the time-weighted average prednisone dose values assumed to be approximately normal. This assumption was satisfactorily tested in the Palace et al. trial of azathioprine plus prednisolone vs. prednisolone alone.13 For 90% power to obtain a significant result at the 5% two-tailed level, the MGTX trial required 60 subjects in each arm. A separate power calculation was not performed for the extension study which enrolled patients based on their preference to continue with trial assessments.
An objective of the extension study was to maximize the amount of information collected to gain better insight into how subjects fared after month 36. Subjects who were enrolled at later stages were not expected to complete all visits through month 60. Statistical analyses adjusted for the amount of follow-up contributed per subject. Time-weighted outcomes were based on the area under the curve averaged up to the final visit available for that patient. For the analysis of achieving MMS, the Cox Proportional hazards model considered the outcome as censored if the event did not happen by the end of the study or the subject dropped out before this outcome was reached.
Time-weighted average QMG, prednisone dose, and MG-ADL analyses were calculated by computing the area under the curve using the trapezoidal rule divided by the number of days from randomisation to the last visit. To compare the two treatment groups with respect to these outcomes, t-tests were performed for the main analyses, while Wilcoxon 2-sample exact test was used for the subgroup analysis. In addition to these tests, 99.5% confidence intervals on the mean difference were constructed. Cox proportional hazards models were used to evaluate the time from month 0 to reach initial MMS, while logistic regression with the treatment group in the model was used to compare the proportion achieving MMS at months 48 and 60.
For the MG-QOL15, we used Wilcoxon 2-sample test at each time point. All analyses were done using SAS version 9.4.
Role of the funding source
The NIH (NINDS) funded the MGTX trial and extension study. Through the review process, NINDS contributed to development of the dual primary outcome. NINDS had no role in study performance, data collection, data analysis or interpretation, or writing of this report. The corresponding author had full access to all data in the study and final responsibility for the decision to submit for publication. MGTX investigators have established a policy for data sharing. Researchers wishing to access the data collected in the MGTX Extension Study are requested to contact Dr. Gil Wolfe at gilwolfe@buffalo.edu.
RESULTS
For the MGTX trial, a total of 6958 patients were assessed for eligibility, with 6727 not meeting inclusion criteria, mainly due to duration of disease beyond 5 years (47%), age limits (42%), use of nonglucocorticoid immunosuppressive agents (29%), and prior thymectomy or chest surgery (28%). Of 231 eligible subjects, 126 patients (55%) were randomised into MGTX between July 26, 2006 and November 28, 2012, and 111 (88% of enrolled subjects) completed the 36-month assessment (see Appendix). Of these 111 subjects, 68 (61%) entered the extension study between September 1, 2009 and August 26, 2015 (Figure 1). Three of 35 (9%) extension study patients who had been randomised to thymectomy refused thymectomy. Three of 33 (9%) extension study patients randomised to prednisone alone insisted on thymectomy which was performed prior to month 36 in two and after month 36 in one. Of the 35 patients in the thymectomy group, 26 (74%) completed the month 60 visit, with 4 patients dropping out and 5 others not reaching the end of year 5 prior to the study terminating. Of the 33 patients in the prednisone alone group, 24 (73%) completed the month 60 visit, with 5 dropping out and 4 others not reaching the end of year 5 at study closure.
Figure 1. Trial profile.

“Did not complete” refers to subjects who did not reach that visit before the study ended.
There were a few notable differences among subjects who entered the extension study and those who did not (appendix). This included a higher proportion of Hispanic patients, a more severe QMG score when first enrolled in MGTX, but better MG-ADL scores and fewer treatment-associated complications when they entered the extension study. Baseline characteristics were similar between groups in the extension study (Table 1).
Table 1.
Demographic and clinical characteristics
| Prednisone Alone (N=33) |
Thymectomy plus Prednisone (N=35) |
|
|---|---|---|
| Gender | ||
| Women | 24 (73) | 27 (77) |
| Men | 9 (27) | 8 (23) |
| Median age in years at enrollment (range) | 35.4 (18.0–63.0) | 34.3 (18.0–63.0) |
| Median disease duration in years at enrollment (range) | 1.49 (0.24–3.97) | 1.22 (0.15–2.95) |
| Ethnicity | ||
| Asian | 3 (9) | 5 (14) |
| Black, African American | 3 (9) | 2 (6) |
| Hispanic | 15 (45) | 12 (34) |
| White, not Hispanic origin | 10 (30) | 13 (37) |
| Other (Mixed, Native American, or Alaskan) | 2 (6) | 3 (9) |
| MG Foundation of America Class at enrollment† | ||
| Class IIa | 12 (36) | 12 (34) |
| Class IIb | 8 (24) | 9 (26) |
| Class III | 12 (36) | 12 (34) |
| Class IV | 1 (3) | 2 (6) |
| Therapy at enrollment | ||
| Pyridostigmine | 32 (97) | 33 (94) |
| Corticosteroids | 24 (73) | 26 (74) |
| Prior intravenous immunoglobulin | 7 (21) | 2 (6) |
| Prior plasma exchange | 4 (12) | 5 (14) |
| Enrollment measures | ||
| Quantitative MG score | 13.00 ± 4.68 | 12.34 ± 5.05 |
| Alternate day prednisone dosage, mg | 48.5 ± 30.7 | 46.3 ± 32.7 |
| MG Activity of Daily Living score | 5.48 ± 2.99 | 5.37 ± 3.46 |
Data are n (%) or mean (SD)
Myasthenia Gravis Foundation of America Class (II mild weakness, III moderate weakness, IV severe weakness, (“a” denotes predominantly limb and axial, “b” denotes predominantly bulbar).
For the primary outcome, patients in the thymectomy plus prednisone group showed significantly improved time-weighted average QMG scores from month 0 to month 60 compared to the prednisone alone group (5.47±3.87 vs. 9.34±5.08; p=0.0007; Table 2, Figure 2). Similarly, the time-weighted average prednisone dose from month 0 to month 60 for the thymectomy plus prednisone group was significantly lower compared to the prednisone alone group, with an average alternate-day dose of 24±21 mg vs. 48±29 mg (p=0.0002; Table 2, Figure 2). Pre-specified subgroup analyses of the time-weighted average QMG score by age at disease onset (less than or ≥ 40 years) favored the thymectomy arm for both age groups (5.87±4.24 vs. 9.53±5.69, p=0.0213 for disease onset <40 years; 4.69±3.05 vs. 8.92±3.53, p=0.0056 for disease onset ≥ 40 years). Likewise, pre-specified subgroup analysis favored thymectomy irrespective of patient sex on the time-weighted average QMG score (6.20±4.02 vs. 9.96±5.34, p=0.0092 for women; 3.00±1.92 vs. 7.70±4.13, p=0.0274 for men). These subgroup analyses also favored the thymectomy group for the time-weighted average prednisone dose except for men, which failed to reach significance possibly due to small sample size (Table 2). For the few patients naïve to prednisone at initial entry into the MGTX trial, only the time-weighted prednisone dose analysis showed a significant difference between the thymectomy plus prednisone vs. prednisone alone arm (Table 2).
Table 2.
Changes in QMG score and prednisone dose between baseline and 60 months
| Prednisone alone (n=33) |
Thymectomy plus prednisone (n=35) |
Estimated Difference (95% CI*)† |
P Value | |||
|---|---|---|---|---|---|---|
| Primary Outcomes | ||||||
| Time-weighted average QMG score | 9.34 ± 5.08 | 33 | 5.47±3.87 | 35 | 3.87 (0.71 to 7.04) | 0.0007 |
| Time-weighted average alternate-day prednisone dose (mg) | 48 ± 29 | 33 | 24 ± 21 | 35 | 24 (12 to 36) | 0.0002 |
| Subgroup Analyses | ||||||
| Time-weighted average QMG score | ||||||
| Prednisone use at month 0 | 0.69& | |||||
| Yes | 9.71 ± 5.25 | 24 (73) | 5.56 ± 3.55 | 26 (74) | 4.16 (0.45 to 7.86) | 0.0022 |
| No | 8.36 ± 4.75 | 9 (27) | 5.21 ± 4.92 | 9 (26) | 3.15 (−4.26 to 10.56) | 0.16 |
| Sex | 0.71& | |||||
| Women | 9.96 ± 5.34 | 24 (73) | 6.20 ± 4.02 | 27 (77) | 3.76 (−0.10 to 7.63) | 0.0092 |
| Men | 7.70 ± 4.13 | 9 (27) | 3.00 ± 1.92 | 8 (23) | 4.70 (−0.55 to 9.95) | 0.0274 |
| Age at disease onset | 0.81& | |||||
| < 40 yr | 9.53 ± 5.69 | 23 (70) | 5.87 ± 4.24 | 23 (66) | 3.66 (−0.72 to 8.03) | 0.0213 |
| ≥ 40 yr | 8.92 ± 3.53 | 10 (30) | 4.69 ± 3.05 | 12 (34) | 4.22 (−0.20 to 8.64) | 0.0056 |
| Time-weighted average alternate-day prednisone dose (mg) | ||||||
| Prednisone use at month 0 | 0.40& | |||||
| Yes | 54 ± 31 | 24 (73) | 26 ± 21 | 26 (74) | 27 (12 to 42) | 0.0005 |
| No | 34 ± 19 | 9 (27) | 18 ± 20 | 9 (26) | 16 (−4 to 35) | 0.0400 |
| Sex | 0.36& | |||||
| Women | 47 ± 26 | 24 (73) | 26 ± 23 | 27 (77) | 21 (7 to 35) | 0.0024 |
| Men | 51 ± 38 | 9 (27) | 17 ± 8 | 8 (23) | 34 (5 to 64) | 0.0592 |
| Age at disease onset | 0.78& | |||||
| < 40 yr | 48 ± 29 | 23 (70) | 26 ± 23 | 23 (66) | 23 (7 to 38) | 0.0031 |
| ≥ 40 yr | 48 ± 31 | 10 (30) | 21 ± 16 | 12 (34) | 26 (5 to 48) | 0.0112 |
Data are n (%) and mean (SD)
CI denotes confidence interval for the mean
We used 95% confidence intervals in all analyses except for analyses involving the QMG score, for which we used 99.5% confidence intervals, per protocol. QMG= Quantitative Myasthenia Gravis Score XX
Figure 2.
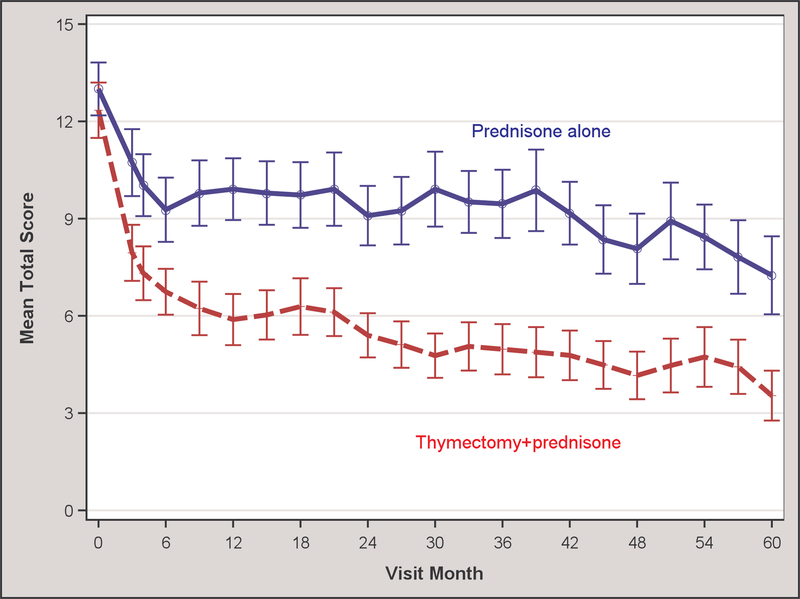
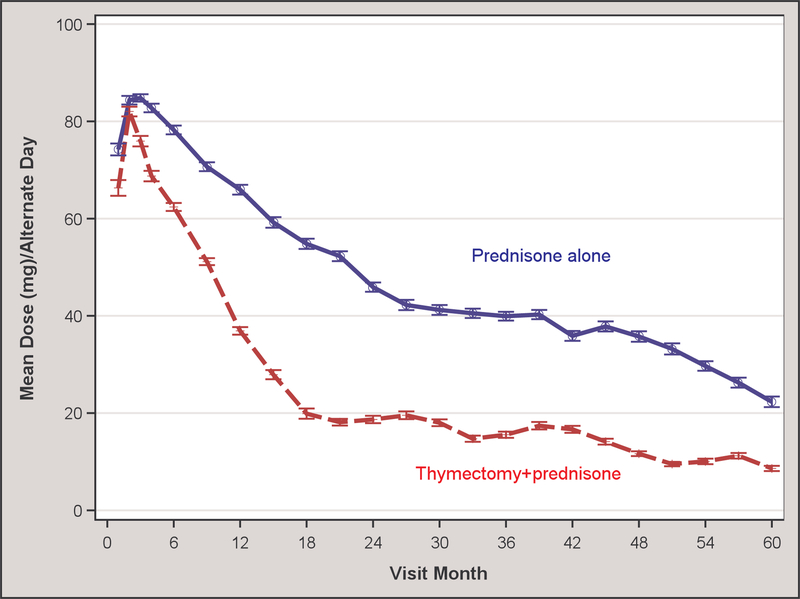
(A) Quantitative MG score and (B) average prednisone dose according to treatment group over 5-year period
For secondary outcomes, the time-weighted average MG-ADL score from month 0 to month 48 favored the thymectomy group (1.10±1.51 vs. 2.55±3.02; p=0.0245) but from month 0 to month 60 there was no significant difference, with both treatment groups exhibiting very low MG-ADL scores in the 1–2 point range by that time point (Table 3). The proportion of patients in MMS at month 60 was significantly higher (23/26, 88% vs. 14/24, 58%; estimated difference 30.1%, with 95% CI −53.4% to 6.9%, p=0.0236) in the thymectomy plus prednisone compared to the prednisone alone group (Table 3). From month 0 to month 60, the proportion of patients requiring azathioprine (7/35, 20% vs. 19/33, 58%; estimated difference 37.6% with 95% CI 16.1% to 59.0%, p=0.0014) or intravenous immunoglobulin (3/35, 9% vs. 11/33, 33%; estimated difference 24.8% with 95% CI 6.2% to 43.3%, p=0.0162) was also significantly reduced by thymectomy (Table 3). There was no significant difference between the two groups in utilization of plasma exchange, with neither group exceeding 14%. Patients who underwent thymectomy had a significantly lower MG-QOL15 score at month 39, indicating less disease burden on quality of life (4.8±9.2 vs. 13.1±14.0; p=0.0029), but no significant differences were found at months 48 or 60 between the two groups (Table 3).
Table 3.
Secondary outcomes at 5 years and earlier time points
| Treatment Group | Estimated Difference (95% CI) |
P Value | ||
|---|---|---|---|---|
| Prednisone Alone | Thymectomy +prednisone | |||
| Time-weighted average prescribed AD prednisone dose (mg)a | 49.0 ± 29.2 (N=33) |
25.9 ± 20.7 (N=35) |
23.1 (10.9 to 35.2) | 0.0003 |
| Penalized time-weighted average AD prednisone dose (mg; Method 1)a,b | 66.2 ± 36.7 (N=33) |
31.0 ± 31.8 (N=35) |
35.2 (18.6 to 51.9) | < 0.0001 |
| Penalized time-weighted AD average prednisone dose (mg; Method 2)a,c | 60.6 ± 34.6 (N=33) |
28.3 ± 27.9 (N=35) |
32.3 (17.2 to 47.5) | < 0.0001 |
| Time-weighted average MG Activities of Daily Living month 0–60a,d | 3.26 ± 2.77 (N=32) |
1.61 ± 1.46 (N=34) |
1.65 (0.54 to 2.75) | 0.0044 |
| at month 48 | 2.55 ± 3.02 (N=29) | 1.10 ± 1.51 (N=31) | 1.45 (0.20 to 2.71) | 0.0245 |
| at month 60 | 2.04 ± 2.63 (N=24) | 1.23 ± 1.75 (N=26) | 0.81 (−0.48 to 2.07) | 0.21 |
| Azathioprine usef | 19/33 (58) | 7/35 (20) | 37.6% (16.1% to 59.0%) | 0.0014 |
| Plasma exchange usef | 4/33 (12) | 5/35 (14) | −2% (−18.2% to 13.9%) | 0.73 |
| Intravenous immunoglobulin usef | 11/33 (33) | 3/35 (9) | 24.8% (6.2% to 43.3%) | 0.0162 |
| Minimal Manifestation Statuse | ||||
| at month 48f | 15/29 (52) | 23/31 (74) | −22.5% (−46.3% to 1.4%) | 0.07 |
| at month 60f | 14/24 (58) | 23/26 (88) | −30.1% (−53.4% to 6.9%) | 0.0216 |
| MG-QOL15g | ||||
| at month 39 | 13.1 ± 14.0 (N=32) | 4.8 ± 9.2 (N=33) | 0.0029 | |
| at month 48 | 9.0 ± 10.1 (N=29) | 4.9 ± 7.9 (N=30) | 0.13 | |
| at month 60 | 7.7 ± 9.24 (N=24) | 7.8 ± 10.9 (N=26) | 0.96 | |
Data are n (%) or mean (SD)
P value based on two sample t-test.
Method 1: penalized using maximum dose before azathioprine.
Method 2: penalized using dose at time of starting azathioprine.
Myasthenia Gravis Activities of Daily Living scores 0,1, 2, 3, where 0=normal and higher score is worse.
P value=0.03 based on the Cox model on modeling time to first Minimal Manifestation Status over the period of 0–60 months.
P values based on logistic regression.
P values based on Wilcoxon 2-sample test.
AD denotes alternate day, ADL Activities of Daily Living, MG myasthenia gravis.
The pre-specified penalties on prednisone dosing for initiating azathioprine revealed significantly lower time-weighted average prednisone requirements from month 0 to month 60 in the thymectomy group (Table 3), irrespective of whether the analysis used the maximum prednisone dose before starting azathioprine (Method 1, 31.0±31.8 mg vs. 66.2±36.7 mg; p<0.0001) or the actual dose at the time of azathioprine initiation (Method 2, 28.3±27.9 mg vs. 60.6±34.6 mg; p<0.0001).
Cumulative days in the hospital by subjects who required hospitalisation for MG exacerbations from month 0 to month 60 were similar between the two groups (Table 4). Hospitalisations using Medical Dictionary for Regulatory Activities (MEDRA) coding were of low frequency (≤6%) through month 60 for all disorder categories except for the nervous system that primarily reflected MG exacerbation (Table 4). Such hospitalisations were three times as common (10 of 33 patients, 30% vs. 3 of 35 patients, 9%) in the prednisone alone group compared to the thymectomy plus prednisone group. In the treatment-associated complications survey we recorded 29 events in the thymectomy group and 37 in the prednisone alone group (Table 4). These events were primarily related to complications of prednisone therapy.
Table 4.
Adverse events from extension study
| Prednisone Alone (N=33) |
Thymectomy+ Prednisone (N=35) |
|
|---|---|---|
| Number of events through month 60 on TAC survey | 37 | 29 |
| Patients having ≥1 event through month 60 on TAC survey | 14 (42) | 12 (34) |
| Classification | ||
| Life threatening | 5 (15) | 1 (3) |
| Disability, Incapacity† | 0 (0) | 6 (17) |
| Required medical or surgical intervention | 2 (6) | 5 (14) |
| Death | 0 (0) | 0 (0) |
| Hospitalisations for all causes | 16 (48) | 5 (14) |
| Cumulative hospital days* | 29.2 ± 22.3 | 26.0 ± 21.2 |
| Hospitalisation by MEDRA codes | ||
| Gastrointestinal disorders | 1 (3) | 1 (3) |
| Hepatobiliary disorders | 1 (3) | 0 (0) |
| Infections and infestations | 2 (6) | 2 (6) |
| Injury, poisoning and procedure complications | 0 (0) | 1 (3) |
| Metabolism and nutrition disorders | 0 (0) | 1 (3) |
| Nervous system disorders | 10 (30) | 3 (9) |
| Respiratory, thoracic and mediastinal disorders | 2 (6) | 0 (0) |
| Surgical and medical procedures | 2 (6) | 0 (0) |
| Vascular disorders | 1 (3) | 0 (0) |
| Hospitalisation for MG exacerbation | ||
| Months 0–60: # of patients | 10/33 (30) | 2/35 (6) |
| cumulative days | 26.4 ± 22.9 | 26.0 ± 21.2 |
Data are n (%) or mean (SD)
Disability/incapacity etiologies: for prednisone alone group, worsening swallowing difficulties and myasthenia gravis; in thymectomy+prednisone group, osteoporotic thoracic fracture, ocular muscle involvement due to relapsing MG, post-thymectomy diaphragmatic hemiparesis, rib fracture, impending myasthenic crisis, Pott’s fracture, tear of left knee meniscus, and low back pain with possible stenosis.
Only for those who had hospitalisation
MEDRA denotes medical dictionary for regulatory activities, MG myasthenia gravis; TAC treatment associated complications survey
Between months 36 and 60, only four patients (2 in the thymectomy plus prednisone group, 2 in the prednisone only group) exhibited a ≥2 point increase in the QMG score, the threshold widely accepted as indicative of clinical worsening.14 There were no deaths during the extension study.
DISCUSSION
The MGTX Extension Study demonstrates a continued benefit for thymectomy plus prednisone versus prednisone alone on time-weighted average QMG scores, a validated measure of clinical status, while reducing time-weighted average prednisone requirements for up to 5 years following thymic resection in acetylcholine receptor antibody-positive generalised MG patients. The extension study reinforces the benefit of thymectomy observed in the randomised, controlled MGTX trial,1 extending the favorable impact beyond 3 years, while dispelling doubts about the procedure’s benefits or the longevity of its impact.3 Furthermore, the proportion of patients in MMS at the end of the extension study was significantly greater for the thymectomy group.
More studies are focusing on long-term outcomes of treatment in MG, and what management strategies early in the disease course might influence better disease control over time. Although full remission after treatment is uncommon,15 two large-scale retrospective studies have shown that MG outcomes have improved markedly over the last half century,16 and that 95% of patients have either no, purely ocular or only mild generalised weakness after several years of treatment.17
An international panel of MG experts convened through the MG Foundation of America recently generated a treatment guidance. The panel defined the goal of MG management as having patients achieve MMS or remission with no greater than mild adverse events.8 In a retrospective survey across Japan, an aggressive treatment strategy in the first month after diagnosis that incorporates plasma exchange, intravenous immunoglobulin or methylprednisolone -- individually or in combination – yielded an outcome of persistent MMS or better in 123 of 249 (49%) patients at a mean of 6 years while on prednisolone doses ≤5 mg/day.18 Of 439 patients treated less aggressively using oral corticosteroids, 185 (42%) also achieved MMS, but over a mean of 11 years. In a retrospective single-centre study, MMS or better outcomes were observed in 60 of 74 (81%) MG patients at a mean follow-up of 6 years using conventional treatments including thymectomy, pyridostigimine, prednisone, mycophenolate mofetil, azathioprine, and cyclosporine.19 Similarly, in a study of 268 MG patients, MMS or better with no more than mild side effects was observed in 155 of 213 (73%) subjects with complete data at 5 years and 87 of 116 (75%) at 10 years.20 Of the many variables tested, only disease onset after 50 years and thymectomy were found predictive of reaching MMS or better at 10 years.20
The studies summarized above included a full range of MG patients with variable antibody status and even thymoma. The proportion of patients who underwent thymectomy ranged from 20%,19 40%,20 and 49%,18 and in only one of the retrospective series was thymectomy clearly recommended as part of management.20 Compared to MGTX, therapeutic options in these series were more liberal, with a variety of immunosuppressive agents utilized. Still, the proportion of MGTX Extension Study subjects achieving a desired outcome defined as MMS or better compares favorably with these series, even exceeding them. For those MGTX patients who reached a 5-year study visit, 88% were in MMS if they had undergone thymectomy.
The extension study has several limitations, and we would exercise caution in predicting such a high likelihood of favorable outcome for all MG patients who undergo thymectomy. The extension study included 68 of 111 patients (61%) who completed the 36-month MGTX trial, and only 50 (45%) reached the month 60 assessment. Compared to the entire MGTX population, the extension study cohort at 36 months had more favorable MG-ADL scores and fewer treatment-associated complications, perhaps predicting a better outcome at month 60 (Appendix). This uncertainty is akin to long-term observations in general, since subjects who are less responsive to or tolerant of study interventions may drop out over time.
When further comparing MGTX trial subjects who entered the extension study to those who did not, extension study subjects had slightly worse QMG scores at month 36 (Appendix). Although MG-ADL scores were lower in extension study subjects, absolute mean scores were very low in both populations, with the difference amounting to just over 1 point on the 24-point scale. Extension study subjects were requiring slightly higher doses of prednisone at month 36 to maintain MMS, possibly indicative of greater disease activity over prior weeks (Appendix). We again cannot exclude the possibility that the extension study enrolled patients who ultimately would have more favorable outcomes than those in the MGTX trial who did not participate or the generalised MG population at large. However, when considering in full the clinical outcomes and prednisone requirements for extension study patients compared to those who did not participate, we believe the extension study enrollment was generally representative of the entire MGTX cohort.
For the QMG, either a 2 or 3-point reduction (depending on the baseline score) has been established by the University of Toronto group as a minimal clinically important difference14 and a reduction of 2.3 points has correlated with observations of clinical improvement by neurologists with expertise in MG.21 In the extension study, thymectomy reduced the time-weighted average QMG by 3.87 points at 5 years versus medical therapy alone, exceeding the estimated difference of 2.85 points seen at 3 years in MGTX.1 Likewise, the proportion of thymectomy patients in MMS at 5 years was significantly higher, with 88% (23 of 26 patients) achieving this milestone versus 58% (14 of 24) in the prednisone alone group. These MMS proportions were 67% (39 of 58) versus 47% (24 of 51), respectively, at the 3-year MGTX outcome for all subjects, including those who did not participate in the extension study. Based on the MMS figures, it is reasonable to conclude that benefits conferred by thymectomy persist beyond a three-year window and may even increase over the next two years. Beyond clinical outcomes, both patient populations experienced reduced prednisone requirements to maintain MMS over the two-year extension study. For the thymectomy arm, the average alternate-day prednisone dose fell to 11.9 mg at month 60 (Figure 2B), essentially reaching a dosing level of 5 mg a day or lower that a Japanese multicentre study has associated with quality-of-life metrics comparable to patients who are in complete stable remission off all therapy.22 A prednisolone dosing level of 5 mg a day or less has been adopted by Japanese experts as a goal of therapy in their national guideline for MG.23, 24
Results of the MGTX Extension Study provide further evidence of the positive impact offered by thymectomy in AChR antibody-positive generalised non-thymomatous MG patients, the single largest subpopulation of the disease.15 This benefit from thymectomy persists at least for 5 years and extends beyond clinical status alone to include significant reductions in immunosuppressive medication requirements and hospitalisations for disease exacerbations. Implications of these results include revisions to formal practice parameters that had previously failed to uncover evidence to argue in favor of the procedure3, 4 and reversing trends showing a marked reduction in MG-related admissions for thymectomy after 2000.25
Supplementary Material
RESEARCH IN CONTEXT.
Evidence before the study
We searched PubMed for English language articles using the terms “randomised,” “thymectomy,” and “myasthenia gravis” to identify 39 articles published between January 1, 2012 to August 28, 2018. This PubMed search overlapped with prior searches conducted in design, performance, and publication stages of the MGTX trial, a prospective, randomised, rater-blinded study of thymectomy in patients with myasthenia gravis (MG) over 3 years. Our search identified the one publication that reported results of the MGTX trial and one publication from our investigator group reporting biomarker results from the MGTX trial. There were 6 letters to the editor or editorials commenting on MGTX trial results. No other randomised thymectomy studies in MG were found. Prior to MGTX, observational studies mostly argued in favor of thymectomy in improving outcomes in non-thymomatous MG. Practice parameters, however, identified numerous flaws in these studies, arguing for a randomised, controlled trial. MGTX determined that extended transsternal thymectomy combined with a standardised prednisone protocol was superior to prednisone alone at 3 years in improving clinical status and lowering medication requirements in generalised non-thymomatous MG.
Added value of this study
The MGTX extension study which followed patients under the same protocol through month 60 (5 years) demonstrates continued benefit conferred by thymectomy in patients with generalised non-thymomatous MG. These benefits include improved disease outcomes and reduced immunosuppressive medication requirements and need for hospitalisations to address disease exacerbations. In addition, when compared to observational long-term outcome studies in MG that tracked minimal manifestation status rates, the extension study results for the thymectomy arm are favorable.
Implications of all the available evidence
Results of the MGTX Extension Study bolster the evidence that thymectomy performed within the first few years of the disease course confers benefits that persist for at least 5 years in patients with generalised, non-thymomatous MG. The benefit extends beyond clinical status alone and includes reduced requirements for medication and hospitalisation. The results from the study provide further support for the use of thymectomy in managing MG.
Acknowledgements
The MGTX investigators would like to acknowledge the efforts and valuable input provided by the MGTX Data Safety Monitoring Board members: Steven Keller, M.D. (Chairman), Alan Dyer, Ph.D., Donald Schotland, M.D., John Winer, M.D., F.R.C.P., and Peter Gilbert, Sc.M. (ex officio).
This work was supported in part by CTSA grants from the National Centre for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH) awarded to the following institutions: University of Alabama at Birmingham (UL1TR001417); University of Kansas Medical Centre for Frontiers: The Heartland Institute for Clinical and Translational Research (UL1TR000001); University at Buffalo/SUNY (UL1TR001412), and University of Texas Health Science Centre at San Antonio (UL1TR001120 and National Centre for Research Resources/NCATS 8UL1TR000149). The Muscular Dystrophy Association and MG Foundation of America provided support for initial planning of the MGTX trial.
The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or NCATS.
Declaration of interests
Drs. Wolfe, Kaminski, Aban, Marx, Ströbel, Oger, Cea, Heckmann, Evoli, Nix, Ciafaloni, Antonini, Witoonpanich, King, Beydoun, Chalk, Barboi, Amato, Shaibani, Katirji, Lecky, Buckley, Dias-Tosta, Yoshikawa, Waddington-Cruz, Pulley, Rivner, Kostera-Pruszczyk, Pascuzzi, Jackson, Verschuuren, Massey, Kissel, Werneck, Benatar, Barohn, Tandan, Mozaffar, Silvestri, Conwit, Sonett, and Cutter, Prof. Vincent, Mr. Minisman and Ms. Kuo report grants from NIH (NINDS), grants from Muscular Dystrophy Association, and grants from the Myasthenia Gravis Foundation of America, during the conduct of this study. In addition, Dr. Benatar reports per patient reimbursement from UCB Pharma, Alexion Pharmaceuticals, and NINDS (NIH) for NeuroNEXT 103 Trial, and personal fees from Ra Pharmaceuticals, outside the submitted work. Dr. Cutter reports data and safety monitoring board activity for AMO Pharmaceuticals, Biolinerx, Horizon Pharmaceuticals,, Merck, Merck/Pfizer, Opko Biologics, Neurim, Orphazyme, Sanofi-Aventis, Reata Pharmaceuticals, Receptos/Celgene, Teva Pharmaceuticals, NHLBI (Protocol Review Committee), NICHD (OPRU oversight committee) outside the submitted work; Dr. Cutter also reports consulting or advisory board activity for Atara Biotherapeutics, Axon, Biogen, Biotherapeutics, Argenix, Brainstorm Cell Therapeutics, Charleston Labs Inc., Click Therapeutics, Genzyme, Genentech, GW Pharma, Klein-Buendel Incorporated, Medimmune, Medday, Novartis, Roche, Scifluor, Somahlution, Teva pharmaceuticals, TG Therapeutics, and UT Houston, outside the submitted work. Dr. Evoli reports personal fees from Grifols, outside the submitted work. Dr. Massey reports consultant activities for QuatroBio LLC outside the submitted work. Dr. Pulley reports personal fees from Grifols and personal fees from CSL-Behring, outside the submitted work. Dr. Verschuuren reports grants from Myasterix, project ID 602420, grants from Princes Beatrix Fonds, other payments from Consultancies, outside the submitted work. Dr. Wolfe reports personal fees from Grifols, personal fees from Shire, grants from CSL Behring, grants from ArgenX, grants and personal fees from Alexion, outside the submitted work.
Funding: NIH (NINDS) U01 NS042685
Footnotes
See Appendix for MGTX Study Group members
Contributor Information
Gil I. Wolfe, Department of Neurology, Univ. at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY, USA
Henry J. Kaminski, Department of Neurology, George Washington University School of Medicine and Health Sciences, Washington, D.C., USA
Inmaculada B. Aban, Dept. of Biostatistics, Univ. of Alabama at Birmingham, Birmingham, AL, USA
Greg Minisman, Dept. of Biostatistics, Univ. of Alabama at Birmingham, Birmingham, AL, USA.
Hui-Chien Kuo, Dept. of Biostatistics, Univ. of Alabama at Birmingham, Birmingham, AL, USA.
Alexander Marx, Institute of Pathology, University Medical Centre Mannheim, University of Heidelberg, Mannheim, Germany.
Philipp Ströbel, Institute of Pathology, University of Göttingen, Göttingen, Germany.
Claudio Mazia, Department of Neurology, University of Buenos Aires, Buenos Aires, Argentina.
Joel Oger, Division of Neurology, University of British Columbia, Vancouver, Canada.
J. Gabriel Cea, Department of Neurology, University of Chile, Santiago, Chile
Jeannine M. Heckmann, Division of Neurology, Department of Medicine, University of Cape Town, Cape Town, South Africa
Amelia Evoli, Department of Neurology, Catholic University, Rome, Italy.
Wilfred Nix, Department of Neurology, Johanes Gutenberg University, Mainz, Germany.
Emma Ciafaloni, Department of Neurology, University of Rochester Medical Centre, Rochester, NY, USA.
Giovanni Antonini, Department of Neurology, Mental Health and Sensory Organs, University of Rome “Sapienza,” Rome, Italy.
Rawiphan Witoonpanich, Department of Neurology, Mahidol University, Bangkok, Thailand.
John O. King, Department of Neurology, University of Melbourne, Melbourne, Australia
Said R. Beydoun, Department of Neurology, University of Southern California, Los Angeles, CA, USA
Colin H. Chalk, Department of Neurology, McGill University, Montreal, Canada
Alexandru C. Barboi, Department of Neurology, Medical College of Wisconsin, Milwaukee, WI, USA
Anthony A. Amato, Department of Neurology, Harvard Medical School, Boston, MA, USA
Aziz I. Shaibani, Nerve and Muscle Centre of Texas, Houston, Texas, USA
Bashar Katirji, Department of Neurology, Case Western Reserve University, Cleveland, OH, USA.
Bryan R. F. Lecky, Walton Centre for Neurology and Neurosurgery, Liverpool, U.K.
Camilla Buckley, Nuffield Department of Clinical Neurosciences, Oxford University, Oxford, U.K..
Angela Vincent, Nuffield Department of Clinical Neurosciences, Oxford University, Oxford, U.K..
Elza Dias-Tosta, Unit of Neurology, Hospital de Base do Distrito Federal, Brasília, Brazil.
Hiroaki Yoshikawa, Department of Neurology, Kanazawa University, Kanazawa, Japan.
Márcia Waddington-Cruz, Department of Neurology, Federal University, Rio de Janeiro, Brazil.
Michael T. Pulley, Department of Neurology, University of Florida, Jacksonville, FL, USA
Michael H. Rivner, Department of Neurology, Georgia Regents University, Augusta, GA, USA
Anna Kostera-Pruszczyk, Department of Neurology, Medical University of Warsaw, Warsaw, Poland.
Robert M. Pascuzzi, Department of Neurology, Indiana University School of Medicine, Indianapolis, IN, USA
Carlayne E. Jackson, Department of Neurology, University of Texas Health Science Centre, San Antonio, TX, USA
Jan J.G.M. Verschuuren, Department of Neurology, Leiden University Medical Centre, Leiden, The Netherlands
Janice M. Massey, Department of Neurology, Duke University Medical Centre, Durham, NC, USA
John T. Kissel, Department of Neurology, The Ohio State University Wexner Medical Centre, Columbus, OH, USA
Lineu C. Werneck, Department of Neurology, Universidade Federal do Parana, Curitiba, Brazil
Michael Benatar, Department of Neurology, University of Miami, Miami, FL, USA.
Richard J. Barohn, Department of Neurology, University of Kansas Medical Centre, Kansas City, KS, USA
Rup Tandan, Department of Neurological Sciences, University of Vermont College of Medicine, Burlington, VT, USA.
Tahseen Mozaffar, Department of Neurology, University of California, Irvine, Orange, CA, USA.
Nicholas J. Silvestri, Department of Neurology, Univ. at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY, USA
Robin Conwit, Division of Extramural Research, NIH/National Institute of Neurological Disorders and Stroke, Bethesda, MD, USA.
Joshua R. Sonett, Section of General Thoracic Surgery, Columbia University Medical Centre, New York, NY, USA
Alfred Jaretzki, III, Section of General Thoracic Surgery, Columbia University Medical Centre, New York, NY, USA.
John Newsom-Davis, Nuffield Department of Clinical Neurosciences, Oxford University, Oxford, U.K..
Gary R. Cutter, Dept. of Biostatistics, Univ. of Alabama at Birmingham, Birmingham, AL, USA
References
- 1.Wolfe GI, Kaminski HJ, Aban IB, et al. Randomised trial of thymectomy in myasthenia gravis. N Engl J Med 2016; 376:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blalock A, Harvey AM, Ford FR, Lilienthal JL. The treatment of myasthenia gravis by removal of the thymus gland. JAMA 1941; 117:1529–1533. [Google Scholar]
- 3.Gronseth GS, Barohn RJ. Thymectomy for non-thymomatous autoimmune myasthenia gravis (an evidence-based review). Neurology 2000; 55:7–15. [DOI] [PubMed] [Google Scholar]
- 4.Cea G, Benatar M, Verdugo RJ, Salinas RA. Thymectomy for non-thymomatous myasthenia gravis (Review). Cochrane Library 2013; 10:1–20. [DOI] [PubMed] [Google Scholar]
- 5.Oosterhuis HJ. Observations of the natural history of myasthenia gravis and the effect of thymectomy. Ann NY Acad Sci 1981; 377:678–690. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez M, Gomez MR, Howard FM, Taylor WF. Myasthenia gravis in children: long-term follow-up. Ann Neurol 1983; 13:504–510. [DOI] [PubMed] [Google Scholar]
- 7.Jaretzki A III, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Neurology 2000; 55:16–23. [DOI] [PubMed] [Google Scholar]
- 8.Sanders DB, Wolfe GI, Benatar M, et al. International consensus guidance for the management of myasthenia gravis. Neurology 2016; 87:419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barohn RJ, McIntire D, Herbelin L, Wolfe GI, Nations S, Bryan WW. Reliability testing of the Quantitative Myasthenia Gravis Score. Ann NY Acad Sci 1998; 841:769–772. [DOI] [PubMed] [Google Scholar]
- 10.Wolfe GI, Herbelin L, Nations SP, Foster B, Bryan WW, Barohn RJ. Myasthenia gravis activities of daily living profile. Neurology 1999; 52:1487–1489. [DOI] [PubMed] [Google Scholar]
- 11.Burns TM, Conaway MR, Cutter GR, Sanders DB, Muscle Study Group. Less is more, or almost as much: a 15-item quality-of-life instrument for myasthenia gravis. Muscle Nerve 2008; 38:957–963. [DOI] [PubMed] [Google Scholar]
- 12.Moons P, De Geest S, Abraham I, Van Cleemput J, Vanhaecke J. Symptom experience associated with maintenance immunosuppression after heart transplantation: patient’s appraisal of side effects. Heart Lung 1998; 27:315–325. [DOI] [PubMed] [Google Scholar]
- 13.Palace J, Newsom-Davis J, Lecky B. A randomised double-blind trial of prednisolone alone or with azathioprine in myasthenia gravis. Neurology 1998; 50:1778–1783. [DOI] [PubMed] [Google Scholar]
- 14.Katzberg HD, Barnett C, Merkies ISJ, Bril V. Minimal clinically important difference in myasthenia gravis: outcomes from a randomised trial. Muscle Nerve 2014; 49:661–665. [DOI] [PubMed] [Google Scholar]
- 15.Gilhus NE. Myasthenia gravis. N Engl J Med 2016; 375:2570–2581. [DOI] [PubMed] [Google Scholar]
- 16.Grob D, Brunner N, Namba T, Pagala M. Lifetime course of myasthenia gravis. Muscle Nerve 2008; 37:141–149. [DOI] [PubMed] [Google Scholar]
- 17.Kawaguchi N, Kuwabara S, Nemoto Y, et al. Treatment and outcome of myasthenia gravis: retrospective multi-centre analysis of 470 Japanese patients, 1999–2000. J Neurol Sci 2004; 224:43–47. [DOI] [PubMed] [Google Scholar]
- 18.Utsugisawa K, Nagane Y, Akaishi T, et al. Early fast-acting treatment strategy against generalised myasthenia gravis. Muscle Nerve 2017; 55:794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salins S, Teter B, Kavak K, Wolfe GI, Silvestri NJ. Low-dose medication and long-term outcome in myasthenia gravis. J Clin Neuromusc Dis 2016; 18:61–66. [DOI] [PubMed] [Google Scholar]
- 20.Andersen JB, Gilhus NE, Sanders DB. Factors affecting outcome in myasthenia gravis. Muscle Nerve 2016; 54:1041–1049. [DOI] [PubMed] [Google Scholar]
- 21.Bedlack RS, Simel DL, Bosworth H, Samsa G, Tucker-Lipscomb B, Sanders DB. Quantitative myasthenia gravis score: Assessment of responsiveness and longitudinal validity. Neurology 2005; 64:1968–1970. [DOI] [PubMed] [Google Scholar]
- 22.Masuda M, Utsugisawa K, Suzuki S, et al. The MG-QOL15 Japanese version: validation and associations with clinical factors. Muscle Nerve 2012; 46:166–173. [DOI] [PubMed] [Google Scholar]
- 23.Utsugisawa K, Suzuki S, Nagane Y, et al. Health-related quality-of-life and treatment targets in myasthenia gravis. Muscle Nerve 2014; 50:493–500. [DOI] [PubMed] [Google Scholar]
- 24.Murai H Japanese clinical guidelines for myasthenia gravis: putting into practice. Clin Exp Neuroimmunol 2015; 6:21–31. [Google Scholar]
- 25.Alshekhlee A, Miles JD, Katirji B, Preston D, Kaminski HJ. Incidence and mortality rates of myasthenia gravis and myasthenic crisis in US hospitals. Neurology 2009; 72:1548–1554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.