

Summary:
Lafora disease (LD) is a fatal childhood epilepsy caused by recessive mutations in either the EPM2A or EPM2B gene. A hallmark of LD is the intracellular accumulation of insoluble polysaccharide deposits known as Lafora bodies (LBs) in the brain and other tissues. In LD mouse models, genetic reduction of glycogen synthesis eliminates LB formation and rescues the neurological phenotype. Therefore LBs have become a therapeutic target for ameliorating LD. Herein, we demonstrate that human pancreatic α-amylase degrades LBs. We fused this amylase to a cell-penetrating antibody fragment, and this antibody-enzyme fusion (VAL-0417) degrades LBs in vitro and dramatically reduces LB loads in vivo in Epm2a−/− mice. Using metabolomics and multivariate analysis we demonstrate that VAL-0417 treatment of Epm2a−/− mice reverses the metabolic phenotype to a wild-type profile. VAL-0417 is a promising drug for the treatment of LD and a putative precision therapy platform for intractable epilepsy.
Graphical Abstract
eTOC blurb
Lafora disease (LD) is a devastating childhood epilepsy caused by intracellular glycogen aggregates called Lafora bodies (LBs) in the brain and other tissues. Herein, Brewer et al. generated a first-in-class antibody-enzyme fusion, VAL-0417, that degrades LBs in vitro and in vivo pre-clinical models, showing promise as a LD drug.
Introduction
The progressive myoclonic epilepsies (PMEs) are a group of inherited disorders characterized by recurrent seizures, myoclonus, and progressive neurological decline. There are currently no treatments for PMEs, and anti-epilepsy drugs are palliative at best (Shahwan et al., 2005). Lafora disease (LD; epilepsy, progressive myoclonus type 2, EPM2) is a severe form of PME that typically manifests with tonic-clonic seizures and myoclonic jerks in the early teen years followed by rapid neurological deterioration, increasingly severe and frequent epileptic episodes, dementia and death within ten years of onset (OMIM: 254780). LD is caused by mutations in the EPM2A or EPM2B genes that encode laforin, a glycogen phosphatase, and malin, an E3 ubiquitin ligase that ubiquitinates enzymes involved in glycogen metabolism (reviewed in (Gentry et al., 2018)). LD is distinguishable from other PMEs by the presence of cytosolic polysaccharide inclusions known as Lafora bodies (LBs) most notably in the brain, where they are found in neuronal cell bodies dendrites, and astrocytic processes, and in other tissues such as muscle, heart, and liver. Among the PMEs, LD is uniquely considered a glycogen storage disease (GSD). Independent studies from multiple groups demonstrated that Epm2a−/− and Epm2b−/−mice, similar to LD patients, develop LBs and exhibit neurodegeneration, neurological impairments, and increased susceptibility to seizures (Criado et al., 2012; DePaoli-Roach et al., 2010; Duran et al., 2014; Ganesh et al., 2002; Garcia-Cabrero et al., 2012; Garcia-Cabrero et al., 2014; Tiberia et al., 2012; Valles-Ortega et al., 2011).
Glycogen and starch are the major carbohydrate storage molecules in mammals and plants, respectively (Brewer and Gentry, in press). Mammalian glycogen, LBs, and starch are all glucose polymers (i.e. polysaccharides) composed of α−1,4-linked linear chains of glucose and α−1,6 linked branch points, but they have different properties. Glycogen consists of linear chains of ~13 glucose units that each give rise to two branches, producing a highly water-soluble molecule with a maximum diameter of 42 nm (Graham et al., 2010). LBs, which form in the absence of functional laforin or malin, are comprised of abnormal polysaccharide referred to as polyglucosan. The term polyglucosan refers to any glucose polymer that is abnormal, and other types of polyglucosan bodies have been described in some GSDs (Cavanagh, 1999). Unlike glycogen, LBs are water-insoluble, hyperphosphorylated, contain long glucose chains, and range in size from a few microns to up to 35 or 40 µm (Nitschke et al., 2013; Sakai et al., 1970; Tagliabracci et al., 2008; Yokoi et al., 1968). Dr. Gonzalo Rodriguez Lafora first described these inclusions in patient autopsies, noting their histochemical similarity to plant starch (Lafora, 1911). Decades later Sakai and colleagues isolated these inclusions from patient tissue and showed that LBs are indeed structurally more similar to starch than to glycogen (Sakai et al., 1970; Yokoi et al., 1968). Plant starch is composed of infrequently branched, longer glucose chains that exclude water and allow for much larger, more energy-dense molecules to form (up to 100 µm in diameter) (Ball et al., 2011; Emanuelle et al., 2016; Tester et al., 2004). Plant starch and LBs both contain significantly more covalently bound phosphate and longer glucan chains than mammalian glycogen (DePaoli-Roach et al., 2015; Gentry et al., 2009; Sakai et al., 1970). Loss of laforin or malin function results in the transformation of glycogen molecules into hyperphosphorylated polyglucosan molecules with an altered chain length distribution, and these aberrant molecules, prone to precipitation, accumulate to form LBs (Sullivan et al., 2017). How laforin and malin prevent this process is still being defined.
Reactive astrocytes and activated microglia upregulated in aged Epm2a−/− and Epm2b−/−mice, particularly in the hippocampus (Duran et al., 2014; López-González et al., 2017; Sanchez-Elexpuru et al., 2017; Turnbull et al., 2011). These glial markers are used to measure neurodegeneration in these models. Results from multiple labs have established that accumulation of polyglucosan drives this gliosis in both LD and non-LD models. Glycogen synthase is the only mammalian enzyme able to catalyze glucose polymerization in vivo. Epm2b−/− mice lacking glycogen synthase (Gys1) in the brain are devoid of cerebral glycogen and LBs, exhibit no gliosis, and have normal electrophysiology (Duran et al., 2014). A similar phenotypic rescue was observed when Gys1−/− mice were crossed with Epm2a−/− mice (Pederson et al., 2013). Furthermore, Epm2b−/− mice lacking just one Gys1 allele in the brain have reduced glycogen and also show near complete rescue of these phenotypes (Duran et al., 2014). Epm2a−/−and Epm2b−/− mice lacking Protein Targeting to Glycogen (PTG), a protein that promotes glycogen synthesis, also exhibit reduced LB accumulation, and neurodegeneration and myoclonic epilepsy are resolved in these animals (Turnbull et al., 2011; Turnbull et al., 2014). These results demonstrate that decreased or complete absence of the glycogen synthesis machinery ablates LB formation and neurodegeneration in LD mouse models. The reverse has also been observed: overexpression of a constitutively active form of glycogen synthase in otherwise wild type animals drives neurodegeneration in both flies and mice (Duran et al., 2012). The accumulating polysaccharide in transgenic animals overexpressing glycogen synthase is a polyglucosan (i.e. abnormal) rather than normal glycogen (Raben et al., 2001). Collectively, the aforementioned studies demonstrate that cerebral LB accumulation is pathogenic. These studies have both elucidated the molecular etiology of LD and have made LBs an obvious therapeutic target.
Efforts to develop a targeted therapy (i.e. precision medicine) for LD are ongoing (Brewer and Gentry, 2018; Brewer et al., 2019). One form of precision medicine that has been utilized for treating GSDs is the introduction of exogenous replacement enzymes. Enzyme replacement therapy has proven effective for Pompe disease (OMIM: 232300), an inherited GSD (van der Ploeg et al., 2010). Pompe patients are deficient in the lysosomal enzyme that degrades glycogen, acid a-glucosidase (GAA), and are currently treated with a recombinant human form of this protein known as rhGAA or alglucosidase alfa (Myozyme®, Lumizyme®, Genzyme) (Kishnani et al., 2007; van der Ploeg et al., 2010). The uptake of rhGAA is a receptor-mediated endocytic process that targets rhGAA to the lysosome. However, there is a significant portion of cytosolic glycogen in Pompe patients, and since rhGAA only targets lysosomal glycogen, those with large pools of cytoplasmic glycogen do not respond well to the current therapy (Thurberg et al., 2006). In LD, LBs are entirely cytosolic. Histological studies from patient tissues report that LBs are not membrane bound, and this observation has been confirmed in mouse models (Berard-Badier et al., 1980; Criado et al., 2012; Ishihara et al., 1987; Van Hoof and Hageman-Bal, 1967). Thus, a therapeutic enzyme degrading LBs must be delivered to the cytosol.
Although cytosolic targets remain challenging for protein therapeutics, antibody-based delivery platforms provide a means for penetrating the cell membrane (reviewed by (Rehman et al., 2016)). The monoclonal anti-DNA autoantibody 3E10 and its antigen-binding (Fab) and variable domain (Fv) fragments can be fused to an enzyme to facilitate cytosolic delivery in multiple cell types (Hansen et al., 2006; Hansen et al., 2005; Weisbart et al., 1998). This antibody-enzyme fusion (AEF) strategy has proven successful for the replacement of a cytosolic enzyme and rescue of X-linked myotubular myopathy in mice (Lawlor et al., 2013). A Fab-GAA fusion has similar efficacy to reduce glycogen in Pompe mice with the additional advantage of targeting both lysosomal and cytosolic glycogen and is now in Phase 1/2 clinical trials (Valerion Therapeutics, 2016; Yi et al., 2017). Rather than utilizing the AEF platform to deliver a replacement enzyme, we generated an AEF designed to penetrate cells and degrade pathogenic LBs. We show that an AEF comprised of the humanized 3E10 Fab fragment and pancreatic α-amylase degrades LBs in vitro, degrades glycogen and polyglucosan in situ, and reduces LB load in vivo. Furthermore, we utilize a metabolomics approach to demonstrate that the drug reverses the physiological effects of LB accumulation.
Results
Construction of an antibody-enzyme fusion (AEF) that degrades starch
An important consideration in designing an LB-degrading enzyme is that LBs are less sensitive to enzymatic hydrolysis than normal glycogen. Resistance to digestion with diastase (a generic term for enzymes that degrade polysaccharides) is a defining feature of LBs and one factor that differentiates LBs from glycogen (Girard et al., 2013; Van Hoof and Hageman-Bal, 1967). However, in routine histological preparations, embedded sections are only incubated with diastase for 15–30 minutes prior to staining. Early studies reported that LBs in tissue sections can be digested by a 5–10 hour incubation with pancreatic α-amylase and γ-amylase (i.e. amyloglucosidase), but they are somewhat resistant to digestion by β-amylase or glycogen phosphorylase (Nikaido et al., 1971; Sakai et al., 1970; Yokoi et al., 1968).
Another consideration in the AEF design is the biochemical structure of the target LBs themselves. Glycogen, starch, and LBs are all polysaccharides with different properties. LBs are considered more similar to starch than glycogen due to their insolubility, elevated phosphate levels, and the increased chain length of their constituent glucose chains. We hypothesized that starch could serve as a proxy for LBs in a screen to identify a candidate therapeutic enzyme. We screened a panel of amylases to determine which possessed robust activity against starch, and therefore would likely degrade LBs. We found that only α-amylases showed significant activity, and pancreatic α-amylase had the highest capacity to degrade starch (Fig. 1A). Pancreatic α-amylase is naturally secreted by the human pancreas for digestion of carbohydrates in the gut, cleaving α−1,4 linkages to yield maltose, maltotriose and low molecular weight oligosaccharides. It has previously been shown to digest LBs in tissue sections (Nikaido et al., 1971). Thus, pancreatic α-amylase is a putative candidate for degrading LBs.
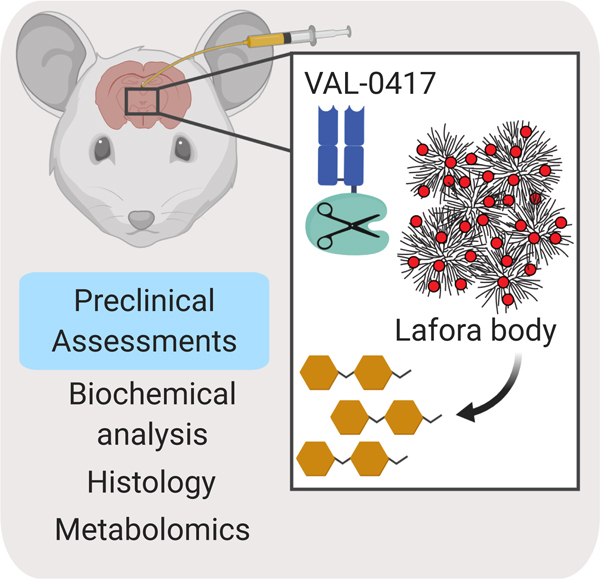
Fig. 1.
The antibody-enzyme fusion VAL-0417 degrades starch, a proxy for LBs. (A) Degradation of starch by a panel of amylases. 1 mg of starch was incubated with 5 µg of amylase overnight with constant agitation, then soluble and insoluble fractions were separated by centrifugation. Degradation product in the soluble fractions was quantified by measuring glucose equivalents (i.e. glucan). Reactions were performed in triplicate and data shown are means ± SD. (B) Schematic representation of VAL-0417. The gene encoding the human IgG1 Fab heavy chain fragment (H) was fused to AMY2A, encoding human pancreatic α-amylase, and coexpressed with the gene encoding the human light chain (L) in HEK293–6E cells. Heavy and light chain signal peptides were included to facilitate proper folding and assembly of the Fab fragment. The predicted molecular weight of each polypeptide is shown. (C) Purity of VAL-0417 assessed by reducing (+BME) and nonreducing (-BME) SDS-PAGE. (D) Chemical structure of the α-amylase-specific substrate E-G7-pNP. Reducing (inner) and non-reducing (outer) ends of the substrate are displayed. (E) Degradation of 1 mg starch after a 2-hour incubation with increasing amounts of VAL-0417, expressed as glucan released in the soluble fraction. Mean ±SD of triplicate reactions are shown. (F) Scanning electron micrographs of starch granules in the absence of treatment and after overnight treatment with VAL-0417. Samples were visualized at 2kV by an FE Quanta 250 scanning electron microscope. See also Figure S1.
An AEF designated as VAL-0417 was generated by fusing the humanized 3E10 IgG1 Fab heavy chain fragment with human pancreatic α-amylase and then coexpressed in HEK293–6E cells with the corresponding light chain as a secreted heterodimer (Fig. 1B). The fusion was purified from HEK293–6E cell conditioned media by affinity chromatography, and the purity was determined by reducing and nonreducing SDS-PAGE (Fig. 1C). The addition of β-mercaptoethanol (BME) dissociated the heavy and light chains, producing two distinct bands of 75 and 25 kDa. In the absence of BME, only a single band was observed at 100 kDa, corresponding to the intact Fab-amylase fusion. The specific activity of an α-amylase was then tested using a substrate that consists of seven glucose moieties and a chromophoric para-nitrophenyl moiety that is capped with an ethylidine group to block the action of other hydrolytic enzymes (Fig. 1D) (Kruse-Jarres et al., 1989). Using this substrate, the specific activity of VAL-0417 was determined to be 10.5 units/mg.
VAL-0417 activity was then tested against plant starch. After a 2-hour incubation, VAL-0417 degraded starch in a dose-dependent manner: 5 µg of enzyme released 61 µg of soluble glucan product (Fig. 1E). We employed scanning electron microscopy (SEM) to visualize the effects of degradation on the starch granules. In the absence of enzymes, starch granules had a smooth, polyhedral or subspherical appearance (Fig. 1F). Overnight treatment with VAL-0417 produced cavities in the granule surface, representing areas of active hydrolysis, and numerous partially degraded granules were visible (Fig. 1F). When the ratio of VAL-4017 to starch was reduced by 10-fold, amylolysis was less extensive: fewer partially degraded particles were visible and most granules were only decorated with small pits (Fig. S1). Other groups have observed similar “pitting” of starch granules with α-amylase and other amylolytic enzymes (Jung et al., 2017; Sujka, 2007).
Isolation and characterization of LBs from LD mice
Polysaccharides (both glycogen and LBs) are typically purified from Epm2a−/− and Epm2b−/−mice based on a method described by Pflüger in 1909 where the tissue is heated in strong KOH and the glycogen is precipitated with alcohol (Good, 1933; Pflüger, 1909). The isolated particles are 15–65 nm in size when visualized by transmission electron microscopy (Tagliabracci et al., 2008), orders of magnitude smaller than the intact LBs observed in fixed tissue sections, and the method does not separate LBs from glycogen. It is well-established that heating changes the physiochemical properties of starch, leads to granule swelling, and enhances its sensitivity to enzymatic degradation (Kulp, 1981). Therefore, it is likely that this protocol disrupts the native structure of LBs, intermixing the individual polyglucosan constituents of LBs with normal glycogen molecules. We developed a procedure for isolating pure and native LBs from tissue so that we could test the sensitivity of LBs to VAL-0417 degradation.
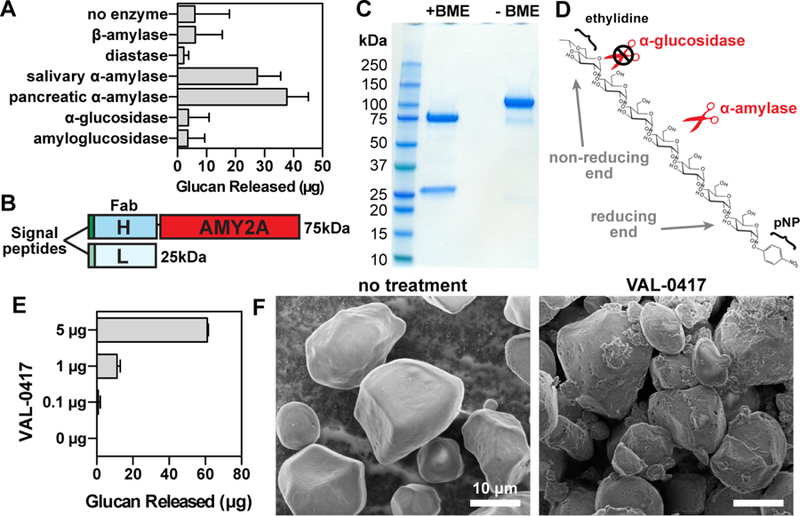
A study from the 1960s showed that native LBs could be isolated from patient brain tissue by centrifugation steps and successive treatments with proteolytic enzymes (Yokoi et al., 1968). We designed a similar method for isolating native LBs from the brain, heart and skeletal muscle of Epm2a−/− mice (Fig. 2A; see STAR Methods). Our procedure separates soluble glycogen (present in the supernatant) from the insoluble LBs (Fig. S2A and B; see STAR Methods). Final LB yields from each tissue type corresponded to ~30% of the total polysaccharide detected in the original tissue homogenate (Fig. 2B and C). Phosphate content of the final LBs from skeletal muscle was 5-fold higher than phosphate in normal muscle glycogen, which is in agreement with published results (Fig. 2D) (Tagliabracci et al., 2008). Iodine spectral scans of the purified LBs were compared to those of commercial glycogen and amylopectin, the major component of plant starch to which LBs are most similar. While glycogen has a peak maximum at ~440 nm when stained with Lugol’s iodine, amylopectin has a right-shifted spectrum and a peak maximum at 560 nm due to its reduced branching (Fig. 2E) (Krisman, 1962). The spectra of the purified LBs were also shifted toward longer wavelengths, with peak maxima at 510–520 nm (Fig. 2E). These data indicate that the purified LBs are less branched than glycogen, consistent with previous reports on Epm2a−/− and Epm2b−/− mice (Tagliabracci et al., 2008; Valles-Ortega et al., 2011).
Fig. 2.
A novel protocol for isolating native LBs from LD mice. (A) LB purification scheme. (B) Polysaccharide was purified at different steps in the protocol via the Pflüger method and quantitated via glucose measurement following hydrolysis. Initial tissue weights and total polysaccharide at each step are shown. (C) Polysaccharide yields normalized to tissue weight. Triplicate samples were removed from each fraction and each measured in triplicate. Mean ±SD are shown. (D) Phosphate content of LBs from skeletal muscle and normal rabbit muscle glycogen. Mean ±SD of triplicate measurements are shown. (E) Normalized iodine spectra of purified LBs compared to commercial liver glycogen and amylopectin. Spectra shown are an average of 3 replicates. (F) Brain, (G) heart, and (H) skeletal muscle LBs stained with Lugol’s solution and visualized using a Zeiss Axioimager Z1. See also Figure S2.
Final LB preparations were also stained with Lugol’s iodine and examined by light microscopy. Minimal tissue debris was observed and the LBs tended to clump (Fig. 2, F to H). LBs from different tissues also had noticeably distinct morphologies. Brain LBs were generally spherical, sub-spherical or irregularly shaped (Fig. 2F), and a range of sizes were observed: the maximum diameter of a single LB ranged from 1–10 µm, most being 2–4 µm (Fig. S2C). These preparations also had a significant amount of granular material that stained less intensely with iodine; small amounts of this material were visible throughout the slide but appeared to form a large aggregate in some fields of view (Fig. S2F). Similar PAS-positive granular material has been reported in fixed tissue sections from patient brain biopsies or LD mice, which are referred to as dust-like particles, or simply “dust” (Edgar, 1963; Machado-Salas et al., 2012; Van Heycop Ten Ham, 1975; Van Hoof and Hageman-Bal, 1967).
Heart LBs were more often ellipsoid or ovoid in shape, frequently with pointed ends, although some were sub-spherical like the brain LBs (Fig. 2G). Heart LBs also ranged in diameter/length from 1–10 µm, most of them being 2–4 µm (Fig. S2D). Dust-like particles were also observed among the heart LBs, although no masses of granular material were observed as with the brain LBs (Fig. S2G). LBs have been reported in LD patient cardiac tissue: in fixed and stained tissue sections, they are irregularly shaped or elongated with pointed ends (Edgar, 1963; Van Heycop ten Ham and Jager, 1963; Yokota et al., 1987; Yokota et al., 1988). Although these reports do not discuss dust-like particles in the heart, they appear to be present in the micrographs, particularly in the studies that involve immunostaining with an LB-specific antibody (Yokota et al., 1987; Yokota et al., 1988). PAS-stained cardiac sections from malin KO mice show LBs of similar shape and size and diastase-resistant granular material (Criado et al., 2012; DePaoli-Roach et al., 2010). LBs from skeletal muscle were the smallest and most homogenous in size and shape: the vast majority were ellipsoid and 1–3 µm long (Fig. 2H and S2E). There appeared to be virtually no dust in these preparations. In LD patients, LBs have been reported to similarly range in diameter from 0.2–2 µm in skeletal muscle (Van Heycop Ten Ham, 1975).
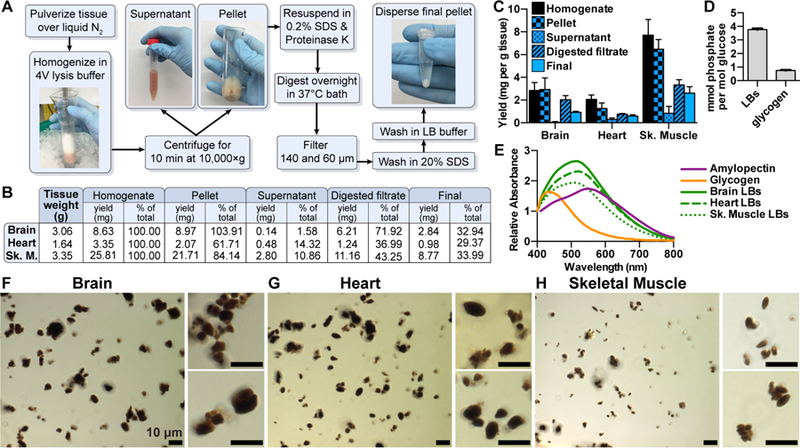
We also utilized SEM to visualize the purified LBs. We found that washing LBs in ethanol and allowing them to dry in a vacuum concentrator, the method used for visualization of starch granules, caused the LBs to aggregate and form very large clumps after being applied to carbon tape (Fig. S2H). It was difficult to distinguish individual LBs within the clumps against the similarly textured carbon tape. As an alternative drying method, LBs were diluted in water, applied to a mounted silicon wafer and lyophilized prior to visualization. This method yielded less aggregated LBs and allowed for their morphological analysis. These micrographs showed that LBs from all three tissues had a similarly textured appearance (Fig. 3), in contrast to the smooth surface of starch (Fig. 1F). As observed via light microscopy, individual brain LBs up to 10 µm in diameter were observed, which were spherical or irregularly shaped (Fig. 3A). Ovoid and ellipsoid LBs from heart and small ellipsoid LBs from skeletal muscle were also consistent with our light micrographic observations (Fig. 3B and C). Visualization of skeletal muscle LBs at very high magnification (30,000×) revealed the f ine texture of the LBs (Fig. 3D).
Fig. 3.
Scanning electron micrographs of isolated LBs. Scanning electron micrographs of LBs purified from brain (A), heart (B), and skeletal muscle (C) displayed using the same scale. LBs from skeletal muscle are also shown at a higher magnification (D). Samples were visualized at 2 kV using an FE Quanta 250. See also Figure S2.
Degradation of LBs by VAL-0417
As described above, native LBs purified from different tissues possess unique morphology. To determine if VAL-0417 could degrade the different types of LBs, we incubated 50 µg of Epm2a−/− LBs from brain, heart and skeletal muscle with VAL-0417. We observed a consistent dose-response effect: 1 µg VAL-0417 released 6–9 µg of glucan (i.e. an oligosaccharide composed of several glucose moieties) into the soluble fraction, indicating 12–18% of the LBs had been degraded (Fig. 4A). Increasing the dose to 10 µg of VAL-0417 released over twice as much, 15–19 µg of glucan, corresponding to 30–38% of the total substrate. A small amount of soluble glucan (5 µg or less) was observed in the samples lacking enzyme, so we visualized the soluble fractions from each treatment by light microscopy. We observed some LB fragments in the soluble fraction that did not sediment with centrifugation for LBs with and without VAL-0417 (Fig. S3A). We consistently observed that mechanical agitation caused some LBs to fragment. However, agitation was required to keep the LBs in suspension.
Fig. 4.
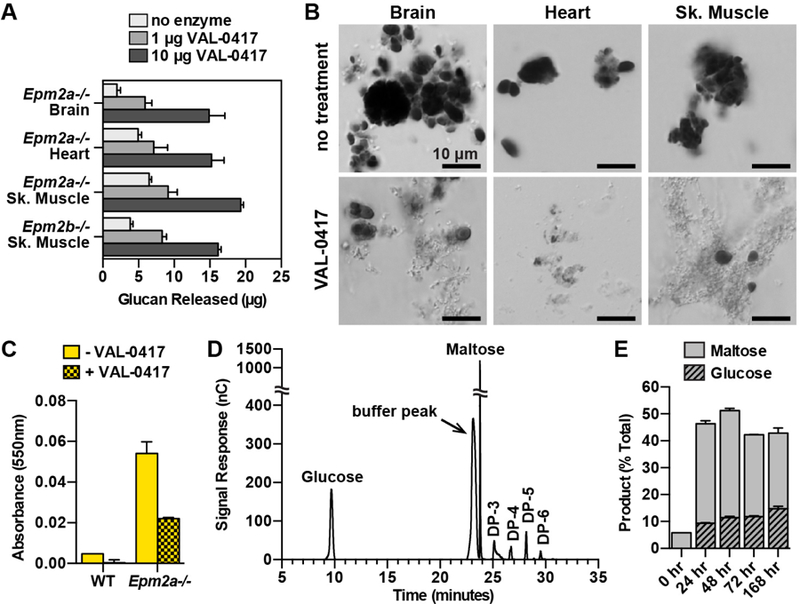
VAL-0417 degrades LBs in vitro. (A) Degradation of 50 µg LBs from different tissues after overnight incubation with 0, 1, or 10 µg VAL-0417. (B) LBs from different tissues after overnight incubation +/− 10 µg VAL-0417. Insoluble fractions were resuspended post-degradation, stained with Lugol’s solution, and visualized using a Nikon Eclipse E600. (C) Iodine absorbance in pellet fractions after incubation of WT and Epm2a−/− skeletal muscle homogenates +/− 25 µg VAL-0417. In (A), (C), and (E) mean ± SD of triplicates are shown. (D) HPAEC-PAD chromatogram of degradation product after incubation with VAL-0417 for 168 hours. Peak identities were determined based on the elution profile of degradation buffer and glucan standards. The chromatogram is representative of triplicate experiments, and glucose/maltose quantification from the replicates is shown in (E). (E) Quantification of glucose and maltose at various time points throughout the degradation reaction of LBs with VAL-0417. Product released are expressed as a percentage of total LBs. See also Figures S3 and S4.
We also purified LBs from skeletal muscle of 12-month-old Epm2b−/− mice and found they were indistinguishable from Epm2a−/− skeletal muscle LBs in appearance, size and iodine spectra (Fig. S3, B to E). We observed a comparable dose-response effect of VAL-0417 on Epm2b−/−LBs: 1 µg and 10 µg VAL-0417 released 16% and 32% of the total, respectively (Fig. 4A). In a similar experiment, we incubated LBs with 10 µg VAL-0417 overnight and then visualized the product by staining the insoluble fraction with Lugol’s iodine. Untreated LBs had no change in appearance and appeared largely intact after the overnight agitation, but VAL-0417 treatment led to fewer iodine-positive LBs and the appearance of a filamentous degradation product (Fig. 4B).
Previous work has shown that various proteins are associated with LBs in vivo (Criado et al., 2012; Machado-Salas et al., 2012). Our purification protocol removes these proteins, so it is possible that an intact protein coat could inhibit LB degradation by VAL-0417. To test this possibility, we measured LB load in crude muscle homogenates after incubation with VAL-0417 using iodine absorbance. While virtually no absorbance was detected at 550nm in the WT samples with or without VAL-0417 treatment, treatment of the Epm2a−/− samples with VAL-0417 reduced iodine absorbance by >50%, indicating substantial LB degradation (Fig. 4C).
Since VAL-0417 is a potential therapeutic, it is important to define the soluble product(s) it releases from LBs. Pancreatic α-amylase is reported to release glucose, maltose, maltotriose and other short oligosaccharides (Robyt and French, 1967). Amylolytic products can be separated and quantified via high-performance anion-exchange chromatography coupled with pulsed amperometric detection (HPAEC-PAD). We digested LBs from Epm2a−/− skeletal muscle with VAL-0417 for a total of 168 hours with additional enzyme added every 24 hours. At 24-, 48- and 72- hour time points, the soluble fractions of LB digestions were removed for HPAEC-PAD analysis. At all 4 time points, maltose and glucose were the major degradation products, followed by small amounts of oligosaccharides with 3, 4, 5 and 6 degrees of polymerization (DP-3, 4, etc., where each DP corresponds to one glucose unit) (Fig. 4D and Fig. S4, B to D). Quantitation of the glucose and maltose peaks showed that maltose levels remained constant throughout degradation and glucose levels increased steadily over the 168-hour experiment (Fig. 4E). Cumulatively, these data demonstrate that VAL-0417 robustly digests brain, heart and muscle LBs in vitro. It is not inhibited by endogenous proteins in muscle homogenates, and its degradation products are primarily glucose and maltose, with a few short oligosaccharides.
VAL-0417 uptake and polysaccharide degradation in situ
To degrade LBs in vivo, VAL-0417 must penetrate and remain active inside cells. Cell penetration of the parent 3E10 mAb and its fragments require functional expression of the membrane equilibrative nucleoside transporter 2 (ENT2), which is involved in a nucleoside salvage pathway and is ubiquitously expressed in rodents and humans (Crawford et al., 1998; Hansen et al., 2007; Lu et al., 2004). VAL-0417 uptake and activity were assessed in two cell lines: fibroblast-derived Rat1 cells accumulating normal glycogen and HEK293 cells engineered to accumulate polyglucosan (HEK293-PTG/PP1Cα) (Fig. S4F; see STAR Methods). After a 20-hour treatment with increasing doses of VAL-0417, cells were washed, lysed, and probed for ENT2 and VAL-0417. ENT2 protein levels were stable in both lines, though Rat1 cells have slightly higher levels, and were unchanged with increasing concentrations of VAL-0417 (Fig. 5A). Western blot analysis of VAL-0417 using an antibody for AMY2A revealed a dose-dependent increase in protein levels (Fig. 5A). Total polysaccharide levels per well (glycogen in Rat1 cells and polyglucosan in HEK293-PTG/PP1Cα cells) were also measured. Both Rat1 and HEK293-PTG/PP1Cα lines displayed a dose-dependent decrease in total polysaccharide after treatment with VAL-0417 (Fig. 5B), with reductions of 30% and 26% of the untreated control, respectively. Thus, VAL-0417 is both taken up by cells and degrades glycogen and polyglucosan in situ.
Fig. 5.
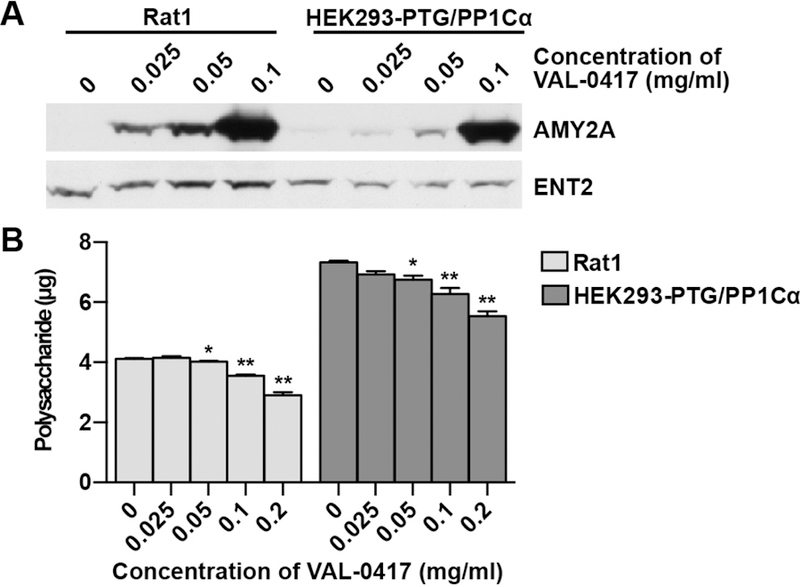
VAL-0417 uptake and polysaccharide reduction in cell culture. (A) Rat1 cells and HEK293 cells stably expressing PTG and PP1Cα after 20-hour treatment with increasing concentrations of VAL-0417. VAL-0417 levels were assessed by Western blotting using an anti-AMY2A antibody. ENT2 levels were also quantified by Western blotting. (B) Polysaccharide levels per well in Rat1 and HEK293-PTG/PP1Cα cells after 20-hour treatment with VAL-0417. Data shown are a mean of 4 measurements ± SE. Statistical significance determined by t-test is indicated: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. See also Figure S4F.
In vivo uptake of VAL-0417 and LB reduction in Epm2a−/− mice
We have shown that VAL-0417 degrades LBs in vitro, penetrates cultured cells, and degrades glycogen and polyglucosan in situ. We next tested VAL-0417 uptake in an in vivo setting and biodistribution after peripheral injections. We performed intramuscular (IM) injections of VAL-0417 to determine if it can be detected in the injected muscle and other tissues and if it is quickly degraded and/or cleared. We injected 0.6 mg VAL-0417 into the left gastrocnemius muscles of WT C57BL/6 mice, then 2 and 24 hours post-injection, euthanized the mice and collected right and left gastrocnemii, right and left quadriceps, heart, liver, and brain. The same tissues were also collected 2 hours post-injection from control mice treated with phosphate-buffered saline (PBS). We designed a sandwich enzyme-linked immunosorbent assay (ELISA) to specifically detect VAL-0417 in tissue homogenates with very low background. At the 2-hour VAL-0417 post-injection time point, 3200 ng of VAL-0417 per mg protein (corresponding to 15% of the injected amount) was detected in the injected (left) gastrocnemius (Fig. 6A). After 24 hours, VAL-0417 levels dropped by 20-fold (to 151 ng per mg protein), corresponding to 0.7% of the initial injected amount. At 2 hours, low levels of VAL-0417 (2–9 ng per mg protein, 0.02–0.1% of the injected amount) were detected in the other muscles, heart, liver, and brain, indicating some VAL-0417 had entered circulation. After 24 hours, VAL-0417 levels nearly returned to baseline in muscles, liver, and brain, but remained constant in the heart. Since blood circulates the entire body of a mouse within minutes, the very high levels of VAL-0417 in the injected muscle 2 and 24 hours post-injection strongly suggest that VAL-0417 is taken up by cells rather than immediately cleared. The low levels of VAL-0417 in non-injected tissues indicate VAL-0417 entered the circulation. In mice, ENT2 expression levels are high in brain, muscle and heart, and low in liver (Lu et al., 2004). It is possible that these tissues took up a small amount of VAL-0417 from the circulation that was mostly cleared by 24 hours. ENT2 levels were probably not limiting since VAL-0417 levels were similar regardless of tissue differences in ENT2 expression. It is notable that VAL-0417 levels remained constant in heart even after 24 hours.
Fig. 6.
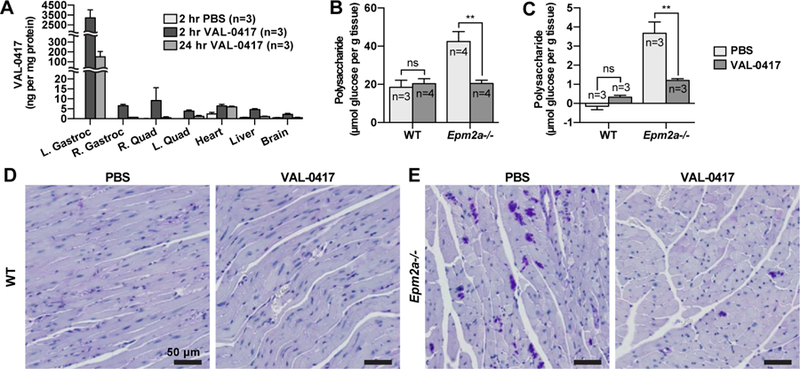
VAL-0417 reduces LB load in vivo after intramuscular (IM) or intravenous (IV) injection. (A) Biodistribution of VAL-0417 levels determined by sandwich ELISA after IM injection of WT mice with PBS or 0.6 mg VAL-0417. (B) Quantification of polysaccharide in the injected gastrocnemii of WT and Epm2a−/− mice after 3 IM injections of PBS or VAL-0417 administered over the course of one week. (C) Quantification of polysaccharide in the heart of WT and Epm2a−/− mice after 4 IV injections of PBS or VAL-0417 administered over a two-week period. Polysaccharides in (B) and (C) were isolated via the Pflüger method and quantified by glucose measurement assays following hydrolysis. Statistical significance is indicated as determined by ANOVA: ** p ≤ 0.01. (D, E) PAS-stained heart tissue of WT (D) and Epm2a−/− (E) mice after IV treatment regimen. Intensely staining PAS-positive deposits are LBs (purple). Tissues were counterstained with hemotoxylin (blue).
We next tested whether IM injections of VAL-0417 reduced polysaccharide levels in vivo in Epm2a−/− mice. To determine polysaccharide levels in individual muscles, we utilized the Pflüger method rather than our native LB protocol b ecause the Pflüger method recovers ~100% of the polysaccharides, enables more accurate and sensitive measurements from small amounts of tissue, and yields both normal glycogen and LBs. 10-month-old WT and Epm2a−/− mice were injected in the right gastrocnemius with either 0.6 mg VAL-0417 or PBS. Three injections were performed over the course of one week, on days 1, 4, and 7. On day 8 the mice were euthanized, and the injected muscles were collected for total polysaccharide quantification. Polysaccharide levels in the WT mice were similar regardless of treatment, and consistent with reported skeletal muscle glycogen levels for C57BL/6 mice (Tagliabracci et al., 2008). In Epm2a−/− animals, VAL-0417 treatment reduced polysaccharide levels by 53% relative to the PBS-treated controls (Fig. 6B). These data indicate that in vivo, VAL-0417 does not decrease normal glycogen levels, but degrades LBs in Epm2a−/− mice.
Intravenous (IV) injections are sometimes preferable to IM injections to achieve rapid and extensive bioavailability of the drug to various tissues, but the preferred route depends on the drug (Jin et al., 2015). We tested whether a high dose of VAL-0417 administered intravenously could reduce LBs in the heart. Like skeletal muscle and brain, heart tissue has a very high LB load, and cardiac abnormalities have been reported in both LD mice and in patients (de Graaf et al., 1989; Oksel et al., 1999; Villalba-Orero et al., 2017). Furthermore, our biodistribution experiment indicated there was less turnover of VAL-0417 in the heart compared to other tissues (Fig. 6A). 8-month-old WT and Epm2a−/− animals were injected via the tail vein with either PBS or VAL-0417 over the course of two weeks, on days 1, 5, 8 and 13. The mice were euthanized on day 14, and hearts were removed and flushed with PBS. A small section of tissue was excised for fixation and periodic acid-Schiff (PAS) staining, and the remaining tissue was flash frozen and analyzed for polysaccharide content via the Pflüger method. The glycogen content of the heart in WT animals is very low in comparison to skeletal muscle (Pederson et al., 2004). Accordingly, we found that polysaccharide levels in WT animals were very low levels irrespective of treatment (Fig. 6C). In contrast, polysaccharide levels in the PBS-treated Epm2a−/− mice were very high in comparison to the WT animals and were reduced by 67% after the two-week treatment regimen with VAL-0417 (Fig. 6C).
Results from the fixed, PAS-stained heart sections were consistent with polysaccharide quantification. PAS-positive deposits, i.e. LBs, were not observed in WT tissue irrespective of treatment and were abundant in tissue of PBS-treated Epm2a−/− mice (Fig. 6D and 6E). Conversely, Epm2a−/− mice treated with VAL-0417 showed a marked reduction in LBs (Fig. 6E). These data collectively indicate that after IM or IV injection, VAL-0417 penetrates cells, remains active, and degrades LBs in vivo.
Cerebral LB accumulation drives epilepsy and neurodegeneration in LD (Duran et al., 2014). Most drugs, particularly antibody-based drugs, do not cross the blood-brain barrier (Kumar et al., 2018). Any therapy targeting cerebral LBs must be delivered into the CNS. There are multiple routes of drug administration to the CNS that bypass the blood-brain barrier. The intrathecal (IT) route involves injection of the drug directly into the spinal cord or the subarachnoid space. Drugs can also be injected directly into the ventricles of the brain via intracerebroventricular (ICV) injection. Both IT and ICV are considered safe and well-tolerated drug delivery routes in both pediatric and adult populations, but the procedures have different risks and benefits (Cohen-Pfeffer et al., 2017; Kumar et al., 2018; Vuillemenot et al., 2016).
We first tested IT versus ICV administration of either PBS or VAL-0417 in WT mice over a period of 3 days. The IT catheter was surgically implanted intrathecally at the lumbar region. The ICV cannula was implanted in the cerebral lateral ventricle. The IT catheter and ICV cannula were each attached to an osmotic pump that delivered a continuous infusion of VAL-0417 (0.08 mg/day) or PBS for 3 days. Mice were sacrificed on the fourth day and brains were removed and sectioned as shown (Fig. 7A). Homogenates from each section were analyzed via a VAL-0417 ELISA assay (Fig. S5A) and amylase assays were used to quantify VAL-0417 activity (Fig. S5B). Negligible ELISA signal was detected in tissue treated with PBS either by IT or ICV, and amylase activity in the tissue was very low. In tissues treated with VAL-0417, VAL-0417 levels were markedly higher with ICV administration compared to IT. In most slices, amylase activity was also markedly higher with ICV treatment of VAL-0417. After ICV administration, VAL-0417 levels and activity were nearly equal in all slices, indicating this route facilitates homogenous distribution of the drug throughout the brain. In contrast, IT administration only resulted in high VAL-0417 levels in the most caudal sections, which are nearest the brain stem. Since LBs are abundant throughout the cortex and cerebellum, a thorough distribution is necessary and thus ICV administration is preferable to IT. Furthermore, these data are indicative of cellular uptake of VAL-0417 in the brain, since the drug was not cleared in the period after infusion (day 3) and before euthanasia (day 4). These data also demonstrate that the drug remains active after uptake.
Fig. 7.
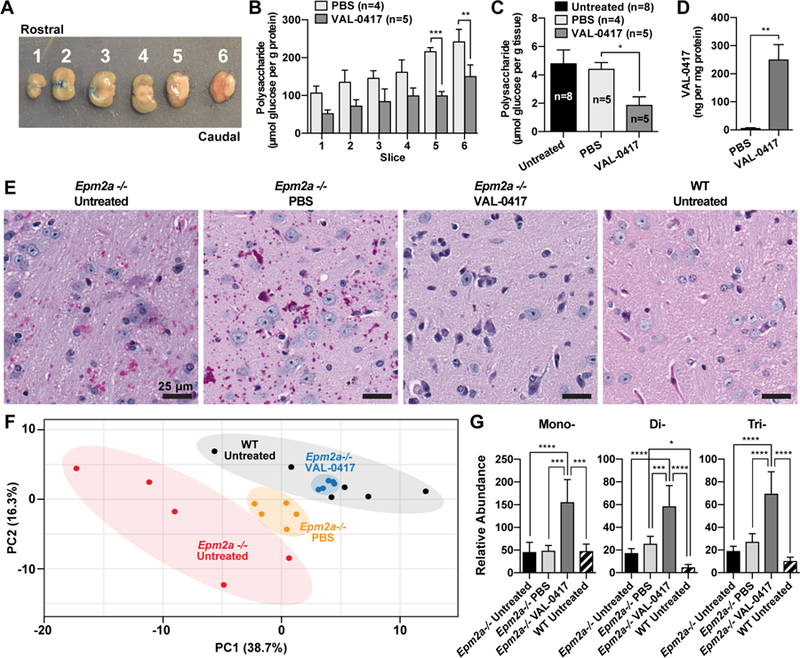
VAL-0417 reduces LB load and reverses metabolic derangement in vivo after continuous ICV infusion. (A) Representative photograph of six 2.0 mm coronal sections. Evans blue dye was injected through the cannula into the lateral ventricle to verify its location within slice 2. (B) Polysaccharide content normalized to protein content in Epm2a−/− brain slices after 28 days of continuous ICV infusion of VAL-0417 or PBS. (C) Total brain polysaccharide normalized to tissue weight from untreated, PBS treated, and VAL-0417 treated Epm2a−/− mice after 8-day ICV infusion. (D) VAL-0417 levels in PBS and VAL-0417 treated animals determined by ELISA. In (B), (C), and (D) at least three technical replicates were performed for each isolated slice to determine an average per slice per animal, and data shown are the mean from each treatment group ± SE. The numbers of animals in each treatment group (n) are shown. Statistical significance is indicated as determined by ANOVA (B) and t-test (C): * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. (E) PAS-stained brain slices of PBS and VAL-0417 treated Epm2a−/− mice after 8-day ICV infusion. LB deposits appear purple; tissue was counterstained with hemotoxylin (blue). (F) Two-dimensional principle component (PC) plots for polar metabolites of PBS and VAL-0417 treated Epma2−/− animals (orange and blue dots, respectively) after 8-day infusion compared to untreated WT and Epm2a−/− controls (black and red dots, respectively). There is a clear separation between the groups, which has been shaded accordingly. (G) Relative abundance of mono-, di- and tri-saccharides determined by GCMS from untreated WT and Epm2a−/− mice and also Epm2a−/− mice treated with PBS or VAL-0417. Statistical significance was determined by two-way ANOVA: * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001, **** p ≤ 0.0001. See also Figures S5, S6 and S7.
To target cerebral LBs, we administered VAL-0417 directly into the cerebral lateral ventricle of 5-month-old Epm2a−/− mice. A continuous infusion of VAL-0417 (0.08 mg/day) or PBS was administered for 28 days via the ICV cannula and pump. Mice were then euthanized, and brains were removed and sectioned as shown (Fig. 7A). Polysaccharides were isolated from each section, quantified, and normalized to protein levels. Polysaccharide levels relative to protein content were lowest in the rostral section and highest in the caudal section (Fig. 7B). This is the first time polysaccharide distribution across different brain regions has been quantitated in Epm2a−/− mice, and is consistent with the high levels of LBs observed in histological sections from brainstem and cerebellum (Criado et al., 2012; Rubio-Villena et al., 2018; Valles-Ortega et al., 2011). Treatment with VAL-0417 reduced the polysaccharide load in all sections, with statistical significance in the two most caudal sections that contain the highest glucan load (Fig. 7B).
To define initial pre-clinical parameters regarding optimal treatment length, we performed a second ICV experiment to determine whether a shorter treatment regimen still reduced the amount of polysaccharide in the brain. 7-month old Epm2a−/− mice received a continuous infusion of VAL-0417 at an equivalent rate (0.08mg/day) or PBS for 7 days. On the 8th day, the mice were euthanized, and the brains were hemisected along the medial longitudinal fissure. The left hemisphere was frozen and pulverized for biochemical analyses, and the right hemisphere was fixed and sectioned for PAS staining. The brains of age-matched untreated cohorts of WT and Epm2a−/− animals were similarly analyzed as surgical controls. Polysaccharide levels relative to tissue weight in untreated and PBS-treated animals were consistent with what has been previously reported for Epm2a−/− animals of this age, i.e. 2–3 times higher than WT levels (<2 µmol glucose/g tissue) (Nitschke et al., 2017; Tagliabracci et al., 2008). Treatment with VAL-0417 reduced brain polysaccharide in the Epm2a−/− mice by 69% compared to untreated and PBS-treated Epm2a−/− mice (Fig. 7C). The amount of VAL-0417 in the treated brains was 249 ng per mg protein (Fig. 7D). This value is slightly higher than the VAL-0417 levels in the injected gastrocnemius 24 hours after IM injections in WT mice (Fig. 6A). Fixed and PAS-stained tissue slices showed an abundance of LBs in the PBS-treated Epm2a−/− brains similar to age-matched untreated Epm2a−/− controls with no observable LBs in age-matched WT animals (Fig. 7E and Fig. S6). In the untreated and PBS-treated Epma2−/− animals, LBs were least abundant in the frontal cortex, more abundant in the thalamus, and most abundant in the cerebellum and brainstem (Fig. S6). This regional distribution is very consistent with the slice-specific quantification we performed via the Pflüge r method (Fig. 7B). Strikingly, the VAL-0417 treated Epm2a−/− brains are virtually devoid of LBs in all regions (Figs. 7E and S6).
Metabolomic profiles as a biomarker for VAL-0417 treatment
It is critical to establish that the reduction of LBs in the brain produces a physiological amelioration of LD pathology. However, like many mouse models of human diseases, LD models do not completely recapitulate the human phenotype (Perlman, 2016). In contrast to patients, LD mice do not have a reduced lifespan, neurophysiological impairments are difficult to detect in young mice, and in older mice, they are often significant only by a small margin (Ganesh et al., 2002; Garcia-Cabrero et al., 2012; Garcia-Cabrero et al., 2014; Sanchez-Elexpuru et al., 2017). Furthermore, although older mice develop myoloclonic jerks, they do not have spontaneous grand mal seizures (Ganesh et al., 2002; Turnbull et al., 2011; Valles-Ortega et al., 2011). Seizure susceptibility must be detected in older mice using kindling agents such as pentylenetetrazol or kainate (Duran et al., 2014; Garcia-Cabrero et al., 2014; Valles-Ortega et al., 2011). The discrepancy between human and mouse phenotypes is a particularly common issue that arises when studying neurological diseases. The extreme differences between the size, lifespan, and complexity of the two species are just a few of the potential causes of this problem (Dawson et al., 2018).
Conversely, metabolic pathways are more highly conserved between rodents and humans. The metabolic signatures of Alzheimer’s disease mouse models parallel those of patients and are therefore readily translatable (Trushina and Mielke, 2014). Metabolomic similarities between humans and mice have also been observed in Huntington’s disease, type 2 diabetes, nonalcoholic fatty liver disease, and colorectal tumors (Barr et al., 2010; Manna et al., 2014; Salek et al., 2007; Underwood et al., 2006). It is likely that the metabolic changes that underlie a disease are similar in the two species, although they may have species-specific physiological manifestations.
Glycogen is formed from UDP-glucose, and its catabolism leads to the release of glucose-1-phosphate, which is rapidly converted to glucose-6-phosphate (Brown and Ransom, 2007). Glucose-6-phosphate is a central molecule in cellular metabolism, supplying carbon for glycolysis, the pentose phosphate pathway, and amino acid synthesis. Therefore, altered brain glycogen metabolism is expected to produce measurable shifts in the cerebral metabolome. We sought to measure these changes in the cerebral metabolome to define the physiological outcome of VAL-0417 treatment. We utilized gas chromatography-mass spectrometry (GCMS) to assess central carbon metabolites of glycolysis and the TCA cycle, as well as amino acids, nucleotides, and mono-, di- and trisaccharides (Fiehn, 2016; Fiehn et al., 2000; Kind et al., 2009). Polar metabolites were extracted from the pulverized brain tissue of the same mice that were treated with PBS or VAL-0417 via ICV for 8 days. Age-matched untreated WT and Epm2a−/− animals were used as controls. We identified over 100 metabolites from all four treatment groups to utilize in a multivariate analysis, which included both principle component analysis (PCA) and heat map clustering. The metabolites analyzed and relative abundance of each are listed in Table S1. The 2-dimensional PCA plot using all available metabolomic data revealed three discrete clusters (Fig. 7F). Metabolites of untreated WT and Epm2a−/− groups segregated into two discrete groups, while metabolites of the PBS-treated Emp2a−/− mice comprised a third group. The metabolic differences between the untreated and PBS-treated animals are likely due to the physiological effects of surgery and ventricular infusion. Strikingly, metabolites of the VAL-0417 treated Epm2a−/− mice clustered tightly within the WT group, indicating that after LB degradation by VAL-0417 the cerebral metabolism returned to normal. We further corroborated this result with clustering heatmap analysis (Fig. S7). While the metabolic profiles of the untreated Epm2a−/− animals were distinct, metabolites of the VAL-0417 treated Epm2a−/−animals were interspersed with the WT animals.
We demonstrated that VAL-0417 degrades LBs in vitro by releasing glucose, maltose and short oligosaccharides (Fig. 4D and 4E). To determine whether oligosaccharides were accumulated in the brain tissue, the relative spectral abundance of mono-, di- and tri-saccharides was determined. There was a significant increase in each oligosaccharide in the brain tissue from the VAL-0417 treated Epm2a−/− animals compared to the other groups (Fig. 7G). These results coupled with the overlapping metabolic profile of the VAL-0417 treated animals with the untreated WT animals demonstrate the effectiveness of VAL-0417 and metabolic profiling as a means to assess treatment efficacy.
Discussion
LD is caused by mutations in one of two genes both encoding enzymes that impact glycogen architecture. Efforts from multiple labs have shown that LBs are the pathogenic epicenter of LD, and LB removal has become a therapeutic approach for the treatment of LD (Brewer and Gentry, 2018; Brewer et al., 2019). In this study, we combine insights from the fields of antibody-based therapeutics, GSDs, carbohydrate metabolism, and LD to design an AEF that degrades LBs in vitro, penetrates cells and reduces polysaccharide levels in situ, and decreases polysaccharide load in vivo via both IM and ICV injection. This is the first time a significant LB reduction has been achieved in adult LD mice by the use of a drug and the first report of overall brain metabolism in LD mice. Importantly, we define the use of metabolomics for monitoring drug efficacy in LD mouse models.
Antibody-based drugs have made a major impact on the treatment of cancer, autoimmune and inflammatory diseases with worldwide revenues of ~$89 billion in 2016 (Carter and Lazar, 2018). Antibody-based drugs are typically whole IgGs designed to bind and block receptors, deplete target cells, downregulate a receptor or initiate signaling. Bispecific antibodies, antibody fragments and antibody-drug conjugates (ADCs) are also being developed across multiple diseases, further expanding the diversity of therapeutics utilizing antibody targeting (Baeuerle and Reinhardt, 2009; Beck et al., 2017; Carter and Lazar, 2018; Moolten and Cooperband, 1970). In this report, we report a novel class of antibody-based drugs with a similar construction to ADCs: antibody-enzyme fusions (AEFs). Rather than delivering a cytotoxin, AEFs deliver an enzyme to a specific target. The antibody or antibody fragment is essential both for targeting and cellular penetration. AEFs have recently been reported for the treatment of multiple congenital disorders, including a muscle wasting disorder known as X-linked myotubular myopathy and the GSD Pompe disease. For X-linked myotubular myopathy, the deficient lipid phosphatase myotubularin fused to an Fv fragment was delivered by intramuscular injection and improved muscle function in a mouse model of myotubular myopathy (Lawlor et al., 2013). For Pompe disease, a Fab fragment fused to GAA delivered intravenously reduced glycogen load and alleviated pathology in GAA-deficient mice (Yi et al., 2017). These two studies illustrate that enzyme replacement therapy, which can be problematic for cytosolic enzymes, can be enhanced by fusion of the replacement enzyme with a cell-penetrating antibody fragment. We now demonstrate that VAL-0417, an AEF comprised of a Fab fragment fused to pancreatic α-amylase, degrades LBs and reduces polysaccharide loads in vivo. Unlike the two studies above, VAL-0417 does not replace a missing enzyme, but rather delivers a natural human enzyme to a new target. Since LBs are structurally similar to plant starch, and a-amylase naturally degrades dietary starch in the human digestive tract, a-amylase is a logical choice for the VAL-0417 payload.
It is important to distinguish normal glycogen from polyglucosan. Polyglucosan bodies (PGBs) have been defined as “small, non-membrane-bound cytoplasmic structures composed largely of unusual glucose polymers” (Raben et al., 2001). They are typically a pathological feature of GSDs, although PGBs known as corpora amylacea are a hallmark of normal aging and neurodegenerative disorders (Augé et al., 2018; Cavanagh, 1999). PGBs are not found in all GSDs, and not all PGBs are identical with respect to physiochemical structure. The polyglucosan found in Cori’s disease is characterized by short outer chains, while the polyglucosan of LD, Anderson disease, Adult Polyglucosan Body Disease, and Tarui disease contain very long chains (Illingworth and Cori, 1952; Raben et al., 2001). The Pflüger method, originally designed to purify normal glycogen, also precipitates polyglucosan. In this report, we refer to Pflüger-isolated material as polysaccharide, since it contains both glycogen and polyglucosan. Furthermore, the Pflüger method includes an alkalin e boiling step that solubilizes polyglucosan and destroys LB superstructure. LBs (and PGBs) may be considered a higher-order structure of polyglucosan. The LB purification protocol we designed does not include the alkaline boil. Rather, it separates native LBs from normal, soluble glycogen.
LBs from the different tissues displayed varied morphologies. Brain LBs were the most irregular in size and shape, included dust-like particles, and reached the largest size. LBs from brain exhibited the greatest variation in size, which is consistent with clinical observations: LBs from patient brain micrographs are 3–40 µm and LB dust-like granules are even smaller (Minassian, 2001; Van Heycop Ten Ham, 1975). A recent microscopic study of Epm2b−/− mice showed that astrocytic LBs are on average smaller than neuronal LBs, more irregularly shaped, and clustered, while neuronal LBs are larger, more spherical, and isolated (Augé et al., 2018). LBs in the heart were also surprisingly large and included some dust-like particles. LBs from skeletal muscle appeared well organized: they were very small and homogenous in shape, and no dust-like particles were present. It is not clear why such variation in LB size and shape exists in the brain and heart, in contrast to the homogeneity found in skeletal muscle. One possibility is that LB size is limited by the dimensions of the cell or subcellular region it occupies. For example, in patients, dogs, and mice, LBs can be found in the cell bodies of Purkinje neurons, which can be up to 40 µm in diameter (Jian et al., 1990; Machado-Salas et al., 2012; Van Heycop Ten Ham, 1975). In astrocytes, LBs are typically found in processes, which can be very long but are only a few microns wide (Augé et al., 2018; Rubio-Villena et al., 2018). However, cell size constraints do not explain the difference between heart and skeletal muscle LBs, since cardiac and skeletal myocytes are similar in size. It is well known that glycogen metabolism is remarkably different in astrocytes, neurons, cardiomyocytes and skeletal myocytes. Skeletal myocytes may sequester LBs more efficiently than cardiomyocytes, consolidating them into smaller, discrete units. In brain and heart, the dust-like particles may represent a pre-aggregated form of polyglucosan, while the larger particles may represent polyglucosan aggregates that have been sequestered and compacted by the cell. Active cellular sequestration of polyglucosan has been proposed to explain the large, dense, and spherical LBs observed in some neurons (Machado-Salas et al., 2012).
Although LBs from all three tissues had iodine spectral peak maxima at similar wavelengths, indicative of similar degrees of branching, the higher amplitude of the spectra of brain and heart LBs suggests that they may contain longer chains compared to LBs from skeletal muscle (Krisman, 1962; Swanson, 1948). Indeed, Nitschke et al. showed using HPAEC-PAD that in Epm2a−/− and Epm2b−/− mice, brain glycogen had a more exacerbated shift in chain length distribution than muscle glycogen (Nitschke et al., 2017). Although the brain is most severely affected in LD and a neurological phenotype predominates, heart failure and arrhythmia have been reported in LD patients, and Epm2a−/− and Epm2b−/− mice have metabolic cardiomyopathy (de Graaf et al., 1989; Oksel et al., 1999; Villalba-Orero et al., 2017). Glycogen metabolism is variable with tissue type: glycogen synthase and glycogen phosphorylase have tissue-specific isoforms that are disparately regulated by cellular signals (Roach, 2002); the ultrastructure of glycogen is different under different metabolic conditions and in different organs (Ryu et al., 2009; Sullivan et al., 2014); subcellular glycogen pools are variable (Graham et al., 2010); and both phosphate levels and average chain length differ with species and tissue type (Illingworth et al., 1952; Lomako et al., 1993). Due to these differences, the LBs may be more pathological in brain and heart than in skeletal muscle. Conversely, different cell types may be more sensitive to the presence of the LBs, and/or the metabolic perturbations induced by the LBs may have distinct consequences in different organs. Tissue-specific differences are important to consider throughout the clinical development of VAL-0417. ICV administration of VAL-0417 may attenuate neurological symptoms, but cardiac, liver and/or muscular pathology may require treatment by intravenous or intramuscular injection of VAL-0417.
We found that VAL-0417 primarily releases maltose and glucose from LBs, and glucose levels gradually increased over time, as expected for an a-amylase. Cytosolic free glucose would likely be phosphorylated to produce glucose-6-phosphate, entering central carbon metabolism. Although maltose is not considered a typical participant in intracellular energy utilization, studies have shown that maltose can be transported into and out of cells and metabolized in cell culture and humans after intravenous infusion (Leong et al., 2017; Young and Weser, 1971). The small oligosaccharides (DP-3, DP-4, etc.) would likely remain cytosolic and inert until they are further metabolized to glucose and maltose. Our metabolomics approach revealed a significant increase in mono-, di- and trisaaccharides in the VAL-0417 treated brains. These in vivo data strongly support our in vitro oligosaccharide profiling of VAL-0417 degradation products. Strikingly, the metabolomic profile of Epm2a−/− mice treated with VAL-0417 is indistinguishable from the WT mouse metabolic profile. These data strongly suggest that the oligosaccharide products are not detrimental and may be metabolically inert.
Importantly, IM and IV injections of VAL-0417 in Epm2a−/− mice reduced the polysaccharide load (i.e. LBs and glycogen) to WT levels but did not decrease glycogen levels in WT skeletal muscle or heart, respectively. Glycogen levels in WT heart tissue is already very low, and the levels remained negligible after IV treatment with VAL-0417. In contrast, we observed that VAL-0417 reduced polysaccharide levels in cell culture. It should be noted that the cell culture studies were performed in high glucose media, known to promote glycogen accumulation and the cells are engineered to increase glycogen levels. Thus, it is possible that a reduction in polysaccharides with VAL-0417 treatment is only detected when polysaccharide levels accumulate above a baseline level, as glycogen accumulates in cultured cells or LB in LD tissues. When glycogen levels drop below a certain threshold due to degradation by VAL-0417, the cellular machinery may respond to restore those levels. Glycogen is likely to be maintained at a certain baseline level in vivo in muscle, and this may explain why no change was observed in WT muscle treated with VAL-0417. The observation that VAL-0417 reduced polysaccharide levels in Epm2a−/− muscle, heart and brain indicates LBs are degraded in vivo.
Although both IT and ICV injections are performed in the clinic, delivery to the CNS remains a significant challenge (Calias et al., 2014; Wolak and Thorne, 2013). Drug distribution is highly dependent on fluid movement in the CNS, which is a relatively nascent field of research (Abbott et al., 2018; Kumar et al., 2018; Plog and Nedergaard, 2018). While the field has defined fluid movement, detailed biodistribution of actual drugs via ICV and IT delivery is ongoing (Kumar et al., 2018). In this study, we demonstrate that a 150 kDa AEF is modestly distributed throughout the brain after IT administration and maximally distributed by after ICV administration (Fig. S5). Similarly, ICV administration of anti-amyloid antibodies for the treatment of Alzheimer’s disease resulted in widespread distribution throughout the murine brain (Chauhan and Siegel, 2004). Our data support a recent study demonstrating that the bioavailability of antibodies to the brain after IT injection is size-dependent (Pizzo et al., 2018). Our results indicate that VAL-0417 can cross cellular barriers between the ventricles and brain parenchyma (e.g. ependyma, perivascular lining cells, and/or pia and glia limitans), they also highlight the differences in drug permeability and diffusion between the ICV and IT routes.
Importantly, we utilized a metabolomics approach to quantify the physiological effects of VAL-0417 treatment. Using principal component analysis, we found that the profile of polar brain metabolites from WT and Epm2a−/− mice cluster into discreet groups. Treatment of Epm2a−/− mice with the VAL-0417 shifted the Epm2a−/− profile to mimic the WT cluster, reflecting a restoration of normal metabolism. These results illustrate a positive physiological response and amelioration of metabolic derangement in LD animals after VAL-0417 treatment. Studies in multiple disease contexts have shown that the metabolic changes in humans and mice are highly correlated (Barr et al., 2010; Manna et al., 2014; Salek et al., 2007; Trushina and Mielke, 2014; Underwood et al., 2006). Furthermore, metabolomics is now being employed to identify biomarkers in human biofluids or tissues for monitoring disease progression and therapy efficacy (Johnson et al., 2016; Spratlin et al., 2009). A particularly relevant example is the use of the tetrasaccharide glucose (Glc4) as a biomarker for Pompe disease (Bobillo Lobato et al., 2016). Elevated levels of Glc4 have also been observed in patients with GSD Ia and III, and urinary excretion of the Glc4 in Pompe patients correlates with response to Myozyme treatment (An et al., 2005; Manwaring et al., 2012). The dramatic upregulation of oligosaccharides in VAL-0417 treated mouse brain tissue suggests these molecules could be utilized as biomarkers for VAL-0417 therapy in humans.
We have demonstrated that VAL-0417 is a putative precision therapy for LD, an intractable epilepsy. Currently, >30% of epilepsies, including LD, are resistant to available antiseizure drugs (ASDs) and are known as refractory epilepsies (Loscher and Schmidt, 2011). More than 35 different iterations of ASDs have been discovered over the past 150 years with most of them acting primarily on ion channels or neurotransmitters. Due to this focus on one mode of action, the high proportion of refractory epilepsies has remained constant since the 1850s (Loscher and Schmidt, 2011; Scharfman, 2015). The underlying causes of these refractory epilepsies must be identified in order to develop more effective therapies. As the molecular basis of LD has been defined, we have now developed an AEF that targets its underlying cause. As molecular etiologies in other epilepsies become clear, precision therapies, possibly also utilizing the AEF platform, become feasible. LD is also a non-classical GSD. There are over 14 different types of GSDs characterized by glucan accumulations, and 1 in 20,000 people have some form of GSD though the individual types are considered rare (Adeva-Andany et al., 2016; Ozen, 2007). While the glycogen in some GSDs is of normal structure like in Pompe disease, other GSDs such as Cori disease and Andersen disease are characterized by polyglucosans, like LD (Adeva-Andany et al., 2016). Two AEF drugs (VAL-0417 and Fab-GAA) have now been shown to target cytosolic polyglucosan (the present study) and normal glycogen in both the cytosol and lysosome, respectively (Yi et al., 2017). These drugs could be repurposed and/or modified to target glycogen in other GSDs. Not only is VAL-0417 the first drug with the potential for providing a significant clinical benefit to LD patients, it is an example of a precision therapy and expands the repertoire of antibody-based drugs that can be used to treat human disease.
Limitations of Study
This study employs LD mouse models and the use of metabolomics to determine the physiological benefit of LB degradation by VAL-0417. Metabolomics is an extremely powerful technique routinely used for biomarker discovery, and it is increasing utilized to provide insights into disease onset, progression and response to therapy (Johnson et al., 2016). Although the metabolic changes in humans and mice are often strongly correlated (Barr et al., 2010; Manna et al., 2014; Salek et al., 2007; Trushina and Mielke, 2014; Underwood et al., 2006), our analysis provides only a general overview of the physiological and metabolic state of the brain. It is not possible to determine the relative contributions of cellular populations (e.g. neurons and glia) or specific pathway aberrations (e.g. oxidative stress and proteostasis) to the metabolic profiles. Finally, like all mouse models of neurological diseases, LD mice do not perfectly phenocopy the human disease. Further pre-clinical studies will be conducted to validate the physiological benefit of VAL-0417 administration and LB degradation for the treatment of LD.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Researchers may obtain VAL-0417 with a material transfer agreement from Valerion Therapeutics. All reasonable requests for collaboration involving materials used in the research will be fulfilled provided that a written agreement is executed in advance between Valerion Therapeutics and the requester (and his or her affiliated institution). Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Matthew S. Gentry (matthew.gentry@uky.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Epm2a−/− (DePaoli-Roach et al., 2012; Ganesh et al., 2002) and Epm2b−/− (DePaoli-Roach et al., 2010) mice have been previously described. C56Bl/6 WT, Epm2a−/− and Epm2b−/− animals were maintained in a 12:12 hr light-dark cycle and were given ad libitum access to food and water. Both male and female mice were used since there are no sex differences associated with LD (Gentry et al., 2018). All procedures were approved by the UK Institutional Animal Care and Use Committee (IACUC) as specified by the 1985 revision of the Animal Welfare Act.
METHOD DETAILS
Expression and purification of VAL-0417
VAL-0417 was designed and produced by Valerion Therapeutics (Concord, MA). The cDNA encoding the human IgG1 Fab-linker-AMY2A heavy chain and light chain were synthetically produced with codon optimization for mammalian cell expression and cloned into pTT5. HEK293 cells expressing a truncated variant of the Epstein Barr Virus nuclear antigen 1 (HEK293–6E) increase the volumetric yield of monoclonal antibodies and fragments and were used for VAL-0417 expression (Jager et al., 2013). 2 1L cultures of HEK293–6E cells in 2L shake flasks were transfected with 1 mg of total plasmid DNA/L (1:1 ratio heavy chain:light chain) culture using PolyPlus linear Q-PEI at a 1:1.5 (w/v) DNA:PEI ratio. Culture parameters were monitored using a ViCell XR (Beckman Coulter) for density and viability. Culture was harvested 5 days post transfection via centrifugation for 5 minutes at 1000×g. The conditioned culture supernatant was clarified by centrifugation for 30 minutes at 9300×g.
Pre-packed CaptureSelect IgG-CH1 affinity columns (Fisher) were equilibrated in PBS (pH 7.2). VAL-0417 from 2 L of exhausted supernatant was top-loaded onto affinity columns (2 × 1 mL columns in tandem) at 4°C overnight. The column was washed with approximately 15 column volumes (CV) of PBS, 15 CV of buffer B (1×PB S with 500 mM NaCl, pH 7.2) and 15 CV of PBS. The resin-bound fusion protein was eluted with 10 CV of Buffer C (30 mM NaOAc, pH 3.5–3.6), collecting the protein in 1 mL fractions diluted in 1/10th volume Buffer D (3 M NaAcetate pH~9.0) to neutralize. To minimize the elution volume, elution was paused for several minutes between each fraction collected. Fractions were analyzed by A280 prior to pooling fractions and pools were analyzed by SDS-PAGE. VAL-0417 remained in the non-bound pool from the first affinity chromatography pass. The above procedure was repeated to capture remaining fusion protein. The affinity pools were combined prior to dialysis. The combined CaptureSelect IgG-CH1 affinity pool (18 mL) was dialyzed against 3 × 1 L of dialysis buffer (20 mM Histidine, 150 mM NaCl, pH 6.5) at 4° C. The dialyzed pool was concentrated to 1 mg/mL using a VivaSpin 20 (10K MWCO, PES membrane) centrifugal device prior to final analysis and storage at −80°C. Purified Fab-AMY was analyzed by size exclusion chromatography (Agilent HP1100) showing a single peak before and after a freeze-thaw cycle, indicative of a single, stable species. Activity of the purified protein was quantified by an amylase activity colorimetric kit (BioVision) utilizing the amylase-specific E-G7-pNP substrate (Kruse-Jarres et al., 1989).
Iodine-based measurements
Lugol’s iodine was prepared as a 20×stock (1.5 M KI and 100 mM I2). To detect LBs in preliminary purifications, 50 µL samples from purification fractions were boiled for 15 minutes on a 95°C heat block, clarified by centrifugation ( 16,000×g for 1 min), and 35 µL of the supernatant was added to 50 µL 1×Lugol’s iodine and 15 µL water for 100 µL total (Fig. S2A). To analyze iodine absorbance of polysaccharide from LB purifications, Pflüger-isolated polyglucosan fractions were added to 50 µL 1×Lugol’s iodine for 100 µL total and absorbance was measured at 550 nm. Absorbance was graphed alongside glucose-based concentrations to illustrate the fractionation of polyglucosan and glycogen into supernatant and pellet fractions, respectively (Fig. S2B). For spectral scans, 50 µg LBs, rabbit liver glycogen (Sigma), and potato amylopectin (Sigma) were solubilized by boiling for 30 min and added to 50 µL 1×Lugol’s iodine for 100 µL total. Absorbance scans were performed at 400–800 nm in 10 nm steps.
Purification of native LBs
19–24 month old Epm2a−/− mice were euthanized by CO2 and decapitation, and brain, heart and hindlimb skeletal muscle were immediately harvested, flash frozen and stored at −80°C. 12 month old Epm2b−/− mice were euthanized by cervical dislocation, and muscle tissues were similarly collected and stored. Tissues were pulverized over liquid N2 using a Freezer/Mill Cryogenic Grinder (SPEX SamplePrep). Powdered tissue was weighed and homogenized on ice in 4 vol. lysis buffer (100 mM Tris-HCl pH 8.0, 200 mM NaCl, 1mM CaCl2, 0.5% sodium azide) using a Dounce tissue grinder. For larger volumes, the grinding pestle was attached to a motorized drill. The homogenate was centrifuged for 10 min at 10,000×g at 4°C in a Ti-70 rotor and Optima XPN-90 ultracentrifuge (Beckman Coulter) and the supernatant was removed. The pellet was resuspended in an equivalent volume of lysis buffer, and 20% SDS was added for a final concentration of 0.2%, and 20 mg/ml Proteinase K was added for a final concentration of 0.4 mg/ml. Proteolytic digestion was performed in a 37°C water bath overnight. The digested samples were then syringe-filtered through 140 µm and 60 µm nylon net filters installed in Swinnex filter holders (Millipore), centrifuged at 16,000×g for 5 min, and the supernatant was removed. The LBs were resuspended in 10% SDS and then washed 5 times in LB buffer (10 mM HEPES-KOH pH 8.0, 0.1% sodium azide) each time with centrifugation at 16,000×g for 5 min. Final LB pellets were gently and thoroughly dispersed in LB buffer with a pipet. Polysaccharide yield at various steps of the purification were determined using the Pflüger method (see below). 2 µL of LB preparations were stained with 5 µL 20× Lugol’s iodine, mounted on glass slides with a glass coverslip, and visualized at 100x using a Zeiss Axioimager Z1 equipped with an AxioCam 1Cc5 color camera. Purified LBs were stored at −20°C.
Pflüger method for polysaccharide isolation
Glycogen and polyglucosan from Epm2a−/− and Epm2b−/− mice have been purified and characterized by multiple groups using the Pflüger method, but a chaotropic salt must be added to enhance the efficiency of polyglucosan precipitation (Tagliabracci et al., 2008). We wanted to clarify that under native conditions, and prior to the Pflüger treatment, LBs in Epm2a−/− and Epm2b−/− tissues are intact, micron-sized structures (Fig. 2 F to H, Fig. 3, and Fig. S3C and D). Pflüger treatment converts the LBs to much smaller polysaccharide molecules that are on the same nanometer scale as glycogen molecules, but they contain elevated phosphate and an altered chain length distribution (Nitschke et al., 2013; Tagliabracci et al., 2008). Thus, the term “LB” refers to the native, micron-sized polysaccharide-containing structures found in LD tissues, and “polyglucosan” refers to the abnormal polysaccharide comprising LBs, which is released with Pflüger treatment.
The Pflüger method is direct and very sensitive an d was used to track polysaccharide yield throughout the native LB purifications or to detect polysaccharide content of mouse tissues for in vivo studies. Aliquots from native LB purifications or tissue homogenates were added to 10 vol. 30% KOH, boiled for 2 hours, and allowed to cool. 2 vol. cold ethanol and 10 µL LiCl (1 M or 20 mM) were added, and samples were precipitated overnight at −20°C. Precipitated samples were centrifuged for 10 min at 16,000×g at 4°C, the supernatant was removed, and the polysaccharide pellet was resuspended in water. Two additional precipitations with cold ethanol and LiCl were carried out, each for 1–2 hours at −20°C. The final pellet was washed in cold ethanol and resuspended with vortexing in 200 µL water. Polysaccharide was quantified by overnight hydrolysis with amyloglucosidase from Aspergillus niger (Sigma) and glucose determination was carried out using the D-glucose kit (Fisher). Fluorescence (ex340/em445) rather than absorbance of the NADPH product (stoichiometric to glucose) was measured for higher sensitivity.
Separation of glycogen and LBs in LB purification
After the initial centrifugation step in the LB purification, a fraction of the heart and muscle glycogen was consistently present in the supernatant (Fig. 2, B and C). In our preliminary purifications, we found that the homogenate and pellet fractions stained brown with Lugol’s iodine after boiling, as did the final LBs and amylopectin, but the supernatants stained yellow like glycogen (Fig. S2A). We also stained the Pflüg er-purified polysaccharide fractions from our large-scale preparations in Figure 2C with Lugol’s iodine, measured absorbance at 550 nm, and compared the values to the polysaccharide concentrations determined by hydrolysis and glucose measurement (Fig. S2B). While the polysaccharide from the supernatant did not absorb at 550 nm, polysaccharide from the pellet, filtrate and final fractions produced a high absorbance relative to concentration. Polysaccharide in the homogenate fraction was less absorbent relative to concentration. These data indicate that the polysaccharide in the heart and muscle tissue homogenates included both glycogen and polyglucosan (in the form of LBs), that were separated into supernatant and pellet fractions after low-speed centrifugation. No polysaccharide was detected in the supernatant fractions from brain (Fig. 2, B and C). This result is consistent with the observation that in normal mice, glycogen levels are lower in brain than in other tissues in part due to its rapid catabolism after euthanasia (Oe et al., 2016).
Determination of LB phosphate content
Muscle glycogen and LBs contain covalent phosphate linked to the C2, C3, and C6 hydroxyls of glucose moieties, and the relative ratios of these modifications are equivalent (DePaoli-Roach et al., 2015; Nitschke et al., 2013; Tagliabracci et al., 2011). Boiling in mild HCl hydrolyzes glycosidic bonds and releases the acid-labile C2- and C3-linked phosphate leaving C6 phosphoesters intact in the form of glucose-6-phosphate (Nitschke et al., 2013). Thus, inorganic phosphate after mild acid hydrolysis represents only C2- and C3-linked phosphate, i.e. two-thirds of total phosphate. To determine phosphate content, skeletal muscle LBs were boiled for 2 hours at 95°C in 1 M HCl. Reactions were neutralized with NaOH and inorganic phosphate in the sample was determined using the Pi ColorLock Gold Phosphate Detection System (Innova Biosciences). Glycogen purified from rabbit skeletal muscle as previously described was used as a positive control, and the levels of phosphate detected using our method are consistent with two-thirds of published total phosphate levels (DePaoli-Roach et al., 2015).
In vitro degradation assays
Corn starch and LB degradation experiments were performed in degradation buffer (30 mM HEPES-KOH pH 7.5, 5 mM MgCl2, 5 mM CaCl2) and a total volume of 100 µL. Reactions were performed in triplicate using PCR strip tubes and moderate agitation on a vortex to keep substrates in suspension, and we found that this level of agitation did not affect enzyme activity. Degradation reactions were allowed to proceed overnight (13–18 hours) unless otherwise indicated, after which tubes were centrifuged to pellet undigested substrate. 50 µL of the supernatant (containing the soluble degradation product) was transferred to a new tube with 50 µL 2 M HCl and boiled for 2 hours at 95°C in a C1000 thermocycler (Bio-Rad) to hydrolyze all glucans to glucose. Samples were neutralized with 50 µL 2 M NaOH, and glucose was determined using the D-glucose kit. For visualization of LB degradation product, degradation reactions were allowed to proceed as above, and the reactions were centrifuged to pellet undigested substrate. After 75 µL of the supernatant was removed, the remaining 25 µL was resuspended, and 5 µL was stained with 5 µL 20× Lugol’s iodine, mounted on glass slides with a glass coverslip, and visualized using a Nikon Eclipse E600 using DIC/Nomarski contrast and an AxioCam MRm camera at 100x. In addition to the D-glucose kit, the absorbance-based PGO assay (Sigma) was also used to quantify glucose in starch degradation assays.
In vitro degradation of LBs in muscle homogenates was performed as follows: 300 mg of skeletal muscle from WT or laforin KO mice was pulverized over liquid N2 and homogenized in 4 vol. degradation buffer. Homogenate was split into 2 × 500 µL aliquots, and 25 µg VAL-0417 was spiked into one aliquot for a final concentration of 0.05 mg/ml. Samples were incubated on a rotator overnight at room temperature and the next morning centrifuged for 5 min at 16,000×g. The supernatant was removed, and the pelleted material containing LBs was resuspended in degradation buffer and boiled for 30 min on a 95°C heat block to solubilize LBs. The boiled samples were clarified by centrifugation (16,000×g for 5 min) and 25 µL of supernatant was added to a microplate. 50 µL of 1×Lugol’s iodine and 25 µL of water were added to samples, and absorbance was measured at 550 nm.
Scanning electron microscopy
Starch granules were incubated with enzymes in degradation buffer overnight while agitating, washed in 1mL 100% ethanol, dried in a vacuum centrifuge, and applied to carbon tape on a pin mount. Samples were coated with gold and platinum to improve sample conductivity, and visualized under high vacuum at 2 kV using an FEI Quanta 250 field emission scanning electron microscope. Washing in ethanol, drying, and manual application to carbon tape did not yield satisfactory results for LB visualization (Fig. S2), so alternatively, diluted LBs were applied to a mounted silicon wafer, lyophilized, coated in gold and platinum, and visualized at 2 kV with the FEI Quanta 250.
Profiling of degradation product by HPAEC-PAD
80 µg of LBs were treated with 2.67 µg of VAL-0417 in degradation buffer to a final concentration of LBs at 1 µg/µL. The enzymatic reactions were done at 37°C in tri plicates. 20 µL samples were removed at 24, 48, 72 hour intervals followed by addition of 2.67 µg of VAL-0417. The aliquots were stored at −20°C until they were run. The final reactions were continued at 37°C for total of 168 hours. All samples were pr ofiled using CarboPac PA-100 column (Thermo-Dionex, 4 × 250 mm) and detected with PAD detector. 5 µL of samples from each time point (corresponding to 5 µg LBs total) were injected on the column. Blank LBs and degradation buffer were injected as controls. Glucose and maltose in the degradation reactions were quantified by comparing with standards of known amount (Fig. S4A). 5 µg Maltrin100 was also included as a standard for oligosaccharide profiling (Fig. S4E).
Cell culture studies of VAL-0417 uptake
Protein Phosphatase 1 (PP1) stimulates glycogen synthesis by both dephosphorylating and activating glycogen synthase and inhibiting glycogen phosphorylase, the primary enzymes catalyzing glycogen synthesis and degradation, respectively (Newgard et al., 2000). Protein targeting to glycogen (PTG) is a regulatory, glycogen-targeting subunit of PP1 that stimulates glycogen synthesis when overexpressed in cell lines (Printen et al., 1997; Worby et al., 2008). Constitutive overexpression of PTG in WT mice has been shown to lead to cerebral polyglucosan accumulation in Epm2b−/− mice (Duran et al., 2014). We sought to design a cell line that accumulates polyglucosan like that which makes up LBs.
HEK293 cells (Graham et al., 1977) were co-transfected with plasmids pCDH-FLAG-PTG and pCDH-(HA)3-PP1Cα-GFP harboring mouse PTG and the human PP1Cα (catalytic alpha-subunit of PP1) cDNAs, respectively. Mixed clones were selected for ~10 days in the presence of 1 µg/ml hygromycin and 0.2 mg/ml puromycin, expanded and stored in liquid N2. Analyses of protein expression and glycogen synthase activity ratio in the absence and presence of glucose-6-phosphate indicated that both proteins were expressed, and the glycogen synthase activity ratio was increased 3-fold, from 0.02 in control cells to 0.06 in transfected cells. Quantitation of the expression of PTG and PP1Cα is difficult because the basal levels are very low, undetectable under our conditions.
For quantitation of polysaccharide levels HEK293, HEK293-PTG/PP1Cα and Rat1fibroblasts (Skurat et al., 2000) were plated in 96-well plates at a density of 40,000 cells/well. After 48 hr, media was changed and the next day, the cells were washed 3 times with PBS followed by overnight incubation at 40°C in 50 µL of 0.2 M sodium acetate pH 4.8 containing 0.2% Triton and 0.3 mg/ml amyloglucosidase (Aspergillus niger; Sigma). Identical cell cultures were lysed in 50 µL of in 0.2 M sodium acetate pH 4.8 containing 0.2% Triton for protein determination by the Bradford procedure.
After overnight digestion, polysaccharide was measured by transferring 40 µL samples to a new 96-well plate. A 160 µL reaction mixture consisting of 0.375 M ethanolamine pH7.6, 5 mM MgCl2, 1.12 mM NADP, 2.5 mM ATP and 0.2 Units of G6PDH (Roche Biochemicals 10127655001) was added and OD340 nm was recorded. Subsequently, 0.75 units of hexokinase (Roche 11426362001) were added, the reaction incubated at room temperature for 30 min and OD340 recorded again. Background absorbance was subtracted from sample absorbance and glucose equivalents were determined based a digested glycogen standard curve. The engineered HEK293 line accumulated >30-fold more polysaccharide than the native HEK293 cells, and twice the level of normal glycogen found in Rat1 cells (Fig. S4F).
To determine the effect of VAL-0417 on polysaccharide degradation, cells were plated in 96-well plates at a density of 40,000 cells/well, and after 4 days various concentrations of VAL-0417 were added for 20 hr to fresh growth media. Cells were then washed 3 times with PBS, fixed for 7 min with 3.7% formaldehyde in 90% ethanol, washed again 3 times with PBS and polysaccharide content was determined by hydrolysis and glucose determination. Statistical significance was assessed by using an unpaired Student t-test.
For Western analyses, 20 µg of total cell lysates were separated on SDS-PAGE and protein transferred to nitrocellulose membranes that were probed with antibodies against human pancreatic α-amylase (AMY2A) and ENT2 followed by appropriate HRP-conjugated secondary antibodies and chemiluminescence.
Enzyme-linked immunosorbent assay
The capture antibody raised against the 3E10 Fab fragment was generated by Valerion Therapeutics (Concord, MA). Wells of a 96-well plate were incubated with 100 µL of capture antibody overnight (~16 hours) at a final concentration of 2 µg/ml in PBS. All incubations were done in a humidified chamber. Wells were then rinsed 3 times with 200 µL PBS followed by incubation for 1 hour with 200 µL of blocking solution (5% non-fat milk in PBS). Wells were then rinsed 3 times with 200 µL PBS. 100 µL of diluted tissue homogenates were added and incubated for 1 hour. Wells were then rinsed 3 times with 200 µL of Tris-buffered saline (TBS) and 100 µL of primary antibody (anti-AMY2A) in 5% non-fat milk and TBS was added and incubated for 1 hour. The wells were then washed (solution added and incubated for 5 minutes before being removed) 3 times with TBS. 100 µL of secondary antibody (anti-rabbit IgG, HRP-linked) in 5% non-fat milk in TBS was added and incubated for 1 hour. Wells were then washed 5 times with TBS. 100 µL of TMB substrate was added and a timer started. After the highest concentration of standard curve saturated or a pre-set time was met, the reaction was stopped by adding 100 µL of stopping solution (0.18% H2SO4). The plate was then read for absorbance at 450 nm.
In vivo mouse studies
For each IM injection, 20 µL VAL-0417 (30 mg/ml; 0.6 mg per injection) or 20 µL PBS was injected into one gastrocnemius. For the IV injections, mice were pretreated intraperitoneally with diphenhydramine (15mg/kg) before each injection to prevent anaphylaxis. After the diphenhydramine, the mice where kept under a heat lamp for twenty minutes. A Mouse Tail Illuminator (BrainTree Scientific, Inc.) was used to restrain the mouse and an insulin syringe/needle was used to inject PBS or VAL-0417 into the dorsal tail vein. 100 µL of PBS was injected into IV control mice on days 1, 5, 8, and 13. For the IV treatment group, 100 µL of 30 mg/ml VAL-0417 (3 mg total) was administered on days 1 and 5, 100 µL of 20 mg/ml VAL-0417 (2 mg total) on day 8, and 130 uL of 23 mg/ml VAL- 0417 (3 mg total) on day 13. At the indicated time points post-injection(s), mice were euthanized by cervical dislocation and decapitation. Tissues were harvested, flash frozen, powdered over liquid N2 using Freezer/Mill Cryogenic Grinder (SPEX SamplePrep), and stored at −80°C. Powdered tissues were weighed, homogenized in assay buffer from the BioVision amylase assay kit and protein concentration was determined using the Pierce BCA Protein Assay Kit. For the ELISA assay, tissue homogenates were diluted in PBS for a final concentration of 0.1 µg protein/µL in 100 µL. Diluted homogenates were added to 96-well plates for the ELISA utilizing a capture antibody binding the 3E10 Fab fragment and anti-AMY2A as the detection antibody. Polysaccharide content was determined using the Pflüger method.
IT and ICV studies were performed by Northern Biomedical Research, Inc. (Spring Lake, MI). Animals were anesthetized with a mixture of oxygen 0.5 to 1 L/min and isoflurane 1 to 5% prior to and during the catheter implantation procedure. The IT catheter attached to an osmotic pump was inserted at the cisterna magna and advanced caudally 2.5 cm to the lumbar region. The ICV cannula attached to an osmotic pump was inserted into the cerebral lateral ventricle and anchored VAL-0417 (30 mg/ml) or PBS was continuously administered via the IT catheter or ICV cannula for 3, 8 or 28 days (0.11 µL/hr). On the day following the last day of infusion, mice were euthanized by isofluorane/oxygen sedation and perfusion via the left cardiac ventricle with 0.001% sodium nitrite in heparinized saline. For the 3- and 28- day experiments, brains were harvested, weighed, sectioned using a Rodent Brain Matrix (RBM-2000C, ASI Instruments) with 2.0 mm coronal section slice intervals, flash frozen and stored at −80°C. Slices were directly homogenized in BioVision assay buffer using polypropylene pellet pestles in microcentrifuge tubes. VAL-0417 levels and amylase activity were determined by ELISA and the BioVision assay and normalized to protein content. Protein and polysaccharide content were determined by BCA assay and the Pflüger method, respectively.
For the 8-day ICV experiment, brains were weighed, separated into two hemispheres (longitudinal dissection) and each hemisphere was weighed. The right hemisphere was rinsed in PBS and submerged in 10% neutral buffered formalin (NBF) for approximately 48 hours at room temperature with periodic agitation. After approximately 48 hours, the 10% NBF was replaced with 70% ethanol and stored at room temperature. Staining was performed on 4-micron thick sections cut from formalin fixed paraffin embedded tissue. Slides were deparaffinized and hydrated stepwise. Combination Periodic Acid-Schiff’s Stain staining was carried out on 4 mm sections, along with appropriate positive controls per standard protocols (Luna, 1968; Sheehan and Hrapchak, 1980). After staining, slides were scanned and analyzed and images were prepared using the HALO image analysis software (PerkinElmer). The left hemisphere was frozen in liquid nitrogen, pulverized using the Freezer/Mill Cryogenic Grinder, and the powdered tissue was stored at −80°C. Aliquots of pulverized tissue were weighed and resuspended in BioVision assay buffer for the ELISA, added directly to 30% KOH for polysaccharide determination via the Pflüger method, or used for m etabolite extraction.
GCMS metabolomics analysis
20 mg pulverized mouse brain were extracted for polar metabolites using 1ml of ice cold 50% methanol. The polar fraction was transferred to a V-shaped GCMS glass vial and dried using a SpeedVac (Thermo) followed by derivatization and GCMS analysis. Dried polar samples were derivatized by sequential addition of 20 mg/ml methoxylamine hydrochloride in pyridine then the trimethylsilylating agent N-methyl-N- trimethylsilyl-trifluoroacetamide (MSTFA) with brief agitation in between at 37 °C. After cooling, the derivatized mixture analyzed by GCMS. GCMS protocols were similar to those described previously (Jung and Oh, 2015; MacRae et al., 2012), except a modified temperature gradient was used for GC: Initial temperature was 130° C, held for 4 minutes, rising at 6° C/minutes to 243° C, ri sing at 60° C/minutes to 280° C, held for 2 minutes. The electron ionization (EI) energy was set to 70 eV. Scan (m/z:50–800) and full scan mode were used for metabolomics analysis. Mass spectra were translated to relative metabolite abundance using the Automated Mass Spectral Deconvolution and Identification System (AMDIS) software matched to the FiehnLib metabolomics library (available through Agilent) for retention time and fragmentation pattern matching with a confidence score of >80 (Fiehn, 2016; Fiehn et al., 2000; Kind et al., 2009). For mono-, di- and trisaccharide quantitation, molecular ions 319 (mono) with a retention time between 17.5 and 18.5 min were used, 361 (di) with a retention time between 24.5 and 26.5 min were used, and 361 (tri) with a retention time between 31 and 32 min were used. Values were first normalized to norvaline within the sample to account for sample loss, then DNA-derived thymine to represent input volume.
QUANTIFICATION AND STATISTICAL ANALYSIS
For quantification of LB size and distribution, individual LBs in 3–5 micrographs were measured manually using ImageJ and a frequency distribution was calculated for LBs from each tissue type using Prism software (GraphPad) and a bin width of 1 µm. Only clearly delineated LBs were measured; clumps and dust-like particles were excluded. Statistical significance of polysaccharide reduction in cultured cells and animal tissues was determined using the Prism software (GraphPad). A t-test was used when two groups were present, and two-way analysis of variation (ANOVA) was used for comparing three groups or more. Data are represented as mean ± standard error of the mean (SE) or standard deviation (SD), as indicated. For in vivo experiments, the numbers of animals (n) per group are indicated. PCA and clustering heatmap analysis were performed using the ClustVis package for R (available through Github, https://github.com/taunometsalu/ClustVis) (Metsalu and Vilo, 2015). All available metabolomics data points were used for multivariate analysis.
Supplementary Material
Supplementary File 1. Figures S1–S7 with legends.
Table S1. List of all metabolites and their relative abundance per animal utilized in PCA and heatmap analyses, related to Figure 7.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Pancreatic α-amylase | Abcam | Cat#: ab21156; RRID: AB_446061 |
| ENT2 | Alomone Labs | Cat#: ANT-052; RRID: AB_11219146 |
| 3E10 | Valerion Therapeutics | N/A |
| Rabbit IgG HRP-linked | Cell Signaling | Cat#: 7074; RRID: AB_2099233 |
| Goat anti-Rabbit HRP-liked | Millipore | Cat#: AP307P, RRID: AB_92641 |
| Biological Samples | ||
| Purified glycogen from rabbit skeletal muscle | Anna DePaoli-Roach (DePaoli-Roach et al., 2015) | N/A |
| Glycogen type III from rabbit liver | Sigma | SKU: G8876 |
| Amylopectin from potato | Sigma | SKU: A8515 |
| Starch from corn | Sigma | SKU: S4126 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pre-packed CaptureSelect IgG-CH1 affinity columns | Fisher | Cat#: 494320001 |
| VivaSpin 20, 10,000 MWCO, PES Membrane | Viva | SKU: VS2002 |
| Proteinase K | Invitrogen | Cat#: 25530–049 |
| Nylon net filters, 140 µm, 25 mm | Millipore | Cat#: NY4H02500 |
| Nylon net filters, 60 µm, 25 mm | Millipore | Cat#: NY6002500 |
| Swinnex filter holders, 25 mm | Millipore | Cat#: SX0002500 |
| Amyloglucosidase from Aspergillus niger, ∼70 U/mg | Sigma | SKU: 10115 |
| Glucose 6 phosphate dehydrogenase (G6PDH) | Roche Biochemicals | SKU: 10127655001 |
| Critical Commercial Assays | ||
| Amylase Activity Colorimetric Assay Kit | BioVision | K711 |
| R Biopharm Inc. D-glucose kit | Fisher Scientific | 10716251035 |
| Pi ColorLock Gold Phosphate Detection System | Innova Biosciences | 303–0125 |
| PGO Enzyme preparation | Sigma | P7119–10CAP |
| TMB Substrate Solution | Fisher Scientific | N301 |
| Pierce BCA Protein Assay Kit | Fisher Scientific | 23227 |
| Experimental Models: Cell Lines | ||
| HEK293–6E | National Research Council Canada (Jager et al., 2013) | N/A |
| HEK293 | DePaoli-Roach Laboratory (Graham et al., 1977) | N/A |
| Rat1 fibroblasts | Roach Laboratory ATCC CRL-2210 (Skurat et al., 2000) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: C56Bl/6 | Gentry Laboratory | N/A |
| Mouse: Epm2a−/− | Gentry Laboratory (DePaoli-Roach et al., 2012; Ganesh et al., 2002) | N/A |
| Mouse: Epm2b−/− | DePaoli-Roach Laboratory (DePaoli-Roach et al., 2010) | N/A |
| Recombinant DNA | ||
| pTT5 | National Research Council Canada | N/A |
| IgG1 Fab-linker-AMY2A Heavy chain cDNA | Proteos, Inc. | N/A |
| IgG1 Light chain cDNA | Proteos, Inc. | N/A |
| pCDH-CMV-MCS-EF1α-Puro | System Biosciences | Cat#: CD510B-1 |
| pCDH-FLAG-PTG | Generated by Anna DePaoli-Roach in pCDH-CMV-Hygro | N/A |
| pCDH-(HA)3-PP1Cα-GFP | Generated by Anna DePaoli-Roach in pCDH-CMV-GFP-Puro | N/A |
| Software and Algorithms | ||
| Prism | GraphPad | N/A |
| HALO Image Analysis Software | PerkinElmer | N/A |
| ClustVis package for R | GitHub | N/A |
| Other | ||
| Freezer/Mill Cryogenic Grinder | SPEX SamplePrep | 6775 |
| FEI Quanta 250 scanning electron microscope | Thermo Fisher Scientific | N/A |
| Dionex CarboPac PA-100 column 4 × 250 mm | Thermo Fisher Scientific | 043055 |
| Single quadrupole gas chromatography coupled to mass spectrometry detector (GS/MSD) system | Agilent | 5977B |
Highlights.
VAL-0417 is an antibody-enzyme fusion comprised of a Fab fragment and an amylase
Purified LBs from LD mouse models are degraded by VAL-0417 in vitro
VAL-0417 administration reduces LBs in vivo in muscle, heart and brain
Aberrant brain metabolism is reversed after VAL-0417 treatment
Context and Significance.
Lafora disease (LD) is a severe form of epilepsy that manifests itself in teenagers and eventually leads to death. A hallmark of LD is the accumulation of carbohydrates as glycogen aggregates in brain and other tissues. Genetic analyses have also identified two genes involved in glycogen storage as causative of the human disease. Although there is currently no treatment for LD, genetic reduction of glycogen aggregates in animal models can reverse LD. Researchers at the University of Kentucky employed a novel approach whereby they engineered an enzyme that can degrade glycogen aggregates to penetrate cells and showed that one week infusion of their compound (VAL-0417) in the brains of LD mice reversed the disease, showing promise as a LD treatment.
Acknowledgments:
We thank Drs. Craig Vander Kooi and Jeffrey Rush of the University of Kentucky (UK) Department of Molecular and Cellular Biochemistry, Deb Ramsdell and Drs. Beth Goad, Rob Schaffer, and Hal Landy of Valerion Therapeutics for valuable feedback and discussions. We also thank Drs. Berge Minassian and Felix Nitschke for fruitful discussions and recommendations. We appreciate the technical support of Dr. Carole Moncman, Azin Akbari, Nico Briot, the UK Electron Microscopy Core, Dr. Thomas Wilkop and the UK Light Microscopy Core, Dana Napier and Karrie Jones. We also thank the UK Sanders-Brown Center on Aging and Adam Bachstetter for assistance with slide scanning and Biswa Choudhury and Sulabha Argade for their assistance with HPAEC-PAD experiments (GlycoAnalytics, University of California, San Diego, CA).
Funding: This work is supported by a sponsored project to M.S.G. from Valerion Therapeutics, NIH R01 NS070899 to M.S.G., P01 NS097197 to M.S.G., and F31 NS093892 to M.K.B. Funding for the UK Alzheimer’s Disease Center (P30 AGO28383) and the Biospecimen Procurement & Translational Pathology Shared Resource Facility of the UK Markey Cancer Center (P30 CA177558) also contributed to this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: D.A. is Chief Scientific Officer and has an equity interest in Valerion Therapeutics. B.L.H. and T.M. are consultants of Valerion Therapeutics. M.S.G. received a sponsored project award from Valerion Therapeutics in accordance with University of Kentucky policies. The other authors declare that they have no competing interests. Valerion Therapeutics has filed one or more patent applications, including WO2018049237A1 of which D.A. is an inventor related to AEF and its use.
References:
- Abbott NJ, Pizzo ME, Preston JE, Janigro D, and Thorne RG (2018). The role of brain barriers in fluid movement in the CNS: is there a ‘glymphatic’system? Acta neuropathologica 135, 387–407. [DOI] [PubMed] [Google Scholar]
- Adeva-Andany MM, González-Lucán M, Donapetry-arcía GC, Fernández-Fernández C, and Ameneiros-Rodríguez E (2016). Glycogen metabolism in humans. BBA Clinical 5, 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An Y, Young SP, Kishnani PS, Millington DS, Amalfitano A, Corzo D, and Chen Y-T (2005). Glucose tetrasaccharide as a biomarker for monitoring the therapeutic response to enzyme replacement therapy for Pompe disease. Molecular genetics and metabolism 85, 247–254. [DOI] [PubMed] [Google Scholar]
- Augé E, Pelegrí C, Manich G, Cabezón I, Guinovart JJ, Duran J, and Vilaplana J (2018). Astrocytes and neurons produce distinct types of polyglucosan bodies in Lafora Disease. Glia in press [DOI] [PMC free article] [PubMed]
- Baeuerle PA, and Reinhardt C (2009). Bispecific T-cell engaging antibodies for cancer therapy. Cancer research 69, 4941–4944. [DOI] [PubMed] [Google Scholar]
- Ball S, Colleoni C, Cenci U, Raj JN, and Tirtiaux C (2011). The evolution of glycogen and starch metabolism in eukaryotes gives molecular clues to understand the establishment of plastid endosymbiosis. J Exp Bot 62, 1775–1801. [DOI] [PubMed] [Google Scholar]
- Barr J, Vázquez-Chantada M, Alonso C, Pérez-ormenzana CM, Mayo R, Galán A, Caballería J, Martín-Duce A, Tran A, and Wagner C (2010). Liquid chromatography− mass spectrometry-based parallel metabolic profiling of human and mouse model serum reveals putative biomarkers associated with the progression of nonalcoholic fatty liver disease. Journal of proteome research 9, 4501–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck A, Goetsch L, Dumontet C, and Corvaia N (2017). Strategies and challenges for the next generation of antibody-drug conjugates. Nature reviews. Drug discovery 16, 315–337. [DOI] [PubMed] [Google Scholar]
- Berard-Badier M, Pellissier JF, Gambarelli D, de Barsy T, Roger J, and Toga M (1980). The retina in Lafora disease: light and electron microscopy. Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. Albrecht von Graefe’s archive for clinical and experimental ophthalmology 212, 285–294. [DOI] [PubMed] [Google Scholar]
- Bobillo Lobato J, Jiménez Hidalgo M, and Jiménez Jiménez L (2016). Biomarkers in lysosomal storage diseases. Diseases 4, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer MK, and Gentry MS (2018). The 3rd International Lafora Epilepsy Workshop: Evidence for a cure. Epilepsy Behav [DOI] [PMC free article] [PubMed]
- Brewer MK, and Gentry MS (in press). Brain glycogen structure and its associated proteins: past, present and future. In Advances in Neurobiology: Brain Glycogen Metabolism Schousboe A, and DiNuzzo M, eds. (Springer; ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer MK, Grossman TR, McKnight TR, Goldberg YP, Landy H, and Gentry MS (2019). The 4th International Lafora Epilepsy Workshop: Shifting paradigms, paths to treatment, and hope for patients. Epilepsy & Behavior 90, 284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, and Ransom BR (2007). Astrocyte glycogen and brain energy metabolism. Glia 55, 1263–1271. [DOI] [PubMed] [Google Scholar]
- Calias P, Banks WA, Begley D, Scarpa M, and Dickson P (2014). Intrathecal delivery of protein therapeutics to the brain: a critical reassessment. Pharmacology & therapeutics 144, 114–122. [DOI] [PubMed] [Google Scholar]
- Carter PJ, and Lazar GA (2018). Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nature reviews. Drug discovery 17, 197–223. [DOI] [PubMed] [Google Scholar]
- Cavanagh JB (1999). Corpora-amylacea and the family of polyglucosan diseases. Brain Res Brain Res Rev 29, 265–295. [DOI] [PubMed] [Google Scholar]
- Chauhan NB, and Siegel GJ (2004). Intracerebroventricular passive immunization in transgenic mouse models of Alzheimer’s disease. Expert review of vaccines 3, 717–725. [DOI] [PubMed] [Google Scholar]
- Cohen-Pfeffer JL, Gururangan S, Lester T, Lim DA, Shaywitz AJ, Westphal M, and Slavc I (2017). Intracerebroventricular Delivery as a Safe, Long-Term Route of Drug Administration. Pediatric neurology 67, 23–35. [DOI] [PubMed] [Google Scholar]
- Crawford CR, Patel DH, Naeve C, and Belt JA (1998). Cloning of the human equilibrative, nitrobenzylmercaptopurine riboside (NBMPR)-insensitive nucleoside transporter ei by functional expression in a transport-deficient cell line. The Journal of biological chemistry 273, 5288–5293. [DOI] [PubMed] [Google Scholar]
- Criado O, Aguado C, Gayarre J, Duran-Trio L, Garcia-Cabrero AM, Vernia S, San Millan B, Heredia M, Roma-Mateo C, Mouron S, et al. (2012). Lafora bodies and neurological defects in malin-deficient mice correlate with impaired autophagy. Hum Mol Genet 21, 1521–1533. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Golde TE, and Lagier-Tourenne C (2018). Animal models of neurodegenerative diseases. Nat. Neurosci 21, 1370–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf AS, Ancker E, Rutherfoord GS, van der Walt JJ, and Rossouw DJ (1989). Lafora-body disease with optic atrophy, macular degeneration and cardiac failure. Journal of the neurological sciences 93, 69–84. [DOI] [PubMed] [Google Scholar]
- DePaoli-Roach AA, Contreras CJ, Segvich DM, Heiss C, Ishihara M, Azadi P, and Roach PJ (2015). Glycogen phosphomonoester distribution in mouse models of the progressive myoclonic epilepsy, Lafora disease. The Journal of biological chemistry [DOI] [PMC free article] [PubMed]
- DePaoli-Roach AA, Segvich DM, Meyer CM, Rahimi Y, Worby CA, Gentry MS, and Roach PJ (2012). Laforin and malin knockout mice have normal glucose disposal and insulin sensitivity. Hum Mol Genet 21, 1604–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaoli-Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, and Roach PJ (2010). Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. The Journal of biological chemistry 285, 25372–25381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran J, Gruart A, Garcia-Rocha M, Delgado-Garcia JM, and Guinovart JJ (2014). Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet 23, 3147–3156. [DOI] [PubMed] [Google Scholar]
- Duran J, Tevy MF, Garcia-Rocha M, Calbo J, Milan M, and Guinovart JJ (2012). Deleterious effects of neuronal accumulation of glycogen in flies and mice. EMBO molecular medicine 4, 719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar GW (1963). Progressive myoclonus epilepsy as an inborn error of metabolism comparable to storage disease. Epilepsia 4, 120–137. [DOI] [PubMed] [Google Scholar]
- Emanuelle S, Brewer MK, Meekins DA, and Gentry MS (2016). Unique carbohydrate binding platforms employed by the glucan phosphatases. Cellular and molecular life sciences : CMLS 73, 2765–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O (2016). Metabolomics by Gas Chromatography-Mass Spectrometry: Combined Targeted and Untargeted Profiling. Current protocols in molecular biology 114, 30.34.31–30.34.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiehn O, Kopka J, Dörmann P, Altmann T, Tre thewey RN, and Willmitzer L (2000). Metabolite profiling for plant functional genomics. Nature biotechnology 18, 1157. [DOI] [PubMed] [Google Scholar]
- Ganesh S, Delgado-Escueta AV, Sakamoto T, Avila MR, Machado-Salas J, Hoshii Y, Akagi T, Gomi H, Suzuki T, Amano K, et al. (2002). Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet 11, 1251–1262. [DOI] [PubMed] [Google Scholar]
- Garcia-Cabrero AM, Marinas A, Guerrero R, de Cordoba SR, Serratosa JM, and Sanchez MP (2012). Laforin and malin deletions in mice produce similar neurologic impairments. Journal of neuropathology and experimental neurology 71, 413–421. [DOI] [PubMed] [Google Scholar]
- Garcia-Cabrero AM, Sanchez-Elexpuru G, Serratosa JM, and Sanchez MP (2014). Enhanced sensitivity of laforin- and malin-deficient mice to the convulsant agent pentylenetetrazole. Frontiers in neuroscience 8, 291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Dixon JE, and Worby CA (2009). Lafora disease: insights into neurodegeneration from plant metabolism. Trends Biochem Sci 34, 628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Guinovart JJ, Minassian BA, Roach PJ, and Serratosa JM (2018). Lafora disease offers a unique window into neuronal glycogen metabolism. The Journal of biological chemistry 293, 7117–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard JM, Turnbull J, Ramachandran N, and Minassian BA (2013). Progressive myoclonus epilepsy. Handbook of clinical neurology 113, 1731–1736. [DOI] [PubMed] [Google Scholar]
- Good CAK, H.; Somogyi M (1933). The determination of glycogen. J. Biol. Chem 100, 485–491. [Google Scholar]
- Graham FL, Smiley J, Russell WC, and Nairn R (1977). Characteristics of a human cell line transformed by DNA from human adenovirus type 5. The Journal of general virology 36, 59–74. [DOI] [PubMed] [Google Scholar]
- Graham TE, Yuan Z, Hill AK, and Wilson RJ (2010). The regulation of muscle glycogen: the granule and its proteins. Acta physiologica (Oxford, England) 199, 489–498. [DOI] [PubMed] [Google Scholar]
- Hansen JE, Sohn W, Kim C, Chang SS, Huang NC, Santos DG, Chan G, Weisbart RH, and Nishimura RN (2006). Antibody-mediated Hsp70 protein therapy. Brain research 1088, 187–196. [DOI] [PubMed] [Google Scholar]
- Hansen JE, Tse CM, Chan G, Heinze ER, Nishimura RN, and Weisbart RH (2007). Intranuclear protein transduction through a nucleoside salvage pathway. The Journal of biological chemistry 282, 20790–20793. [DOI] [PubMed] [Google Scholar]
- Hansen JE, Weisbart RH, and Nishimura RN (2005). Antibody mediated transduction of therapeutic proteins into living cells. TheScientificWorldJournal 5, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth B, and Cori GT (1952). Structure of glycogens and amylopectins. III. Normal and abnormal human glycogen. The Journal of biological chemistry 199, 653–660. [PubMed] [Google Scholar]
- Illingworth B, Larner J, and Cori GT (1952). Structure of glycogens and amylopectins. I. Enzymatic determination of chain length. The Journal of biological chemistry 199, 631–640. [PubMed] [Google Scholar]
- Ishihara T, Yokota T, Yamashita Y, Takahashi M, Kawano H, Uchino F, Kamei T, Matsumoto N, Kusunose Y, and Yamada M (1987). Comparative study of the intracytoplasmic inclusions in Lafora disease and type IV glycogenosis by electron microscopy. Acta pathologica japonica 37, 1591–1601. [DOI] [PubMed] [Google Scholar]
- Jager V, Bussow K, Wagner A, Weber S, Hust M, Frenzel A, and Schirrmann T (2013). High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC biotechnology 13, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jian Z, Alley MR, Cayzer J, and Swinney GR (1990). Lafora’s disease in an epileptic Basset hound. New Zealand veterinary journal 38, 75–79. [DOI] [PubMed] [Google Scholar]
- Jin J.-f., Zhu L.-l., Chen M, Xu H. m., Wang H. f., Feng X. q., Zhu X. p., and Zhou Q (2015). The optimal choice of medication administration route regarding intravenous, intramuscular, and subcutaneous injection. Patient preference and adherence 9, 923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CH, Ivanisevic J, and Siuzdak G (2016). Metabolomics: beyond biomarkers and towards mechanisms. Nature reviews Molecular cell biology 17, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J-Y, and Oh M-K (2015). Isotope labeling pattern study of central carbon metabolites using GC/MS. Journal of Chromatography B 974, 101–108. [DOI] [PubMed] [Google Scholar]
- Jung YS, Lee BH, and Yoo SH (2017). Physical structure and absorption properties of tailor-made porous starch granules produced by selected amylolytic enzymes 12, e0181372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind T, Wohlgemuth G, Lee DY, Lu Y, Palazoglu M, Shahbaz S, and Fiehn O (2009). FiehnLib: mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Analytical chemistry 81, 10038–10048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishnani PS, Corzo D, Nicolino M, Byrne B, Mandel H, Hwu WL, Leslie N, Levine J, Spencer C, McDonald M, et al. (2007). Recombinant human acid [alpha]-glucosidase: major clinical benefits in infantile-onset Pompe disease. Neurology 68, 99–109. [DOI] [PubMed] [Google Scholar]
- Krisman CR (1962). A method for the colorimetric estimation of glycogen with iodine. Analytical biochemistry 4, 17–23. [DOI] [PubMed] [Google Scholar]
- Kruse-Jarres JD, Kaiser C, Hafkenscheid JC, Hohenwallner W, Stein W, Bohner J, Klein G, Poppe W, and Rauscher E (1989). Evaluation of a new alpha-amylase assay using 4.6-ethylidene-(G7)-1–4-nitrophenyl-(G1)-alpha-D-maltoheptaoside as substrate. Journal of clinical chemistry and clinical biochemistry. Zeitschrift fur klinische Chemie und klinische Biochemie 27, 103–113. [PubMed] [Google Scholar]
- Kulp KL, K. (1981). Heat-Moisture Treatment of Starches. I. Physiochemical Properties. Cereal Chemistry 58, 46–48. [Google Scholar]
- Kumar NN, Pizzo ME, Nehra G, Wilken-Resman B, Boroumand S, and Thorne RG (2018). Passive Immunotherapies for Central Nervous System Disorders: Current Delivery Challenges and New Approaches. Bioconjugate chemistry 29, 3937–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafora GR (1911). Uber des Vorkommen amyloider KJrperchen im innern der Ganglienzellen. Virchows Arch. f. Path. Anat 205, 295. [Google Scholar]
- Lawlor MW, Armstrong D, Viola MG, Widrick JJ, Meng H, Grange RW, Childers MK, Hsu CP, O’Callaghan M, Pierson CR, et al. (2013). Enzyme replacement therapy rescues weakness and improves muscle pathology in mice with X-linked myotubular myopathy. Hum Mol Genet 22, 1525–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong DS, Tan JG, Chin CL, Mak SY, Ho YS, and Ng SK (2017). Evaluation and use of disaccharides as energy source in protein-free mammalian cell cultures. Scientific reports 7, 45216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Whelan WJ, and Marchase RB (1993). Glycogen contains phosphodiester groups that can be introduced by UDPglucose: glycogen glucose 1-phosphotransferase. FEBS Lett 329, 263–267. [DOI] [PubMed] [Google Scholar]
- López-González I, Viana R, Sanz P, and Ferrer I (2017). Inflammation in Lafora disease: Evolution with disease progression in laforin and malin knock-out mouse models. Molecular neurobiology 54, 3119–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loscher W, and Schmidt D (2011). Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia 52, 657–678. [DOI] [PubMed] [Google Scholar]
- Lu H, Chen C, and Klaassen C (2004). Tissue distribution of concentrative and equilibrative nucleoside transporters in male and female rats and mice. Drug metabolism and disposition: the biological fate of chemicals 32, 1455–1461. [DOI] [PubMed] [Google Scholar]
- Luna LG (1968). Manual of histologic staining methods of the Armed Forces Institute of Pathology
- Machado-Salas J, Avila-Costa MR, Guevara P, Guevara J, Duron RM, Bai D, Tanaka M, Yamakawa K, and Delgado-Escueta AV (2012). Ontogeny of Lafora bodies and neurocytoskeleton changes in Laforin-deficient mice. Experimental neurology 236, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae James I., Sheiner L, Nahid A, Tonkin C, Striepen B, and McConville Malcolm J. (2012). Mitochondrial Metabolism of Glucose and Glutamine Is Required for Intracellular Growth of Toxoplasma gondii. Cell Host & Microbe 12, 682–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manna SK, Tanaka N, Krausz KW, Haznadar M, Xue X, Matsubara T, Bowman ED, Fearon ER, Harris CC, and Shah YM (2014). Biomarkers of coordinate metabolic reprogramming in colorectal tumors in mice and humans. Gastroenterology 146, 1313–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manwaring V, Prunty H, Bainbridge K, Burke D, Finnegan N, Franses R, Lam A, Vellodi A, and Heales S (2012). Urine analysis of glucose tetrasaccharide by HPLC; a useful marker for the investigation of patients with Pompe and other glycogen storage diseases. Journal of Inherited Metabolic Disease: Official Journal of the Society for the Study of Inborn Errors of Metabolism 35, 311–316. [DOI] [PubMed] [Google Scholar]
- Metsalu T, and Vilo J (2015). ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic acids research 43, W566–W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian BA (2001). Lafora’s disease: towards a clinical, pathologic, and molecular synthesis. Pediatric neurology 25, 21–29. [DOI] [PubMed] [Google Scholar]
- Moolten FL, and Cooperband SR (1970). Selective destruction of target cells by diphtheria toxin conjugated to antibody directed against antigens on the cells. Science (New York, N.Y.) 169, 68–70. [DOI] [PubMed] [Google Scholar]
- Newgard CB, Brady MJ, O’Doherty RM, and Saltiel AR (2000). Organizing glucose disposal: emerging roles of the glycogen targeting subunits of protein phosphatase-1. Diabetes 49, 1967–1977. [DOI] [PubMed] [Google Scholar]
- Nikaido T, Austin J, and Stukenbrok H (1971). Studies in myoclonus epilepsy. 3. The effects of amylolytic enzymes on the ultrastructure of Lafora bodies. J Histochem Cytochem 19, 382–385. [DOI] [PubMed] [Google Scholar]
- Nitschke F, Sullivan MA, Wang P, Zhao X, Chown EE, Perri AM, Israelian L, Juana-Lopez L, Bovolenta P, Rodriguez de Cordoba S, et al. (2017). Abnormal glycogen chain length pattern, not hyperphosphorylation, is critical in Lafora disease 9, 906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke F, Wang P, Schmieder P, Girard JM, Awrey DE, Wang T, Israelian J, Zhao X, Turnbull J, Heydenreich M, et al. (2013). Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy Lafora disease. Cell metabolism 17, 756–767. [DOI] [PubMed] [Google Scholar]
- Oe Y, Baba O, Ashida H, and Nakamura KC (2016). Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns 64, 1532–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksel F, Tekgul H, Genc S, Ozyurek R, Akalin T, and Tututuncuoglu S (1999). A case of Lafora’s disease associated with cardiac arrhythmia. J Child Neurol 14, 745–746. [DOI] [PubMed] [Google Scholar]
- Ozen H (2007). Glycogen storage diseases: new perspectives. World journal of gastroenterology 13, 2541–2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson BA, Chen H, Schroeder JM, Shou W, DePaoli-Roach AA, and Roach PJ (2004). Abnormal cardiac development in the absence of heart glycogen. Molecular and cellular biology 24, 7179–7187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson BA, Turnbull J, Epp JR, Weaver SA, Zhao X, Pencea N, Roach PJ, Frankland PW, Ackerley CA, and Minassian BA (2013). Inhibiting glycogen synthesis prevents Lafora disease in a mouse model. Ann Neurol 74, 297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlman RL (2016). Mouse models of human disease: An evolutionary perspective. Evolution, medicine, and public health 2016, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pflüger E (1909). Meine Methode der quantitativen Analyse des Glykogenes und die Arteigenthümlichkeit der Substanzen des Thierleibes. Archiv für die gesamte Physiologie des Menschen und der Tiere 129, 362–378. [Google Scholar]
- Pizzo ME, Wolak DJ, Kumar NN, Brunette E, Brunnquell CL, Hannocks MJ, Abbott NJ, Meyerand ME, Sorokin L, and Stanimirovic DB (2018). Intrathecal antibody distribution in the rat brain: surface diffusion, perivascular transport and osmotic enhancement of delivery. The Journal of physiology 596, 445–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plog BA, and Nedergaard M (2018). The glymphatic system in central nervous system health and disease: past, present, and future. Annual Review of Pathology: Mechanisms of Disease 13, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Printen JA, Brady MJ, and Saltiel AR (1997). PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science (New York, N.Y.) 275, 1475–1478. [DOI] [PubMed] [Google Scholar]
- Raben N, Danon M, Lu N, Lee E, Shliselfeld L, Skurat AV, Roach PJ, Lawrence JC Jr., Musumeci O, Shanske S, et al. (2001). Surprises of genetic engineering: A possible model of polyglucosan body disease. Neurology 56, 1739–1745. [DOI] [PubMed] [Google Scholar]
- Rehman K, Hamid Akash MS, Akhtar B, Tariq M, Mahmood A, and Ibrahim M (2016). Delivery of Therapeutic Proteins: Challenges and Strategies. Current drug targets 17, 1172–1188. [DOI] [PubMed] [Google Scholar]
- Roach PJ (2002). Glycogen and its Metabolism. Current Molecular Medicine 2, 101–120. [DOI] [PubMed] [Google Scholar]
- Robyt JF, and French D (1967). Multiple attach hypothesis of alpha-amylase action: action of porcine pancreatic, human salivary, and Aspergillus oryzae alpha-amylases. Archives of biochemistry and biophysics 122, 8–16. [DOI] [PubMed] [Google Scholar]
- Rubio-Villena C, Viana R, Bonet J, Garcia-Gimeno MA, Casado M, Heredia M, and Sanz P (2018). Astrocytes: new players in progressive myoclonus epilepsy of Lafora type. Hum Mol Genet [DOI] [PMC free article] [PubMed]
- Ryu JH, Drain J, Kim JH, McGee S, Gray-Weale A, Waddington L, Parker GJ, Hargreaves M, Yoo SH, and Stapleton D (2009). Comparative structural analyses of purified glycogen particles from rat liver, human skeletal muscle and commercial preparations. Int J Biol Macromol 45, 478–482. [DOI] [PubMed] [Google Scholar]
- Sakai M, Austin J, Witmer F, and Trueb L (1970). Studies in myoclonus epilepsy (Lafora body form). II. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology 20, 160–176. [DOI] [PubMed] [Google Scholar]
- Salek RM, Maguire ML, Bentley E, Rubtsov DV, Hough T, Cheeseman M, Nunez D, Sweatman BC, Haselden JN, and Cox R (2007). A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiological genomics 29, 99–108. [DOI] [PubMed] [Google Scholar]
- Sanchez-Elexpuru G, Serratosa JM, and Sanchez MP (2017). Sodium selenate treatment improves symptoms and seizure susceptibility in a malin-deficient mouse model of Lafora disease. Epilepsia [DOI] [PMC free article] [PubMed]
- Scharfman HE (2015). Neuroscience. Metabolic control of epilepsy. Science (New York, N.Y.) 347, 1312–1313. [DOI] [PubMed] [Google Scholar]
- Shahwan A, Farrell M, and Delanty N (2005). Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. The Lancet. Neurology 4, 239–248. [DOI] [PubMed] [Google Scholar]
- Sheehan DC, and Hrapchak BB (1980). Theory and practice of histotechnology (Cv Mosby; ). [Google Scholar]
- Skurat AV, Dietrich AD, and Roach PJ (2000). Glycogen synthase sensitivity to insulin and glucose-6-phosphate is mediated by both NH2-and COOH-terminal phosphorylation sites. Diabetes 49, 1096–1100. [DOI] [PubMed] [Google Scholar]
- Spratlin JL, Serkova NJ, and Eckhardt SG (2009). Clinical applications of metabolomics in oncology: a review. Clinical cancer research 15, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sujka MJ, J. (2007). Starch granule porosity and its changes by means of amylolysis. International Agrophysics 27. [Google Scholar]
- Sullivan MA, Aroney ST, Li S, Warren FJ, Joo JS, Mak KS, Stapleton DI, Bell-Anderson KS, and Gilbert RG (2014). Changes in glycogen structure over feeding cycle sheds new light on blood-glucose control. Biomacromolecules 15, 660–665. [DOI] [PubMed] [Google Scholar]
- Sullivan MA, Nitschke S, Steup M, Minassian BA, and Nitschke F (2017). Pathogenesis of Lafora Disease: Transition of Soluble Glycogen to Insoluble Polyglucosan. International journal of molecular sciences 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson MA (1948). Studies on the structure of polysaccharides; relation of the iodine color to the structure. The Journal of biological chemistry 172, 825–837. [PubMed] [Google Scholar]
- Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, Minassian BA, Depaoli-Roach AA, and Roach PJ (2008). Abnormal metabolism of glycogen phosphate as a cause for lafora disease. The Journal of biological chemistry 283, 33816–33825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Heiss C, Karthik C, Contreras CJ, Glushka J, Ishihara M, Azadi P, Hurley TD, DePaoli-Roach AA, and Roach PJ (2011). Phosphate incorporation during glycogen synthesis and Lafora disease. Cell metabolism 13, 274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tester RF, Karkalas J, and Qi X (2004). Starch—composition, fine structure and architecture. Journal of Cereal Science 39, 151–165. [Google Scholar]
- Thurberg BL, Lynch Maloney C, Vaccaro C, Afonso K, Tsai AC, Bossen E, Kishnani PS, and O’Callaghan M (2006). Characterization of pre- and post-treatment pathology after enzyme replacement therapy for Pompe disease. Laboratory investigation; a journal of technical methods and pathology 86, 1208–1220. [DOI] [PubMed] [Google Scholar]
- Tiberia E, Turnbull J, Wang T, Ruggieri A, Zhao XC, Pencea N, Israelian J, Wang Y, Ackerley CA, Wang P, et al. (2012). Increased laforin and laforin binding to glycogen underlie Lafora body formation in malin-deficient Lafora disease. The Journal of biological chemistry 287, 25650–25659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trushina E, and Mielke MM (2014). Recent advances in the application of metabolomics to Alzheimer’s Disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1842, 1232–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Depaoli-Roach AA, Zhao X, Cortez MA, Pencea N, Tiberia E, Piliguian M, Roach PJ, Wang P, Ackerley CA, et al. (2011). PTG Depletion Removes Lafora Bodies and Rescues the Fatal Epilepsy of Lafora Disease. PLoS Genet 7, e1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Epp JR, Goldsmith D, Zhao X, Pencea N, Wang P, Frankland PW, Ackerley CA, and Minassian BA (2014). PTG protein depletion rescues malin-deficient Lafora disease in mouse. Ann Neurol 75, 442–446. [DOI] [PubMed] [Google Scholar]
- Underwood BR, Broadhurst D, Dunn WB, Ellis DI, Michell AW, Vacher C, Mosedale DE, Kell DB, Barker RA, and Grainger DJ (2006). Huntington disease patients and transgenic mice have similar pro-catabolic serum metabolite profiles. Brain 129, 877–886. [DOI] [PubMed] [Google Scholar]
- Valerion Therapeutics LLC (2016). VAL-1221 Delivered Intravenously in Ambulatory and Ventilator-free Patients With Late-Onset Pompe Disease (Clinicaltrials.gov Identifier ).
- Valles-Ortega J, Duran J, Garcia-Rocha M, Bosch C, Saez I, Pujadas L, Serafin A, Canas X, Soriano E, Delgado-Garcia JM, et al. (2011). Neurodegeneration and functional impairments associated with glycogen synthase accumulation in a mouse model of Lafora disease. EMBO molecular medicine 3, 667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, Herson S, Kishnani PS, Laforet P, Lake SL, et al. (2010). A randomized study of alglucosidase alfa in late-onset Pompe’s disease. The New England journal of medicine 362, 1396–1406. [DOI] [PubMed] [Google Scholar]
- Van Heycop Ten Ham MW (1975). Lafora disease, a form of progressive myoclonus epilepsy. In Handbook of clinical neurology Vinken PJ, and Bruyn GW, eds. (Holland, Amsterdam: North Holland Publishing Company; ), pp. 382–422. [Google Scholar]
- Van Heycop ten Ham MW, and Jager DH (1963). Progressive Myoclonus Epilepsy with Lafora Bodies. Clinical-Pathological Features. Epilepsia 4, 95–119. [DOI] [PubMed] [Google Scholar]
- Van Hoof F, and Hageman-Bal M (1967). Progressive familial myoclonic epilepsy with Lafora bodies. Electron microscopic and histochemical study of a cerebral biopsy. Acta Neuropathol 7, 315–336. [DOI] [PubMed] [Google Scholar]
- Villalba-Orero M, Sanchez-Elexpuru G, Lopez-Olaneta M, Campuzano O, Bello-Arroyo E, Garcia-Pavia P, Serratosa JM, Brugada R, Sanchez MP, and Lara-Pezzi E (2017). Lafora Disease Is an Inherited Metabolic Cardiomyopathy. J Am Coll Cardiol 69, 3007–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemenot BR, Korte S, Wright TL, Adams EL, Boyd RB, and Butt MT (2016). Safety evaluation of CNS administered biologics—stu dy design, data interpretation, and translation to the clinic. Toxicological Sciences 152, 3–9. [DOI] [PubMed] [Google Scholar]
- Weisbart RH, Stempniak M, Harris S, Zack DJ, and Ferreri K (1998). An autoantibody is modified for use as a delivery system to target the cell nucleus: therapeutic implications. Journal of autoimmunity 11, 539–546. [DOI] [PubMed] [Google Scholar]
- Wolak DJ, and Thorne RG (2013). Diffusion of macromolecules in the brain: implications for drug delivery. Molecular pharmaceutics 10, 1492–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby CA, Gentry MS, and Dixon JE (2008). Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). The Journal of biological chemistry 283, 4069–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Sun T, Armstrong D, Borneman S, Yang C, Austin S, Kishnani PS, and Sun B (2017). Antibody-mediated enzyme replacement therapy targeting both lysosomal and cytoplasmic glycogen in Pompe disease [DOI] [PubMed]
- Yokoi S, Austin J, Witmer F, and Sakai M (1968). Studies in myoclonus epilepsy (Lafora body form). I. Isolation and preliminary characterization of Lafora bodies in two cases. Arch Neurol 19, 15–33. [DOI] [PubMed] [Google Scholar]
- Yokota T, Ishihara T, Kawano H, Yamashita Y, Takahashi M, Uchino F, Kamei T, Kusunose Y, Yamada M, and Matsumoto N (1987). Immunological homogeneity of Lafora body, corpora amylacea, basophilic degeneration in heart, and intracytoplasmic inclusions of liver and heart in type IV glycogenosis. Acta pathologica japonica 37, 941–946. [DOI] [PubMed] [Google Scholar]
- Yokota T, Ishihara T, Yoshida H, Takahashi M, Uchino F, and Hamanaka S (1988). Monoclonal antibody against polyglucosan isolated from the myocardium of a patient with Lafora disease. Journal of neuropathology and experimental neurology 47, 572–577. [DOI] [PubMed] [Google Scholar]
- Young JM, and Weser E (1971). The metabolism of circulating maltose in man. The Journal of clinical investigation 50, 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1. Figures S1–S7 with legends.
Table S1. List of all metabolites and their relative abundance per animal utilized in PCA and heatmap analyses, related to Figure 7.